

Abstract
Examples of clinal variation in phenotypes and genotypes across latitudinal transects have served as important models for understanding how spatially varying selection and demographic forces shape variation within species. Here, we examine the selective and demographic contributions to latitudinal variation through the largest comparative genomic study to date of Drosophila simulans and Drosophila melanogaster, with genomic sequence data from 382 individual fruit flies, collected across a spatial transect of 19 degrees latitude and at multiple time points over 2 years. Consistent with phenotypic studies, we find less clinal variation in D. simulans than D. melanogaster, particularly for the autosomes. Moreover, we find that clinally varying loci in D. simulans are less stable over multiple years than comparable clines in D. melanogaster. D. simulans shows a significantly weaker pattern of isolation by distance than D. melanogaster and we find evidence for a stronger contribution of migration to D. simulans population genetic structure. While population bottlenecks and migration can plausibly explain the differences in stability of clinal variation between the two species, we also observe a significant enrichment of shared clinal genes, suggesting that the selective forces associated with climate are acting on the same genes and phenotypes in D. simulans and D. melanogaster.
Keywords: comparative genomics, Drosophila, latitudinal cline, latitudinal variation, parallelism, population genomics
Introduction
Latitudinal transects have been studied across the tree of life, in a large number of bacteria, plant and animal species, revealing phenotypic and genetic clines (Feder & Bush 1989; Weber & Schmid 1998; Salgado & Pennings 2005; Fuhrman et al. 2008; Baumann & Conover 2011). A correlation between phenotypic variation and latitude is suggestive of local adaptation. For example, local adaptation to temperature is implicated in the correlation between decreased lifespan and latitude in ectotherms (Munch & Salinas 2009), and local adaptation to photoperiod is implicated in the correlation between flowering time and latitude in plants (Keller et al. 2011). However, neutral demographic processes also generate clinal variation. For example, ‘isolation by distance’, where gene flow is decreased between geographically distant populations, can produce patterns of variation similar to those resulting from local adaptation (Endler 1977). Strong patterns of clinal variation can also be generated by introgression between separate invading populations (Cruzan 2005) or range expansion of a single founding population (Excoffier et al. 2009). These demographic processes can be coincident with selective processes. Although disentangling selective and demographic scenarios is challenging, genomic data sets have the power to identify patterns associated either with selection or with demography. We perform a genomic study across two closely related Drosophila species, allowing us to elucidate general patterns that are shared between the species as well as refine our understanding of how the processes underlying clinal variation differ between these species.
The genus Drosophila represents a powerful system for the study of selection and demography. This group is composed of several species with broad distribution and represents Old and more recent New World colonizations. Drosophila melanogaster has been studied extensively in a latitudinal context. Several phenotypic traits and genetic loci vary with latitude in D. melanogaster (Vigue & Johnson 1973; Mettler et al. 1977; Voelker et al. 1977; Knibb et al. 1981; Oakeshott et al. 1982; Singh et al. 1982; David et al. 1985; Coyne & Beecham 1987; James et al. 1995; Munjal et al. 1997; Karan et al. 1998; Schmidt et al. 2000, 2005, 2008; Gockel et al. 2001; Mitrovski & Hoffmann 2001; Hoffmann et al. 2002; Sezgin et al. 2004; Pool & Aquadro 2007; Emerson et al. 2009; Paaby et al. 2010), and D. melanogaster latitudinal variation has been studied in a genomic context and on multiple continents (North America, Australia, Europe, Asia and Africa) (Turner et al. 2008; Kolaczkowski et al. 2011; Fabian et al. 2012; Bergland et al. 2015; Reinhardt et al. 2014). One advantage to using D. melanogaster for the study of adaptation to latitude is that it is a relatively recent colonizer of temperate climates (10 000–20 000 years since expansion out of central Africa; Lachaise et al. 1988; Li & Stephan 2006). Temperate-adapted characters such as cold tolerance and starvation resistance are more pronounced at higher latitudes in D. melanogaster, suggesting that clinal variation in D. melanogaster is a result of local adaptation to temperate climates (Karan et al. 1998; Hoffmann et al. 2002; Schmidt et al. 2008). Additionally, there is some parallelism in clinal allele frequency patterns along the North American and Australian latitudinal clines, suggesting that there has been convergent adaptation to latitude (Turner et al. 2008; Fabian et al. 2012; Reinhardt et al. 2014). The D. melanogaster latitudinal clines are also subject to confounding demographic effects. Both North American and Australian populations seem to be a result of admixture (either pre- or postcolonization) between European and African populations (Duchen et al. 2013; Bergland et al. 2015; Kao et al. 2015). Although the D. melanogaster latitudinal clines are robust and some do seem to result from local adaptation, demography complicates the inference of selection.
Comparative studies can help us understand general patterns of latitudinal variation. The sister species D. simulans and D. melanogaster (~3×106 years diverged; Hey & Kliman 1993) represent a powerful system for comparative study. These species are similar in their range, ecology and evolutionary history (Cariou 1987; Hey & Kliman 1993). They have experienced parallel expansions out of Africa, adaptation to temperate climates and development of human commensalism (David & Capy 1988; Lachaise et al. 1988; Lachaise & Silvain 2004). Unfortunately, the limited amount of research on clinal variation in D. simulans has made a large comparative study of latitudinal variation impossible.
While D. simulans exhibits clinal variation in some of the same traits as D. melanogaster (pigmentation: David et al. 1985; body size: Arthur et al. 2008), D. simulans also seems less temperate adapted (McKenzie & Parsons 1974; Gibert et al. 2004; Arthur et al. 2008). For example, D. simulans has less physiological tolerance to cold and starvation (reviewed in Hoffmann & Harshman 1999). Another key clinal trait in D. melanogaster is a reproductive diapause, which is hypothesized to be important for survival through the high-latitude winters (Saunders et al. 1989; Schmidt & Conde 2006). Reproductive diapause has not been observed in D. simulans. Certain phenotypes that are clinal in both species vary less with latitude in D. simulans than in D. melanogaster (starvation: Arthur et al. 2008; desiccation: McKenzie & Parsons 1974), supporting the hypothesis of a more shallow cline in D. simulans (reviewed in Gibert et al. 2004).
While local adaptation could explain the above patterns, a shallow cline in D. simulans could also result from demographic patterns. Contemporary demographic patterns such as seasonal bottlenecks and migration may contribute to clinal variation. Although the true demographic patterns in D. simulans are not known, D. simulans has been hypothesized to experience strong bottlenecks and/or employ migratory behaviour in response to seasonal fluctuations. This is supported by the temporal abundance patterns found along latitudinal clines in Europe and North America (Boulétreau-Merle et al. 2003; Fleury et al. 2004; Schmidt 2011; Behrman et al. 2015). Specifically, D. simulans tends to be in greater relative abundance in the more equatorial populations and does not appear at the higher latitudes until later in the year than D. melanogaster. Additionally, in temperate North America, there are distinct differences between D. melanogaster and D. simulans in the population age structure across seasonal time that are also indicative of different overwintering strategies (Behrman et al. 2015). In D. melanogaster, the earliest observed spring populations have a uniformly young age distribution, shifting to a heterogeneous age distribution over time. This pattern is consistent with populations that overwinter locally. In D. simulans the earliest observed postwinter populations are already age heterogeneous, which is more consistent with annual recolonization from a refugia (either local or more distant) than with in situ overwintering. The D. simulans relative abundance and age distribution patterns can be explained by either (i) annual extirpation and recolonization of high-latitude populations, (ii) in situ overwintering and maintenance of a small resident population or (iii) both a strong annual bottleneck and subsequent input of migrants with the maintenance of a small resident population. Each of these scenarios would also contribute to a shallow cline.
Genomic analyses of latitudinal variation have been performed in D. melanogaster; however, no such studies exist for D. simulans. Genomic data sets are critical to understanding general patterns of clinal variation. With genomic data, we can statistically differentiate subtle patterns, such as the enrichment of functional genic classes and parallelism in clinal variants between species. Here, we present a multiyear, multiseason, genomewide analysis of population differentiation and latitudinal variation in D. simulans and D. melanogaster. We directly compare the amount of clinal variation in D. simulans and D. melanogaster using these genomic data and confirm that, in line with phenotypic observations, D. simulans has less clinal variation than D. melanogaster. We find evidence for a strong contribution of annual variation to D. simulans population genetic structure, which is not found in D. melanogaster. The strong, stable cline in D. melanogaster is a stark contrast to the weak cline seen across D. simulans populations, where we see greater evidence of processes that increase differentiation from year-to-year, such as migration and bottlenecks. We also observe signatures of spatially varying selection in D. melanogaster and to a lesser extent in D. simulans, and evidence for convergent evolution of clinal variation across genes.
Materials and methods
Sequence data
D. simulans
We sampled individuals from four D. simulans populations along the East Coast of North America, spanning 19 degrees latitude (Table S1, Supporting information). From north to south, the population and year of collection are as follows: Maine 2011 (ME), Pennsylvania 2011 (PA), Virginia 2010 (VA) and Florida 2011 (FL). Three separate samples of the PA population were taken, one each in August, September and November (named PA8.2011, PA9.2011 and PA11.2011, respectively). Populations were sampled by direct aspiration of flies from substrates and by collection with banana and yeast baited traps. We extracted DNA from a total of 267 female flies (an average of ~50 files per sample) using Favorgen 96-well genomic DNA extraction kits and quantified the DNA with a Picogreen fluorescence assay. Moleculo (now Illumina TruSeq) performed the per-individual library preparation and sequenced paired-end 100-bp reads on an Illumina HiSeq 2000. Depth of sequencing coverage per individual varied from 0.01× to 5×. We aligned reads to the D. simulans v2 reference genome (Hu et al. 2013) with BWA version 0.6.2 aln and sampe functions (default parameters; Li et al. 2009). We performed PCR duplicate removal with SAMTOOLS version 0.1.19 (Li et al. 2009) and indel realignment with GATK version 2.4 (McKenna et al. 2010). A total of ~3.2 M single nucleotide polymorphisms (SNPs) were called with GATK Unified Genotyper version 2.4 using all reads combined (DePristo et al. 2011). Genotype calls were made per individual for each SNP (ploidy 2). For each individual and SNP, we randomly chose a single chromosome of the diploid genotype for use in the final analysis, to avoid bias from individuals with higher coverage. SNPs were then filtered for ~2.5 M nucleotides of repetitive DNA identified using REPEATMASKER (Smit et al. 2013). We also filtered out high coverage sites (upper 95th quantile) and low coverage sites (<5× per collection site). For consistency with the D. melanogaster data, we filtered out sites within 5 bp of an indel, with low minor allele frequency (MAF) sites (mean MAF <10% across the four collection sites) and that were non-bi-allelic. Allele frequencies for each population were calculated relative to the reference genome. The filtered data set had a mean coverage of 20× per population, with 2.2 M SNPs on autosomes and 0.3 M SNPs on the X chromosome. For functional analysis, we used a D. simulans cDNA-guided genome annotation (Rogers et al. 2014) and the SNP functional annotator SNPEFF V4.0 (Cingolani et al. 2012).
