

Abstract
The hypothalamus is a center of food intake and energy metabolism regulation. Information signals from peripheral organs are mediated through the circulation or the vagal afferent pathway and input into the hypothalamus, where signals are integrated to determine various behaviors, such as eating. Numerous appetite‐regulating peptides are expressed in the central nervous system and the peripheral organs, and interact in a complex manner. Of such peptides, gut peptides are known to bind to receptors at the vagal afferent pathway terminal that extend into the mucosal layer of the digestive tract, modulate the electrical activity of the vagus nerve, and subsequently send signals to the solitary nucleus and furthermore to the hypothalamus. All peripheral peptides other than ghrelin suppress appetite, and they synergistically suppress appetite through the vagus nerve. In contrast, the appetite‐enhancing peptide, ghrelin, antagonizes the actions of appetite‐suppressing peptides through the vagus nerve, and appetite‐suppressing peptides have attenuated effects in obesity as a result of inflammation in the vagus nerve. With greater understanding of the mechanism for food intake and energy metabolism regulation, medications that apply the effects of appetite‐regulating peptides or implantable devices that electrically stimulate the vagus nerve are being investigated as novel treatments for obesity in basic and clinical studies.
Keywords: Gut peptides, Hypothalamus, Vagus nerve
Introduction
The number of obese individuals is increasing globally as well as domestically in Japan, and the number of obese patients with multiple obesity‐related diseases, such as diabetes, hypertension, cardiovascular diseases and sleep apnea syndrome, is also increasing, leading to a large issue both socially and economically. Although obesity complications often improve if obese individuals are able to successfully lose weight, in reality it is not an easy task to lose weight and to maintain a reduced weight. It is therefore essential to elucidate the full mechanistic picture of appetite and energy metabolism regulation for the purpose of establishing an effective and safe method for weight loss. In the present article, we reviewed the mechanism of appetite regulation by the central nervous system and the peripheral organs, as well as the vagal afferent pathway that exists between these two entities.
Appetite Regulation in the Central Nervous System
The hypothalamus is the center of appetite regulation, and neuropeptide Y (NPY)/agouti‐related regulatory peptide (AgRP) neurons that enhance appetite and pro‐opiomelanocortin (POMC) neurons that suppress appetite are both present in the hypothalamic arcuate nucleus (Figure 1)1, 2. The α‐melanocyte‐stimulating hormone produced by POMC neurons suppresses appetite by acting as an agonist of the melanocortin 4 receptor (MC4R)3. MC4R‐deficient mice show hyperphagia and obesity4, and humans carrying the MC4R mutation present with hyperphagia and obesity5, 6, 7. NPY and AgRP enhance appetite through the NPY receptor and MC4R, respectively. AgRP was reported to be an inverse agonist of MC4R, and competes with α‐melanocyte‐stimulating hormone to increase appetite3, 8. In addition to this mechanism, AgRP also acts as an agonist to increase K+ efflux through inwardly rectifying potassium channel Kir7.1, and produces a strong hyperpolarization in MC4R‐expressing neurons in the paraventricular nucleus of mice9. Clinical studies examining the effects of MC4R agonists as an anti‐obesity medication are currently underway10.
Figure 1.
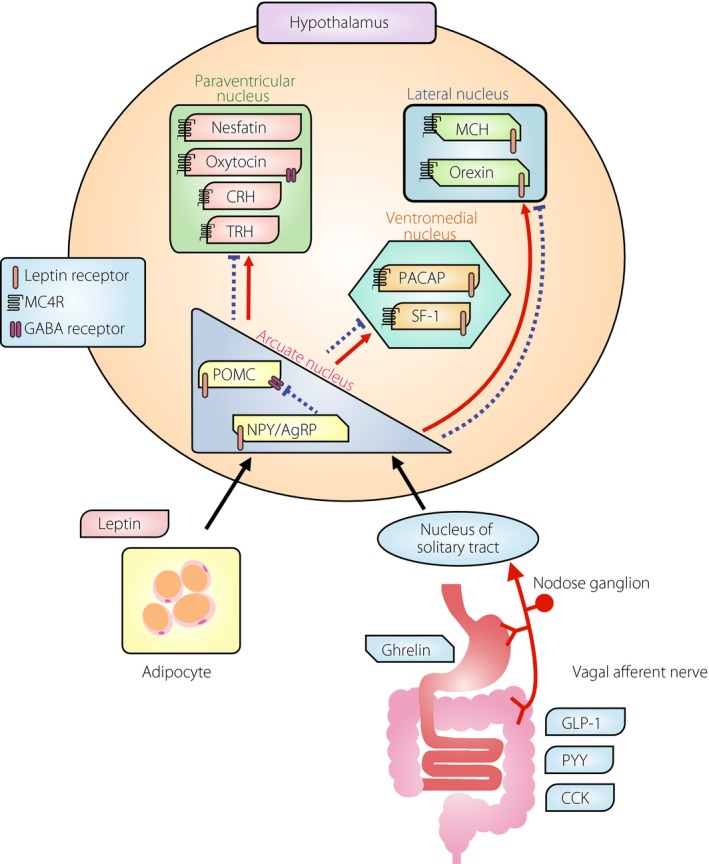
