

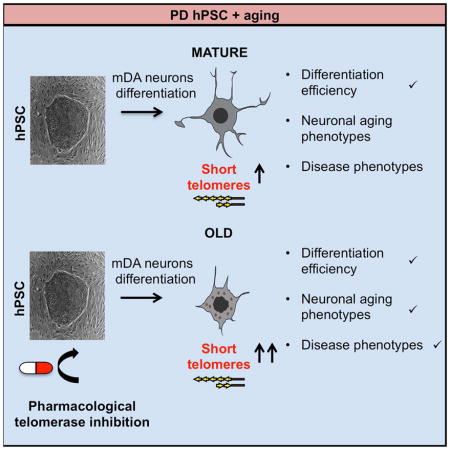
SUMMARY
Modeling late onset disorders such as Parkinson’s disease (PD) using iPSC technology remains a challenge as current differentiation protocols yield cells with the properties of fetal-stage cells. Here we tested whether it is possible to accelerate aging in vitro to trigger late onset disease phenotypes in an iPSC model of PD. In order to manipulate a factor that is involved in natural aging as well as in premature aging syndromes, we used telomere shortening as an age-inducing tool. We show that shortened telomeres result in age-associated as well as potentially disease-associated phenotypes in human pluripotent stem cell (hPSC) derived midbrain dopamine (mDA) neurons. Our approach provides proof of concept for the further validation of telomere shortening as an induced aging tool for late onset diseases modeling.
ETOC BLURB
Vera et al. follow the induction of age- and potentially disease-related phenotypes in hPSC-derived midbrain dopamine (mDA) neurons following telomere shortening. The study provides a proof of concept for manipulating telomere length as a strategy to model late onset disease in hiPSC-derived lineages.
INTRODUCTION
Induced pluripotent stem cell (iPSC) lines derived from patients affected by genetic disease represent a powerful tool for disease modeling and drug discovery. Modeling of early developmental disorders of the nervous system has been successful for several disorders such as spinal muscular atrophy, familial dysautonomia or primary herpes simplex encephalitis (Ebert et al., 2009; Lafaille et al., 2012; Lee et al., 2009) (Ebert et al., 2009) among others. In contrast, the modeling of late-onset disorders, such as Alzheimer’s (AD) and Parkinson’s (PD) raises additional challenges such as the ability to faithfully recapitulate disease phenotypes that occur only late in life (Srikanth and Young-Pearse, 2014). A potential reason underlying this challenge is the reset of donor age in human pluripotent stem cells during somatic cell reprogramming (Mahmoudi and Brunet, 2012). Thus, the ability to induce “age” in iPSC based models may be an important tool for the study of late-onset disease. Our lab recently presented a strategy to artificially induce aging in vitro for late-onset disease modeling. Using a model of PD we engineered the expression of the protein progerin in iPSC-derived midbrain dopamine (mDA) neurons, the cell predominantly affected in PD (Miller et al., 2013). We showed that PROGERIN expression induces both general aging-associated phenotypes such as abnormal nuclear morphologies and accumulation of DNA damage and ROS as well as features more specific to neuronal aging such as shorter dendrites. Adding the “aging” factor to the genetic vulnerability of PD iPSC also enhances relevant phenotypes of PD such as the progressive loss of tyrosine hydroxylase (TH) expression, which is the rate-limiting enzyme in the synthesis of dopamine (DA) (Miller et al., 2013). However, it remains unclear whether the aging phenotype induced by progerin mimics physiological or pathological aging. With the objective of manipulating a factor more closely associated to physiological aging we propose here telomere shortening as an alternative aging inducing tool.
Telomere attrition is one of the best known mechanisms of aging, both in human (Harley et al., 1990) and mice (Flores et al., 2008). Telomeres are special nucleoprotein structures at the ends of eukaryotic chromosomes (Wellinger and Sen, 1997) that protect them from degradation and DNA damage (Chan and Blackburn, 2002; Palm and de Lange, 2008). When cells divide, the telomeres are not fully replicated, leading to telomere shortening with every replication. The main mechanism to counteract shortening is telomere elongation via the enzyme telomerase, a reverse transcriptase that can elongate telomeres de novo after each cell division (Greider and Blackburn, 1985). Telomerase is activated during fetal development (Wright et al., 1996). However, after birth, telomerase is repressed in most somatic tissue (Blasco et al., 1995), and as a consequence, progressive telomere erosion occurs in most somatic cells throughout life. Some cell types, such as stem cells and germ cells, retain moderate telomerase activity levels. However, those levels are often not sufficient to prevent telomere shortening with aging (Flores et al., 2008). Critically short telomeres can trigger a persistent DNA damage response, which leads to cellular senescence and/or apoptosis (Blasco, 2005). Those cellular changes compromise tissue function and the capacity for regeneration, factors that contribute to organismal aging (Canela et al., 2007). Progressive telomere shortening has been proposed to represent a “molecular clock” that underlies organism aging. Several well known premature ageing disorders in humans such as dyskeratosis congenita (DC) are characterized by a faster rate of telomere attrition. DC patients carry mutations in components of the telomerase complex, which result in decreased telomerase stability and telomere length (Mitchell et al., 1999). DC patients develop multiple pathologies over time such as defects of the skin and the hematopoietic system, bone marrow failure, and premature death (Blasco, 2005).