We also sequenced two 2010 temporal samples (July and September) from the Pennsylvania population (named PA7.2010 and PA9.2010, respectively). These population samples consisted of male files sequenced using pooled population sequencing (pool-seq), where individuals from each sample were pooled prior to DNA extraction and sequencing. We extracted DNA from these two samples using a lithium chloride precipitation. Sequencing library construction followed the protocol described in Bergland et al. (2014). The samples were sequenced with paired-end 100-bp reads on an Illumina HiSeq 2000. The effective number of chromosomes (NC) represented in the pooled samples was calculated as
| (1) |
where N is the number of chromosomes in the pool, and R is the read depth at that site (Kolaczkowski et al. 2011; Feder et al. 2012; Bergland et al. 2014). This adjusts for the additional error introduced by sampling of the pool at the time of sequencing. Sequencing reads mapped to autosomes were down-sampled to match the NC of the X chromosome.
D. melanogaster
We compared D simulans data to published D. melanogaster data from a study conducted by Bergland et al. (2014). Three of the four D. melanogaster collection sites (FL, PA, ME) were the same as the D. simulans collection sites. The fourth D. simulans site (VA) was imperfectly matched to a D. melanogaster Georgia (GA) collection site (Table S1, Supporting information). We used two D. melanogaster temporal samples of the PA site (November 2009 and November 2010). The other sites were sampled once each; FL in 2010, GA in 2008 and ME in 2009. Bergland et al. (2014) produced sequence data by pooling males files within each population and sequencing on an Illumina HiSeq 2000. We mapped the raw reads to the D. melanogaster genome version 5.5 using BWA version 0.7.9 aln and sampe algorithms, with default parameters (Li & Durbin 2009). Reads mapping to autosomes were down-sampled to match the NC of the X chromosome for each population. Allele frequency was calculated relative to the reference allele for each SNP used in Bergland et al. (2014) (~600 K SNPs). SNP calling in Bergland et al. (2014) differed from the D. simulans SNP calling. The data in Bergland et al. (2014) were exclusively pool-seq data, for which SNPs were called using the program CRISP (Bansal 2010). Additional filtering also took place, notably, the exclusion of SNPs not also identified in the Drosophila Genetic Reference Panel (DGRP). The differences in SNP calling and filtering, along with real differences in genetic diversity between the two species, account for the smaller number of SNPs in the D. melanogaster data set.
Pool-seq error model
In this study, we compare pooled D. melanogaster population samples with (primarily) nonpooled D. simulans population samples. Pool-seq is known to have inherent errors in allele frequency estimation; therefore, we must take care to model this variance appropriately (Kofler et al. 2011; Zhu et al. 2012; Lynch et al. 2014). This is particularly important for our analysis of the relative proportion of clinal variation in D. simulans and D. melanogaster. As all of the D. melanogaster samples are pooled, these samples inherently have an additional source of error that is not accounted for, resulting in an overestimate of the sample size. As clinal patterns are expected to be more pronounced in D. melanogaster, a perceived increase in clinal variation in D. melanogaster could be attributed to the pool-seq variance. To arrive at a conservative estimate of clinal variation in D. melanogaster a liberal estimate of pool-seq error should be used.
Two methods for accounting for extra variance in pool-seq data are (i) modifying the statistical tests used (e.g. modification of the null expectation, as in Bastide et al. 2013) and (ii) translating the additional variance into an effective sample size. We chose the latter, using our comparable barcoded data set to assess the additional pool-seq error. To model the pool-seq error, we compared the level of genetic differentiation among barcoded temporal samples (D. simulans PA8.2011, PA9.2011, PA11.2011) with differentiation between pooled temporal samples (D. simulans PA7.2010, PA9.2010), with the assumption that within-population samples should have similar amounts of month-to-month variation from one year to the next. This assumption is reasonable, as we observe this to be the case (see below) for populations within a few months of each other. Note that this may not be the case for certain months, particularly for those during or directly following a winter bottleneck. For this analysis, we use the proportion of SNPs found to be at significantly different allele frequencies as a measure of genetic differentiation (Fisher’s exact test for each SNP). Only SNPs with a total of 40 chromosomes between the two samples being tested were used to ensure equal power between data sets. The range of chromosomes per population varied (PA8.2011: 8-35; PA9.2011: 8-36; PA11.2011: 4-32; PA7.2010: 18-22; PA9.2010: 22-18). The Fisher’s exact test provides a test of the deviation from panmixia (i.e. variation above binomial sampling error) with a standard expectation of a uniform P-value distribution and is robust to small and unequally distributed sample sizes. Panmixia is rejected if there is enrichment of differentiated SNPs above the expectation. For pool-seq data, we do not expect a uniform P-value distribution under panmixia for two reasons: (i) to account for the two levels of sampling (chromosomes and reads), we use a single effective sample size (NC), which is close to but not exactly the same as correctly using the convolution of two binomials, and (ii) the average error in allele frequency estimation for pool-seq data may be greater than binomial, even with the effective NC calculation. We use Fisher’s exact test on pooled and non-pooled data to estimate this second error component.
We first tested the differentiation among the three barcoded PA 2011 samples. We found that each of the three comparisons had similar levels of differentiation (between 0.99% and 1.05% of SNPs differentiated at P < 0.01, an average of 0.01% over expected; Fig. S1, Supporting information), representing near-uniform P-value distributions (the null distribution). In contrast, the pooled temporal samples (PA7.2010 and PA9.2010) showed an enrichment of differentiated SNPs (1.26% of SNPs differentiated at P < 0.01). This is consistent with additional sampling error being introduced in the process of pooled DNA extraction, amplification, sequencing and mapping, resulting in an overestimate of the effective number of chromosomes sampled.
To determine how much additional variance is introduced by pool-seq, above what is accounted for by the NC(N, R) correction already implemented, we tested two models of pooled error. We used the data from all three barcoded PA temporal comparisons to perform a linear regression of differentiation with increasing NC, providing the barcoded null model. We then found the additional variance component, which we call ε, that results in the best fit of the pooled PA comparison to the barcoded null (lowest sum of square deviations from the null). The first model tested fits an ε that is independent of R:
| (2) |
The second model tested fits an ε that is inversely proportional to R (greater error at lower read depth):
| (3) |
We found Model 2 (with the R dependence) to be a better fit to the data than Model 1, with a best-fit value of ε to be 0.1 (Fig. S5, Supporting information). Using this error model, we can calculate a more conservative NC, which we use for the calculation of NC for all pooled samples (D. simulans PA7.2010 and PA9.2010 samples; all D. melanogaster samples). Applying this NC(N, R, ε) correction to the pooled D. simulans PA7.2010 and PA9.2010 samples, we find a slight depletion of significantly differentiated SNPs compared to the barcoded samples (0.84% at P < 0.01, compared with 1.01%). This indicates that our correction for pooled-error results in a conservative estimate of the effective number of chromosomes in a pooled sample.
Use of this correction for pooled error also decreases the average coverage per population. However, even with the use of our pool-seq error correction, our pool-seq libraries are still more efficient in estimating population allele frequency than our barcoded libraries (per raw sequencing read). For example, from 898 M raw barcoded reads, we retrieved a total of 115× coverage across all populations, which is an average of 8.1 M reads per 1× coverage. This is compared to 2.7 M and 4.3 M reads per 1× coverage for the D. simulans PA7.2010 and PA9.2010 pool-seq libraries, respectively. In summary, our two D. simulans pool-seq libraries were 39–53% more efficient in population allele frequency estimation per raw sequence read than our barcoded libraries. This increased pool-seq efficiency may be particularly pronounced in our study, as our barcoded libraries had high heterogeneity in coverage across individuals.
Measures of genetic variation, genetic differentiation and isolation by distance
We calculated two measures of within-population genetic variation – mean expected heterozygosity (H) and Watterson’s theta (θS). For these analyses, we considered only sites covered by exactly 20 chromosomes in a given population, to avoid any biases resulting from differences in coverage among populations. Mean heterozygosity was calculated as
| (4) |
where N is the number of sites (polymorphic and monomorphic) and p is the allele frequency of each site. θS was measured as the proportion of polymorphic SNPs, divided by the sample size correction:
| (5) |
where S is the proportion of SNPs in the genome, and n is the number of chromosomes (i.e. 20).
We measured between-population genetic differentiation with the FST statistic (Weir & Cockerham 1984, equations 1:4). FST calculations were performed for each pairwise population comparison, for each SNP. As sample size affects the results of the FST statistic, we consider only SNPs with a total depth of coverage of 40–44 chromosomes between the two populations, with a minimum of 5 per population. In D. simulans, the maximum number of chromosomes per population ranged from 36–39 (FL: 39; VA: 39; PA8.2011: 37; PA9.2011: 39; PA11.2011: 36; ME: 37; PA7.2010: 39; PA9.2010: 39). In D. melanogaster, the maximum ranged from 31–39 (FL: 31; GA: 39; PA8.2011: 39; PA.2009: 39; PA.2010: 31; ME: 39). As the variance in the pool-seq allele frequency estimates is accounted for by the measure of effective number of chromosomes, NC(N; R; ε), no additional pool-seq correction is necessary for FST or genetic variation calculations.
We assessed isolation by distance with a linear regression of FST with geographic distance between populations (degrees latitude). We incorporated into a multiple linear regression model the effect of comparison with the Maine population (vs. comparison between two non-Maine populations) and within-year (vs. between-year) comparison. The final regression model is of the form:
| (6) |
where yi is the pairwise FST, d is the distance (degrees latitude) between two populations, m is whether or not one of the two populations of the comparison is Maine, y is between vs. within-year comparison, and εi is the gaussian error at the ith SNP.
