

Summary
Caspase-8 activation can be triggered by death receptor-mediated formation of the death-inducing signaling complex (DISC) and by the inflammasome adaptor ASC. Caspase-8 assembles with FADD at the DISC and with ASC at the inflammasome through its tandem death effector domain (tDED), which is regulated by the tDED-containing cellular inhibitor cFLIP and the viral inhibitor MC159. Here we present the caspase-8 tDED filament structure determined by cryo-electron microscopy. Extensive assembly interfaces not predicted by the previously proposed linear DED chain model were uncovered, and further confirmed by structure-based mutagenesis in filament formation in vitro, and Fas-induced apoptosis and ASC-mediated caspase-8 recruitment in cells. Structurally, the two DEDs in caspase-8 use quasi-equivalent contacts to enable assembly. Using the tDED filament structure as a template, structural analyses reveal the interaction surfaces between FADD and caspase-8, and the distinct mechanisms of regulation by cFLIP and MC159 through comingling and capping, respectively.
Graphical abstract
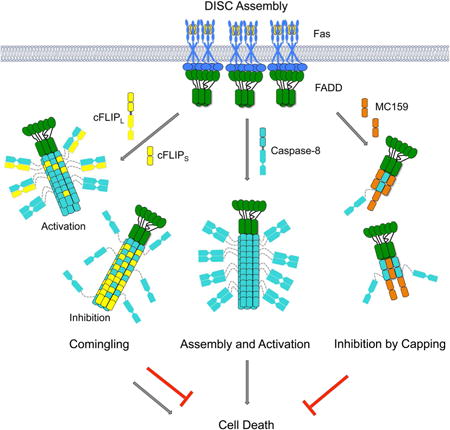
Introduction
Apoptosis, a physiological form of cell death, has classically been divided into intrinsic and extrinsic pathways. While the intrinsic pathway relies on mitochondrial damage, cytochrome c release and apoptosome formation, the extrinsic pathway is mediated by death receptors (DRs) in the tumor necrosis factor (TNF) receptor (TNFR) superfamily (Green, 2003; Strasser et al., 2009). DRs contain an intracellular death domain (DD) and are composed of 8 members, Fas (CD95 or APO-1), TNFR1, DR3, DR4, DR5, DR6, EDA-R and NGF-R (French and Tschopp, 2003; Wu and Hymowitz, 2009). These receptors are type I transmembrane proteins, with an extracellular region composed of three to four cysteine-rich domain segments for ligand recognition. DR signaling is critically important for maintenance of lymphocyte homeostasis (Locksley et al., 2001). Genetic mutations and abnormal expression of DRs, their ligands and signaling proteins have been associated with many human diseases such as the autoimmune lymphoproliferative syndrome, cancer, multiple sclerosis, and stroke (French and Tschopp, 2003; Siegel et al., 2004).
Fas is a prototypical member of the DR family (Kischkel et al., 1995). Upon interaction with Fas ligand (FasL), its intracellular DD recruits the adaptor FADD through DD/DD interactions. FADD also has a death effector domain (DED), which in turn recruits caspase-8 (Casp-8) and/or Casp-10 via the tandem DEDs (tDEDs) in these caspases (Figure 1A). DDs, DEDs, caspase recruitment domains (CARDs) and Pyrin domains (PYDs) all belong to the death domain fold superfamily with characteristic six-helix bundle structures (Ferrao and Wu, 2012). The ternary complex of Fas or a Fas-like DR, FADD and a caspase constitute the DISC. It is presumed that Casp-8 and Casp-10 become activated in the DISC through proximity induced dimerization and auto-proteolytic processing (Boatright et al., 2003). Active Casp-8/-10 cleave effector caspases to initiate apoptosis. TNFR1 is another prototypical DR, which directly recruits the adaptor TRADD via DD/DD interactions upon ligand stimulation (Micheau and Tschopp, 2003). Apoptosis induction by TNFR1 or a TNFR1-like DR requires formation of secondary complexes including the cytosolic TRADD/FADD/Casp-8 complex (Micheau and Tschopp, 2003) and the RIP1/FADD/Casp-8 complex (Feoktistova et al., 2011; Tenev et al., 2011). Therefore, DED-mediated interaction between FADD and Casp-8 is the converging downstream element critical for cell death induction (Figure 1A). In addition, recent studies suggested that the inflammasome adaptor ASC recruits Casp-8 through a PYD/tDED interaction to induce apoptosis (Sagulenko et al., 2013; Vajjhala et al., 2015) (Figure 1A).
Figure 1. The Fas/FADD Complex, FADDDED and ASCPYD Promote Casp-8tDED Filament Formation.

(A) Domain composition and interaction hierarchy of Casp-8 activation in the death receptor pathway and the inflammasome pathway.
(B) A gel filtration profile of GFP-Casp-8tDED purification. An SDS-PAGE of the major peaks is shown. Peak 1 at the void position contained the target protein.
(C) An electron micrograph of GFP-Casp-8tDED filaments.
(D) Fluorescence polarization (FP) assay of FasDD/FADD, FADD and ASCPYD nucleated Casp-8tDED filament formation.
(E) Streptavidin-gold labeling of the FasDD/FADD-biotin/Casp-8tDED filaments, showing the end localization of the FasDD/FADD-biotin complex.
(F, G) Nucleation of Casp-8tDED filament formation by the FasDD/FADD complex (F) and FADDDED (G). Calculated apparent dissociation constants (Kapp), with fitting errors, are shown above the plots.
See also Figure S1.
Tandem DEDs are also present in FLICE/Casp-8-like inhibitory proteins (FLIP) of both cellular and viral origin (cFLIP and vFLIP) (Tschopp et al., 1998) (Figure 1A). The long form of cFLIP, cFLIPL, has an inactive caspase-like domain and hetero-dimerizes with the caspase domain of Casp-8 and cFLIPL has been shown to promote Casp-8 activation (Yu et al., 2009). In contrast, the shorter form of cFLIP, cFLIPS, can inhibit Casp-8 activation (Hughes et al., 2016). Thus cFLIPL and cFLIPS may play differential roles in modulating DR signal transduction. The vFLIP protein MC159 from the Molluscum contagiosum virus inhibits Casp-8 activation (Yang et al., 2005). In contrast, the vFLIP protein K13 from the Kaposi's sarcoma herpes virus does not engage the DR pathway but interacts with NEMO to activate the NF-κB pathway (Bagneris et al., 2008).
It has been shown that ligand binding induces aggregation of DRs and formation of DISC clusters on the cell membrane to initiate apoptotic signaling (Kischkel et al., 1995; Siegel et al., 2004). These clusters should rely on both DD/DD and DED/DED interactions among the DISC component proteins. Structural and biochemical studies on Fas/FADD and RIP1/FADD DD complexes have implicated helical assembly in DD/DD oligomerization (Esposito et al., 2010; Jang et al., 2014; Wang et al., 2010). However, despite structures of isolated DEDs, how DEDs oligomerize and interact with each other remained elusive due to lack of oligomeric structures (Bagneris et al., 2008; Eberstadt et al., 1998; Shen et al., 2015; Yang et al., 2005). Yet, DED/DED interactions are particularly important because of the over-abundance of Casp-8 in the DISC (Dickens et al., 2012; Schleich et al., 2012). In the absence of direct structural information, a linear DED chain was modeled to account for Casp-8 self-association and interactions with other DED-containing proteins (Dickens et al., 2012; Schleich et al., 2012).
By visualizing the structure of the Casp-8tDED filament using cryo-electron microscopy (cryo-EM), we here provide a framework for understanding DISC assembly and regulation. In contrast to the linear DED chain model, the interactions in the filament structure span three directions to form the three types of asymmetric interfaces. Structure-based mutagenesis confirmed the importance of these interfacial residues in the Casp-8 filament for FADD-mediated apoptotic cell death and ASC-mediated Casp-8 recruitment, demonstrating that interactions beyond those predicted by the DED chain are equally important. Therefore, the Casp-8tDED filament structure corrected the oversimplification of the DED chain model that may misguide the understanding of DISC functions. Moreover, the structural information allowed analysis on the interaction of Casp-8 with other DED-containing proteins FADD, cFLIP and MC159, revealing distinct, unexpected modes of regulation in the DISC.