Mechanism of appetite regulation by the hypothalamus and peripheral tissues. Leptin secreted by adipocytes travels through the circulation and acts on the hypothalamic arcuate nucleus, and gut peptides such as ghrelin and glucagon‐like peptide‐1 (GLP‐1) act on the hypothalamus through the vagus nerve. Black letters indicate appetite‐enhancing peptides and white letters indicate appetite‐suppressing peptides; the interactions of these peptides are mediated by the melanocortin 4 (MC4R), gamma amino butyric acid (GABA) and leptin receptors. The α‐melanocyte‐stimulating hormone produced by the pro‐opiomelanocortin (POMC) neuron is an MC4R agonist, and the agouti‐related regulatory peptide (AgRP) is an inverse agonist of MC4R, and these antagonistically regulate appetite. Suppression of POMC neurons by NPY/AgRP neurons through the GABA receptors is also important. Solid lines and dashed lines show the representative transmission of enhancing and suppressing actions, respectively, in the hypothalamus. CCK, cholecystokinin; CRH, corticotropin‐releasing hormone; MCH, melanin‐concentrating hormone; NPY, neuropeptide Y; PACAP, pituitary adenylate cyclase‐activating peptide; PYY, peptide YY; SF‐1, steroidogenic factor 1; TRH, thyrotropin‐releasing hormone.
Adipocytes‐derived leptin passes through the sparse blood–brain barrier near the hypothalamus, and suppresses appetite by activating POMC neurons and inhibiting NPY/AgRP neurons in the arcuate nucleus (Figure 1)1, 11, 12. In contrast, gastric‐derived ghrelin acts on the arcuate nucleus primarily through the vagal afferent pathway and nuclei tractus solitary (NTS), and enhances appetite by activating NPY/AgRP neurons and suppressing POMC neurons (Figure 1)13, 14, 15. The axons of POMC neurons and NPY/AgRP neurons are projected to the paraventricular nucleus, ventromedial nucleus, lateral nucleus and dorsomedial nucleus, and to non‐hypothalamic brain regions, thereby controlling the neuronal activity at the projection target. Furthermore, the NPY/AgRP neurons also release gamma amino butyric acid (GABA) and suppress POMC neurons through the GABA receptor16. Indeed, specific deletion of the vesicular GABA transporter gene in AgRP neurons of mice showed a lean phenotype under both normal chow and high‐fat diet. The orexigenic effect of ghrelin was robustly attenuated in these mice, indicating that GABA release from AgRP neurons is required for ghrelin‐stimulated food intake17.
In the hypothalamic paraventricular nucleus, specific neurons for the appetite suppressants, corticotropin‐releasing hormone, nesfatin, oxytocin and thyrotropin‐releasing hormone, are present, and neuronal inputs from the hypothalamic lateral nucleus and arcuate nucleus are observed (Figure 1)18, 19. Neuronal projections to histamine neurons that suppress appetite are also observed. In addition, NPY/AgRP neurons enhance appetite by suppressing oxytocin neurons through GABA receptors expressed on oxytocin neurons in the paraventricular nucleus20. Prader–Willi syndrome, a condition in which hyperphagia and obesity is present from the childhood, exhibits a partial disappearance of oxytocin neurons21. Oxytocin has also been studied as an anti‐obesity medication, and peripherally administered oxytocin activated vagal afferent neurons and suppressed food intake through the NTS22. A double‐blind single‐dose study of intranasal oxytocin in humans showed that the energy intake through voluntary food consumption was significantly reduced in the oxytocin group compared with placebo23.
Neurons containing appetite‐enhancing orexin or melanin‐concentrating hormone are present in the hypothalamic lateral nucleus, and MC4Rs expressed on each of these neurons mediate the actions of POMC and NPY/AgRP neurons (Figure 1)24, 25, 26. Although melanin‐concentrating hormone receptor antagonists have been tried as an anti‐obesity medication27, 28, there are currently no clinical trials in progress investigating such drugs. The ventromedial nucleus of the hypothalamus has been classically referred to as the ‘satiety center,’ and expresses high levels of the leptin receptor and MC4R; furthermore, neurons containing steroidogenic factor 1 or pituitary adenylate cyclase‐activating peptide are also present in this region (Figure 1)29, 30. Steroidogenic factor 1 is a transcription factor that regulates energy homeostasis by modulating energy expenditure and leptin receptor expression in the ventromedial nucleus31. Mechanisms of central anorectic action of pituitary adenylate cyclase‐activating peptide are the activation of the POMC neuron and the increase of POMC messenger ribonucleic acid expression in the arcuate nucleus32, 33. Pituitary adenylate cyclase‐activating peptide was also required for leptin's effects including the reduction of food intake and increase in sympathetic nerve activity in white adipose tissue34, 35.