Measures of clinal variation
To identify clinal SNPs, we used a generalized linear model (conducted in R version 3.1.0; R Core Team 2014) of allele frequency and population latitude, using a binomial error model and weights proportional to the effective number of chromosomes at each site (NC):
| (7) |
where yi is the allele frequencies at the ith SNP, and εi is the binomial error given the NC at the ith SNP. This type of regression is particularly appropriate for the analysis of clinal variation of allele frequencies, as it takes into account precision (number of chromosomes sampled per population) and the curve-linear behaviour at low allele frequencies. For each species, we used five population measurements sampled from the four populations – one sample from each population, with an additional year’s sample for PA (for D. simulans, we used PA7.2010 and PA8.2011). Each year of Pennsylvania samples was treated as a separate datapoint in the regression analysis, with a single timepoint for each year.
The average NC across the populations used in the clinal regression varied little from chromosome to chromosome, ranging from 21.2–21.4 in D. melanogaster and 20.9–21.8 in D. simulans. There was no significant difference between the two species in mean NC (t-test P = 0.19) or total N summed over all five populations (t-test P =0.13) (Fig. S2, Supporting information). This equality of sample sizes is important because it allows us to compare the two data sets without confounding differences in power.
We identified two sets of clinal SNPs based on the results of the clinal regressions – SNPs that were statistically significant at P < 0.01, and SNPs that were statistically significant at false discovery rate (FDR) of Q < 0.2. FDR Q values represent the proportion of false positives in a set of tests and were calculated with the R package qvalue (Storey 2015). We use the P < 0.01 set to estimate the relative proportion of clinal loci in D. melanogaster and D. simulans, allowing us account for the number of false positives due to multiple testing (using the null expectation) in a way that does not skew the false-negative rates. The proportion of clinal loci (SNPs) is calculated as:
| (8) |
where L is the number of SNPs tested, obsP<0.01 is the observed number of tests with P < 0.01, and expP<0.01 is the expected number of tests with P < 0.01 under the null expectation (L·0.01). For the remainder of the analyses (i.e. clinal consistency, functional genic classes, shared clinal genes), we use FDR Q-values, ensuring equal proportions of false positives in the D. melanogaster and D. simulans data sets.
To test the consistency of clinal patterns of allele frequency across years, we measured how well the regression coefficient from one year predicts the directionality in a second year. Allele frequency measures from three D. simulans sites from 2011 and two from 2010 were available. We performed a logistic regression across the three 2011 sites (FL, PA2011, ME) and asked whether the same trend of either increasing or decreasing frequency with latitude was observed in 2010. Specifically, we asked if the sign of the regression coefficient agreed with the sign of the difference between the 2010 populations (VA, PA2010). If there was agreement, these SNPs were deemed to be ‘consistently clinal’. If SNPs truly are clinal from year to year, it is expected that the proportion of SNPs found to be consistently clinal will increase with the stringency of the regression test (lower Q-value). We then performed a similar analysis in D. melanogaster, comparing the regression of the three 2008/2009 sites (GA2008, PA2009, ME2009) with two 2010 sites (FL2010, PA2010). As the inclusion of sites from two different years in the regression might bias towards identifying sites that truly are persistently clinal, thereby increasing the amount of clinal consistency detected, we compared this analysis with a mixed-year analysis of D. simulans. For this analysis, we performed a regression of D. simulans VA2010, PA2010 and ME2011, compared with the difference between the FL2011 and PA2011 sites. This provided a comparison that was liberal to finding clinal consistency in D. simulans. Results from the D. simulans mixed-year analysis were not significantly different from the single-year analysis (within two standard deviation), with the exception of chromosome 2L, for which the mixed-year analysis shows a decrease in clinal consistency (Fig. S3, Supporting information).
Enrichment tests
To test for enrichment of genic categories and of polymorphisms shared between D. melanogaster and D. simulans in sets of clinal SNPs (Q < 0.2), we compared our data sets with 100 bootstrap control data sets matched for mean allele frequency across the populations (by 20th quantile bin), inversion status (within the same inversion or outside inversions, applicable to D. melanogaster only; by 7th quantile bin), chromosome and effective sample size NC (by 10th quantile bin). The sizes of matching bins were chosen to result in the most well-matched controls that were also independent of one another. Genic categories for each species were identified with SNPeff (Cingolani et al. 2012), except for short introns. We used the set of D. melanogaster short introns identified in (Lawrie et al. 2013) and identified short introns in D. simulans as those <68 bp in the annotation by Rogers et al. (2014). We used the same D. melanogaster inversion breakpoints as in (Corbett-Detig & Hartl 2012).
We tested for an enrichment of genes identified as clinal in both D. melanogaster and D. simulans. We identified a gene as clinal if it had at least one clinal genic SNP (i.e. in the CDS, UTR or intronic regions). We measured the per cent of shared clinal genes as the overlap of D. simulans clinal genes with D. melanogaster clinal genes (contains at least one SNP with Q < 0.2). This was performed for five sets of D. simulans clinal genes, ranging in stringency from Q < 0.5 to Q < 0.1. For each set of D. simulans clinal genes, we produced 100 control sets of D. simulans genes matched for gene length (by 10th quantile bin) and SNP density (by 10th quantile bin) and measured the proportion of control genes shared with D. melanogaster clinal genes. Genes were omitted if <85 unique control genes could be identified. The distributions of gene length and SNP density for the clinal compared with the control gene sets overlapped well, and the majority (87%) of control genes were unique across permutations (Fig. S4, Supporting information).
Results
D. simulans SNPs across space and time
Here, we study D. simulans population genetic variation using genomic sequence data from 382 individual fruit flies (267 individually barcoded and 115 in pooled samples). Samples represented a spatial transect of four populations over 19 degrees latitude and a temporal transect of multiple time points over the course of two years (Table S1, Supporting information). We identified 2.5×106 bi-allelic D. simulans single nucleotide polymorphisms (SNPs) across the four major autosomal chromosome arms and the X chromosome (see Methods for filtering parameters). We utilized a matched D. melanogaster data set of pooled population sequence data (~6×105 SNPs; Bergland et al. 2014) to compare patterns of within, between, interannual and latitudinal population genetic variation. For all pool-seq samples, we applied a stringent pool-seq error correction that accounted for finite sampling and additional pool-seq variance (see Methods), allowing us to confidently compare the D. melanogaster data set with the D. simulans data set.
Larger proportion of clinal variants in D. melanogaster than D. simulans
We found a larger proportion of latitudinally clinal variants in D. melanogaster (3.7%) than in D. simulans (2.5%) (P < 0.01; Fig. 1D). The difference in the proportion of clinal variants was even greater when we considered only autosomal SNPs (4.3% in D. melanogaster compared with 2.1% in D. simulans; Fig. 2). As major chromosomal inversions in D. melanogaster show clinal patterns in frequency (Mettler et al. 1977), we asked whether inversions account for the difference between species. We found an elevated proportion of clinal SNPs in D. melanogaster inversions; however, D. melanogaster had a higher proportion of clinal SNPs than D. simulans in noninverted regions as well (Table S2, Supporting information). Similarly, although we did see an enrichment of clinal SNPs in low-recombination regions for D. melanogaster, the proportion of clinal SNPs outside low-recombination regions was still greater for D. melanogaster than D. simulans (Table S2, Supporting information).
Fig. 1.
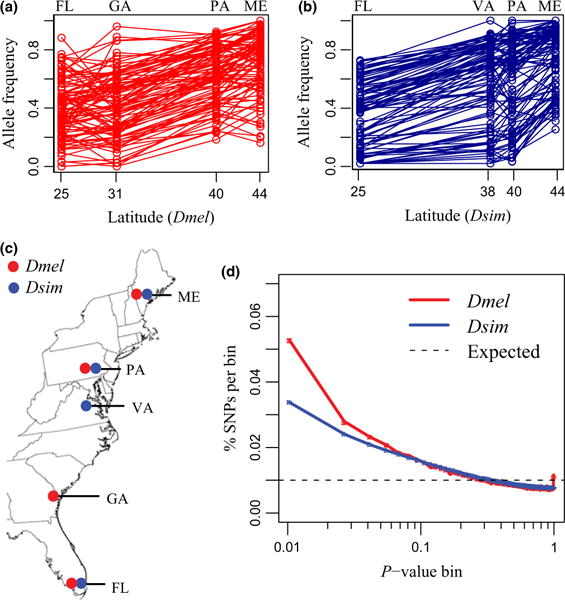
Clinal genetic variation with latitude. (a, b): Allele frequency trajectories for clinal SNPs (P < 0.01, sample of 100). Allele frequencies are polarized such that FL < ME. (c) Distribution of populations used to assess clinal variation. (d) P-value distributions from logistic regressions of allele frequency with latitude (bins of 0.01). Error bars are two standard error (not visible).
Fig. 2.
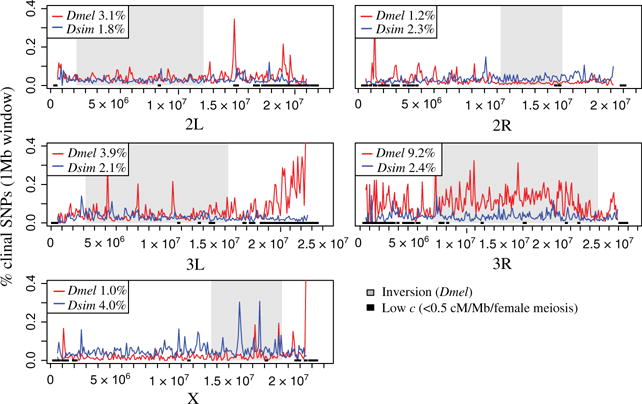
The distribution of clinal SNPs across the genome. The mean proportion of clinal SNPs (P < 0.01) per 1Mb window is plotted across the Drosophila melanogaster genome. Shaded areas represent the D. melanogaster major inversions. Black along the x-axis represents low-recombination rate regions (<0.5 cM/Mb/female meiosis, 100-kb bins). The proportion of SNPs clinal on each chromosome is listed in the legends.
We found substantial variation in clinality among chromosomes. The most striking pattern in D. melanogaster was the strong enrichment of clinal variants on chromosome 3R (9% clinal; Fig. 2). In D. melanogaster, much of the 3R chromosome is covered by three large cosmopolitan inversions. These inversions, particularly In(3R)P, have previously been found to be strongly clinal (Mettler et al. 1977; Kapun et al. 2014). On the X chromosome D. melanogaster and D. simulans had the opposite patterns of clinal variation. D. melanogaster had less clinal variation on the X chromosome (1% clinal) than any of the autosomes, whereas D. simulans had more clinal variation on the X chromosome (4% clinal) than any of the autosomes. Lower levels of clinal variation on the D. melanogaster X chromosome have been observed in previous studies (David & Capy 1988; Fabian et al. 2012; Kolaczkowski et al. 2011).