Results
Formation of Filaments, Not Chains, by Casp-8tDED Represents DISC Assembly
Biochemical reconstitution of the DISC has been hampered by the poor solubility of Casp-8tDED. Using the monomeric GFP as the fusion partner, we obtained highly soluble His-GFP-Casp-8tDED, which eluted from the void position of a Superdex 200 gel filtration column (Figure 1B). Visualization by negative staining EM revealed filamentous structures with an apparent diameter of ∼20 nm (Figure 1C), which is significantly larger than the ∼3 nm width of its tDED (Shen et al., 2015) in the proposed DED chain model for FADD and Casp-8 oligomerization (Dickens et al., 2012; Schleich et al., 2012).
To assess Casp-8tDED filament formation, we needed a monomeric Casp-8tDED, but WT Casp-8tDED aggregated even when tagged at both termini with the solubility tags MBP and Sumo, respectively (His-MBP-Casp-8tDED-Sumo) (Figure S1A-B). Searching existing mutagenesis data led us to the partially defective Casp-8tDED-Y8A mutant (Majkut et al., 2014; Yang et al., 2005), which we purified in a monomeric form but still formed filaments when the His-MBP tag was removed by the TEV protease (Figure S1C-D). These filaments are indistinguishable from the WT Casp-8tDED filaments (Figure 1C).
His-MBP-Casp-8tDED-Y8A-Sumo labeled with the TAMRA fluorophore was used in a fluorescence polarization (FP) assay to monitor filament formation. Upon TEV cleavage, the rate of polymerization of the Casp-8tDED-Y8A mutant alone was minimal at the concentration used (Figure 1D). Upon addition of a substoichiometric amount of FADD, the polymerization rate was significantly enhanced (Figure 1D), demonstrating nucleation of Casp-8tDED-Y8A by the upstream adaptor. ASC also enhanced Casp-8tDED polymerization. In the Fas DISC, it is the ternary complex of Fas, FADD and Casp-8 that results in apoptosis. To recapitulate this signaling process, we used the purified complex of FasDD and full-length FADD, which together nucleated Casp-8tDED-Y8A filament formation most robustly (Figure 1D). Labeling biotinylated FADD by 10 nm streptavidin-gold particles showed that the FasDD/FADD complex is localized at one end of the filaments (Figure 1E), supporting that the Fas/FADD complex nucleates directional polymerization of Casp-8. Titration of the FasDD/FADD complex into Casp-8tDED gave an apparent KD of 175 ± 1.4 nM for the interaction (Figure 1F, S1E). FADDDED alone also promoted Casp-8tDED-Y8A polymerization, albeit less efficiently, with an apparent KD of 823 ± 1.8 nM (Figure 1G, S1F), suggesting that multiple oligomerization domains reinforce each other during DISC assembly.
Cryo-EM Structure Determination of the Casp-8tDED Filament
To facilitate cryo-EM structure determination of the Casp-8tDED filament, we first solved the crystal structure of a monomeric Casp-8tDED mutant (F122G/L123G), which was nearly identical to the structure of another Casp-8tDED mutant (Shen et al., 2015) and highly similar to the structures of vFLIP MC159 and K13 (Table 1, Figure 2A-B, S2A-B). We collected EM data on both negatively stained and cryo-vitrified His-GFP-Casp-8tDED filament samples (Figure 2C). We first determined that the outer region of the filament from His-GFP did not contribute significantly to the power spectra (Figure S2C) because His-GFP is likely flexibly attached to the core tDED filaments and the Casp-8tDED-Sumo filament gave an essentially identical power spectrum (Figure S2D-F). Using a radius of ∼45 Å for the inner tDED region of the filament, the main layer lines in the power spectra from the three different EM data sets were consistent with point group symmetry of C3 and helical symmetry of ∼53° right-h anded rotation and ∼14.0 Å axial rise per subunit (Figure S2D), which is similar to that of the ASCPYD filaments (Lu et al., 2014). Because the asymmetric unit of the ASCPYD filament structure contains only one domain, C3 symmetry of the Casp-8tDED filament would need to be a pseudo-symmetry in which DED1 and DED2 are treated as equivalent (Figure 2D).
Table 1. Crystallographic Statistics.
| Data collection | |
|---|---|
| Beam line | 24ID-E, Advanced Photon Source |
| Wavelength (Å) | 0.9768 |
| Space group | P64 |
| Cell dimensions (Å) | 131.2, 131.2, 67.5 |
| Resolution (Å) | 50 - 3.1 |
| Rmerge (%) | 6.5 (74.9)* |
| l/σl | 21.6 (1.8)* |
| Completeness (%) | 99.2 (99.7)* |
| Refinement | |
| Resolution (Å) | 43 - 3.1 |
| No. reflections | 11,891 |
| Rwork / Rfree (%) | 21.6 / 27.1 |
| No. atoms | 4,328 |
| Average B-factors (Å2) | 61.5 |
| R.M.S. Deviations Bond lengths (Å) / angles (°) | 0.007 / 1.032 |
| Ramachandran plot Most favored / allowed (%) | 93.9 / 100.0 |
Numbers in parentheses are for the highest resolution shell.
Figure 2. Crystal Structure of MBP-Casp-8tDED-F122G/L123G and Cryo-EM Reconstruction of the GFP-Casp-8tDED Filament.
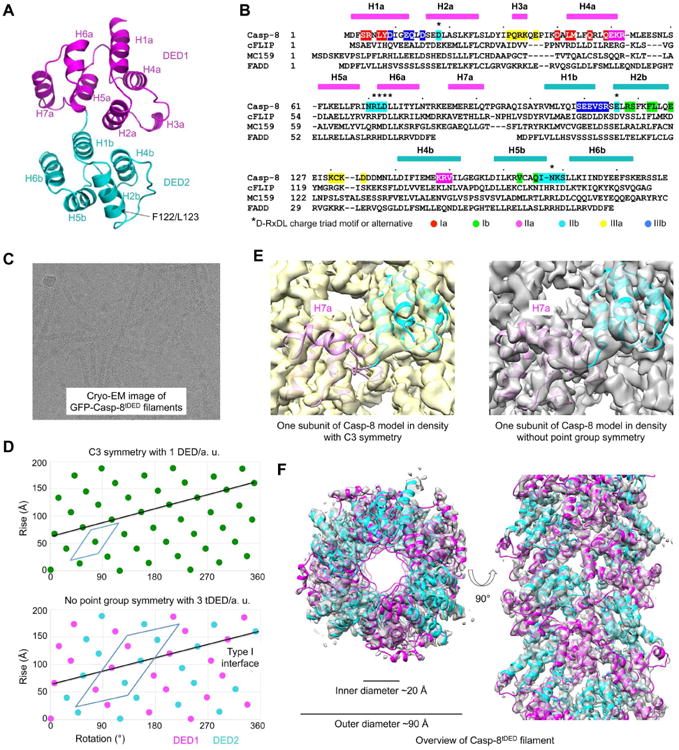
(A) A ribbon diagram of the Casp-8tDED (F122G/L123G) crystal structure labeled with secondary structures, and locations of the mutations.
(B) Sequence alignment among Casp-8, cFLIP, MC159 and FADD. Secondary structures are labeled on the top. Key residues involved in filament assembly are highlighted with type Ia in red, type Ib in green, type IIa in magenta, type IIb in cyan, type IIIa in yellow and type IIIb in blue.
(C) A cryo-electron micrograph of GFP-Casp-8 tDED filaments.
(D) Helical net plots showing the asymmetric units of the filament in C3 symmetry (top) and without point group symmetry (bottom), and the direction of the type I interaction that connects DED1 and DED2 in tDED.
(E) One subunit of the Casp-8tDED model fitted into cryo-EM densities reconstructed in C3 symmetry (left) and without point group symmetry (right). The density for H7a is absent in the C3 map.
(F) Top and side views of the GFP-Casp-8tDED filament structure superimposed with the final cryo-EM density.
