

Abstract
Kidney disease affects millions of people worldwide and it is now widely accepted that many pathological processes may persist after AKI that can cause the progression to CKD. Tubulointerstitial fibrosis manifests soon after injury and while many cellular and molecular components of kidney fibrosis have been discovered, largely in animal models, new therapeutic strategies are still desperately needed. The renal endothelium has emerged as important in progression of fibrosis through regulation of hypoxia, inflammation, and cellular crosstalk. This review aims to highlight our current understanding of the role of the endothelium in interstitial fibrosis and to identify potential therapeutic targets.
Keywords: endothelium, kidney, myofibroblasts, pericytes, chronic kidney disease
Introduction
Progressive kidney fibrosis, a hallmark of chronic kidney disease, can be a consequence of maladaptive repair processes after acute kidney injury (AKI). The severity and type of injury dictates the ability for the kidney to recover function or progressively decline and activates the fibrotic pathway as part of the wound healing response. The clinical and pathophysiological phases of acute kidney injury (AKI) begin with early events that are initiated by endothelial dysfunction, microvascular injury, leukocyte adhesion, inflammation, oxidative injury, and tubular apoptosis/necrosis[1]. Renewal of endothelial function heralds the recovery phase of AKI and is necessary for tubule regeneration. Thus, the endothelium contributes to both the initiation of and recovery from AKI and preserving endothelial function could be key to attenuating progressive kidney damage and prevention of fibrosis.
Insults to the kidney trigger the endothelium to switch from a quiescent to an activated state which can lead to a cascade of pathways that contribute to fibrosis[2,3] (Figure 1) in mice and humans. Endothelial cell dysfunction can lead to increased vascular permeability, activation of complement, inflammation, vasoconstriction and capillary rarefaction. Microvascular congestion by leukocytes and RBCs, vasoconstriction, and capillary rarefaction can all result in tissue hypoxia. Hypoxia directly induces myofibroblast differentiation or may indirectly induce a pro-fibrotic response in tubule epithelial cells and/or leukocytes, all leading to progression of fibrosis[2]. Endothelial dysfunction (vasoconstriction and leukocyte adhesion molecule expression) is associated with renal tubule cell stress[4], which can lead to production of profibrotic factors. Additionally, endothelial dysfunction may direct pericytes (PC) or other interstitial cells to differentiate into myofibroblasts[5,6], promoting fibrosis. Conversely, accumulating evidence suggests that endothelial dysfunction can lead to pericyte activation[5], resulting in vascular instability and microvascular rarefaction. This review will summarize recent developments on the role of the endothelium in regulating vascular functions in health and in interstitial fibrosis following kidney injury.
Figure 1.
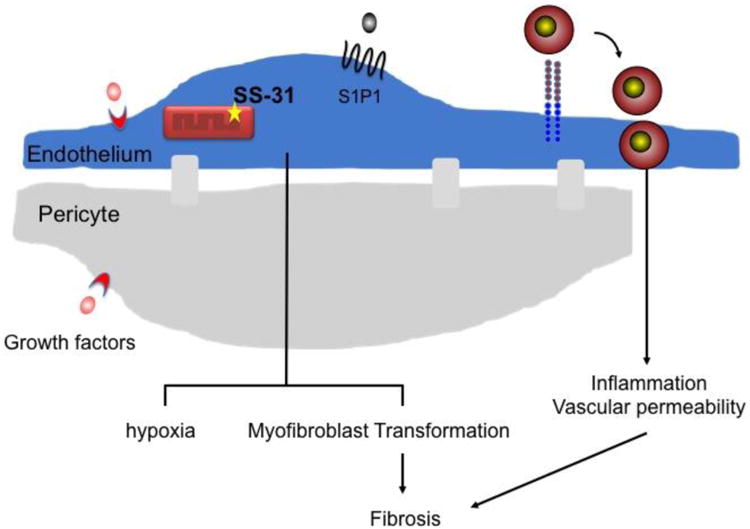
Potential targets to prevent endothelial dysfunction and renal interstitial fibrosis. During progressive kidney fibrosis peritubular capillaries recruit inflammatory cells, have increased vascular permeability and vasoconstriction, and undergo rarefaction. Endothelial dysfunction may also lead to pericyte detachment, proliferation, migration and transformation to myofibroblasts. Strategies to enhance endothelial function by promoting survival or angiogenesis (VEGF121), preserving mitochondrial function (SS31), preventing leukocyte adhesion molecule expression and enhancing vascular barrier function (S1P1, IGF1R) may reduce fibrosis.