The concept of telomeres as a mitotic clock in proliferating somatic cells has been supported by several studies, which strongly argue for a major role of telomeres in replicative aging. However, the role of telomeres in non-dividing cells such as neurons remains obscure. There is considerable evidence for a link between short telomeres or changes in telomere-associated proteins with neurodegeneration. Of notice, mutations in the telomere-associated proteins (TRF2 interacting proteins) ATM, WRN, and NBS1 are responsible for human neurological disorders characterized by developmental abnormalities. Those include increased risk for various cancers, signs of premature aging and telomere shortening below a critical length. Interestingly, these pathologies present in human patients have been modeled in mice only when studied in combination with telomerase deficiency and short telomeres in the context of the telomerase-deficient mouse model (Blasco, 2005). These data support the idea that short telomeres can trigger premature aging as well as neurodegeneration (Barlow et al., 1996; Chang et al., 2004; Lebel and Leder, 1998; Ranganathan et al., 2001; Wong et al., 2003). In the other hand, treatment of old mice with adeno associated virus (AAV) expressing mouse TERT delays physiological aging and extends longevity by improving health and fitness (Bernardes de Jesus et al., 2012). Furthermore, it has been shown that reactivation of telomerase in old, advanced generation (G4) mice of an inducible-TERT knock in mouse model can reverse neurodegeneration phenotypes associated with the accelerated aging phenotype in those mice (Jaskelioff et al.). Finally, individuals with short telomeres in peripheral blood mononuclear cells (PBMC) appear to be more prone to neurodegenerative disorders such as AD, PD and dementia (Grodstein et al., 2008; Guan et al., 2012; Hochstrasser et al., 2012; Honig et al., 2012; Honig et al., 2006; Jenkins et al., 2006; Kume et al., 2012; Maeda et al., 2012; Martin-Ruiz et al., 2006; Panossian et al., 2003; Thomas et al., 2008; von Zglinicki et al., 2000; Watfa et al., 2011). Considering that telomere length is thought to be proportional within individuals across different organs, these data support an association between telomere length and neurodegeneration.
In the current study we aim to test telomere shortening as an induced aging tool in neurons by testing whether iPSC lines and iPSC-derived mDA with shortened telomeres exhibit age or disease related phenotypes in a model of Parkinson’s disease.
RESULTS
Telomere dynamics in neural differentiation
As a first step toward generating neural cells with reduced telomere length by telomerase downregulation we set out to define the best time point for downregulating telomerase. Therefore we measured telomere length and the dynamics of telomerase activity during neuronal differentiation in vitro. We initially measured telomerase activity at different time points during mDA neuron differentiation of the human embryonic stem cell (hESC) line H9 using a well-established protocol (Kriks et al., 2011). Telomerase activity was measured by the Telomerase Repeat Amplification Protocol (TRAP). Using this method, we show that telomerase is almost completely silenced by day 18 of the mDA differentiation protocol (Fig. 1A–B). We also measured telomerase mRNA levels by qRT-PCR, showing a very small decrease in TERC levels but a dramatic loss of TERT during differentiation (Fig. 1C). In parallel to the downregulation of telomerase, we also observed telomere shortening during mDA neuronal differentiation, as measured by High Throughput Q-FISH (HT Q-FISH) (Fig. 1D–F). There was both a decrease in mean telomere length (Fig. 1D) and an increase in the percentage of cells with short telomeres (Fig. 1E).
Figure 1. Telomere dynamics in neural differentiation.

(A) TRAP polyacrylamide gel representing the telomerase activity at different time points of the mDA differentiation protocol. Two dilutions for each sample (1 and 0.2μg of protein) and a negative control treated with RNAse are included as well as an internal PCR control (IC) (B) Quantification of telomerase activity measured by TRAP assay on cells at different time points of mDA neurons differentiation protocol. Bars represent mean ± SEM of the two dilutions per sample (C) mRNA levels of TERC and TERT measured by quantitative PCR. Data are represented as mean ± SEM (D) Quantification of the mean ± SEM telomere length measured by HT Q-FISH. Numbers above bars indicate the number of telomere spots quantified (E) Percentage of short telomeres (<8kb) measured by HT QFISH. Numbers above bars indicate the number of short telomeres out of the total number of telomeres (F) Representative HT Q-FISH images. Nuclei are stained with DAPI (blue). Telomeres are stained with CY3 (yellow).