See also Figure S2.
Cryo-EM reconstruction using the C3 symmetry resulted in a map with recognizable secondary structures. However, the use of the C3 symmetry averaged DED1 and DED2 domains and is likely the reason that the connection helix H7a between DED1 and DED2 was absent in the map (Figure 2E). In addition, the C3 symmetry failed to account for additional layer lines seen in the averaged power spectra (Figure S2D-2F), indicating that it could not be the correct symmetry. Fitting of the map with the crystal structure of either DED1 or DED2 of Casp-8tDED revealed the location of the type I interface that is supposed to connect DED1 and DED2 (Figure 2D). The power spectra suggested helical symmetry with 99.4° right-handed rotation and 27.1 Å axial rise per asymmetric unit, without point group symmetry. The asymmetric unit contains three Casp-8tDED molecules, which is six DEDs (Figure 2D). Reconstruction using the correct symmetry yielded a map that contained the connecting helix H7a and fit well with the Casp-8tDED crystal structure (Figure 2E). There is no obvious density for the His-GFP region. The resulting filament model (Table 2) displayed an outer diameter of ∼90 Å with a central hole of ∼20 Å in diameter (Figure 2F). Fourier shell correlation (FSC) curves between independently reconstructed half maps, and between the final map and the model are consistent with ∼4.6 Å in resolution (Figure S2G-H).
Table 2. Casp-8tDED Filament Structure Statistics.
| Cryo-EM data collection | |
|---|---|
| Voltage (kV) | 200 |
| Defocus range (μm) | 1.1 - 2.6 |
| Number of images | 388 |
| Number of particles | 33,969 |
|
| |
| Final model (containing 9 subunits) | |
|
| |
| Resolution (°) | 4.6 |
| Number of protein residues | 1,629 |
| Number of atoms | 27,414 |
| Bad bonds (%) | 0 |
| Bad angles (%) | 0 |
| Ramachandran statistics | |
| Favored regions (%) | 99.16 |
| Allowed regions (%) | 0.65 |
| Disallowed regions (%) | 0.19 |
| Rotamer outliers (%) | 0.06 |
| MolProbity score | 1.56 |
| Clash score | 6.49 |
Unique Quasi-Equivalence in the Casp-8tDED Filament
The Casp-8tDED filament structure utilizes the three types of interactions that have been described for helical assembly by other members of the DD superfamily (Ferrao and Wu, 2012; Lu et al., 2014) (Figure 3A). Casp-10tDED may form a similar structure as shown from the sequence conservation of the interaction surfaces (Figure S3A). An extensive surface area of ∼ 3,150 Å2 is buried for each Casp-8 molecule upon filament formation. The three tDED molecules in the asymmetric unit of the filament form three parallel helical strands by interacting with adjacent tDED molecules using the type I interface (Figure 3A). This interaction is mediated by DED2 of one tDED (type Ib) and DED1 of a neighboring tDED (type Ia), mainly composed of H2b and H5b of DED2 and H1a and H4a of DED1 (Figure 3B). In particular, R118, F122, L123 at H2b of DED2 pack against Y8 at H1a and L42 and L44 at H4a of DED1, forming dominant hydrophobic interactions (Figure 3B). In addition, E126 at H2b of DED2 may form polar interactions with S4 at H1a of DED1, and Q168 of H5b of DED2 interacts with R5 of H1a of DED1 (Figure 3B). Within each tDED, the DED1 type Ib surface interacts with the DED2 type Ia surface (Figure S3B). The chain of tDED molecules linked by the type I interaction provides the structural basis for the proposed DED chain model (Dickens et al., 2012; Majkut et al., 2014; Schleich et al., 2012), which represents a linear sub-structure of these filaments.
Figure 3. Quasi-Equivalent Interactions in the Casp-8tDED Filament.
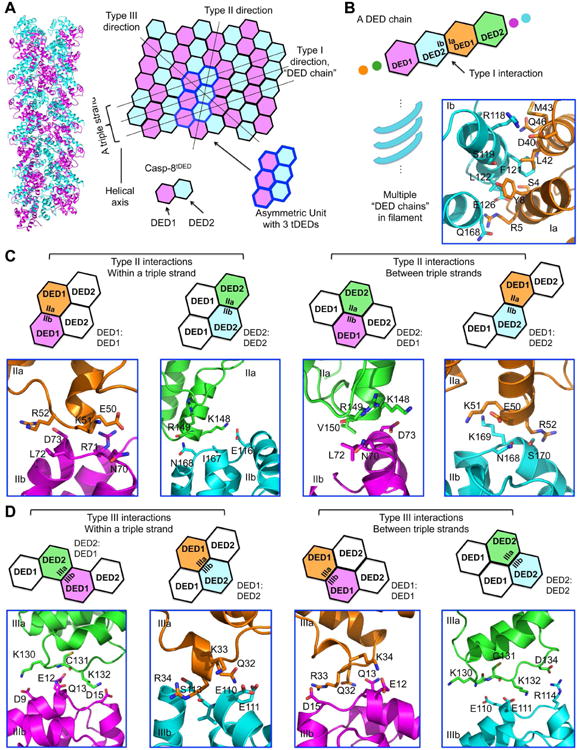
(A) Ribbon and schematic diagram of Casp-8tDED helical assembly. Each DED is shown as a hexagon and the three tDED molecules in the asymmetric unit are encircled in blue lines.
(B) The type I interface in the Casp-8tDED filament, with residues involved shown as stick models. DED1 is colored in magenta or orange and DED2 is colored in cyan or green.
(C, D) The four quasi-equivalent type II (C) and type III (D) interactions, with residues involved shown as stick models. The specifically contacts are illustrated by the schematics above.
See also Figure S3.
As previous DD superfamily filament structures contain single domains, such as those of ASC PYD, MAVS CARD and Casp-1 CARD (Lu et al., 2016; Lu et al., 2014; Wu et al., 2014), it is intriguing how two DEDs with limited sequence identity of 20-30% can assemble into the same filament. The interstrand interactions between the different DED chains, which are of type II and type III, display this conundrum, with four possibilities of DED1: DED1, DED1: DED2, DED2: DED1 and DED2: DED2 interactions. Within a triple strand, the type II interactions are homotypic, DED1: DED1 and DED2: DED2, while the type III interactions are heterotypic, DED1: DED2 and DED2: DED1 (Figure 3A, 3C-D). In contrast, between adjacent triple strands, the type II interactions are heterotypic and the type III interactions are homotypic (Figure 3A, 3C-D). Because tDEDs in the filament use all the combinations of interactions, the apparently quasi-equivalent type II and III interactions must be critically important for Casp-8 assembly in the DISC.
Examining the different pairs of type II interactions revealed that the IIa surface is invariably formed mainly by H4 in DED1 or DED2, while the opposing IIb surface by the H5-H6 region, involving the conserved charge triad in DEDs (Yang et al., 2005) (Figure 2B, S3C-D). For type III interactions, the IIIa surface is mainly composed of residues from H3 in DED1 or the loop between H2 and H4 in DED2, while the type IIIb by the H1-H2 region (Figure 2B, S3D). Unlike the largely hydrophobic nature of the type I interaction, type II and III interactions are predominantly hydrophilic and charged (Figure 3C-D). In particular, although the interfacial residues are quite different between DED1 and DED2 in these interactions, there appeared to be conserved electrostatic complementation between IIa and IIb surfaces, and between IIIa and IIIb surfaces (Figure S3D). Examples include the R52: D73 and K148: D73 type II interactions (Figure 3C), and the D134: R114 and D15: R33 type III interactions (Figure 3D). This match of charges may explain how one interface is able to engage two different interfaces to achieve the quasi-equivalence in these interactions, and suggests the importance of charge complementarity in Casp-8tDED filament assembly.