Role of endothelium in pathophysiology of renal fibrosis
Hypoxia
Early studies highlighted the role of hypoxia in murine kidney fibrosis models. Vasoconstriction, in part regulated by the vascular endothelium, in response to injury can result in localized or general hypoxia in kidneys of humans and mice[3]. These hemodynamic disturbances, along with microvascular plugging of RBCs and leukocytes, lead to a concept described as the ‘no-reflow’ phenomenon in renal ischemia reperfusion injury (IRI), whereby areas of the peritubular capillary network exhibit a loss of blood flow during the reperfusion phase, further contributing to regional reductions in blood flow and presumed hypoxia[7]. Although total renal blood flow can return to baseline levels within 5 hrs of IRI, vascular reactiveness is altered for at least one week[8] suggesting that endothelial dysfunction may persist well after IRI and contribute to long term maladaptive processes.
Vascular rarefaction can also lead to hypoxia. Human kidney biopsies from patients with chronic tubule interstitial disease had a reduction in peritubular capillary density that was associated with tubule atrophy and increased interstitial fibrosis compared to normal kidneys[7]. Mouse models of interstitial fibrosis observed vascular rarefaction occurring as early as day 1 after severe IRI and 3 days after unilateral ureteral obstruction (UUO)[9]. Microvascular loss is apparent at 4 weeks after IRI[10], but can apparently return to sham levels 6 to 9 weeks after IRI when renal function was restored[11]. The extent and timing of vascular rarefaction likely depends on the type and severity of fibrosis model. The renal endothelium lacks regenerative capacity early after IRI, unlike the tubule epithelium, potentially due to a low level of angiogenic signals and endothelial proliferation[2,12]. Approaches to preserve the renal endothelium such as infusion of VEGF in pigs after established renovascular disease, prevented microvascular rarefaction, attenuated fibrosis & normalized renal blood flow and kidney function compared to controls[13], suggesting that targeting therapeutic interventions to promote endothelial function may be an effective strategy to reduce fibrosis.
Tissue inflammation
Endothelial cells are the gatekeepers to tissue inflammation. Hypoxia/reoxygenation, mechanical and cytokine stimuli can induce leukocyte adhesion molecule expression on the endothelium promoting rolling, adhesion and transmigration of immune cells, and in particular innate immune cells such as neutrophils & monocytes. The precise role of the endothelium in specifically modulating adaptive immune cell infiltration and its consequence on fibrosis is unknown. Monocytes can differentiate to tissue macrophages which can promote inflammation, tubule repair and/or fibrosis[14]. Due to their cellular plasticity, the macrophage phenotype may switch depending on environmental milieu. Alternatively, resident macrophages may also have a role tissue recovery and progression of fibrosis. While many studies focus on the secreted factors by macrophages that can drive tissue fibrosis, less is understood about intracellular interactions within the renal microenvironment.
Crosstalk to pericytes
Emerging evidence demonstrates an important role for endothelial and pericyte crosstalk[15] in renal vascular homeostasis and disease. Under normal homeostatic conditions, a basement membrane separates the endothelium and pericytes, but through direct contact or indirect paracrine factors, communication is maintained between these two cell types[5]. In disease states, endothelial dysfunction may lead to pericyte detachment, proliferation and migration and vasculature lacking pericytes is unstable, prone to leakiness and can lead to microvascular rarefaction[16]. Activation of pericytes can also contribute to microvascular loss[16]. The specific cell-cell interactions and paracrine signaling pathways between endothelial cells and pericytes important for homeostasis as well as during disease states in the renal microvasculature have yet to be defined. Blockade of prosurvival factors such as VEGFR2 or PDGFRβ has been shown to reduce pericyte proliferation and differentiation, microvascular rarefaction, inflammation and fibrosis[16]. The apparent discrepancy between these findings and that administration of VEGF protects against fibrosis[13] may be due to the complex signaling pathway including three VEGF receptors and the regulated expression of four distinct VEGF genes and multiple splice variants. Therefore, these studies cannot simply be explained by a binary ligand receptor interaction.