Other molecules, such as serotonin and noradrenaline, are also involved in appetite suppression, and anti‐obesity medications that target these amines have been developed. One such example is mazindol, which is the only anti‐obesity medication presently available in Japan. Mazindol inhibits the re‐uptake of noradrenaline at the presynapse, and shows its effects by paraventricular nucleus excitation and lateral nucleus suppression36. However, caution against drug dependency is necessary, because its pharmacological action is similar to that of amphetamine37. Furthermore, it is contraindicated in severe hypertension and cerebrovascular disorders, and careful administration is essential in diabetes. Lorcaserin, an anti‐obesity medication approved in Western countries, is a serotonin 2C receptor agonist, and suppresses appetite through the serotonin 2C receptor expressed on POMC neurons38, 39, 40, 41.
Vagus Nerve that Links the Digestive Tract and the Central Nervous System
The vagus nerve is the 10th cranial nerve that transmits various information from the digestive tract to the central nervous system. The vagus nerve is composed of both afferent fibers (visceral sensory nerve) and efferent fibers (motor nerves), although more than 70% of the subdiaphragmatic trunks are composed of afferent fibers42. The vagus afferent nerve is a bipolar neuron with the cell body located in the nodose ganglion, exhibiting a projection to the NTS and one to various peripheral organs (Figure 1). A part of the peripheral nerve terminals of the vagal afferent fibers is distributed along the mucosal and submucosal layers of the digestive tract, and sends information from molecules, such as gut peptides, to the central nervous system through the NTS43. Meal‐induced stretching signals through gastric mechanoreceptors are also sent by the vagal afferent fibers. Nesfatin neurons also contribute to this mechanism, at least in part, because gastric distension activates nesfatin‐expressing neurons in the NTS of rats44. In addition, the glucagon‐like peptide‐1 (GLP‐1) receptor in the NTS plays a role in appetite suppression by detecting mechanical‐stretching stimuli of the stomach45.
Gut Peptides and the Vagus Nerve
Although there are many types of gut peptides, ghrelin, which is produced and secreted by the stomach, alone enhances appetite13, 14, 15. All other peptides, such as cholecystokinin (CCK), peptide YY (PYY) and GLP‐1, play a role in appetite suppression46. Peptides produced in the digestive tract bind to their respective receptors located at the vagus nerve fiber terminals, which extend to the mucosal layer of the digestive tract. Each of these receptors is synthesized in the nodose ganglion, and is subsequently transported to the terminals on the digestive tract end. When the gut peptide binds with its receptor, the vagal afferent discharge is suppressed in the case of appetite‐enhancing peptide ghrelin, and is increased for appetite‐suppressing peptides CCK, peptide YY and GLP‐115. As described, gut peptide information is converted to electrical signals and reaches the brainstem (NTS), thereby controlling neurotransmitter release from the brainstem and sending information to the superior neuron towards the hypothalamus (Figure 1). For ghrelin, information that ascends the vagal afferent pathway is transmitted to noradrenaline neurons at NTS, and appetite‐enhancing actions are exhibited through the promotion of noradrenaline secretion from the hypothalamus14. Peptide YY promotes the electrical activity of vagal afferent fibers, sends satiety signals to the hypothalamus, and activates appetite‐suppressing POMC neurons and suppresses appetite‐enhancing NPY neurons in the hypothalamic arcuate nucleus47. Nesfatin is expressed in the hypothalamus and the peripheral organs including the stomach and duodenum. Peripherally administered nesfatin reduced food intake through activation of vagal afferent fibers and subsequently post‐synaptic neurons in the NTS48, 49, 50.