Interfacial Residues are Important for Casp-8tDED Filament Formation in Vitro and in Cells
We carried out structure-guided mutagenesis to validate the Casp-8 filament model using the MBP-Casp-8tDED-Sumo construct (Figure 4A). Consistent with the results shown earlier (Figure S1A, S1C), WT Casp-8tDED eluted from the void fraction of a gel filtration column while the Y8A mutant (on type I interface) shifted the elution profile mostly to the monomeric fraction (Figure 4A). Additional type I mutants including F122E and F122G/L123G were almost completely impaired in filament formation, eluting as monomers (Figure 4A). The F122G/L123G mutant was used for the crystal structure determination. The S4D mutant was partially defective, but when combined with Y8A, was entirely impaired in filament formation. Notably, mutants of the type II and III interfaces, including the K148D/R149E and R52E type II interaction mutants and the D15R and C131D type III interaction mutants, also caused severe defects in filament formation.
Figure 4. Structure-Based Mutants Disrupt Casp-8tDED Filament Formation in Vitro and in Cells.
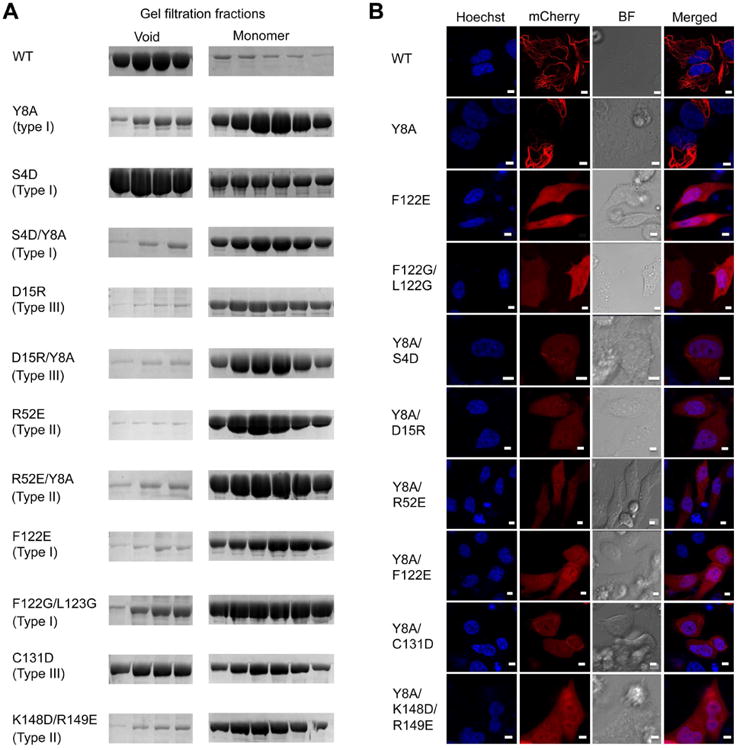
(A) Gel filtration profiles of WT and mutant Casp-8tDED showing the filamentous, void fraction and the monomeric fraction from a Superdex 200 column.
(B) Morphology of transfected mCherry-fused WT and mutant Casp-8tDED visualized by fluorescence microscopy. Hoechst was used to stain nuclei.
See also Figure S4.
We further tested structure-guided mutants in filament formation by transfecting mCherry-fused Casp-8tDED constructs into HeLa cells and performing confocal and fluorescence microscopy (Figure 4B, S4). Because some mutants were more defective in vitro when combined (Figure 4A), we used only two single mutants but mainly double and triple mutants. Consistent with the residual filament formation ability, the Y8A mutant alone failed to abolish filament formation. The F122E mutant, as well as the double and triple mutants tested, disrupted filament formation (Figure 4B). Collectively, these mutational data support the Casp-8 filament structure in vitro and in cells. The defective nature of type II and III interface mutants demonstrated that Casp-8 self-association is not limited to type I interaction-mediated DED chain formation, but require all three types of interactions for filament formation.
Casp-8tDED Filament Defective Mutants are Impaired in FasL-Induced Cell Death
We hypothesized that assembly of Casp-8 filaments at the DISC triggers Casp-8 activation and cell death. Therefore, filament-defective mutants of Casp-8 should have reduced ability to induce cell death at the DISC. To test this hypothesis, we chose Casp-8 mutants that were the most defective in filament formation based on the biochemical and cellular characterizations (Figure 4), and performed a FasL-induced cell death assay. WT and mutant Casp-8 were transfected into the Casp-8 deficient Jurkat cell line I9.2. Transfection of WT or mutant Casp-8 led to similar expression levels and did not induce significant cell death (Figure S5A). FasL stimulation at increasing concentrations progressively induced enhanced cell death (Figure 5A). In comparison with WT Casp-8, the filament-defective mutants reduced the number of dead cells, shown either to graded doses of FasL (Figure 5A) or at a single FasL concentration (Figure 5B). The F122E and K148D/R149E mutants, which were both completely defective in filament assays in vitro and in cells, did not cause any significant cell death in comparison with the non-transfected control or the catalytic mutant C360S. Other mutants showed reduced FasL-induced cell death despite the similar expression levels. The cellular data demonstrated the requirement for Casp-8 filament assembly in caspase activation and apoptosis at the DISC.
Figure 5. Filament Formation-Defective Mutants of Casp-8 Compromised Death Receptor- and Inflammasome-Mediated Signaling.

(A) Cell death in Casp-8 deficient Jurkat cell line I9.2C reconstituted with WT and mutant Casp-8 induced by isoleucine zipper-fused FasL (FasL-LZ).
(B) Histogram of cell death in I9.2C reconstituted with WT and mutant Casp-8 in the presence of 50 ng/ml Fas-LZ.
(C) Analysis of ASC-mediated Casp-8 speck formation using flow cytometry. All Casp-8-Myc constructs contain the C360S mutation in addition to the stated mutations. WT refers to Casp-8 C360S without any mutation in the tDED. Cells were stained with Myc and ASC antibodies and analyzed by flow cytometry. Cells with an elevated peak height to area ratio for Casp-8 show concentration of Casp-8 on the ASC speck (Sester et al., 2015) and are boxed.
(D) Casp-8 speck formation in cells transfected with the indicated Casp-8 plasmids, with or without ASC. Mean and range of duplicate transfections in a representative experiment are shown.
(E) ASC speck formation in cells transfected with ASC and the indicated Casp-8 plasmids. Mean and range of duplicate transfections in a representative experiment are shown.
See also Figure S5.
Casp-8tDED Filament Defective Mutants are Impaired in Recruitment to ASC Specks
We showed previously that the inflammasome adaptor ASC is able to recruit Casp-8 through a PYD/tDED interaction (Vajjhala et al., 2015). Inflammasome activation or ASC expression leads to formation of a single speck per cell (Hornung et al., 2009) and ASC interaction localizes Casp-8 into these specks. To address if the Casp-8 filament-forming residues are important for its interaction with ASC specks, we assessed the effects of Casp-8 mutations in ASC-mediated Casp-8 speck formation using flow cytometry (Sester et al., 2015) (Figure 5C). The C-terminally Myc-tagged WT and mutant Casp-8 were expressed in HEK293 cells with or without co-expression of ASC, followed by anti-Myc antibody and anti-ASC antibody staining, and analysis of fluorescence pulse shape to indicate protein distribution in the cell. The expression levels of WT and mutant Casp-8 were equivalent either in the presence or in the absence of ASC (Figure S5B). The expression levels of ASC were also equivalent under different conditions (Figure S5C). When the percentages of cells with Casp-8 or ASC specks were assessed by flow cytometry, the F122E single mutant and all double mutants significantly reduced the level of Casp-8 speck formation (Figure 5D). Examination of transfected cells by immunofluorescence microscopy showed that the flow cytometric data provided a good indication of the tendency of Casp-8 to colocalize with ASC (Figure S5D). These data suggest that recruitment of Casp-8 by ASC and its formation of filaments at the ASC speck (Vajjhala et al., 2015) use interfaces similar to its self-association in the DISC. The conclusion is consistent with the similar helical symmetry between the ASCPYD and the Casp-8tDED filaments (Lu and Wu, 2014). The Casp-8 specks were completely abolished in the absence of ASC (Figure 5D), indicating that ASC is required for Casp-8 clustering in the cell. Interestingly, the frequency of ASC specks was reliably increased in the presence of WT Casp-8 suggesting that overexpression of Casp-8 can reinforce ASC self-association. Mutants that were not recruited to ASC specks (Figure 5D) were unable to reinforce ASC speck formation (Figure 5E).