We asked whether the increased amount of clinal variation observed in D. melanogaster could be explained by greater D. melanogaster population structure. We looked at the effect of population structure by comparing genomewide mean pairwise FST. First, we noticed that on average (across all SNPs) D. simulans had a greater mean FST than D. melanogaster, indicating that a net increase in population structure was not driving the increased proportion of clinal variants in D. melanogaster. To look at the effect population structure had on the magnitude of clinal variation, we asked how mean FST scaled with the clinal effect size β (regression coefficient). We found that D. melanogaster had a stronger relationship between FST and β than D. simulans (Fig. S5, Supporting information), indicating that in D. melanogaster more of the observed population structure was due to clinal genetic differentiation.
Consistency of clinal variants from year to year
To assess the stability of clinal variation over time, we measured how well the clinal regression coefficient in 1 year predicted the allele frequency directionality in a second year. To ensure equal power and noise for the D. melanogaster and D. simulans analyses, we used false discovery rate (FDR) corrected Q-value significance thresholds for the clinal regressions and down-sampled the number of SNPs to the same number in each species and chromosome. We found evidence for clinal consistency from year to year in both species, with the proportion of clinal consistency increasing with Q-value stringency to 67% and 54% for D. melanogaster and D. simulans, respectively (at clinal Q < 0.3; Fig. 3; all chromosomes). Note that the Q-values are generally higher in this analysis than in the full clinal regression, as we use three populations instead of five. We found that D. melanogaster had significantly greater clinal consistency from year to year than D. simulans for each chromosome (Fisher’s exact test P<10−14) except the X chromosome (P = 0.3).
Fig. 3.
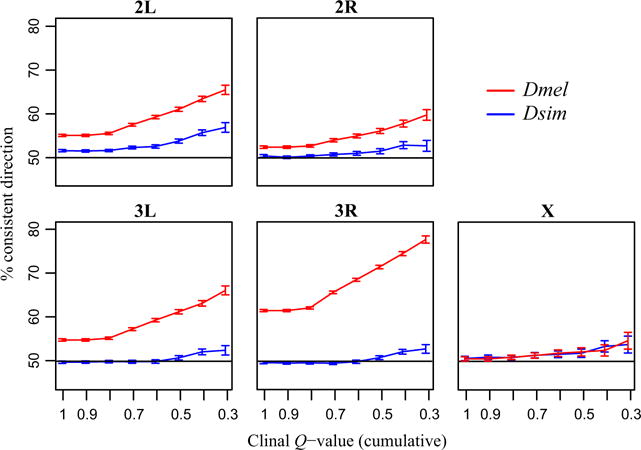
Consistency of clinal variation across years. The proportion of SNPs for which the clinal regression coefficient from 1 year predicts the directionality in a second year is plotted for sets of clinal SNPs of increasing clinal stringency (decreasing Q-value). Error bars are two standard error.
Selection and parallelism in clinal variants
If clinal SNPs have phenotypic effects that are under spatially varying selection, we expect functional sites to be over-represented in the sets of clinal SNPs. Our expectation is that intergenic regions, short introns and synonymous sites are less likely to be functional than UTR’s, nonsynonymous sites and long introns. We used a constant FDR (Q < 0.2) and number of SNPs per species (25 134 autosomal and 805 X chromosome SNPs) to ensure equal noise and power for the D. melanogaster and D. simulans analyses. For the set of SNPs clinal in D. melanogaster autosomes, we found a significant enrichment of all genic classes (UTR’s, long intron, synonymous coding and nonsynonymous coding) except short introns and found a depletion of intergenic regions, compared with 100 bootstrap control data sets matched for chromosome, mean minor allele frequency, sample size, recombination rate and inversion status (see Methods; Fig. 4A). Additionally, we found a marginal increase in the proportion of nonsynonymous SNPs compared with synonymous SNPs (P = 0.1). Conversely, the D. melanogaster X chromosome was enriched for intergenic SNPs and depleted for long introns and nonsynonymous SNPs (Fig. S6, Supporting information). The set of D. simulans clinal SNPs showed a marginal enrichment (P < 0.1) of 5′UTR SNPs and a marginal depletion of intergenic SNPs (autosomes; Fig. 4B).
Fig. 4.
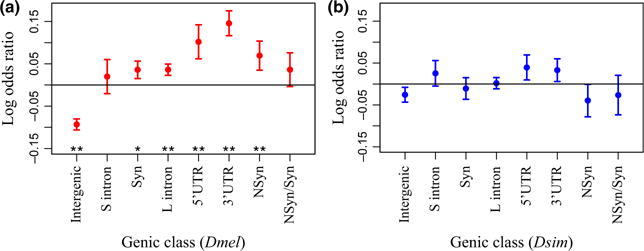
Enrichment of clinal autosomal SNPs (Q < 0.2, down sampled to 25134 SNPs) in each functional genic class. Plotted is the log of the odds ratio of the proportion of each genic class in the set of clinal SNPs compared with 100 matched controls. Error bars are one standard deviation. Bootstrap P-value *P ≤ 0.05; **P ≤ 0.01.
If selection is acting similarly on both species, we might find evidence of convergent evolution of clinal variants. We asked whether there was an enrichment for SNPs or genes that are clinal in both D. simulans and D. melanogaster. We found no significant enrichment for shared clinal SNPs (61 shared clinal polymorphisms of 32 136 shared polymorphisms total). However, we did observe an enrichment of shared clinal genes (Fig. 5). We compared the proportion of shared clinal genes with the proportion for 100 bootstrap control sets of genes, matched for D. simulans gene length and SNP density (see Methods). Of the genes with at least one clinal SNP (Q < 0.2; 5559 D. simulans genes and 5556 D. melanogaster genes), 56% were clinal in both species, compared to a mean of 45% across the bootstrap replicates (P = 0.01). This enrichment became even more pronounced at more stringent D. simulans clinal regression thresholds (for D. simulans clinal regression Q < 0.1, observed: 65%, control: 46%; Fig. 5). We did not find the shared clinal genes to be enriched in SNPs that were also clinally consistent (Fisher’s exact test; P > 0.3 for both species).
Fig. 5.
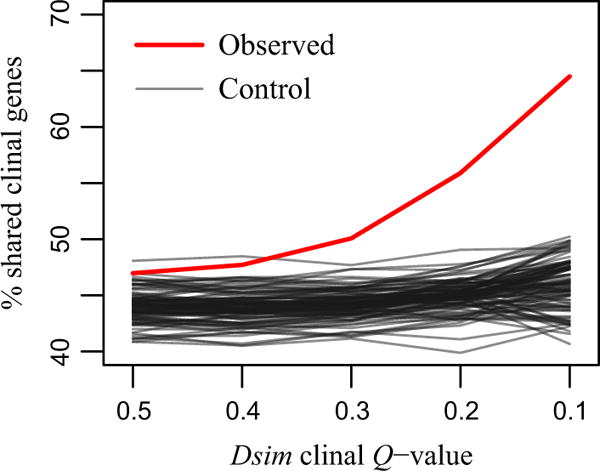
Per cent overlap of Drosophila simulans clinal genes with Drosophila melanogaster clinal genes (Q < 0.2), over increasing stringency of D. simulans clinal regression.
We next queried the list of 3342 shared clinal genes for its overlap with a set of 13 genes previously found to be clinal in D. melanogaster. To arrive at a set of putatively clinal genes, we gathered genes from targeted studies of clinal variation (rather than genomic scans). The result was 13 genes with strong support in the literature and was comprised of the seven metabolism genes Pgm (Verrelli & Eanes 2001; Sezgin et al. 2004), G6pd (Oakeshott et al. 1983), Gpdh (Oakeshott et al. 1982), UGP (Sezgin et al. 2004), Treh (Sezgin et al. 2004), Pgd (Oakeshott et al. 1983), and Hex-C (Duvernell & Eanes 2000) and the six nonmetabolism genes sgg (Rand et al. 2010), mth (Schmidt et al. 2000; Duvernell et al. 2003), cpo (Schmidt et al. 2008), per (Costa et al. 1992), Adh (Vigue & Johnson 1973; Berry & Kreitman 1993) and InR (Paaby et al. 2010). All except one of these genes (mth) were analysed in both species, leaving a final set of 12 genes. Of these 12 genes, 10 were clinal in both species. The two genes that were not found to be clinal in both species were Pgd and Hex-C.
We also compared our results to a recent study of gene expression in D. melanogaster and D. simulans low- (Panama) and high- (Maine) latitude populations (Zhao et al. 2015). For each population, gene expression was measured at 21°C and 29°C. Zhao and colleagues identified sets of 76 and 106 genes with latitude-specific expression in both species, at 21°C and 29°C, respectively (Zhao et al. 2015, Table S8, Supporting information). We compared the intersection of these data sets and our shared clinal genes data set with the intersection for 100 bootstrap control data sets matched for D. simulans gene length and SNP density (see Methods). We found only a marginal (P = 0.1) enrichment of latitude-specific genes at 29°C, and no enrichment of latitude-specific genes at 21°C, in our set of shared clinal genes. Zhao and colleagues also identified sets of genes with differential expression between temperatures (21°C and 29°C) in both species- 375 genes in the Maine populations and 861 in the Panama populations (Table S10, Supporting information, Zhao et al.). Also controlling for gene length and SNP density, we did find an enrichment of temperature-responsive genes in our set of shared clinal genes; however, this was only true for the Panama populations (P = 0.02) and not the Maine populations (P = 0.18).
Population genetic patterns in space
Visual inspection of frequency trajectories along the cline showed a more monotonic increase in allele frequency with latitude in D. melanogaster than D. simulans (Fig. 1). To further investigate this, we asked whether genetic differentiation between populations increased monotonically with physical distance between populations, a pattern know as ’isolation by distance’. We found that D. simulans had a weaker pattern of isolation by distance than D. melanogaster (Fig. 6). While in D. melanogaster the regression of genetic differentiation (FST) and physical distance between populations (degrees latitude) was significant (P<10−5, R2 = 0:94), in D. simulans this was only significant (P = 0.001) in a regression model that included Maine (ME) as an explanatory variable (Table S3, Supporting information). In D. melanogaster, there was no effect of ME comparison. The significant effect of ME comparison in D. simulans was due to the disproportionate amount of divergence of ME from the other populations. Interestingly, we also found less genetic diversity in the D. simulans ME population than the other D. simulans populations (Fig. S7, Supporting information). In addition, the level of differentiation among the three southern D. simulans populations was considerably lower than for the three southern D. melanogaster populations (Fig. 6).
Fig. 6.