Interaction between Multiple Peptides
In the vagus nerve, multiple receptors are co‐expressed on one cell body, and affect food intake in an additive, synergistic or antagonistic manner. For example, both leptin and CCK receptors are expressed on the vagus nerve. Cultured vagus nerve ganglion cells treated with CCK alone showed activation at 10 nmol/L, which is approximately 1,000‐fold the physiological concentration; however, cells simultaneously treated with both leptin and CCK showed activation at 10 pmol/L, which is the physiological concentration of CCK51. In vivo experiments have also shown that the combination of CCK and leptin exhibits a greater appetite‐suppressing effect than CCK treatment alone52. The mechanism of CCK action promotes the translocation of early growth response‐1 (EGR1), an immediate‐early gene in the vagal afferent pathway, into the nucleus, thereby upregulating the expression of appetite‐suppressing cocaine‐ and amphetamine‐regulated transcript, and consequently suppressing appetite51. The mechanism of leptin action upregulates EGR1 expression through signal transducer and activator of transcription 3, and synergistically suppresses appetite with CCK. In contrast, ghrelin blocks EGR1 nuclear translocation and signal transducer and activator of transcription 3, and also competes with both CCK and leptin, thereby enhancing appetite51. CCK activates nesfatin‐expressing neurons in the paraventricular nucleus and NTS in rats53. In contrast, the orexin and CCK receptors are expressed in vagal afferent neurons in humans and rats. The vagal afferent discharge increased by CCK was attenuated by prior administration of orexin54. This is an example of the antagonistic interaction between anorectic and orexigenic peptides through the vagus nerve.
Although ghrelin and GLP‐1 exhibit opposite effects on food intake and the electrical activity of the vagus nerve, we showed that appetite suppression and increased vagus nerve discharge observed with GLP‐1 administration are canceled when rats are pretreated with ghrelin 30 min before GLP‐1 (unpublished data). Similarly, appetite enhancement and suppressed vagus nerve discharge observed with ghrelin administration are canceled when rats are pretreated with GLP‐1 30 min before ghrelin. Similar results were also shown in a study that investigated CCK and ghrelin55. However, when the two peptides are administered 60 min apart, the interaction disappears, and the normal effects of the peptide administered secondarily are observed (unpublished data). This indicates that satiety signals after eating are canceled when time passes to some extent, playing an essential role in the time‐dependent control of satiety and hunger. It has also been reported in humans that postprandial CCK and GLP‐1 secretion peaks earlier when eating quickly, as compared with eating slowly56. Ghrelin reaches peak circulating concentrations in a fasting state immediately before a meal57, thereby showing appetite‐enhancing effects; however, because elevation in the circulating levels of CCK and GLP‐1 occurs early with fast eating, appetite suppression by CCK and GLP‐1 is not fully shown, potentially leading to loss of satiety and hyperphagia. Furthermore, it has been reported that ghrelin inhibits GLP‐1 receptor translocation to the cell membrane in the vagus nerve58, and when the circulating ghrelin concentration decreases postprandially, the GLP‐1 receptor is translocated to the cell membrane, facilitating the GLP‐1 actions.
Vagus Nerve in Obesity and Diabetes
It has been reported that there are alterations in vagus nerve responses toward appetite‐regulating peptides in an obese state. In high‐fat diet‐fed obese mice, the vagus nerve response to CCK and leptin is known to decrease59. As a result, the effects of appetite‐suppressing peptides are attenuated in obesity, and this is thought to contribute to the further promotion of obesity. In addition, the intestinal flora is altered (decreases in phylum Bacteroidetes and increases in phylum Firmicutes), and lipopolysaccharide production is increased in obesity60. Lipopolysaccharide upregulates the expression of suppressor of cytokine signaling‐3 that elicits leptin resistance through Toll‐like receptor 4 expressed in the vagal afferent pathway61. Consequently, responses to CCK decline as previously described, leading to disrupted appetite suppression. It has been reported that such leptin resistance occurs at the vagus nerve before it occurs at the hypothalamus in an obese animal model61. We also showed that mice fed a high‐fat diet develop inflammation in the intestinal tract as a result of changes in the intestinal flora, and that this inflammation spreads to the vagus nerve ganglion cells and the hypothalamus through the vagal afferent pathway distributed along the intestinal tract62. Furthermore, inflammation also spreads to the vagus nerve ganglia and hypothalamus similarly in mice given a high‐fat diet for only 1 day63. In contrast, although plasminogen activator inhibitor‐1 is known to increase in obesity, plasminogen activator inhibitor‐1 is reported to antagonize the appetite‐suppressing effects of CCK by blocking the translocation of EGR1 into the nucleus in vagus nerve ganglion cells64. Therefore, a vicious cycle ensues in obesity, where the actions of appetite‐suppressing peptides are decreased through changes in the intestinal flora, leading to the development of inflammation and increased plasminogen activator inhibitor‐1.
Autonomic neuropathy is one of the complications of diabetes, and conditions such as orthostatic hypotension, gastric atony or neurogenic bladder are sometimes observed. In diabetes, demyelination of the vagus nerve, and damage to the axon and dendrite of the sympathetic ganglion are also observed at times65. This might lead to inadequate transmission of food intake signals through the vagus nerve from the aforementioned gut peptides to the central nervous system and the consequent development of appetite regulation abnormalities, such as hyperphagia. However, there are no appropriate methods at the present time to functionally evaluate the vagal afferent pathway of the digestive tract in humans. Future investigation is therefore necessary.