FADD-Mediated Unidirectional Casp-8 Polymerization Using Three Types of Interactions
FADDDED is responsible for recruitment of Casp-8 through the DED/tDED interaction. Since FADDDED is able to nucleate Casp-8tDED filament assembly, we hypothesized that FADDDED adopts a similar helical assembly as Casp-8tDED. We found that recombinant GFP-FADDDED showed a filamentous morphology similar to Casp-8tDED (Figure 6A). We generated a structure model for the FADDDED filament by superimposing the FADDDED structure (Eberstadt et al., 1998) onto DED1 and DED2 in the Casp-8tDED filament structure. This exercise produced FADD/FADD interactions that are similar in nature to Casp-8/Casp-8 interactions, with type I interactions mainly hydrophobic and type II and III interactions largely hydrophilic and electrostatic (Figure S6A-D), suggesting the validity of the FADD filament model.
Figure 6. Insights into DED/DED interactions among FADDDED, Casp-8tDED and cFLIPtDED.
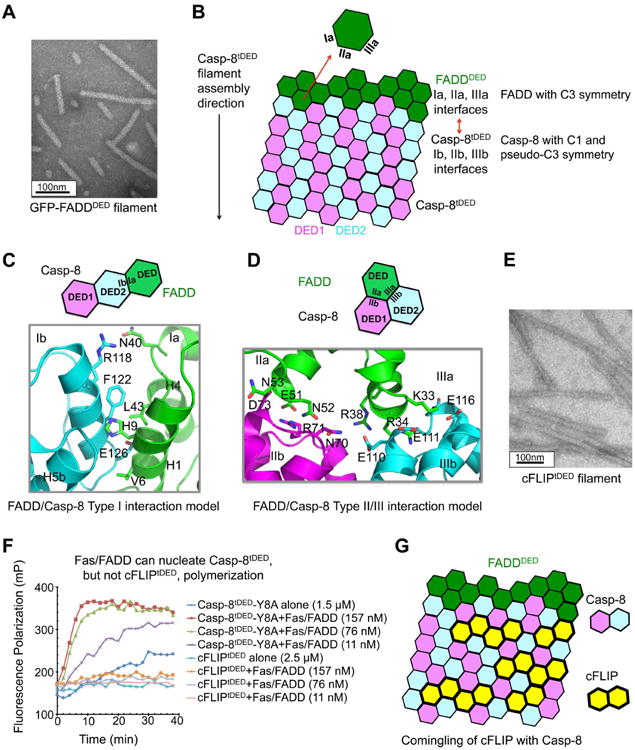
(A) An electron micrograph of GFP-FADDDED filament.
(B) Schematic diagram of FADDDED-nucleated Casp-8tDED filament formation.
(C) Predicted type I interface of the FADDDED/Casp-8tDED interaction, showing the essentially hydrophobic surfaces.
(D) Predicted type II and type III interfaces between FADDDED and Casp-8tDED, showing the charge complementarity.
(E) An electron micrograph of cFLIPtDED filament.
(F) The FasDD/FADD complex potentiated Casp-8tDED, but not cFLIP polymerization.
(G) Schematic diagram for the predicted comingling of cFLIP with Casp-8 in the FADDDED/Casp-8tDED complex.
See also Figure S6.
To elucidate how filamentous FADDDED may nucleate Casp-8 filament formation, we took into account that the Fas/FADD complex resides on one end of Casp-8 filaments in the ternary DISC complex (Figure 1E). Consistently, the type Ia, IIa and IIIa surfaces of FADD appear structurally compatible with the Casp-8tDED Ib, IIb and IIIb surfaces respectively (Figure 6B-D). While the type I interface is mostly hydrophobic (Figure 6C), the type II and III interactions are electrostatically complementary, such as in the interactions of E51 of FADD with R71 of Casp-8DED1 and of K33, R34 and R38 of FADD with E110, E111 and E116 of Casp-8DED2 (Figure 6D). These interactions are supported by previous mutagenesis studies in which mutations of FADD residues E51, K33 and R38 disrupted interaction with Casp-8tDED (Carrington et al., 2006). In contrast, the type IIb surface of FADD and the type IIa surface of Casp-8 are both positively charged with electrostatic repulsion, which unlikely interact with each other (Figure S6E). Therefore, both the structural data and the gold labeling result suggest a unidirectional FADD-mediated Casp-8 polymerization and signal propagation using all three types of interactions (Figure 6B).
In contrast to our model, a previous mutagenesis-based study suggested that Casp-8 preferentially uses its type Ia surface (H1/H4) to interact with FADD type Ib surface (H2/H5) to initiate the DED chain (Majkut et al., 2014). However, because mutations on the type Ia or type Ib surface also affect FADD or Casp-8 self-association, the disruptive phenotypes do not distinguish the FADD/Casp-8 interaction from self-associations. Given our model above, we hypothesized that the disruptive phenotype of Casp-8Y8G (Ia) and the non-disruptive phenotype of Casp-8F122A (Ib) in the earlier study (Majkut et al., 2014) were dependent on the mutant residues rather than mutation sites. We therefore used alternative mutant residues: caspase-8Y8A (Ia), which is likely less disruptive than Y8G, and caspase-8F122E (Ib), which is likely more defective than F122A. In co-immunoprecipitation assays, we found that indeed the F122E mutant was defective and the Y8A mutant intact in the interaction with FADD (Figure S6F), further demonstrating the significance of the filament structure.
cFLIP Comingles with Casp-8
cFLIP plays an important role in regulating Casp-8 recruitment and activation at the DISC, but the molecular mechanism remains unclear. Earlier studies have suggested that cFLIP directly interacts with FADD (Majkut et al., 2014) and inhibits Casp-8 activation by competing with Casp-8 for recruitment to FADD (Yang et al., 2005). However, recent studies showed that cFLIP alone only weakly interacted with FADD but assembled effectively into the DISC in a co-operative manner that is dependent on Casp-8, suggesting that DISC assembly is a hierarchical process from FADD to Casp-8, then to cFLIP (Hughes et al., 2016; Schleich et al., 2016).
We found that recombinant cFLIPtDED is filamentous in vitro (Figure 6E), and therefore used a cFLIPtDED polymerization assay to investigate if the Fas/FADD complex could directly interact with cFLIPtDED to nucleate its filament formation (Figure 1F). The assay used recombinant monomeric MBP-cFLIPtDED-Sumo tagged with the TAMRA fluorophore (Figure S6G), which formed filaments upon removal of the N-terminal MBP tag (Figure S6H). In agreement with hierarchical assembly (Hughes et al., 2016; Schleich et al., 2016), the Fas/FADD complex did not significantly enhance cFLIPtDED polymerization, but promoted Casp-8tDED polymerization under similar conditions (Figure 6F), supporting the weak interaction between FADD and cFLIP.
We generated a structure model of cFLIPtDED using the MC159 structure as template (Li et al., 2006; Yang et al., 2005) on the Swiss-Model server (Biasini et al., 2014). A structural analysis revealed that the different interaction surfaces of cFLIPtDED and Casp-8tDED are essentially similar (Figure S6I), suggesting that cFLIP may comingle with Casp-8 (Figure 6G). Therefore, incorporation of cFLIPS into the Casp-8 filament may reduce the local concentration of the Casp-8 caspase domain to inhibit its dimerization and auto-processing. For cFLIPL, its caspase-like domain is a preferred heterodimerization partner of the caspase domain of Casp-8, which leads to catalytic activation of Casp-8 (Yu et al., 2009). However, because the Casp-8/cFLIP heterodimer is not fully active but an attenuated species that processes only selected natural substrates (Pop et al., 2011), high cFLIP concentration leads to reduced Casp-8 activity. Therefore, the effect of cFLIPL on Casp-8 may be dependent on its expression level, with enhancement at a lower expression and inhibition at a higher expression.