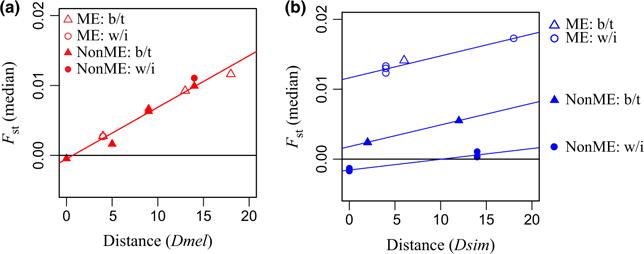
Isolation by distance. Between-population genetic differentiation (median FST) is plotted against geographic distance (degrees latitude). (a) Drosophila melanogaster. (b) Drosophila simulans. For D. simulans, regression lines are plotted separately for population comparisons without ME within a year, without ME between years and with ME within a year, reflecting the significant effect of distance, ME vs. non-ME comparison, and within- vs. between-year comparison in the regression model. ME: FST between one non-ME population and ME; Non-ME: FST between two non-ME populations; b/t: FST between two samples taken between years; w/i: FST between two samples taken within a year.
Population genetic patterns in time
The analysis of isolation by distance incorporated data from different years. We used this to determine whether there was a difference in the amount of interannual variation between D. melanogaster and D. simulans. As D. simulans has low clinal consistency, we might expect to also find a greater amount of interannual variation in D. simulans. We can test this with the isolation by distance regression model and ask whether there is a significant effect of between- vs. within-year comparison. Specifically, between-year comparisons should have greater FST than predicted by a regression of within-year comparisons. In D. simulans, we did indeed find that the effect of within- vs. between-year comparison was significant in the regression model (P = 0.002), with between-year comparisons showing greater genetic differentiation (Fig. 6; Table S3, Supporting information). The significant effect of between-year sampling implies that there was a detectable level of interannual variation in D. simulans. In contrast, in D. melanogaster, there was no effect of between-year comparison.
Although much of the clinal variation in D. simulans is not maintained from year to year (low clinal consistency) and there is interannual variation, can we still find evidence of genetic continuity in a population from year to year? We assessed the level of genetic continuity across years by comparing the level of differentiation (FST) among populations within a year to the level of differentiation within a population across years. We asked whether the PA.2010 samples were most similar to the PA.2011 samples (genetic continuity between years) or to the VA.2010 sample (genetic similarity between sites, within a year). We found significantly lower within-population differentiation (PA.2010/PA.2011) than between-site within-year differentiation (PA.2010/VA.2010) (chisquared P < 0.0001; Fig. S4, Supporting information), indicating that a given D. simulans population does maintain some degree of genetic similarity from year to year.
Increased X chromosome differentiation and clinal variation in D. simulans
The X chromosome in D. simulans showed two patterns not observed in D. melanogaster- an increased proportion of clinal variants and increased population genetic differentiation compared with the autosomes. The increased level of X chromosome differentiation was particularly pronounced in any comparisons with ME (Fig. 7). We asked whether the increased differentiation on the X chromosome was consistent with its reduced effective population size resulting from hemizygosity in males. We used the formula proposed by Ramachandran et al. (2004) that predicts the relationship between autosomal FST and X chromosome FST, given a particular sex ratio. To perform this analysis, we calculated pairwise FST for autosomal loci and the corresponding expected X chromosome FST values, assuming equal proportions of breeding males and females. Only in the ME comparisons were the X chromosome FST values significantly greater than expected when accounting for decreased effective population size (Fig. 7). With regard to the proportion of clinal variants, it is impossible to say whether the increased level of clinal variation on the X chromosome was due to the general pattern of increased X chromosome differentiation because the two signals are both strongly affected by increased ME differentiation.
Fig. 7.
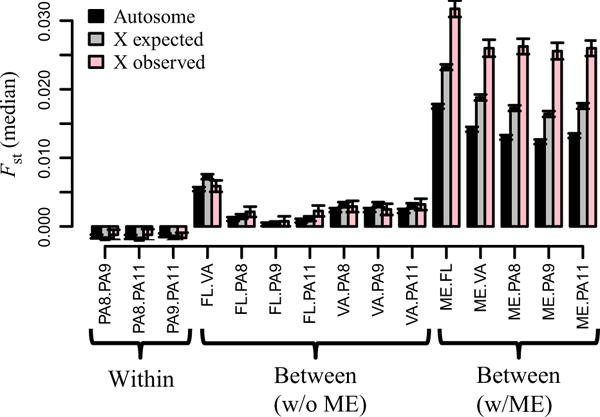
Expected vs. observed X chromosome FST in Drosophila simulans. An expectation of X chromosome median FST is calculated from the autosomal FST values. Within-population FST measures are from the three PA samples taken over the course of 2011. Between-population FST measures are divided up into comparisons that include ME and those that do not include ME. Error bars are 2 standard error.
Discussion
Our study is the first to conduct a comparative genomic analysis of D. simulans and D. melanogaster latitudinal variation. We expect D. melanogaster to have a larger proportion of clinal genetic variants than D. simulans, as D. melanogaster has been documented to have more strongly clinal phenotypes (Gibert et al. 2004; Arthur et al. 2008). The absence of D. simulans at high latitudes early in the year (Boulétreau-Merle et al. 2003; Fleury et al. 2004; Behrman et al. 2015) can be explained by either a stronger D. simulans winter bottleneck or population extinction and recolonization, both of which would result in a less stable cline from year to year. Our experimental design focuses on testing these predictions of less clinal variation and less clinal stability in D. simulans, as compared with D. melanogaster.
Less clinal variation in D. simulans than D. melanogaster
We find strong support for a larger proportion of clinal variants in D. melanogaster than in D. simulans, particularly for D. melanogaster autosomes, which harbour twice as much clinal variation as D. simulans autosomes (4.3% and 2.1%, respectively). We have ensured that this result is not confounded by differences in power or the additional sampling error of pool-seq. With a greater sample size (i.e. additional populations), it is possible that we would find an even greater proportion of clinal variants. For example, using deeper coverage and additional populations, Bergland et al. (2014) identified approximately one-third of common D. melanogaster SNPs as clinal. Our study design of four populations along a latitudinal transect makes our measurements of clinal variation sensitive to outlier allele frequencies at the Florida and Maine populations. In D. simulans, we do find that Maine is a genetic outlier, which could be contributing to the lower proportion of clinal variation identified. However, multiple lines of evidence from this study do support the conclusion of a more robust cline in D. melanogaster than in D. simulans, including increased clinal consistency, stronger isolation by distance, and more clear signatures of selection in D. melanogaster than D. simulans.
The strong pattern of clinal consistency in D. melanogaster, where clinal SNPs tend to show the same allele frequency pattern from year to year, indicates that the D. melanogaster cline is stable, rather than transient and re-established on an annual basis. Not only does D. simulans have a smaller proportion of clinal variants, the variants that are clinal are much less likely to be clinal from year to year than D. melanogaster. This indicates that the D. simulans cline is less stable, with a greater proportion of clinal variants due to processes operating on annual timescales.
The strong pattern of isolation by distance in D. melanogaster is also indicative of a robust cline. The pattern of isolation by distance in D. melanogaster is independent of whether or not the population pair was sampled in the same or different years. In contrast, in the D. simulans isolation by distance regression model, there is a significant effect of within- vs. between-year sampling of population pairs, indicating that interannual variation drives a detectable amount of population genetic variation. One important note is that in D. simulans, the genetic continuity at a collection site (i.e. across years) is still greater that the genetic similarity between collection sites (within a year), indicating that there is a balance between the processes resulting in these two patterns. For D. melanogaster, the pattern of isolation by distance is unperturbed by interannual variation, possibly indicating low effective migration rate between populations or a balance between selection and migration not seen in D. simulans.
Although a demography-driven pattern of isolation by distance can result in stable clinal variation, stability can also result from local adaptation to variable conditions along a transect. We find that D. melanogaster clinal SNPs are significantly enriched for functional genic classes, including UTR’s, coding regions and long introns and have a marginally elevated proportion of nonsynonymous to synonymous sites. This suggests that D. melanogaster clinal variants are under selection. We see weak evidence for selection in D. simulans, which shows a marginal enrichment for 5′UTR’s and no enrichment for other genic classes, suggesting that neutral processes play a stronger role.
Our comparisons of clinal variation in these two species reveal robust patterns of allele frequency with latitude in D. melanogaster, and weaker patterns in D. simulans. D. melanogaster not only harbours a larger proportion of clinal SNPs, but allele frequency patterns of clinal variants persist more from year to year, and there is evidence that clinal variants are under increased spatially varying selection. These results are consistent with previous studies that suggest less clinality in D. simulans. Specifically, some characters show no clinality in D. simulans (weight, wing length: Gibert et al. 2004; hexokinases: Duvernell & Eanes 2000; absence of diapause: Schmidt et al. 2005), while others show a decreased amplitude of clinality (wing length, thorax length, ovariole number: Gibert et al. 2004, cold tolerance, starvation tolerance: Hoffmann & Harshman 1999).
Shared clinal genes
A given selection pressure may act on the same genes in closely related species. As selection pressures along the latitudinal cline are expected to vary in the same manner for D. melanogaster as for D. simulans, the two species may exhibit similar genetic responses. We find a significant enrichment for genes that are clinal in both species. Fifty-six per cent of the 5559 D. simulans clinal genes were also clinal in D. melanogaster, compared to 45% in the matched controls. The enrichment of shared clinal genes increases with increasing stringency of the clinal regression. This supports the hypothesis of convergent evolution in these species due to the action of similar selection pressures on similar genetic backgrounds. This result is also consistent with the finding of parallel latitudinal gene expression in D. melanogaster and D. simulans (Zhao et al. 2015).
Although there is a significant enrichment of shared clinal genes (~20% more shared clinal genes than expected), we still cannot say which of the ~3000 shared clinal genes are true positives. However, we can ask whether genes previously identified as clinal tend to be shared clinal genes in our data set. When we look at a set of 12 genes with substantial literature support for latitudinal variation in D. melanogaster, 10 are clinal in both D. melanogaster and D. simulans. These genes include Pgm (Verrelli & Eanes 2001; Sezgin et al. 2004), G6pd (Oakeshott et al. 1983), Gpdh (Oakeshott et al. 1982), UGP (Sezgin et al. 2004), Treh (Sezgin et al. 2004), sgg (Rand et al. 2010), mth (Schmidt et al. 2000; Duvernell et al. 2003), cpo (Schmidt et al. 2008), per (Costa et al. 1992), Adh (Vigue & Johnson 1973; Berry & Kreitman 1993) and InR (Paaby et al. 2010).