Weight Loss Therapy Targeting the Vagus Nerve
The vagus nerve is also currently the focus of study as a target for weight loss. During the time‐period when vagotomy was carried out to treat peptic ulcers, postoperative weight loss and decreased appetite were frequently observed66. However, these effects were not long‐term, and were attributed to the physical adaptation that occurs after vagotomy. In recent years, clinical studies in which the vagus nerve is electrically stimulated with an implantable device have been ongoing67. In an open‐label observational study, an implantable device with electrodes positioned at the anterior and posterior vagal trunks was evaluated for 6 months, and a 14.2% (mean) decrease in excess weight (total bodyweight – ideal bodyweight) was observed in study participants with a mean body mass index of 41.267. In a study that followed obese patients with type 2 diabetes for 1 year, the mean excess weight loss was 25%, HbA1c declined by 1.0% and systolic blood pressure was reduced by 8 mmHg68. In a recently reported double‐blind controlled trial that included 239 morbidly obese individuals (mean body mass index of 41.0), a 8.6 × 7.1 × 1.6‐cm device was ligated to the vagus nerve by a laparoscopic procedure under general anesthesia, and a controller was implanted subcutaneously69. Both the treatment and control groups received diet and exercise counseling, as well as weight management education, and were followed for 1 year. Device manipulations, such as periodic battery recharging, were required for both groups. Participants in the treatment group received at least 12 h of vagal blocking by electrical stimulation during waking hours per day, whereas those in the control group received a sham device that did not deliver electrical stimuli. Although a 6% initial bodyweight loss was observed in the control group after 1 year, a significantly greater weight loss of 9.2% was observed in the treatment group. At 1 year, 52% and 38% of the treatment group achieved an excess weight loss of at least 20% and at least 25%, respectively, and both of these levels were significantly greater than that of the control group. Although serious adverse events were not observed, mild to moderate heartburn, dyspepsia and abdominal pain were observed in the treatment group69. The 18‐month follow‐up results from this trial were recently reported; the treatment group weight loss was maintained at 8.8% (similar to the level at 12 months), whereas the control group rebounded to 3.8% weight loss70. There are several unclear points regarding the mechanism of weight loss induced by electrical stimulation to the vagus nerve. Although conditions, such as reduced gastric motility, reduced gastric excretion, increased postprandial satiety and decreased hunger between meals, are presumed to play a role in such a mechanism, future investigations regarding the role of gut peptides are anticipated.
Conclusion
The vagus nerve serves to link and exchange signals between the periphery and central nervous system, and is an essential pathway in the regulation of food intake and energy metabolism. It has become clear that abnormalities, such as inflammation, are elicited in the vagus nerve in obesity, leading to disrupted transmission of signals for appetite regulation. In addition, although the vagus nerve has become a target for weight loss therapy, it is anticipated that the mechanism of food intake and energy metabolism regulation involving the vagus nerve will be clarified, and that the application of such findings will lead to the development of novel therapeutics in the future.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
This study was supported by a Grant for Clinical Research from Miyazaki University Hospital. We thank C Tanaka for technical assistance.
J Diabetes Investig 2016; 7: 812–818
References
- 1. Cowley MA, Pronchuk N, Fan W, et al Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron 1999; 24: 155–163. [DOI] [PubMed] [Google Scholar]
- 2. Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron 1999; 22: 221–232. [DOI] [PubMed] [Google Scholar]
- 3. Ollmann MM, Wilson BD, Yang YK, et al Antagonism of central melanocortin receptors in vitro and in vivo by agouti‐related protein. Science 1997; 278: 135–138. [DOI] [PubMed] [Google Scholar]
- 4. Huszar D, Lynch CA, Fairchild‐Huntress V, et al Targeted disruption of the melanocortin‐4 receptor results in obesity in mice. Cell 1997; 88: 131–141. [DOI] [PubMed] [Google Scholar]