MC159 Inhibits Casp-8tDED Filament Assembly by a Surprising Capping Mechanism
MC159 is one of the first vFLIPs identified to be involved in inhibiting apoptosis through interactions with both FADD and Casp-8 (Yang et al., 2005). In agreement, we found that MC159 inhibited the FasDD/FADD-mediated Casp-8tDED filament assembly, with an apparent Ki of 48.5 ± 1.4 nM (Figure 7A). We further showed that MC159 also inhibited Casp-8 polymerization nucleated by preformed Casp-8 filaments (Figure S7A), indicating that MC159 is able to directly interact with Casp-8 to block its filament formation. Previous in vitro reconstitution indicated direct interaction between FADD and MC159 (Yang et al., 2005).
Figure 7. Capping as the Inhibition Mechanism of Casp-8tDED Filament Formation by MC159.
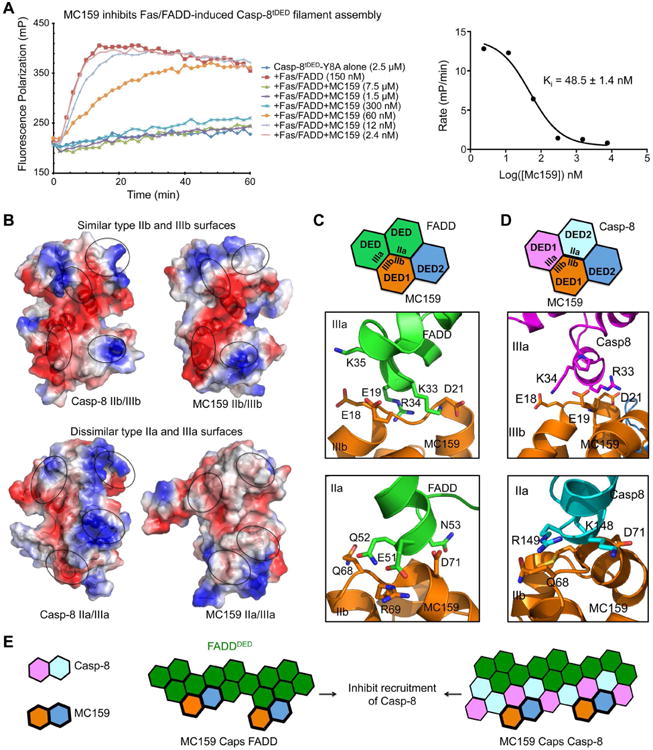
(A) Inhibition of FasDD/FADD-mediated Casp-8tDED polymerization by MC159. The apparent inhibitory constant (Ki), with fitting error, is shown.
(B) Comparison of Casp-8tDED surface with MC159. Type IIb and type IIIb interfaces of Casp-8tDED are similar to those of MC159, while type IIa and type IIIa interfaces of Casp-8tDED are dissimilar from those of MC159.
(C) Predicted type II and type III interfaces between FADDDED and MC159.
(D) Predicted type II and type III interfaces between Casp-8tDED and MC159.
(E) Schematic diagram illustrating the capping mechanism of MC159 for inhibition of FADDDED-mediated Casp-8tDED filament assembly.
See also Figure S7.
Although both cFLIP and MC159 can inhibit apoptosis, the biochemical behaviors of these two proteins are very different; cFLIP forms filament while MC159 is monomeric (Yang et al., 2005). Analysis of the MC159 surface in comparison with Casp-8tDED revealed that only the type IIb and type IIIb surfaces of both DED1 and DED2 are preserved for their interaction with Casp-8 and FADD (Figure 7B). Hence, MC159 is only likely to interact with filamentous FADD (Figure 7C) and Casp-8 (Figure 7D) via these interfaces. Because of the defective type IIa and IIIa interfaces (Figure 7B), the bound MC159 is unable to further recruit Casp-8, capping these filaments to inhibit polymerization and signaling (Figure 7E). Consistent with our model, mapping of previous mutational data on MC159 showed that the disruptive mutation sites are localized at the type IIb and IIIb surfaces, including E18, E19, D21 and R69, which disrupted recruitment to FADD when mutated to alanines (Yang et al., 2005) (Figure 7C-D, 2B). The unique mechanism of capping allows MC159 to inhibit FADD and Casp-8 polymerization at the DISC at a low stoichiometric ratio (Figure 7A).
Discussion
Quasi-Equivalence in DD Superfamily Assemblies
Almost a decade ago the first oligomeric structure in the DD superfamily, the PIDD/RAIDD DD complex, was determined by X-ray crystallography (Park et al., 2007). Since then, helical assembly in oligomerization of the DD superfamily complexes has been established from a plethora of crystallographic and EM studies on DD, CARD and PYD (Lin et al., 2010; Lu et al., 2016; Lu et al., 2014; Qiao et al., 2013; Wu et al., 2014). However, no structural information on DED assembly was available. With the Casp-8tDED structure presented here, oligomeric structures of all four types of domains in the DD superfamily are now represented, demonstrating the conserved helical assembly mechanism.
A striking feature of the Casp-8tDED filament structure is that its DED1 and DED2 domains are equivalent in the type II and III interactions despite the low sequence identity. Because DED1 and DED2 are rigidly linked to each other via the type Ia (DED2): type Ib (DED1) interaction, only one type I interaction, Ia (DED1): Ib (DED2), is available for the interaction between Casp-8tDED molecules. The quasi-equivalence of type II and III interactions is different from homotypic filament structures assembled from single DD superfamily domains, in which each of the three types of asymmetric interactions only has one interaction partner. Our structural analysis revealed that the ability for Casp-8tDED to accommodate different partners in the filament is at least partly due to the conservation of the electrostatic properties of the interfaces (Figure 2B, S3C).
The quasi-equivalence is also manifested in the interaction of Casp-8 with its binding partners of limited sequence identity, including FADDDED, ASCPYD, cFLIPtDED and MC159. In these cases, it appears that the nature of the interactions is largely preserved, but not the amino acid residues. This mechanism of preserving an interaction is frequently observed in protein/protein interactions. A classical example is represented by the interaction of human growth hormone (hGH) with both the hGH receptor and the human prolactin receptor, together with conformational adaptability (Somers et al., 1994). At the modest resolution of the Casp-8tDED filament structure, we do not yet know the precise local structural changes that may also be essential for achieving the quasi-equivalence. However, we may argue that the large interaction surfaces in these filamentous structures may tolerate amino acid changes more readily, enabling evolution of homo- and hetero-oligomerization. We predict that other tandem DD superfamily domains such as the tandem CARD in Nod2 may also use quasi-equivalence for its assembly (Fridh and Rittinger, 2012).
Activation and Inhibition of Casp-8 Filament Formation at the DISC
The studies presented here reveal a filamentous molecular template for understanding Casp-8 recruitment, oligomerization, activation and regulation at the DISC formed by the Fas receptor directly or by the cytoplasmic proteins RIP1 and TRADD (Figure S7B). Consistently, cytoplasmic filaments were observed upon overexpression of FADDDED and Casp-8tDED in cells (Siegel et al., 1998). The Casp-8 filament structure corrects the DED chain model proposed previously, which exclusively relies on the type I interaction, by illustrating the importance of type II and III interactions in FasL-induced apoptosis and in recruitment to ASC specks. The filamentous model is consistent with previous studies, in which both DED domains of Casp-8 were shown to be required for FADD recruitment and Casp-8 self-association (Siegel et al., 1998; Tsukumo and Yonehara, 1999). Similar to the DED chain model, the filamentous DED interactions explain the unequal and variable stoichiometry among the component proteins in the DISC, in particular with Casp-8 overstoichiometric to FADD (Dickens et al., 2012; Schleich et al., 2012). However, in contrast to the linear chain model, the filamentous structures also predict a cooperative assembly process that leads to a threshold response in death receptor activation.