We also find that our set of shared clinal genes is enriched for genes recently identified by Zhao et al. (2015) to have temperature-dependent expression in both D. melanogaster and D. simulans (Panama populations). Interestingly, we find only a marginal enrichment (P = 0.1) for genes with latitude-specific expression (Panama vs. Maine) in both species. One explanation for the lack of enrichment is the difference in sampling schemes. We sampled four populations along a continuous transect and identified loci that vary consistently with latitude. Zhao et al. (2015) sampled two populations from separate continents and identified gene expression differences between these two diverged groups.
Demographic implications of Drosophila clinal patterns
It is possible that D. simulans and D. melanogaster differ in both the initial establishment of clinal variation and the potential for that variation to be maintained. There is evidence that some of the latitudinal variation that we see in D. melanogaster is due to introgression between founding European and African populations (Duchen et al. 2013; Bergland et al. 2015; Kao et al. 2015). There is currently no evidence that this occurred in D. simulans. Additionally, the potential for maintenance of clinal variation might be diminished in D. simulans. As we discuss below, D. simulans population structure may be disproportionately affected by processes such as bottlenecks and migration.
Drosophila simulans overwintering
Drosophila populations experience a contraction as a result of temperate winters (Ives 1970). The decreased genetic diversity observed in high- relative to low-latitude populations of both D. melanogaster (Reinhardt et al. 2014) and D. simulans (Fig. S3, Supporting information) is consistent with stronger bottlenecks at high latitudes. D. simulans seems to be physiologically less winter-adapted than D. melanogaster (Hoffmann & Harshman 1999) and D. simulans is not observed at high latitudes until later in the year (Boulétreau-Merle et al. 2003; Fleury et al. 2004; Schmidt 2011; Behrman et al. 2015), suggesting a stronger bottleneck for D. simulans high-latitude populations than for D. melanogaster high-latitude populations. In addition to the decreased genetic variation we observe in the high-latitude D. simulans Maine population, we find that this population is much more genetically differentiated from the other three populations, a result that could be explained by strong bottlenecks or by complete extirpation and recolonization. Alternatively, these genetic patterns could be explained by selective sweeps in the Maine population or by effects due to the Maine population existing at the edge of the D. simulans range. Although we find evidence of year-to-year genetic continuity of the lower-latitude Pennsylvania population, indicating that there is not complete annual extirpation at the Pennsylvania site, additional sampling is needed to determine whether D. simulans is able to overwinter at latitudes as high as Maine (45° latitude).
Migration
While D. melanogaster has a strong, clear pattern of genetic isolation by distance, this is not true of D. simulans. A weak pattern of isolation by distance can be indicative of substantial gene flow among populations (Endler 1977). Genetic differentiation is particularly low among the three southern D. simulans populations (median FST 0.001–0.006, compared with 0.003–0.012 in D. melanogaster). The low level of differentiation indicates that there is a stronger effect of migration among these populations. Such a contribution of migration to D. simulans population genetic patterns is consistent with the reduced amount of clinal variation in D. simulans, as migration can disrupt clinal patterns resulting from demographic processes or local adaptation. A strong effect of migration in D. simulans and not in D. melanogaster could also contribute to the increased interannual variation observed in D. simulans, as evidenced by the significant effect of between-year comparison in the isolation by distance regressions (between-year comparisons show increased differentiation) and by the reduced level of clinal consistency (the same variants are not clinal from year to year). The effect of annual migration would be more acute in D. simulans than in D. melanogaster if D. simulans does indeed experience stronger annual bottlenecks, such that migrants overwhelm the local population. An additional contributor to weaker population structure in D. simulans than D. melanogaster could be the lack of large cosmopolitan inversions, which could act as a barrier to gene flow among D. melanogaster populations (Mettler et al. 1977; Knibb et al. 1981; Noor et al. 2001; Hoffmann & Weeks 2007).
One caveat to each of the analyses that utilize interannual data is the reliance of the conclusions on few between-year comparisons. For example, if the Virginia sample from 2010 was aberrant in its genetic composition, such as might occur with human-mediated migration from a distant population, our conclusions of low clinal consistency and the interaction of sampling year with isolation by distance in D. simulans might change. Further temporal sampling could bolster these findings.
Increased differentiation on the D. simulans X chromosome
We find more population genetic differentiation on the X chromosome than on autosomes in D. simulans. This pattern is opposite of what we find in D. melanogaster and is particularly pronounced for any comparisons with Maine. Additionally, only in the Maine comparisons are the X chromosome FST values significantly greater than expected when accounting for decreased effective population size (Fig. 7). In contrast, we see a lack of differentiation on the D. melanogaster X chromosome, consistent with previous findings of a drop in X chromosome diversity relative to autosomal diversity in non-African populations (Andolfatto 2001). There are multiple evolutionary processes that can affect the relative rates of divergence of the X and the autosomal chromosomes. Examples of a ‘faster-X’ effect are found across various taxa, including in D. simulans, and to a lesser extent in D. melanogaster (Begun et al. 2007). Certain classes of genes, such as those with greater expression in males than females (Baines et al. 2008), have shown faster-X patterns in Drosophila, as have certain classes of genomic sites, such as nonsynonymous sites, UTR and long introns (in D. melanogaster and D. simulans; Hu et al. 2013). In addition, gene expression differences have accumulated faster between Drosophila species on the X than on autosomes (Meisel et al. 2012). Further evidence for the contribution of selection to faster-X evolution in Drosophila includes the increased selection on tandem duplication on the X chromosome (in D. simulans; Rogers et al. 2015) and faster-X evolution in nonsynonymous sites, UTR and long introns, but not found in synonymous sites and short introns (Hu et al. 2013). The latter study again finds the effect present in both D. simulans and D. melanogaster, but is more marked in D. simulans.
The increased divergence of the D. simulans Maine X chromosome could be due to Maine suffering more extreme winter population bottlenecks. This is consistent with our findings of decreased genetic diversity and high levels of divergence on the autosomes as well as the X chromosome. Strong drift and divergence of the Maine population could also be driving clinal variation. A demographic explanation for the observed clinal variation is consistent with the weak evidence for selection on clinal variants in D. simulans. Another process that could contribute to X chromosome divergence is that of unequal sex ratios. Although we do not have sex ratio data for our populations, multiple sex-distorter systems have been found in other D. simulans populations (Bastide et al. 2013).
Conclusions
We have presented genomic evidence that D. melanogaster has a greater proportion of latitudinally varying loci than D. simulans. In D. simulans, we observe a weak pattern of isolation by distance, with a significant effect of between-year differentiation, low consistency of clinal SNPs from year to year and less evidence for selection on clinal variants than in D. melanogaster. In D. melanogaster, we observe the opposite patterns-strong isolation by distance, strong clinal consistency, low interannual variation and clear evidence for selection acting on clinal variants. We argue that one contributing factor to these differences is the ability of the two species to overwinter in temperate climates, causing differences in bottlenecks and migration. However, despite differences in demography, we do see an enrichment of shared clinal genes between the two species, suggesting that climate-associated selection might act on similar genes and phenotypes in the two taxa.
Supplementary Material
Table S1 Populations sampled along the east coast of the North America.
Table S2 Proportion of clinal SNPs.
Table S3 Isolation by distance linear model.
Table S4 Annotation of D. simulans genome and SNPs by genic category.
Fig. S1 Levels of differentiation between barcoded samples compared with differentiation between pooled samples.
Fig. S2 Sample size (NC) for clinal regressions. (a) Average NC by chromosome. Error bars are 2 standard error (all <0.03). (b) Distribution of total NC (sum over populations).
Fig. S3 Clinal consistency across years.
Fig. S4 Distribution of D. simulans (a) coding sequence (CDS) length and (b) SNP density (number of SNPs divided by the gene length), for the observed clinal genes (Q < 0.2; pink) and the matched control genes (blue).
Fig. S5 Relationship between population structure (mean FST) and clinal effect size (β).
Fig. S6 Enrichment of clinal X chromosome SNPs in each functional genic class.
Fig. S7 D. simulans autosomal diversity across populations.
Fig. S8 FST between the two PA 2010 samples and each of the other population samples in D. simulans.
Acknowledgments
The authors would like to thank Marc Feldman and Jamie Blundell for helpful discussions and Alison Feder, Nandita Garud, David Enard and Zoe Assaf for comments on the manuscript. This work was supported by the National Institute of Health (http://www.nih.gov) grants R01 GM097415, R01 GM089926 to DAP, R01 GM100366 to DAP and PS, and F32 GM097837 to AOB, and by the National Science Foundation (http://www.nsf.gov) grant DEB 0921307 to PS.
Footnotes
Data accessibility
Drosophila simulans sequence fastq files and alignment bam files: NCBI SRA: SRP063680 D. melanogaster sequence fastq files (Bergland et al. 2014): NCBI SRA: PRJNA256231 Allele frequency data: Dryad doi: 10.5061/dryad.3hf2q. FST and latitude matrices for isolation by distance analyses: Dryad doi: 10.5061/dryad.3hf2q. GLM results for clinal regressions: Dryad doi: 10.5061/dryad.3hf2q. D. melanogaster and D. simulans shared clinal genes: Dryad doi: 10.5061/dryad.3hf2q.
Supporting information
Additional supporting information may be found in the online version of this article.
P.S., D.A.P., A.O.B. and H.E.M. designed the research. P.S., K.R.O. and E.L.B. contributed samples. H.E.M. performed experiments. H.E.M. analysed the data. P.S., D.A.P., A.O.B., K.R.O., E.L.B. and H.E.M. discussed conclusions. H.E.M. wrote the manuscript.