Pharmacological telomerase downregulation in hPSC
The strategy selected to downregulate telomerase was its pharmacological inhibition with the compound 2-[(E)-3-naphtalen-2-yl-but-2-enoylamino]-benzoic acid (BIBR1532), a small molecule inhibitor of telomerase catalytic activity. First, we tested whether the telomerase inhibitor BIBR1532 can inhibit the activity of telomerase in undifferentiated H9 human ES cells. Using the TRAP assay, we confirmed that at concentrations of 10μM or higher the activity of telomerase was reduced to 50 % of its original activity (Fig. 2A–B). Based on these results, we used 10μM and 40μM concentration of BIBR1532 for treating hPSCs with DMSO as a control. We treated H9 human ES cells and two PD iPSC cell lines with homozygous mutations in PINK1 (Q456X) and PARKIN (V324A) respectively with the telomerase inhibitor. A two week treatment induced telomere shortening in all three hPSC lines, as measured by the HT Q-FISH method. We observed both a decrease of the mean telomere length (Fig. 2C) and an increase in the percentage of short telomeres (Fig. 2D) in the cells treated with the inhibitor. Telomere shortening induced by the telomerase inhibitor was also confirmed by Southern blot of telomeric restriction fragments (TRF) in the three hPSC cell lines tested (Fig. S1A–C). The increased abundance of shorter telomeric fragments in treated cells can be easily observed by comparing the profile lane of treated and untreated cells (Fig. S1A–C). We did not observe dramatic differences in pluripotency marker expression following treatment with the inhibitor (Fig. S2A–C). Although the cells treated with both concentrations of the inhibitor showed significantly less SSEA3 positive cells, with a decrease of around 20% compared to DMSO treated control (Fig. S2A). No significant differences in the percentage of NANOG positive cells were observed between treated cells and control (Fig. S2B). In the case of OCT4 (POU5F1), only the higher concentration of the inhibitor significantly reduced the percentage of positive cells compared to control cells (Fig. S2C). In addition, cell cycle analysis showed a slight decrease in the percentage of cells in S phase in BIBR1532 treated cells (Fig. S2D). These results indicate that pharmacological downregulation of telomerase at the pluripotent stage results in telomere shortening with some functional consequences for pluripotent cells including a downregulation of some pluripotency markers and reduction in proliferating cells.
Figure 2. Pharmacological telomerase downregulation in hPSCs.

(A) Quantification of telomerase activity levels resulting from the treatment of H9 cells with different concentrations of the telomerase inhibitor BIBR1532, as determined using the TRAP assay. Bars represent mean ± SEM. For comparative purposes, TRAP values are represented as fold-decrease relative to those of the untreated sample (0 μM of BIBR1532), which showed the highest telomerase activity (B) Corresponding TRAP polyacrylamide gel. Two dilutions for each sample (1 and 0.2μg of protein) and a negative control treated with RNAse are included as well as an internal PCR control (IC) (C–D) H9 cells were treated with the telomerase inhibitor BIBR1532 for 14–16 days. Two different concentrations were included (10 and 40 μM) and DMSO was used as a control. (C) Quantification of the mean ± SEM telomere length measured by HT Q-FISH. Numbers above bars indicate the number of telomere spots quantified (D) Percentage of short telomeres (<8kb) measured by HT QFISH. Numbers above bars indicate the number of short telomeres out of the total number of telomeres.
The impact of shortened telomeres on aging and disease-related phenotypes in hPSC-derived neurons
In order to generate hPSC-derived neurons with shortened telomeres, we decided to pre-treat hPSC lines (H9, PINK1 and PARKIN) with the telomerase inhibitor at the pluripotent stage for two weeks and to maintain treatment during the differentiation protocol until day 18. Cells were exposed to the telomerase inhibitor BIBR1532, at either 10μM or 40μM with DMSO treatment serving as a control. To address whether the drug treatment could interfere with mDA neuron differentiation efficiency, we quantified the mDA neuron marker FOXA2 at day 30 of differentiation (Fig. S3A). Differentiation into FOXA2+ putative mDA neurons occurred with high efficiencies independent of the drug treatment (Fig. S3A). Next, we measured telomere length in the resulting neurons. The treatment with the telomerase inhibitor resulted in neurons with shorter telomeres assessed by HT Q-FISH, reflected both by a decrease in the mean telomere length (Fig. 3A) and by an increase in the percentage of short telomeres (Fig. 3B) at day 65 of differentiation. In the PARKIN line, the 10μM treatment group only increased the percentage of short telomeres, while no decrease in the mean telomere length was observed at this concentration (Fig. 3A–B). Those data are compatible with the notion that the percentage of short telomeres is a more sensitive parameter than the mean telomere length in individual cells (Hemann et al., 2001; Vera et al., 2012; Vera and Blasco, 2012). Telomere length of hPSC-derived neurons from treated versus untreated cultures was also measured by Sothern blot in the Parkin line to further validate the Q-FISH data (Fig. S1D). We observed an increase of short telomeres in the neurons treated with the telomerase inhibitor by analyzing the blot and comparing the lane profiles (Fig. S1D).
Figure 3. Shortened telomeres effect on the aging phenotype of hPSC-derived neurons.

Different aging-associated phenotypes were measured in H9 and two PD iPSC lines, PINK1 and PARKIN-derived neurons at day 65 of mDA differentiation protocol. Cells were treated with two different concentrations (10 and 40 μM) of the telomerase inhibitor BIBR1532 and DMSO was used as a control (A) Quantification of the mean ± SEM telomere length measured by HT Q-FISH in hPSC-derived neurons. Numbers above bars indicate the number of telomere spots quantified (B) Percentage of short telomeres (<8kb) measured by HT QFISH in hPSC-derived neurons. Numbers above bars indicate the number of short telomeres out of the total number of telomeres. (C) Quantitative analysis of DNA damage measured by the mean gamma H2AX intensity per nuclei expressed in arbitrary units of fluorescence (AUF). Bars are represented as mean ± SEM. Numbers above bars indicate the number of nuclei quantified (D) Representative images of gamma H2AX immunofluorescence (orange) (E) Mitochondrial ROS quantification, measured by MitoSOX assay, analyzed by FACS. Numbers above bars indicate the number of MitoSOX positive cells out of the total number of cells.