Other kidney cells have also been shown to regulate capillary function. Atrophic epithelial cells can release profibrotic signals which may disrupt endothelial/pericyte interactions in peritubular capillaries, leading to disintegration microvascular rarefaction[17]. Endothelial crosstalk to other cells in the microenvironment, such as epithelial cells and immune cells, is largely unknown.
EndoMT
Endothelial to mesenchymal transition has gained some interest in kidney fibrosis yet; mouse models used for understanding the role of the endothelium in renal fibrosis can be non-specific, confounding results. The renal endothelium is thought to constitute a minor population to the myofibroblast pool[6]. However, the VE-cadherin-Cre (Cdh5-Cre) mouse used for lineage tracing endothelial cells in this study has been shown to have reporter expression in a subset of hematopoietic cells throughout embryonic and adult organs[18]. Many studies exploring the role of EndoMT in renal fibrosis have used the constitutive Tie2-Cre for linage tracing or deletion of specific factors in endothelial cells. For example, the Tie2-Cre reporter colocalized with SMA after IRI[12] and inactivation of endothelial TGFβR signaling reduced fibrosis[19]. However, in the constitutive Tie2-Cre mouse, Tie2 has been shown to be non-specific for the endothelium and is expressed early in hematopoietic cell development[20,21] and potentially on pericyte precursors[22]. The lack of a truly specific marker for the endothelium for lineage tracing makes interpreting these studies difficult[23,24].
Targets to enhance endothelial recovery
Molecular mechanisms of endothelial recovery after AKI and prevention of fibrosis remain largely unknown in murine models of fibrosis. Insulin-like growth factor-1 receptor, a receptor important for cellular proliferation and differentiation, has been shown to be necessary for prevention of UUO induced fibrosis by preserving endothelial barrier integrity[25]. Administration of SS-31, a compound that preserves mitochondrial integrity, at the time of IRI protected against early endothelial injury, microvascular congestion and kidney injury. At 4 weeks after IRI, there was reduced capillary rarefaction and fibrosis in mice treated with SS-31[10] suggesting that preservation of endothelial mitochondrial integrity can prevent kidney injury and fibrosis. Recently, we used an inducible Tie2-Cre to delete sphingosine 1-phosphate 1 receptor (S1P1), a sphingolipid receptor that is essential for vascular function, in endothelial cells in a temporal manner during IRI[26]. With this approach, Cre activation is specific to the endothelium and is not activated in WBC or other kidney cells[20,26]. When the deletion of endothelial S1P1 was delayed either in the injury phase or recovery phase after a mild renal IRI, mice were unable to recover kidney function and exhibited severe injury and fibrosis within 9 days and lasted to at least 14 days. There was marked renal inflammation including neutrophils and macrophages. Furthermore, we demonstrated that S1P1 has a novel role in directly regulating endothelial leukocyte adhesion molecule expression, and suppression of these molecules is crucial to dampening inflammation and allowing for renal recovery. As FTY720, a FDA approved drug that activates S1P1, prevents endothelial dysfunction and kidney fibrosis in rats[27], these data suggest that activation of the S1P1 signaling pathway after AKI may help to prevent fibrosis.
Conclusion
The endothelium plays a critical role in maintaining normal vascular homeostasis and following injury endothelium contributes to repair processes leading to renal recovery. However following severe injury, maladaptive repair ensues leading to progressive fibrosis and loss of renal function. Angiogenic growth factors, activating the sphingolipid receptor signaling pathway or preserving mitochondrial function may be attractive physiological and pathophysiological endothelial processes targetable for therapeutic intervention.