- 5. Yeo GS, Farooqi IS, Aminian S, et al A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet 1998; 20: 111–112. [DOI] [PubMed] [Google Scholar]
- 6. Vaisse C, Clement K, Guy‐Grand B, et al A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet 1998; 20: 113–114. [DOI] [PubMed] [Google Scholar]
- 7. Farooqi IS, Keogh JM, Yeo GS, et al Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. N Engl J Med 2003; 348: 1085–1095. [DOI] [PubMed] [Google Scholar]
- 8. Srinivasan S, Lubrano‐Berthelier C, Govaerts C, et al Constitutive activity of the melanocortin‐4 receptor is maintained by its N‐terminal domain and plays a role in energy homeostasis in humans. J Clin Invest 2004; 114: 1158–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ghamari‐Langroudi M, Digby GJ, Sebag JA, et al G‐protein‐independent coupling of MC4R to Kir7.1 in hypothalamic neurons. Nature 2015; 520: 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen KY, Muniyappa R, Abel BS, et al RM‐493, a melanocortin‐4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. J Clin Endocrinol Metab 2015; 100: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y, Proenca R, Maffei M, et al Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372: 425–432. [DOI] [PubMed] [Google Scholar]
- 12. Pinto S, Roseberry AG, Liu H, et al Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science 2004; 304: 110–115. [DOI] [PubMed] [Google Scholar]
- 13. Nakazato M, Murakami N, Date Y, et al A role for ghrelin in the central regulation of feeding. Nature 2001; 409: 194–198. [DOI] [PubMed] [Google Scholar]
- 14. Date Y, Shimbara T, Koda S, et al Peripheral ghrelin transmits orexigenic signals through the noradrenergic pathway from the hindbrain to the hypothalamus. Cell Metab 2006; 4: 323–331. [DOI] [PubMed] [Google Scholar]
- 15. Date Y, Murakami N, Toshinai K, et al The role of the gastric afferent vagal nerve in ghrelin‐induced feeding and growth hormone secretion in rats. Gastroenterology 2002; 123: 1120–1128. [DOI] [PubMed] [Google Scholar]
- 16. Wu Q, Boyle MP, Palmiter RD. Loss of GABAergic signaling by AgRP neurons to the parabrachial nucleus leads to starvation. Cell 2009; 137: 1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tong Q, Ye CP, Jones JE, et al Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci 2008; 11: 998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maejima Y, Sedbazar U, Suyama S, et al Nesfatin‐1‐regulated oxytocinergic signaling in the paraventricular nucleus causes anorexia through a leptin‐independent melanocortin pathway. Cell Metab 2009; 10: 355–365. [DOI] [PubMed] [Google Scholar]
- 19. Arletti R, Benelli A, Bertolini A. Influence of oxytocin on feeding behavior in the rat. Peptides 1989; 10: 89–93. [DOI] [PubMed] [Google Scholar]
- 20. Atasoy D, Betley JN, Su HH, et al Deconstruction of a neural circuit for hunger. Nature 2012; 488: 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Swaab DF, Purba JS, Hofman MA. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader‐Willi syndrome: a study of five cases. J Clin Endocrinol Metab 1995; 80: 573–579. [DOI] [PubMed] [Google Scholar]
- 22. Iwasaki Y, Maejima Y, Suyama S, et al Peripheral oxytocin activates vagal afferent neurons to suppress feeding in normal and leptin‐resistant mice: a route for ameliorating hyperphagia and obesity. Am J Physiol Regul Integr Comp Physiol 2015; 308: R360–R369. [DOI] [PubMed] [Google Scholar]
- 23. Lawson EA, Marengi DA, DeSanti RL, et al Oxytocin reduces caloric intake in men. Obesity (Silver Spring) 2015; 23: 950–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shimada M, Tritos NA, Lowell BB, et al Mice lacking melanin‐concentrating hormone are hypophagic and lean. Nature 1998; 396: 670–674. [DOI] [PubMed] [Google Scholar]
- 25. Date Y, Ueta Y, Yamashita H, et al Orexins, orexigenic hypothalamic peptides, interact with autonomic, neuroendocrine and neuroregulatory systems. Proc Natl Acad Sci USA 1999; 96: 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sakurai T, Amemiya A, Ishii M, et al Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein‐coupled receptors that regulate feeding behavior. Cell 1998; 92: 573–585. [DOI] [PubMed] [Google Scholar]
- 27. Borowsky B, Durkin MM, Ogozalek K, et al Antidepressant, anxiolytic and anorectic effects of a melanin‐concentrating hormone‐1 receptor antagonist. Nat Med 2002; 8: 825–830. [DOI] [PubMed] [Google Scholar]
- 28. Ito M, Ishihara A, Gomori A, et al Melanin‐concentrating hormone 1‐receptor antagonist suppresses body weight gain correlated with high receptor occupancy levels in diet‐induced obesity mice. Eur J Pharmacol 2009; 624: 77–83. [DOI] [PubMed] [Google Scholar]