Finally, we tested whether neurons with shorter telomeres exhibit other age-related markers. We measured several candidate age-associated phenotypes such as increased DNA damage and a reduction in the number of dendrites per cells. DNA damage, measured by gamma H2AX intensity was increased in the cells treated with the higher concentration of the inhibitor across all three hPSC lines compared (Fig. 3C–D). Pilot studies suggest that mitochondrial ROS, as measured by the percentage of MitoSox positive cells, may also be increased in the PINK and PARKIN lines treated with the telomerase inhibitor, (Fig. 3E). In addition, reduced dendrite numbers, labeled with MAP2 dendrite marker, were observed in H9 for both BIBR1532 concentrations and in the PARKIN line when treated at the higher concentration (Fig. 4A–B). No reduction in dendrite number was observed in PINK cell line (Fig. 4A). One potential confounding factor for interpreting such age-related marker expression in vitro is the possibility of negative selection for those neurons with increased mitochondrial stress or reduced dendrite number. However, in support of our data on dendrite loss, in vitro differentiated neurons derived from the neural stem cells (NSC) of a G3 Terc KO mouse, also have fewer neurites. Therefore, the ability of neurons to extend neurites may be linked to telomere function and chromosomal integrity. Moreover, this study shows that this effect could be rescued by P53 deletion indicating that it is P53 dependent (Ferron et al., 2009). All together, our data support the idea that neurons with short telomeres show an “older” cell phenotype.
Figure 4. Shortened telomeres effect on the neuronal aging, differentiation efficiency and disease-related phenotype of hPSC-derived neurons.

Neuronal aging, differentiation efficiency and disease associated phenotypes were measured in H9 and two PD iPSC lines, PINK1 and PARKIN-derived neurons at day 65 of mDA differentiation protocol. Cells were treated with two different concentrations (10 and 40 μM) of the telomerase inhibitor BIBR1532 and DMSO was used as a control (A) Quantitative analysis of MAP2 dendritic marker measured by the number of dendrites per cell. Bars represent mean ± SEM. Numbers above bars indicate the number of nuclei quantified (B) Representative images of MAP2 (orange) marker of hPSC-derived neurons (C) Quantitative analysis of FOXA2 (mDA marker) immunofluorescence. Numbers above bars represent the number of positive nuclei over the total number of nuclei analyzed (D) Quantitative analysis of FOXA2 and TH (mDA marker lost in early stage of Parkinson’s disease) double positive cells in immunofluorescence images. Numbers above bars represent the number of positive nuclei over the total number of nuclei analyzed (E) Representative images of TH (green) and FOXA2 (red) staining of hPSC-derived mDA neurons.
We next wanted to test whether hES cells with shortened telomeres also exhibit age-related marker expression upon differentiation into alternative lineages such as cardiomyocytes. Similar to the neuronal differentiation studies, telomerase was pharmacologically inhibited for the cardiomyocyte study using the telomerase inhibitor drug BIBR1532. Cells were treated with the telomerase inhibitor for two weeks previous to starting the differentiation protocol and treatment was continued throughout the cardiomyocytes differentiation protocol. Interestingly, telomerase inhibition in cardiomyocytes resulted in a lower percentage of cardiomyocytes, as shown by a decrease in the percentage of TBX5 positive cells (Fig. S4A). The fact that telomerase inhibition resulted in fewer cardiomyocytes may indicate that telomerase activity is needed for cardiomyocyte differentiation. Supporting this idea, Bednarek et al. showed in zebrafish that the absence of telomerase results in fewer cardiomyocytes and impaired heart regeneration (Bednarek et al., 2015).
Furthermore, the differentiated cardiomyocytes showed evidence of increased DNA damage, as measured by gamma H2AX intensity (Fig. S4C), and shorter telomeres (Fig. S4 E–G) as shown by a decrease in mean telomere length (Fig. S4E) and an increase in the percentage of short telomeres by HT Q-FISH (Fig. S4F). The cardiomyocyte data support our findings in neurons that telomere shortening can trigger age-related marker expression in cells with no or minimal capacity for cell proliferation.
In addition to the aging phenotypes observed, neurons with shortened telomeres also show preliminary evidence for disease specific phenotypes. In particular, neurons derived from cells treated with the telomerase inhibitor exhibited a loss of TH while maintaining other neuronal markers such as FOXA2 at day 30 (Fig. S3 A–B) and day 65 of differentiation (Fig. 4C–E). Progressive loss of TH expression is a characteristic feature of early PD at a stage when DA neurons are still alive (Chaudhuri et al., 2006). These data are reminiscent of our previous findings in progerin expressing PD-iPSC derived neurons that show a loss of TH at 3 months after transplantation into the adult mouse striatum (Miller et al., 2013). In conclusion, our current study yields preliminary evidence that shortened telomeres in PD iPSC derived mDA trigger age- and disease-related phenotypes. However, future studies will be required to further validate those findings using genetic strategies of telomere shortening and testing larger cohorts of patient-specific and control lines.