Acknowledgments
Research conducted for this publication was supported by the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health (NIH) under award numbers R01DK085259, R01DK062324 (M.D.O), T32DK 72922 (H.M.P), and by the American Society of Nephrology Ben Lipps Fellowship Program (H.M.P).
References
- 1.Sutton TA, Fisher CJ, Molitoris BA. Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney Int. 2002;62:1539–1549. doi: 10.1046/j.1523-1755.2002.00631.x. [DOI] [PubMed] [Google Scholar]
- 2.Molitoris BA. Therapeutic translation in acute kidney injury: The epithelial/endothelial axis. J Clin Invest. 2014;124:2355–2363. doi: 10.1172/JCI72269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after aki leading to accelerated kidney ageing and ckd. Nat Rev Nephrol. 2015;11:264–276. doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu L, Tiwari MM, Messer KJ, Holthoff JH, Gokden N, Brock RW, Mayeux PR. Peritubular capillary dysfunction and renal tubular epithelial cell stress following lipopolysaccharide administration in mice. Am J Physiol Renal Physiol. 2007;292:F261–268. doi: 10.1152/ajprenal.00263.2006. [DOI] [PubMed] [Google Scholar]
- 5.Kramann R, Humphreys BD. Kidney pericytes: Roles in regeneration and fibrosis. Semin Nephrol. 2014;34:374–383. doi: 10.1016/j.semnephrol.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19:1047–1053. doi: 10.1038/nm.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guerrot D, Dussaule JC, Kavvadas P, Boffa JJ, Chadjichristos CE, Chatziantoniou C. Progression of renal fibrosis: The underestimated role of endothelial alterations. Fibrogenesis Tissue Repair. 2012;5:S15. doi: 10.1186/1755-1536-5-S1-S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Conger JD, Robinette JB, Hammond WS. Differences in vascular reactivity in models of ischemic acute renal failure. Kidney Int. 1991;39:1087–1097. doi: 10.1038/ki.1991.138. [DOI] [PubMed] [Google Scholar]
- 9.Ehling J, Babickova J, Gremse F, Klinkhammer BM, Baetke S, Knuechel R, Kiessling F, Floege J, Lammers T, Boor P. Quantitative micro-computed tomography imaging of vascular dysfunction in progressive kidney diseases. J Am Soc Nephrol. 2016;27:520–532. doi: 10.1681/ASN.2015020204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu S, Soong Y, Seshan SV, Szeto HH. Novel cardiolipin therapeutic protects endothelial mitochondria during renal ischemia and mitigates microvascular rarefaction, inflammation, and fibrosis. Am J Physiol Renal Physiol. 2014;306:F970–980. doi: 10.1152/ajprenal.00697.2013. [DOI] [PubMed] [Google Scholar]
- 11.Khairoun M, van der Pol P, de Vries DK, Lievers E, Schlagwein N, de Boer HC, Bajema IM, Rotmans JI, van Zonneveld AJ, Rabelink TJ, van Kooten C, Reinders ME. Renal ischemia-reperfusion induces a dysbalance of angiopoietins, accompanied by proliferation of pericytes and fibrosis. Am J Physiol Renal Physiol. 2013;305:F901–910. doi: 10.1152/ajprenal.00542.2012. [DOI] [PubMed] [Google Scholar]
- 12.Basile DP, Friedrich JL, Spahic J, Knipe N, Mang H, Leonard EC, Changizi-Ashtiyani S, Bacallao RL, Molitoris BA, Sutton TA. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol. 2011;300:F721–733. doi: 10.1152/ajprenal.00546.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chade AR, Tullos NA, Harvey TW, Mahdi F, Bidwell GL., 3rd Renal therapeutic angiogenesis using a bioengineered polymer-stabilized vascular endothelial growth factor construct. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2015040346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao Q, Harris DC, Wang Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology (Bethesda) 2015;30:183–194. doi: 10.1152/physiol.00046.2014. [DOI] [PubMed] [Google Scholar]