References
- Andolfatto P. Contrasting patterns of X-linked and autosomal nucleotide variation in Drosophila melanogaster and Drosophila simulans. Molecular Biology and Evolution. 2001;18:279–290. doi: 10.1093/oxfordjournals.molbev.a003804. [DOI] [PubMed] [Google Scholar]
- Arthur AL, Weeks AR, Sgró CM. Investigating latitudinal clines for life history and stress resistance traits in Drosophila simulans from eastern Australia. Journal of Evolutionary Biology. 2008;21:1470–1479. doi: 10.1111/j.1420-9101.2008.01617.x. [DOI] [PubMed] [Google Scholar]
- Baines JF, Sawyer SA, Hartl DL, Parsch J. Effects of X-linkage and sex-biased gene expression on the rate of adaptive protein evolution in Drosophila. Molecular Biology and Evolution. 2008;25:1639–1650. doi: 10.1093/molbev/msn111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal V. A statistical method for the detection of variants from next-generation resequencing of DNA pools. Bioinformatics. 2010;26:i318–i324. doi: 10.1093/bioinformatics/btq214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastide H, Gerard PR, Ogereau D, Cazemajor M, Montchamp-Moreau C. Local dynamics of a fast-evolving sex-ratio system in Drosophila simulans. Molecular Ecology. 2013;22:5352–5367. doi: 10.1111/mec.12492. [DOI] [PubMed] [Google Scholar]
- Baumann H, Conover DO. Adaptation to climate change: contrasting patterns of thermal-reaction-norm evolution in Pacific versus Atlantic silversides. Proceedings of the Royal Society of London B: Biological Sciences. 2011;278:2265–2273. doi: 10.1098/rspb.2010.2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun DJ, Holloway AK, Stevens K, et al. Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biology. 2007;5:e310. doi: 10.1371/journal.pbio.0050310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrman EL, Watson SS, O’Brien KR, Heschel SM, Schmidt PS. Seasonal variation in life history traits in two Drosophila species. Journal of Evolutionary Biology. 2015;28:1691–1704. doi: 10.1111/jeb.12690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergland AO, Behrman EL, O’Brien KR, Schmidt PS, Petrov DA. Genomic evidence of rapid and stable adaptive oscillations over seasonal time scales in Drosophila. PLoS Genetics. 2014;10:e1004775. doi: 10.1371/journal.pgen.1004775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergland AO, Tobler R, Gonzalez J, Schmidt P, Petrov D. Secondary contact and local adaptation contribute to genome-wide patterns of clinal variation in Drosophila melanogaster. Molecular Ecology. 2015 doi: 10.1111/mec.13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry A, Kreitman M. Molecular analysis of an allozyme cline: alcohol dehydrogenase in Drosophila melanogaster on the east coast of North America. Genetics. 1993;134:869–893. doi: 10.1093/genetics/134.3.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulétreau-Merle J, Fouillet P, Varaldi J. Divergent strategies in low temperature environment for the sibling species Drosophila melanogaster and D. simulans: overwintering in extension border areas of France and comparison with African populations. Evolutionary Ecology. 2003;17:523–548. [Google Scholar]
- Cariou M. Biochemical phylogeny of the 8 species in the Drosophila melanogaster subgroup, including D. sechellia and D. orena. Genetical Research. 1987;50:181–185. doi: 10.1017/s0016672300023673. [DOI] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett-Detig RB, Hartl DL. Population genomics of inversion polymorphisms in Drosophila melanogaster. PLoS Genetics. 2012;8:e1003056. doi: 10.1371/journal.pgen.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa R, Peixoto AA, Barbujani G, Kyriacou CP. A latitudinal cline in a Drosophila clock gene. Proceedings of the Royal Society of London B: Biological Sciences. 1992;250:43–49. doi: 10.1098/rspb.1992.0128. [DOI] [PubMed] [Google Scholar]
- Coyne JA, Beecham E. Heritability of two morphological characters within and among natural populations of Drosophila melanogaster. Genetics. 1987;117:727–737. doi: 10.1093/genetics/117.4.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruzan MB. Patterns of introgression across an expanding hybrid zone: analysing historical patterns of gene flow using nonequilibrium approaches. The New Phytologist. 2005;167:267–278. doi: 10.1111/j.1469-8137.2005.01410.x. [DOI] [PubMed] [Google Scholar]
- David JR, Capy P. Genetic variation of Drosophila melanogaster natural populations. Trends in Genetics. 1988;4:106–111. doi: 10.1016/0168-9525(88)90098-4. [DOI] [PubMed] [Google Scholar]
- David J, Capy P, Payant V, Tsakas S. Thoracic trident pigmentation in Drosophila melanogaster: differentiation of geographical populations. Génétique, Sélection, Évolution. 1985;17:211–224. doi: 10.1186/1297-9686-17-2-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen P, Zivkovic D, Hutter S, Stephan W, Laurent S. Demographic inference reveals African and European admixture in the North American Drosophila melanogaster population. Genetics. 2013;193:291–301. doi: 10.1534/genetics.112.145912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvernell DD, Eanes WF. Contrasting molecular population genetics of four hexokinases in Drosophila melanogaster, D. simulans and D. yakuba. Genetics. 2000;156:1191–1201. doi: 10.1093/genetics/156.3.1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvernell DD, Schmidt PS, Eanes WF. Clines and adaptive evolution in the methuselah gene region in Drosophila melanogaster. Molecular Ecology. 2003;12:1277–1285. doi: 10.1046/j.1365-294x.2003.01841.x. [DOI] [PubMed] [Google Scholar]
- Emerson KJ, Uyemura AM, McDaniel KL, Schmidt PS, Bradshaw WE, Holzapfel CM. Environmental control of ovarian dormancy in natural populations of Drosophila melanogaster. Journal of Comparative Physiology A, Neuroethology, Sensory, Neural, and Behavioral Physiology. 2009;195:825–829. doi: 10.1007/s00359-009-0460-5. [DOI] [PubMed] [Google Scholar]
- Endler JA. Geographic Variation, Speciation, and Clines. Princeton University Press; Princeton, New Jersey: 1977. [PubMed] [Google Scholar]
- Excoffier L, Foll M, Petit RJ. Genetic consequences of range expansions. Annual Review of Ecology, Evolution, and Systematics. 2009;40:481–501. [Google Scholar]
- Fabian DK, Kapun M, Nolte V, et al. Genome-wide patterns of latitudinal differentiation among populations of Drosophila melanogaster from North America. Molecular Ecology. 2012;21:4748–4769. doi: 10.1111/j.1365-294X.2012.05731.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder JL, Bush GL. Gene frequency clines for host races of Rhagoletis pomonella in the Midwestern United States. Heredity. 1989;63:245–266. [Google Scholar]
- Feder AF, Petrov DA, Bergland AO. LDx: estimation of linkage disequilibrium from high-throughput pooled resequencing data. PLoS One. 2012;7:e48588. doi: 10.1371/journal.pone.0048588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleury F, Ris N, Allemand R, Fouillet P, Carton Y, Boulétreau M. Ecological and genetic interactions in Drosophila-parasitoids communities: a case study with D. melanogaster, D. simulans and their common Leptopilina parasitoids in south-eastern France. Genetica. 2004;120:181–194. doi: 10.1023/b:gene.0000017640.78087.9e. [DOI] [PubMed] [Google Scholar]
- Fuhrman JA, Steele JA, Hewson I, et al. A latitudinal diversity gradient in planktonic marine bacteria. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:7774–7778. doi: 10.1073/pnas.0803070105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibert P, Capy P, Imasheva A, et al. Comparative analysis of morphological traits among Drosophila melanogaster and D. simulans: genetic variability, clines and phenotypic plasticity. Genetica. 2004;120:165–179. doi: 10.1023/b:gene.0000017639.62427.8b. [DOI] [PubMed] [Google Scholar]
- Gockel J, Kennington W, Hoffmann A, Goldstein D, Partridge L. Nonclinality of molecular variation implicates selection in maintaining a morphological cline of Drosophila melanogaster. Genetics. 2001;158:319–323. doi: 10.1093/genetics/158.1.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey J, Kliman RM. Population genetics and phylogenetics of DNA sequence variation at multiple loci within the Drosophila melanogaster species complex. Molecular Biology and Evolution. 1993;10:804–822. doi: 10.1093/oxfordjournals.molbev.a040044. [DOI] [PubMed] [Google Scholar]
- Hoffmann AA, Harshman LG. Desiccation and starvation resistance in Drosophila: patterns of variation at the species, population and intrapopulation levels. Heredity. 1999;83(Pt 6):637–643. doi: 10.1046/j.1365-2540.1999.00649.x. [DOI] [PubMed] [Google Scholar]
- Hoffmann AA, Weeks AR. Climatic selection on genes and traits after a 100 year-old invasion: a critical look at the temperate-tropical clines in Drosophila melanogaster from eastern Australia. Genetica. 2007;129:133–147. doi: 10.1007/s10709-006-9010-z. [DOI] [PubMed] [Google Scholar]
- Hoffmann AA, Anderson A, Hallas R. Opposing clines for high and low temperature resistance in Drosophila melanogaster. Ecology Letters. 2002;5:614–618. [Google Scholar]
- Hu TT, Eisen MB, Thornton KR, Andolfatto P. A second-generation assembly of the Drosophila simulans genome provides new insights into patterns of lineage-specific divergence. Genome Research. 2013;23:89–98. doi: 10.1101/gr.141689.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ives PT. Further genetic studies of the south amherst population of Drosophila melanogaster. Evolution. 1970;24:507–518. doi: 10.1111/j.1558-5646.1970.tb01785.x. [DOI] [PubMed] [Google Scholar]
- James AC, Azevedo RB, Partridge L. Cellular basis and developmental timing in a size cline of Drosophila melanogaster. Genetics. 1995;140:659–666. doi: 10.1093/genetics/140.2.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao JY, Zubair A, Salomon MP, Nuzhdin SV, Campo D. Population genomic analysis uncovers African and European admixture in Drosophila melanogaster populations from the south-eastern United States and Caribbean Islands. Molecular Ecology. 2015;24:1499–1509. doi: 10.1111/mec.13137. [DOI] [PubMed] [Google Scholar]
- Kapun M, van Schalkwyk H, McAllister B, Flatt T, Schlötterer C. Inference of chromosomal inversion dynamics from Pool-Seq data in natural and laboratory populations of Drosophila melanogaster. Molecular Ecology. 2014;23:1813–1827. doi: 10.1111/mec.12594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karan D, Dahiya N, Munjal AK, et al. Desiccation and starvation tolerance of adult Drosophila: opposite latitudinal clines in natural populations of three different species. Evolution. 1998;52:825. doi: 10.1111/j.1558-5646.1998.tb03706.x. [DOI] [PubMed] [Google Scholar]