DISCUSSION
Age is the primary risk factor for many neurodegenerative disorders including PD. A late disease-onset indicates that the genetic mutation provides susceptibility but is not sufficient to trigger the disease. Accordingly, most iPSC models show evidence of biochemical changes that are directly linked to the disease-specific mutation but lack age-associated features such as spontaneous neuronal degeneration. Taking one additional step forward, we have recently presented a strategy to genetically trigger age-like features in iPSC derivatives towards modeling of a late onset disease by engineering the overexpression of progerin in iPSC-derived mDA neurons (Miller et al., 2013). However, the question of whether targeting pathways that trigger progeroid syndromes mimics pathological over physiological aging, remains to be resolved.
Here we present an alternative, potentially more physiological approach to induce aging by altering one of the classic “hallmarks” of aging, telomere shortening. In the present study we show that telomerase is downregulated and telomeres get shorter during neuronal differentiation and that the telomeres of hPSC and hPSC derivatives can be further shortened using a pharmacological approach. We then demonstrate that hPSC derived neurons with shorter telomeres present age-like features such as increased DNA damage, mitochondrial ROS, and reduced dendrite numbers and preliminary disease related features such as TH loss.
While telomere shortening by pharmacological inhibition was reproducible across experiments, as shown both by HT Q-FISH and by Southern blot (Fig. 2C–D, Fig. 3A–B and Fig. S1A–D), we observed some variability in the readout of absolute telomere length within the same cell line across experiments. This variability in absolute telomere length is inherent to any biological cell population and can fluctuate in undifferentiated hPSCs depending on passage and culture conditions. Interestingly, we also observed some variability in telomere length among the various hPSC cell lines (Fig. 2C–D; Fig. 3A–B and Fig. S1A–C) which may reflect the high interindividual variation of telomere length across human populations (Canela et al., 2007). Importantly, as shown in (Vera et al., 2012) the rate of increase in the percentage of short telomeres over time (slope), and not the absolute telomere length, is a significant predictor of lifespan in mice. These results indicate that the measure of telomere shortening most relevant to aging may be the rate of change in each individual and consequently in each cell line. Based on this argument, we propose that a key feature of the current study is the effort to “age” each cell line independently, triggering a relative increase in the percentage of short telomeres.
There are several major limitations for the use of small molecules in regulating telomerase activity. For example telomerase activity is reduced rather than being shut down completely. Therefore, the extent of telomere shortening is fairly modest, even though it was enough to trigger age- and disease-related phenotypes. Furthermore, the impact and mechanism of telomere shortening in postmitotic cells remains to be explored. Our current study represents a proof of concept for the use of telomere shortening as an “induced aging” strategy suitable for iPSC-based disease modeling. However, it will be important in future studies to test the full impact on age- and disease-related phenotypes using more precise genetic strategies of manipulating telomere length. Finally, it will be interesting to directly compare various emerging “induced aging” technologies (Studer et al., 2015) to determine those most suitable and effective at modeling late onset disease in iPSC-based studies.
EXPERIMENTAL PROCEDURES
HT-QFISH
Cells were plated on a clear-bottom, black-walled, 96-well plate, including 4 well replicates per sample, and HT-QFISH was performed as previously described (Canela et al., 2007). Images were captured with the Operetta high throughput automated microscope (Perkin Elmer). Telomere length values were analyzed using individual telomere spots (>1800 per sample) and fluorescence intensities were converted into kilobases as described previously (Canela et al., 2007; McIlrath et al., 2001).
Telomerase assay
The telomerase activity was measured with a modified TRAP (Garcia-Cao et al., 2002). As control for PCR efficiency, an internal control (IC) was included in each reaction (TRAPeze kit, Oncor). The main modification is that radioactive-labeled TS primer was substituted by a fluorescent CY5-labeled TS primer. Protean II (Bio-Rad) electrophoresis chamber was used to run the Accrilamide: Bisacrilamide 19:1 gels. Fluorescent gels were not dried and were imaged wet instead in Typhoon FLA700 laser scanner. Digitalized 15 images were quantified by using ImageJ software. The ‘sum intensity’ was calculated for each lane. Two dilutions were made for each sample (1 ug and 0.2 ug). The RNAse treated sample was used to quantify the background and subtracted to each sample value. Telomerase activity values, expressed in arbitrary units (a.u.) were then multiplied by the dilution factor and normalized by the Day 0 (Fig. 1B) or 0 uM treated (Fig. 2A) sample. For testing telomerase inhibition by BIBR1532 (Fig. 2B), the protein extracts were preincubated with the indicated concentration of BIBR1532 for 15 min on ice before the telomerase reaction was performed.