The current data show that the DED filamentous scaffold is a versatile platform not only for self-association, but also for recruitment and regulation through interactions with other DEDs. Upon ligand-induced receptor activation, the upstream adaptor FADD nucleates Casp-8 filament via the interaction between DED and tDED, both in filamentous forms (Figure S7B). In this recruitment, an interaction surface of FADD that directly contacts Casp-8 is also required for self-association, which may have led to contradictory conclusions (Majkut et al., 2014). Our structural analysis is consistent with the observed hierarchy in DED interactions in which FADD first recruits Casp-8, and Casp-8 further recruits cFLIP (Hughes et al., 2016; Schleich et al., 2016). The predicted comingling of Casp-8 and cFLIPL in the same filament may facilitate the heterodimerization of their caspase domains to promote caspase activation when cFLIPL concentration is relatively low (Figure S7B). Because of the reduced and more restricted activity of the Casp-8/cFLIPL heterodimer (Pop et al., 2011), higher concentration of cFLIPL may suppress Casp-8 activation (Figure S7B). In contrast, cFLIPS is always inhibitory by decreasing the local concentration of the caspase domain of Casp-8 for dimerization and activation (Figure S7B).
Unlike cFLIP, which can form filaments, the vFLIP MC159 was shown to be a monomer (Li et al., 2006; Yang et al., 2005), and to interfere with Casp-8 polymerization. Our structural analysis indicated that MC159 uses its type IIb and IIIb surfaces to cap a FADD or Casp-8 filament, leading to robust inhibition of DISC assembly (Figure S7B). We predict that capping may be a general mechanism for interfering with filament formation at substoichiometric ratios by endogenous inhibitors in diverse immune pathways.
In the Casp-8tDED filament structure, the C-terminus of DED2 protrudes from the filament core, allowing the attached caspase domain to decorate the central filament and to be brought into proximity for dimerization and catalytic activation (Figure S7B). The local concentration of the caspase domain is calculated to be ∼1.5 mM given an extended linker size of ∼40 residues in Casp-8 between its tandem DED and the caspase domain. In cells, the lengths of the filaments are likely limited by the availability of the components. Additionally, crosslinking between multiple oligomerization domains, including the DDs of Fas, RIP1 and FADD, may coalesce the DISC into discrete puncta visible under light microscopy (Siegel et al., 2004). Fas-induced apoptosis may be triggered by as few as two trimers of FasL (Holler et al., 2003). The Casp-8 filament provides an elegant molecular platform that may ‘lock in’ the oligomerization state of Fas and initiate the apoptotic cascade at relatively low concentration or oligomerization state of the ligand.
Experimental Procedures
Casp-8tDED Filament, FADDDED Filament, and cFLIPtDED Filament Preparation
Casp-8tDED and FADDDED were expressed as His-GFP fusions and purified using Ni-NTA affinity and gel filtration chromatography. The void fractions contained the respective filament samples. cFLIPtDED was expressed as a His-MBP fusion and similarly purified. cFLIPtDED filaments were generated upon removal of His-MBP by the TEV protease.
Cryo-EM Data Collection and Image Processing
Cryo-EM data collection was performed on a Tecnai F20 electron microscope (FEI) operated at an acceleration voltage of 200 kV using a K2 Summit direct electron detection camera (Gatan) operated in super-resolution mode with dose-fractionation. A total of 388 images were used for analysis and processing. Helical averaging was performed using the iterative helical real space reconstruction (IHRSR) algorithm (Egelman, 2010).
Fluorescence Polarization Assay
His-MBP-Casp-8tDED-Sumo (Y8A) or His-MBP-cFLIP-Sumo was purified as a monomer and labeled with TAMRA through a sortase reaction. FP readings were taken on a SpectraMax M5e (Molecular Devices) using excitation and emission wavelengths of 561 nm and 585 nm.
Cell Death Assay Induced by Fas Ligand
Casp-8 deficient Jurkat cells were transfected with wild type and mutant Casp-8 constructs in the pcDNA3.1-YFP vector. YFP+ cells were treated with FasL fused to an isoleucine zipper (FasL-LZ) and analyzed for cell death via Annexin V and Live/Dead staining (Invitrogen).
Recruitment of Caspase-8 to ASC Specks
Co-expressed Casp-8 and ASC were immunostained and speck formation was assessed using changes in the ratio of fluorescence peak height to area.
Supplementary Material
Acknowledgments
We thank Drs. Narayanasami Sukumar, Kay Perry, Ryan Ferrao, Gunes Bozkurt, and Li Wang for their assistance in X-ray diffraction data collection at the Advanced Photon Source. The work was supported by funding from the National Institutes of Health (DP1-HD087988, R37-AI050872 and R01-AI045937 to HW, R01-EB001567 to EHE, and GM60635 to PAP) and the National Health and Medical Research Council of Australia (1050651, 1109738, and 1059729 to KJS).
Footnotes
Accession Numbers: The accession numbers for the data reported in this paper are 5JQE for the crystal structure of the Casp-8tDED F122G/L123G mutant, 5L08 for the cryo-EM structure of the Casp-8tDED filament, and EMD-8300 for the cryo-EM map of the Casp-8tDED filament.
Supplemental Information: Supplemental experimental procedures, seven figures and two tables can be found with this article online.
Author Contributions: T.-M.F., A.L., D.B.S., A.C.C. and P.R.V. performed the biochemical and cell biological experiments. Y.L., T.-M.F., E.H.E., Z.L., F.D. and P.A.P. collected diffraction and electron microscopy data, and determined the structures. H.W., R.M.S., and K.J.S. supervised the project. H.W. and T.-M.F. analyzed the structures and wrote the manuscript with contributions from other authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bagneris C, Ageichik AV, Cronin N, Wallace B, Collins M, Boshoff C, Waksman G, Barrett T. Crystal structure of a vFlip-IKKgamma complex: insights into viral activation of the IKK signalosome. Mol Cell. 2008;30:620–631. doi: 10.1016/j.molcel.2008.04.029. [DOI] [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42:W252–258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, et al. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- Carrington PE, Sandu C, Wei Y, Hill JM, Morisawa G, Huang T, Gavathiotis E, Werner MH. The structure of FADD and its mode of interaction with procaspase-8. Mol Cell. 2006;22:599–610. doi: 10.1016/j.molcel.2006.04.018. [DOI] [PubMed] [Google Scholar]
- Dickens LS, Boyd RS, Jukes-Jones R, Hughes MA, Robinson GL, Fairall L, Schwabe JW, Cain K, Macfarlane M. A Death Effector Domain Chain DISC Model Reveals a Crucial Role for Caspase-8 Chain Assembly in Mediating Apoptotic Cell Death. Mol Cell. 2012;47:291–305. doi: 10.1016/j.molcel.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberstadt M, Huang B, Chen Z, Meadows RP, Ng SC, Zheng L, J LM, Fesik SW. NMR structure and mutagenesis of the FADD (Mort1) death-effector domain. Nature. 1998;392:941–945. doi: 10.1038/31972. [DOI] [PubMed] [Google Scholar]
- Egelman EH. Reconstruction of helical filaments and tubes. Methods Enzymol. 2010;482:167–183. doi: 10.1016/S0076-6879(10)82006-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito D, Sankar A, Morgner N, Robinson CV, Rittinger K, Driscoll PC. Solution NMR investigation of the CD95/FADD homotypic death domain complex suggests lack of engagement of the CD95 C terminus. Structure. 2010;18:1378–1390. doi: 10.1016/j.str.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, Hupe M, Cain K, Macfarlane M, Hacker G, Leverkus M. cIAPs Block Ripoptosome Formation, a RIP1/Caspase-8 Containing Intracellular Cell Death Complex Differentially Regulated by cFLIP Isoforms. Mol Cell. 2011;43:449–463. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrao R, Wu H. Helical assembly in the death domain (DD) superfamily. Curr Opin Struct Biol. 2012;22:241–247. doi: 10.1016/j.sbi.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French LE, Tschopp J. Protein-based therapeutic approaches targeting death receptors. Cell Death Differ. 2003;10:117–123. doi: 10.1038/sj.cdd.4401185. [DOI] [PubMed] [Google Scholar]