- Keller SR, Levsen N, Ingvarsson PK, Olson MS, Tiffin P. Local selection across a latitudinal gradient shapes nucleotide diversity in balsam poplar, Populus balsamifera L. Genetics. 2011;188:941–952. doi: 10.1534/genetics.111.128041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knibb W, Oakeshott J, Gibson J. Chromosome inversion polymorphisms in Drosophila melanogaster. I. Latitudinal clines and associations between inversions in Australasian populations. Genetics. 1981;98:833–847. doi: 10.1093/genetics/98.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler R, Orozco-terWengel P, De Maio N, et al. PoPoolation: a toolbox for population genetic analysis of next generation sequencing data from pooled individuals. PLoS One. 2011;6:e15925. doi: 10.1371/journal.pone.0015925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolaczkowski B, Kern AD, Holloway AK, Begun DJ. Genomic differentiation between temperate and tropical Australian populations of Drosophila melanogaster. Genetics. 2011;187:245–260. doi: 10.1534/genetics.110.123059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachaise D, Silvain JF. How two Afrotropical endemics made two cosmopolitan human commensals: the Drosophila melanogaster-D. simulans palaeogeographic riddle. Genetica. 2004;120:17–39. doi: 10.1023/b:gene.0000017627.27537.ef. [DOI] [PubMed] [Google Scholar]
- Lachaise D, Cariou M, David J, Lemeunier F, Tsacas L, Ashburner M. Historical biogeography of the Drosophila melanogaster species subgroup. BMC Evolutionary Biology. 1988;22:159–225. [Google Scholar]
- Lawrie DS, Messer PW, Hershberg R, Petrov DA. Strong purifying selection at synonymous sites in D. melanogaster. PLoS Genetics. 2013;9:e1003527. doi: 10.1371/journal.pgen.1003527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics (Oxford, England) 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Stephan W. Inferring the demographic history and rate of adaptive substitution in Drosophila. PLoS Genetics. 2006;2:e166. doi: 10.1371/journal.pgen.0020166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics (Oxford, England) 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Bost D, Wilson S, Maruki T, Harrison S. Population-genetic inference from pooled-sequencing data. Genome Biology and Evolution. 2014;6:1210–1218. doi: 10.1093/gbe/evu085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie JA, Parsons PA. The genetic architecture of resistance to desiccation in populations of Drosophila melanogaster and D. simulans. Australian Journal of Biological Sciences. 1974;27:441–456. doi: 10.1071/bi9740441. [DOI] [PubMed] [Google Scholar]
- Meisel RP, Malone JH, Clark AG. Faster-X evolution of gene expression in Drosophila. PLoS Genetics. 2012;8:e1003013. doi: 10.1371/journal.pgen.1003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettler LE, Voelker RA, Mukai T. Inversion clines in populations of Drosophila melanogaster. Genetics. 1977;87:169–176. doi: 10.1093/genetics/87.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovski P, Hoffmann AA. Postponed reproduction as an adaptation to winter conditions in Drosophila melanogaster: evidence for clinal variation under semi-natural conditions. Proceedings of the Royal Society of London B: Biological Sciences. 2001;268:2163–2168. doi: 10.1098/rspb.2001.1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munch SB, Salinas S. Latitudinal variation in lifespan within species is explained by the metabolic theory of ecology. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13860–13864. doi: 10.1073/pnas.0900300106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munjal A, Karan D, Gibert P, Moreteau B, Parkash R, David J. Thoracic trident pigmentation in Drosophila melanogaster: latitudinal and altitudinal clines in Indian populations. Genetics Selection Evolution. 1997;29:601. [Google Scholar]
- Noor MA, Grams KL, Bertucci LA, Reiland J. Chromosomal inversions and the reproductive isolation of species. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:12084–12088. doi: 10.1073/pnas.221274498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakeshott JG, Gibson JB, Anderson PR, Knibb WR, Anderson DG, Chambers GK. Alcohol dehydrogenase and glycerol-3-phosphate dehydrogenase clines in Drosophila melanogaster on different continents. Evolution. 1982;36:86. doi: 10.1111/j.1558-5646.1982.tb05013.x. [DOI] [PubMed] [Google Scholar]
- Oakeshott JG, Chambers GK, Gibson JB, Eanes WF, Willcocks DA. Geographic variation in G6pd and Pgd allele frequencies in Drosophila melanogaster. Heredity. 1983;50(Pt 1):67–72. doi: 10.1038/hdy.1983.7. [DOI] [PubMed] [Google Scholar]
- Paaby AB, Blacket MJ, Hoffmann AA, Schmidt PS. Identification of a candidate adaptive polymorphism for Drosophila life history by parallel independent clines on two continents. Molecular Ecology. 2010;19:760–774. doi: 10.1111/j.1365-294X.2009.04508.x. [DOI] [PubMed] [Google Scholar]
- Pool JE, Aquadro CF. The genetic basis of adaptive pigmentation variation in Drosophila melanogaster. Molecular Ecology. 2007;16:2844–2851. doi: 10.1111/j.1365-294X.2007.03324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna: 2014. [Google Scholar]
- Ramachandran S, Rosenberg NA, Zhivotovsky LA, Feldman MW. Robustness of the inference of human population structure: a comparison of X-chromosomal and autosomal microsatellites. Human Genomics. 2004;1:87–97. doi: 10.1186/1479-7364-1-2-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand DM, Weinreich DM, Lerman D, Folk D, Gilchrist GW. Three selections are better than one: clinal variation of thermal QTL from independent selection experiments in Drosophila. Evolution: International Journal of Organic Evolution. 2010;64:2921–2934. doi: 10.1111/j.1558-5646.2010.01039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt JA, Kolaczkowski B, Jones CD, Begun DJ, Kern AD. Parallel geographic variation in Drosophila melanogaster. Genetics. 2014;197:361–373. doi: 10.1534/genetics.114.161463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RL, Shao L, Sanjak JS, Andolfatto P, Thornton KR. Revised annotations, sex-biased expression, and lineage-specific genes in the Drosophila melanogaster group. G3 (Bethesda, MD) 2014;4:2345–2351. doi: 10.1534/g3.114.013532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RL, Cridland JM, Shao L, Hu TT, Andolfatto P, Thornton KR. Tandem duplications and the limits of natural selection in Drosophila yakuba and Drosophila simulans. PLoS One. 2015;10:e0132185. doi: 10.1371/journal.pone.0132184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado C, Pennings S. Latitudinal variation in palatability of salt-marsh plants: are differences constitutive? Ecology. 2005;86:1571–1579. [Google Scholar]
- Saunders DS, Henrich VC, Gilbert LI. Induction of diapause in Drosophila melanogaster: photoperiodic regulation and the impact of arrhythmic clock mutations on time measurement. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:3748–3752. doi: 10.1073/pnas.86.10.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt PS. Evolution and mechanisms of insect reproductive diapause: a plastic and pleiotropic life history syndrome. In: Flatt T, Heyland A, editors. Mechanisms of Life History Evolution: The Genetics and Physiology of Life History Traits and Trade-Offs. Oxford University Press; Oxford: 2011. p. 478. [Google Scholar]
- Schmidt PS, Conde DR. Environmental heterogeneity and the maintenance of genetic variation for reproductive diapause in Drosophila melanogaster. Evolution: International Journal of Organic Evolution. 2006;60:1602–1611. [PubMed] [Google Scholar]
- Schmidt PS, Duvernell DD, Eanes WF. Adaptive evolution of a candidate gene for aging in Drosophila. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:10861–10865. doi: 10.1073/pnas.190338897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt PS, Matzkin L, Ippolito M, Eanes WF. Geographic variation in diapause incidence, life-history traits, and climatic adaptation in Drosophila melanogaster. Evolution: International Journal of Organic Evolution. 2005;59:1721–1732. [PubMed] [Google Scholar]
- Schmidt PS, Zhu CT, Das J, Batavia M, Yang L, Eanes WF. An amino acid polymorphism in the couch potato gene forms the basis for climatic adaptation in Drosophila melanogaster. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:16207–16211. doi: 10.1073/pnas.0805485105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sezgin E, Duvernell DD, Matzkin LM, et al. Single-locus latitudinal clines and their relationship to temperate adaptation in metabolic genes and derived alleles in Drosophila melanogaster. Genetics. 2004;168:923–931. doi: 10.1534/genetics.104.027649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RS, Hickey DA, David J. Genetic differentiation between geographically distant populations of Drosophila melanogaster. Genetics. 1982;101:235–256. doi: 10.1093/genetics/101.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit A, Hubley R, Green P. RepeatMasker Open-4.0 2013 [Google Scholar]
- Storey JD. qvalue: Q-value estimation for false discovery rate control. R package version 2.0.0. 2015 Available from: http://qva.
- Turner TL, Levine MT, Eckert ML, Begun DJ. Genomic analysis of adaptive differentiation in Drosophila melanogaster. Genetics. 2008;179:455–473. doi: 10.1534/genetics.107.083659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrelli BC, Eanes WF. Clinal variation for amino acid polymorphisms at the Pgm locus in Drosophila melanogaster. Genetics. 2001;157:1649–1663. doi: 10.1093/genetics/157.4.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigue CL, Johnson FM. Isozyme variability in species of the genus Drosophila. VI. Frequency-property-environment relationships of allelic alcohol dehydrogenases in D. melanogaster. Biochemical Genetics. 1973;9:213–227. doi: 10.1007/BF00485735. [DOI] [PubMed] [Google Scholar]
- Voelker RA, Mukai T, Johnson FM. Genetic variation in populations of Drosophila melanogaster from the western United States. Genetica. 1977;47:143–148. [Google Scholar]
- Weber E, Schmid B. Latitudinal population differentiation in two species of Solidago (Asteraceae) introduced into Europe. American Journal of Botany. 1998;85:1110. [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- Zhao L, Wit J, Svetec N, Begun DJ. Parallel gene expression differences between low and high latitude populations of Drosophila melanogaster and D. simulans. PLoS Genetics. 2015;11:e1005184. doi: 10.1371/journal.pgen.1005184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Bergland AO, González J, Petrov DA. Empirical validation of pooled whole genome population re-sequencing in Drosophila melanogaster. PLoS One. 2012;7:e41901. doi: 10.1371/journal.pone.0041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Populations sampled along the east coast of the North America.
Table S2 Proportion of clinal SNPs.
Table S3 Isolation by distance linear model.
Table S4 Annotation of D. simulans genome and SNPs by genic category.
Fig. S1 Levels of differentiation between barcoded samples compared with differentiation between pooled samples.
Fig. S2 Sample size (NC) for clinal regressions. (a) Average NC by chromosome. Error bars are 2 standard error (all <0.03). (b) Distribution of total NC (sum over populations).
Fig. S3 Clinal consistency across years.
Fig. S4 Distribution of D. simulans (a) coding sequence (CDS) length and (b) SNP density (number of SNPs divided by the gene length), for the observed clinal genes (Q < 0.2; pink) and the matched control genes (blue).
Fig. S5 Relationship between population structure (mean FST) and clinal effect size (β).
Fig. S6 Enrichment of clinal X chromosome SNPs in each functional genic class.
Fig. S7 D. simulans autosomal diversity across populations.
Fig. S8 FST between the two PA 2010 samples and each of the other population samples in D. simulans.