Pluripotent stem cell culture and iPSC Differentiation
Undifferentiated hESCs (H9/WA-09), and iPSCs were cultured on mitotically inactivated mouse embryonic fibroblasts (MEFs) in ESC media containing 20% knockout serum replacement (KSR) as previously described (Kriks et al., 2011). MDA neuron differentiations were performed as previously described (Kriks et al., 2011). Cardiomyocyte differentiation was done following a modified version of the previously described protocol (Laflamme et al., 2007). Briefly, the hESCs were dissociated into single cells using 0.05% trypsin and cultured in ultra low cluster 6 well plates (2 million cells/well) in hESC media containing Y-drug. The cells aggregate into EBs and grow for 6 days before they are plated on gelatin coated wells. At this point, the hESC medium is replaced with RPMI medium supplemented with B27 and Activin A (100nM). The media is replaced the next day with RPMI supplemented with B27 and BMP4 (10nM). The cells are cultured in BMP4 containing medium for a total of four days and mature further in the same RPMI+B27 medium without any additional growth factors.
Flow Cytometry, mtROS and Cycle Analyses
Cells were dissociated with Accutase (Innovative Cell Technologies) and stained with directly conjugated antibodies (BD Biosciences). The antibody used for flow cytometry was SSEA3 (Sigma). For mtROS assessment, cells were stained with MitoSOX Red (Life Technologies) at a final concentration of 20 μM in cell culture medium. Cell sorting was performed on a FACSAria (BD Biosciences). For cell cycle analysis, cells were collected in PBS and fixed in cold 70% ethanol. Followed by RNase A (Ambion) treatment, cells were stained with propidium iodide (50 μg/ml, Invitrogen) in PBS and subjected to FACS analysis according to standard procedures. For post-sort analysis, data was processed using FlowJo (Tree Star) software.
Quantitative RT–PCR analysis
Cells were lysed with TRIzol (Life Technologies), treated for DNA contamination, and reverse-transcribed using QuantiTect RT kit (Qiagen). The mRNA levels were assayed using the SYBR Green PCR kit (Qiagen) on a Master- Cycler RealPlex2 (Eppendorf). All results were normalized to a GAPDH control. The primers used are detailed in Supplemental Experimental Procedures section.
Immunostaining
Cells were fixed by incubation in 4% paraformaldehyde for 15 minutes and incubated in blocking buffer (0.3% Triton-X100 in PBS) for 30 minutes. Cells were stained with primary antibodies in PBS supplemented with 1% BSA at 4°C overnight, washed and stained with secondary antibodies in PBS supplemented with 0.1% BSA for 1 hour at room temperature, in the dark. Nuclei were stained by DAPI (Invitrogen). The following primary antibodies were used: TH (1:1000; Pel Freeze), FOXA2 (Santa Cruz; 1:200), OCT4 (1:100; Santa Cruz), NANOG (1:50; R&D), and phospho-Histone H2A.X (Ser139; clone 20E3; 1:400; Cell Signaling).
Telomere Restriction Fragment (TRF) Analysis
Cells were harvested by trypsin treatment, washed with PBS, pelleted and frozen at −80°C for genomic DNA collection. Genomic DNA was extracted using QIAamp DNA Mini Kit (QIAGEN). DNA was digested with MboI and AluI, quantitated by fluorometry using Hoechst 33258 and 100ng was loaded on a 0.5% agarose gel run in 0.5X TBE. The gels were depurinated with 0.25N HCl, denatured and neutralized using standard Southern blotting procedures. The DNA was transferred to Hybond-N membrane (Amersham) in 20xSSC and cross-linked by UV exposure in a Stratalinker (Stratagene). Membranes were hybridized overnight at 65°C in Church mix (0.5M sodium phosphate buffer pH 7.2, 1mM EDTA, 7% SDS, 1% BSA) with a 800bp TTAGGG repeat fragment Klenow-labeled using a 5′CCCTAACCCTAA3′ primer and α-32P-dCTP/dATP. Post-hybridization washes were performed in 40mM Na-phosphate buffer pH 7.2, 1 mM EDTA, 1% SDS at 65°C. Membranes were exposed to PhosphorImager screens. To generate the lane profile graph, grey values were obtained in ImageJ and plotted as the percentage of the maximum intensity value of the lane.
Supplementary Material
HIGHLIGHTS.
Downregulation of telomerase and telomere shortening during neural differentiation.
Telomerase inhibition in hPSCs results in derivatives with shorter telomeres.
Neurons with short telomeres present aging-associated phenotypes.
Neurons with short telomeres present potential disease related phenotypes.
Acknowledgments
E.V. was supported by a NYSTEM postdoctoral fellowship. The work of the authors described in this review was supported by grants from the Starr Foundation, NYSTEM (C028503 and C026447), NINDS/NIH (NS072381), and NCI/NIH (P30CA008748).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and four figures.