- 15.Campanholle G, Ligresti G, Gharib SA, Duffield JS. Cellular mechanisms of tissue fibrosis. 3. Novel mechanisms of kidney fibrosis. Am J Physiol Cell Physiol. 2013;304:C591–603. doi: 10.1152/ajpcell.00414.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kida Y, Duffield JS. Pivotal role of pericytes in kidney fibrosis. Clin Exp Pharmacol Physiol. 2011;38:467–473. doi: 10.1111/j.1440-1681.2011.05531.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Venkatachalam MA, Weinberg JM, Kriz W, Bidani AK. Failed tubule recovery, aki-ckd transition, and kidney disease progression. J Am Soc Nephrol. 2015;26:1765–1776. doi: 10.1681/ASN.2015010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alva JA, Zovein AC, Monvoisin A, Murphy T, Salazar A, Harvey NL, Carmeliet P, Iruela-Arispe ML. Ve-cadherin-cre-recombinase transgenic mouse: A tool for lineage analysis and gene deletion in endothelial cells. Dev Dyn. 2006;235:759–767. doi: 10.1002/dvdy.20643. [DOI] [PubMed] [Google Scholar]
- 19.Xavier S, Vasko R, Matsumoto K, Zullo JA, Chen R, Maizel J, Chander PN, Goligorsky MS. Curtailing endothelial tgf-beta signaling is sufficient to reduce endothelial-mesenchymal transition and fibrosis in ckd. J Am Soc Nephrol. 2015;26:817–829. doi: 10.1681/ASN.2013101137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forde A, Constien R, Grone HJ, Hammerling G, Arnold B. Temporal cre-mediated recombination exclusively in endothelial cells using tie2 regulatory elements. Genesis. 2002;33:191–197. doi: 10.1002/gene.10117. [DOI] [PubMed] [Google Scholar]
- 21.Schlaeger TM, Mikkola HK, Gekas C, Helgadottir HB, Orkin SH. Tie2cre-mediated gene ablation defines the stem-cell leukemia gene (scl/tal1)-dependent window during hematopoietic stem-cell development. Blood. 2005;105:3871–3874. doi: 10.1182/blood-2004-11-4467. [DOI] [PubMed] [Google Scholar]
- 22.Lamagna C, Bergers G. The bone marrow constitutes a reservoir of pericyte progenitors. J Leukoc Biol. 2006;80:677–681. doi: 10.1189/jlb.0506309. [DOI] [PubMed] [Google Scholar]
- 23.Falke LL, Gholizadeh S, Goldschmeding R, Kok RJ, Nguyen TQ. Diverse origins of the myofibroblast-implications for kidney fibrosis. Nat Rev Nephrol. 2015;11:233–244. doi: 10.1038/nrneph.2014.246. [DOI] [PubMed] [Google Scholar]
- 24.Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest. 2014;124:2299–2306. doi: 10.1172/JCI72267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liang M, Woodard LE, Liang A, Luo J, Wilson MH, Mitch WE, Cheng J. Protective role of insulin-like growth factor-1 receptor in endothelial cells against unilateral ureteral obstruction-induced renal fibrosis. Am J Pathol. 2015;185:1234–1250. doi: 10.1016/j.ajpath.2015.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perry HM, Huang L, Ye H, Liu C, Sung SJ, Lynch KR, Rosin DL, Bajwa A, Okusa MD. Endothelial sphingosine 1 phosphate receptor 1 mediates protection and recovery from acute kidney injury. J Am Soc Nephrol. 2016 doi: 10.1681/ASN.2015080922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ni H, Chen J, Pan M, Zhang M, Zhang J, Chen P, Liu B. Fty720 prevents progression of renal fibrosis by inhibiting renal microvasculature endothelial dysfunction in a rat model of chronic kidney disease. J Mol Histol. 2013;44:693–703. doi: 10.1007/s10735-013-9521-8. [DOI] [PubMed] [Google Scholar]