- 29. Matsuda K, Azuma M, Maruyama K, et al Neuroendocrine control of feeding behavior and psychomotor activity by pituitary adenylate cyclase‐activating polypeptide (PACAP) in vertebrates. Obes Res Clin Pract 2013; 7: e1–e7. [DOI] [PubMed] [Google Scholar]
- 30. Choi YH, Fujikawa T, Lee J, et al Revisiting the ventral medial nucleus of the hypothalamus: The roles of SF‐1 neurons in energy homeostasis. Front Neurosci 2013; 7: 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim KW, Zhao L, Donato J Jr, et al Steroidogenic factor 1 directs programs regulating diet‐induced thermogenesis and leptin action in the ventral medial hypothalamic nucleus. Proc Natl Acad Sci USA 2011; 108: 10673–10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Resch JM, Boisvert JP, Hourigan AE, et al Stimulation of the hypothalamic ventromedial nuclei by pituitary adenylate cyclase‐activating polypeptide induces hypophagia and thermogenesis. Am J Physiol Regul Integr Comp Physiol 2011; 301: R1625–R1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mounien L, Do Rego JC, Bizet P, et al Pituitary adenylate cyclase‐activating polypeptide inhibits food intake in mice through activation of the hypothalamic melanocortin system. Neuropsychopharmacology 2009; 34: 424–435. [DOI] [PubMed] [Google Scholar]
- 34. Tanida M, Hayata A, Shintani N, et al Central PACAP mediates the sympathetic effects of leptin in a tissue‐specific manner. Neuroscience 2013; 238: 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hawke Z, Ivanov TR, Bechtold DA, et al PACAP neurons in the hypothalamic ventromedial nucleus are targets of central leptin signaling. J Neurosci 2009; 29: 14828–14835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Inoue S, Egawa M, Satoh S, et al Clinical and basic aspects of an anorexiant, mazindol, as an antiobesity agent in Japan. Am J Clin Nutr 1992; 55: 199S–202S. [DOI] [PubMed] [Google Scholar]
- 37. Samanin R, Bendotti C, Bernasconi S, et al Role of brain monoamines in the anorectic activity of mazindol and d‐amphetamine in the rat. Eur J Pharmacol 1977; 43: 117–124. [DOI] [PubMed] [Google Scholar]
- 38. Smith BM, Smith JM, Tsai JH, et al Discovery and structure‐activity relationship of (1R)‐8‐chloro‐2,3,4,5‐tetrahydro‐1‐methyl‐1H‐3‐benzazepine (Lorcaserin), a selective serotonin 5‐HT2C receptor agonist for the treatment of obesity. J Med Chem 2008; 51: 305–313. [DOI] [PubMed] [Google Scholar]
- 39. Martin CK, Redman LM, Zhang J, et al Lorcaserin, a 5‐HT2C receptor agonist, reduces body weight by decreasing energy intake without influencing energy expenditure. J Clin Endocrinol Metab 2011; 96: 837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fidler MC, Sanchez M, Raether B, et al A one‐year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab 2011; 96: 3067–3077. [DOI] [PubMed] [Google Scholar]
- 41. Smith SR, Weissman NJ, Anderson CM, et al Multicenter, placebo‐controlled trial of lorcaserin for weight management. N Engl J Med 2010; 363: 245–256. [DOI] [PubMed] [Google Scholar]
- 42. Berthoud HR, Neuhuber WL. Functional and chemical anatomy of the afferent vagal system. Auton Neurosci 2000; 85: 1–17. [DOI] [PubMed] [Google Scholar]
- 43. Berthoud HR. Vagal and hormonal gut‐brain communication: from satiation to satisfaction. Neurogastroenterol Motil 2008; 20(Suppl 1): 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bonnet MS, Ouelaa W, Tillement V, et al Gastric distension activates NUCB2/nesfatin‐1‐expressing neurons in the nucleus of the solitary tract. Regul Pept 2013; 187: 17–23. [DOI] [PubMed] [Google Scholar]
- 45. Vrang N, Phifer CB, Corkern MM, et al Gastric distension induces c‐Fos in medullary GLP‐1/2‐containing neurons. Am J Physiol Regul Integr Comp Physiol 2003; 285: R470–R478. [DOI] [PubMed] [Google Scholar]
- 46. Crawley JN, Corwin RL. Biological actions of cholecystokinin. Peptides 1994; 15: 731–755. [DOI] [PubMed] [Google Scholar]
- 47. Batterham RL, Cowley MA, Small CJ, et al Gut hormone PYY(3‐36) physiologically inhibits food intake. Nature 2002; 418: 650–654. [DOI] [PubMed] [Google Scholar]
- 48. Shimizu H, Oh IS, Hashimoto K, et al Peripheral administration of nesfatin‐1 reduces food intake in mice: the leptin‐independent mechanism. Endocrinology 2009; 150: 662–671. [DOI] [PubMed] [Google Scholar]
- 49. Iwasaki Y, Nakabayashi H, Kakei M, et al Nesfatin‐1 evokes Ca2 + signaling in isolated vagal afferent neurons via Ca2 + influx through N‐type channels. Biochem Biophys Res Commun 2009; 390: 958–962. [DOI] [PubMed] [Google Scholar]