AUTHOR CONTRIBUTIONS
E.V. performed the experimental work, analyzed the data and wrote the paper. N.B performed the Telomeric Southern blot experiments. L.S. directed the project and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell. 1996;86:159–171. doi: 10.1016/s0092-8674(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Bednarek D, Gonzalez-Rosa JM, Guzman-Martinez G, Gutierrez-Gutierrez O, Aguado T, Sanchez-Ferrer C, Marques IJ, Galardi-Castilla M, de Diego I, Gomez MJ, et al. Telomerase Is Essential for Zebrafish Heart Regeneration. Cell reports. 2015;12:1691–1703. doi: 10.1016/j.celrep.2015.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardes de Jesus B, Vera E, Schneeberger K, Tejera AM, Ayuso E, Bosch F, Blasco MA. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO molecular medicine. 2012;4:691–704. doi: 10.1002/emmm.201200245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nature reviews Genetics. 2005;6:611–622. doi: 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- Blasco MA, Funk W, Villeponteau B, Greider CW. Functional characterization and developmental regulation of mouse telomerase RNA. Science. 1995;269:1267–1270. doi: 10.1126/science.7544492. [DOI] [PubMed] [Google Scholar]
- Canela A, Vera E, Klatt P, Blasco MA. High-throughput telomere length quantification by FISH and its application to human population studies. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:5300–5305. doi: 10.1073/pnas.0609367104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SW, Blackburn EH. New ways not to make ends meet: telomerase, DNA damage proteins and heterochromatin. Oncogene. 2002;21:553–563. doi: 10.1038/sj.onc.1205082. [DOI] [PubMed] [Google Scholar]
- Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D, Pathak S, Guarente L, DePinho RA. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nature genetics. 2004;36:877–882. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- Chaudhuri KR, Healy DG, Schapira AH National Institute for Clinical E. Non-motor symptoms of Parkinson’s disease: diagnosis and management. The Lancet Neurology. 2006;5:235–245. doi: 10.1016/S1474-4422(06)70373-8. [DOI] [PubMed] [Google Scholar]
- Ebert AD, Yu J, Rose FF, Jr, Mattis VB, Lorson CL, Thomson JA, Svendsen CN. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277–280. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron SR, Marques-Torrejon MA, Mira H, Flores I, Taylor K, Blasco MA, Farinas I. Telomere shortening in neural stem cells disrupts neuronal differentiation and neuritogenesis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2009;29:14394–14407. doi: 10.1523/JNEUROSCI.3836-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores I, Canela A, Vera E, Tejera A, Cotsarelis G, Blasco MA. The longest telomeres: a general signature of adult stem cell compartments. Genes & development. 2008;22:654–667. doi: 10.1101/gad.451008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cao M, Gonzalo S, Dean D, Blasco MA. A role for the Rb family of proteins in controlling telomere length. Nature genetics. 2002;32:415–419. doi: 10.1038/ng1011. [DOI] [PubMed] [Google Scholar]
- Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- Grodstein F, van Oijen M, Irizarry MC, Rosas HD, Hyman BT, Growdon JH, De Vivo I. Shorter telomeres may mark early risk of dementia: preliminary analysis of 62 participants from the nurses’ health study. PloS one. 2008;3:e1590. doi: 10.1371/journal.pone.0001590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JZ, Guan WP, Maeda T, Makino N. The Subtelomere of Short Telomeres is Hypermethylated in Alzheimer’s Disease. Aging and disease. 2012;3:164–170. [PMC free article] [PubMed] [Google Scholar]
- Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- Hemann MT, Strong MA, Hao LY, Greider CW. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell. 2001;107:67–77. doi: 10.1016/s0092-8674(01)00504-9. [DOI] [PubMed] [Google Scholar]
- Hochstrasser T, Marksteiner J, Humpel C. Telomere length is age-dependent and reduced in monocytes of Alzheimer patients. Exp Gerontol. 2012;47:160–163. doi: 10.1016/j.exger.2011.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig LS, Kang MS, Schupf N, Lee JH, Mayeux R. Association of shorter leukocyte telomere repeat length with dementia and mortality. Arch Neurol. 2012;69:1332–1339. doi: 10.1001/archneurol.2012.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig LS, Schupf N, Lee JH, Tang MX, Mayeux R. Shorter telomeres are associated with mortality in those with APOE epsilon4 and dementia. Ann Neurol. 2006;60:181–187. doi: 10.1002/ana.20894. [DOI] [PubMed] [Google Scholar]
- Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadinanos J, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 469:102–106. doi: 10.1038/nature09603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins EC, Velinov MT, Ye L, Gu H, Li S, Jenkins EC, Jr, Brooks SS, Pang D, Devenny DA, Zigman WB, et al. Telomere shortening in T lymphocytes of older individuals with Down syndrome and dementia. Neurobiology of aging. 2006;27:941–945. doi: 10.1016/j.neurobiolaging.2005.05.021. [DOI] [PubMed] [Google Scholar]
- Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011;480:547–551. doi: 10.1038/nature10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume K, Kikukawa M, Hanyu H, Takata Y, Umahara T, Sakurai H, Kanetaka H, Ohyashiki K, Ohyashiki JH, Iwamoto T. Telomere length shortening in patients with dementia with Lewy bodies. Eur J Neurol. 2012;19:905–910. doi: 10.1111/j.1468-1331.2011.03655.x. [DOI] [PubMed] [Google Scholar]
- Lafaille FG, Pessach IM, Zhang SY, Ciancanelli MJ, Herman M, Abhyankar A, Ying SW, Keros S, Goldstein PA, Mostoslavsky G, et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature. 2012;491:769–773. doi: 10.1038/nature11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, Reinecke H, Xu C, Hassanipour M, Police S, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nature biotechnology. 2007;25:1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- Lebel M, Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:13097–13102. doi: 10.1073/pnas.95.22.13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461:402–406. doi: 10.1038/nature08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T, Guan JZ, Koyanagi M, Higuchi Y, Makino N. Aging-associated alteration of telomere length and subtelomeric status in female patients with Parkinson’s disease. Journal of neurogenetics. 2012;26:245–251. doi: 10.3109/01677063.2011.651665. [DOI] [PubMed] [Google Scholar]
- Mahmoudi S, Brunet A. Aging and reprogramming: a two-way street. Curr Opin Cell Biol. 2012;24:744–756. doi: 10.1016/j.ceb.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Ruiz C, Dickinson HO, Keys B, Rowan E, Kenny RA, Von Zglinicki T. Telomere length predicts poststroke mortality, dementia, and cognitive decline. Ann Neurol. 2006;60:174–180. doi: 10.1002/ana.20869. [DOI] [PubMed] [Google Scholar]
- McIlrath J, Bouffler SD, Samper E, Cuthbert A, Wojcik A, Szumiel I, Bryant PE, Riches AC, Thompson A, Blasco MA, et al. Telomere length abnormalities in mammalian radiosensitive cells. Cancer Res. 2001;61:912–915. [PubMed] [Google Scholar]
- Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, Mandal PK, Vera E, Shim JW, Kriks S, et al. Human iPSC-Based Modeling of Late-Onset Disease via Progerin-Induced Aging. Cell stem cell. 2013;13:691–705. doi: 10.1016/j.stem.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999;402:551–555. doi: 10.1038/990141. [DOI] [PubMed] [Google Scholar]
- Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- Panossian LA, Porter VR, Valenzuela HF, Zhu X, Reback E, Masterman D, Cummings JL, Effros RB. Telomere shortening in T cells correlates with Alzheimer’s disease status. Neurobiology of aging. 2003;24:77–84. doi: 10.1016/s0197-4580(02)00043-x. [DOI] [PubMed] [Google Scholar]
- Ranganathan V, Heine WF, Ciccone DN, Rudolph KL, Wu X, Chang S, Hai H, Ahearn IM, Livingston DM, Resnick I, et al. Rescue of a telomere length defect of Nijmegen breakage syndrome cells requires NBS and telomerase catalytic subunit. Curr Biol. 2001;11:962–966. doi: 10.1016/s0960-9822(01)00267-6. [DOI] [PubMed] [Google Scholar]
- Srikanth P, Young-Pearse TL. Stem cells on the brain: modeling neurodevelopmental and neurodegenerative diseases using human induced pluripotent stem cells. J Neurogenet. 2014;28:5–29. doi: 10.3109/01677063.2014.881358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer L, Vera E, Cornacchia D. Programming and Reprogramming Cellular Age in the Era of Induced Pluripotency. Cell stem cell. 2015;16:591–600. doi: 10.1016/j.stem.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas P, NJOC, Fenech M. Telomere length in white blood cells, buccal cells and brain tissue and its variation with ageing and Alzheimer’s disease. Mech Ageing Dev. 2008;129:183–190. doi: 10.1016/j.mad.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Vera E, Bernardes de Jesus B, Foronda M, Flores JM, Blasco MA. The rate of increase of short telomeres predicts longevity in mammals. Cell reports. 2012;2:732–737. doi: 10.1016/j.celrep.2012.08.023. [DOI] [PubMed] [Google Scholar]
- Vera E, Blasco MA. Beyond average: potential for measurement of short telomeres. Aging. 2012;4:379–392. doi: 10.18632/aging.100462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Zglinicki T, Serra V, Lorenz M, Saretzki G, Lenzen-Grossimlighaus R, Gessner R, Risch A, Steinhagen-Thiessen E. Short telomeres in patients with vascular dementia: an indicator of low antioxidative capacity and a possible risk factor? Lab Invest. 2000;80:1739–1747. doi: 10.1038/labinvest.3780184. [DOI] [PubMed] [Google Scholar]
- Watfa G, Dragonas C, Brosche T, Dittrich R, Sieber CC, Alecu C, Benetos A, Nzietchueng R. Study of telomere length and different markers of oxidative stress in patients with Parkinson’s disease. J Nutr Health Aging. 2011;15:277–281. doi: 10.1007/s12603-010-0275-7. [DOI] [PubMed] [Google Scholar]
- Wellinger RJ, Sen D. The DNA structures at the ends of eukaryotic chromosomes. Eur J Cancer. 1997;33:735–749. doi: 10.1016/S0959-8049(97)00067-1. [DOI] [PubMed] [Google Scholar]
- Wong KK, Maser RS, Bachoo RM, Menon J, Carrasco DR, Gu Y, Alt FW, DePinho RA. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature. 2003;421:643–648. doi: 10.1038/nature01385. [DOI] [PubMed] [Google Scholar]
- Wright WE, Piatyszek MA, Rainey WE, Byrd W, Shay JW. Telomerase activity in human germline and embryonic tissues and cells. Dev Genet. 1996;18:173–179. doi: 10.1002/(SICI)1520-6408(1996)18:2<173::AID-DVG10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.