- Fridh V, Rittinger K. The tandem CARDs of NOD2: intramolecular interactions and recognition of RIP2. PLoS One. 2012;7:e34375. doi: 10.1371/journal.pone.0034375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DR. Overview: apoptotic signaling pathways in the immune system. Immunol Rev. 2003;193:5–9. doi: 10.1034/j.1600-065x.2003.00045.x. [DOI] [PubMed] [Google Scholar]
- Holler N, Tardivel A, Kovacsovics-Bankowski M, Hertig S, Gaide O, Martinon F, Tinel A, Deperthes D, Calderara S, Schulthess T, et al. Two adjacent trimeric Fas ligands are required for Fas signaling and formation of a death-inducing signaling complex. Mol Cell Biol. 2003;23:1428–1440. doi: 10.1128/MCB.23.4.1428-1440.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, Latz E, Fitzgerald KA. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458:514–518. doi: 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MA, Powley IR, Jukes-Jones R, Horn S, Feoktistova M, Fairall L, Schwabe JW, Leverkus M, Cain K, MacFarlane M. Co-operative and Hierarchical Binding of c-FLIP and Caspase-8: A Unified Model Defines How c-FLIP Isoforms Differentially Control Cell Fate. Mol Cell. 2016;61:834–849. doi: 10.1016/j.molcel.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang TH, Zheng C, Li J, Richards C, Hsiao YS, Walz T, Wu H, Park HH. Structural study of the RIPoptosome core reveals a helical assembly for kinase recruitment. Biochemistry. 2014;53:5424–5431. doi: 10.1021/bi500585u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. Embo J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FY, Jeffrey PD, Yu JW, Shi Y. Crystal structure of a viral FLIP: insights into FLIP-mediated inhibition of death receptor signaling. J Biol Chem. 2006;281:2960–2968. doi: 10.1074/jbc.M511074200. [DOI] [PubMed] [Google Scholar]
- Lin SC, Lo YC, Wu H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- Lu A, Li Y, Schmidt FI, Yin Q, Chen S, Fu TM, Tong AB, Ploegh HL, Mao Y, Wu H. Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat Struct Mol Biol. 2016;23:416–425. doi: 10.1038/nsmb.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schroder GF, Fitzgerald KA, Wu H, Egelman EH. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193–1206. doi: 10.1016/j.cell.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Wu H. Structural mechanisms of inflammasome assembly. The FEBS journal. 2014 doi: 10.1111/febs.13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majkut J, Sgobba M, Holohan C, Crawford N, Logan AE, Kerr E, Higgins CA, Redmond KL, Riley JS, Stasik I, et al. Differential affinity of FLIP and procaspase 8 for FADD's DED binding surfaces regulates DISC assembly. Nature communications. 2014;5:3350. doi: 10.1038/ncomms4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- Park HH, Logette E, Raunser S, Cuenin S, Walz T, Tschopp J, Wu H. Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell. 2007;128:533–546. doi: 10.1016/j.cell.2007.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop C, Oberst A, Drag M, Van Raam BJ, Riedl SJ, Green DR, Salvesen GS. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J. 2011;433:447–457. doi: 10.1042/BJ20101738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Q, Yang C, Zheng C, Fontan L, David L, Yu X, Bracken C, Rosen M, Melnick A, Egelman EH, et al. Structural Architecture of the CARMA1/Bcl10/MALT1 Signalosome: Nucleation-Induced Filamentous Assembly. Mol Cell. 2013;51:766–779. doi: 10.1016/j.molcel.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, Roberts TL, Schroder K, Vince JE, Hill JM, et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013;20:1149–1160. doi: 10.1038/cdd.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleich K, Buchbinder JH, Pietkiewicz S, Kahne T, Warnken U, Ozturk S, Schnolzer M, Naumann M, Krammer PH, Lavrik IN. Molecular architecture of the DED chains at the DISC: regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain. Cell Death Differ. 2016;23:681–694. doi: 10.1038/cdd.2015.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleich K, Warnken U, Fricker N, Ozturk S, Richter P, Kammerer K, Schnolzer M, Krammer PH, Lavrik IN. Stoichiometry of the CD95 Death-Inducing Signaling Complex: Experimental and Modeling Evidence for a Death Effector Domain Chain Model. Mol Cell. 2012;47:306–319. doi: 10.1016/j.molcel.2012.05.006. [DOI] [PubMed] [Google Scholar]
- Sester DP, Thygesen SJ, Sagulenko V, Vajjhala PR, Cridland JA, Vitak N, Chen KW, Osborne GW, Schroder K, Stacey KJ. A novel flow cytometric method to assess inflammasome formation. J Immunol. 2015;194:455–462. doi: 10.4049/jimmunol.1401110. [DOI] [PubMed] [Google Scholar]
- Shen C, Yue H, Pei J, Guo X, Wang T, Quan JM. Crystal structure of the death effector domains of caspase-8. Biochem Biophys Res Commun. 2015;463:297–302. doi: 10.1016/j.bbrc.2015.05.054. [DOI] [PubMed] [Google Scholar]
- Siegel RM, Martin DA, Zheng L, Ng SY, Bertin J, Cohen J, Lenardo MJ. Death-effector filaments: novel cytoplasmic structures that recruit caspases and trigger apoptosis. J Cell Biol. 1998;141:1243–1253. doi: 10.1083/jcb.141.5.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RM, Muppidi JR, Sarker M, Lobito A, Jen M, Martin D, Straus SE, Lenardo MJ. SPOTS: signaling protein oligomeric transduction structures are early mediators of death receptor-induced apoptosis at the plasma membrane. J Cell Biol. 2004;167:735–744. doi: 10.1083/jcb.200406101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somers W, Ultsch M, De Vos AM, Kossiakoff AA. The X-ray structure of a growth hormone-prolactin receptor complex. Nature. 1994;372:478–481. doi: 10.1038/372478a0. [DOI] [PubMed] [Google Scholar]
- Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. 2009;30:180–192. doi: 10.1016/j.immuni.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenev T, Bianchi K, Darding M, Broemer M, Langlais C, Wallberg F, Zachariou A, Lopez J, Macfarlane M, Cain K, et al. The Ripoptosome, a Signaling Platform that Assembles in Response to Genotoxic Stress and Loss of IAPs. Mol Cell. 2011;43:432–448. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Tschopp J, Irmler M, Thome M. Inhibition of fas death signals by FLIPs. Curr Opin Immunol. 1998;10:552–558. doi: 10.1016/s0952-7915(98)80223-9. [DOI] [PubMed] [Google Scholar]
- Tsukumo SI, Yonehara S. Requirement of cooperative functions of two repeated death effector domains in caspase-8 and in MC159 for induction and inhibition of apoptosis, respectively. Genes Cells. 1999;4:541–549. doi: 10.1046/j.1365-2443.1999.00280.x. [DOI] [PubMed] [Google Scholar]
- Vajjhala PR, Lu A, Brown DL, Pang SW, Sagulenko V, Sester DP, Cridland SO, Hill JM, Schroder K, Stow JL, et al. The inflammasome adaptor ASC induces procaspase-8 death effector domain filaments. J Biol Chem. 2015 doi: 10.1074/jbc.M115.687731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Yang JK, Kabaleeswaran V, Rice AJ, Cruz AC, Park AY, Yin Q, Damko E, Jang SB, Raunser S, et al. The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat Struct Mol Biol. 2010;17:1324–1329. doi: 10.1038/nsmb.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Peisley A, Tetrault D, Li Z, Egelman EH, Magor KE, Walz T, Penczek PA, Hur S. Molecular Imprinting as a Signal-Activation Mechanism of the Viral RNA Sensor RIG-I. Mol Cell. 2014;55:511–523. doi: 10.1016/j.molcel.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Hymowitz SG. Structure and function of tumor necrosis factor (TNF) at the cell surface. In: Bradshaw RA, Dennis EA, editors. Handbook of cell signaling. Oxford: Academic Press; 2009. pp. 265–275. [Google Scholar]
- Yang JK, Wang L, Zheng L, Wan F, Ahmed M, Lenardo MJ, Wu H. Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol Cell. 2005;20:939–949. doi: 10.1016/j.molcel.2005.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JW, Jeffrey PD, Shi Y. Mechanism of procaspase-8 activation by c-FLIPL. Proc Natl Acad Sci U S A. 2009;106:8169–8174. doi: 10.1073/pnas.0812453106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.