- 50. Shimizu H, Ohsaki A, Oh IS, et al A new anorexigenic protein, nesfatin‐1. Peptides 2009; 30: 995–998. [DOI] [PubMed] [Google Scholar]
- 51. de Lartigue G, Lur G, Dimaline R, et al EGR1 is a target for cooperative interactions between cholecystokinin and leptin, and inhibition by ghrelin, in vagal afferent neurons. Endocrinology 2010; 151: 3589–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Peters JH, Simasko SM, Ritter RC. Modulation of vagal afferent excitation and reduction of food intake by leptin and cholecystokinin. Physiol Behav 2006; 89: 477–485. [DOI] [PubMed] [Google Scholar]
- 53. Noetzel S, Stengel A, Inhoff T, et al CCK‐8S activates c‐Fos in a dose‐dependent manner in nesfatin‐1 immunoreactive neurons in the paraventricular nucleus of the hypothalamus and in the nucleus of the solitary tract of the brainstem. Regul Pept 2009; 157: 84–91. [DOI] [PubMed] [Google Scholar]
- 54. Burdyga G, Lal S, Spiller D, et al Localization of orexin‐1 receptors to vagal afferent neurons in the rat and humans. Gastroenterology 2003; 124: 129–139. [DOI] [PubMed] [Google Scholar]
- 55. Date Y, Toshinai K, Koda S, et al Peripheral interaction of ghrelin with cholecystokinin on feeding regulation. Endocrinology 2005; 146: 3518–3525. [DOI] [PubMed] [Google Scholar]
- 56. Karl JP, Young AJ, Montain SJ. Eating rate during a fixed‐portion meal does not affect postprandial appetite and gut peptides or energy intake during a subsequent meal. Physiol Behav 2011; 102: 524–531. [DOI] [PubMed] [Google Scholar]
- 57. Shiiya T, Nakazato M, Mizuta M, et al Plasma ghrelin levels in lean and obese humans and the effect of glucose on ghrelin secretion. J Clin Endocrinol Metab 2002; 87: 240–244. [DOI] [PubMed] [Google Scholar]
- 58. Ronveaux CC, Tome D, Raybould HE. Glucagon‐like peptide 1 interacts with ghrelin and leptin to regulate glucose metabolism and food intake through vagal afferent neuron signaling. J Nutr 2015; 145: 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kentish S, Li H, Philp LK, et al Diet‐induced adaptation of vagal afferent function. J Physiol 2012; 590: 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. DiBaise JK, Zhang H, Crowell MD, et al Gut microbiota and its possible relationship with obesity. Mayo Clin Proc 2008; 83: 460–469. [DOI] [PubMed] [Google Scholar]
- 61. de Lartigue G, Barbier de la Serre C, Espero E, et al Diet‐induced obesity leads to the development of leptin resistance in vagal afferent neurons. Am J Physiol Endocrinol Metab 2011; 301: E187–E195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Naznin F, Toshinai K, Waise TM, et al Diet‐induced obesity causes peripheral and central ghrelin resistance by promoting inflammation. J Endocrinol 2015; 226: 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Waise TM, Toshinai K, Naznin F, et al One‐day high‐fat diet induces inflammation in the nodose ganglion and hypothalamus of mice. Biochem Biophys Res Commun 2015; 464: 1157–1162. [DOI] [PubMed] [Google Scholar]
- 64. Kenny S, Gamble J, Lyons S, et al Gastric expression of plasminogen activator inhibitor (PAI)‐1 is associated with hyperphagia and obesity in mice. Endocrinology 2013; 154: 718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Guy RJ, Dawson JL, Garrett JR, et al Diabetic gastroparesis from autonomic neuropathy: surgical considerations and changes in vagus nerve morphology. J Neurol Neurosurg Psychiatry 1984; 47: 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Irving AD, Smith G, Coubrough H. Long‐term metabolic effects of truncal vagotomy and gastrojejunostomy for chronic duodenal ulcer. Clin Nutr 1985; 4: 129–133. [DOI] [PubMed] [Google Scholar]
- 67. Camilleri M, Toouli J, Herrera MF, et al Intra‐abdominal vagal blocking (VBLOC therapy): clinical results with a new implantable medical device. Surgery 2008; 143: 723–731. [DOI] [PubMed] [Google Scholar]
- 68. Shikora S, Toouli J, Herrera MF, et al Vagal blocking improves glycemic control and elevated blood pressure in obese subjects with type 2 diabetes mellitus. J Obes 2013; 2013: 245683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ikramuddin S, Blackstone RP, Brancatisano A, et al Effect of reversible intermittent intra‐abdominal vagal nerve blockade on morbid obesity: the ReCharge randomized clinical trial. JAMA 2014; 312: 915–922. [DOI] [PubMed] [Google Scholar]
- 70. Shikora SA, Wolfe BM, Apovian CM, et al Sustained weight loss with vagal nerve blockade but not with dsam: 18‐month results of the ReCharge trial. J Obes 2015; 2015: 365604. [DOI] [PMC free article] [PubMed] [Google Scholar]