

Abstract
Aims/Introduction
An elevated level of plasma homocysteine has long been suspected as a metabolic risk factor for the development of atherosclerotic vascular diseases in diabetes. Berberine (BBR) has several preventive effects on cardiovascular diseases. The effects of BBR on atherosclerotic plaque stability increased by homocysteine thiolactone (HTL) remain unknown.
Materials and Methods
The model of atherosclerotic vulnerable plaque was induced by placing a collar around the carotid artery in Apoe −/− mice. Endothelium‐dependent relaxation was assayed by organ chamber.
Results
Homocysteine thiolactone (50 mg/kg/day, 8 weeks) reduced the atherosclerotic plaque stability in the carotid artery of Apoe −/− mice, which was reversed by BBR administration (1.0 g/kg/day). In vivo and ex vivo experiments showed that HTL dramatically reduced acetylcholine‐induced endothelium‐dependent relaxation and superoxide dismutase activity, and increased malondialdehyde content, which were inhibited by BBR. Importantly, all effects induced by BBR were abolished by GW9662, an antagonist of peroxisome proliferator‐activated receptor‐γ. Incubation of cultured endothelial cells with HTL significantly reduced cell viabilities and enhanced production of reactive oxygen species. Pretreatment of cells with BBR dose‐dependently reversed HTL‐induced detrimental effects, which were GW9662‐reversible.
Conclusions
Berberine increases atherosclerotic plaque stability in hyperhomocysteinemia mice, which is related to the activation of peroxisome proliferator‐activated receptor‐γ and subsequent suppression of oxidative stress in endothelial cells.
Keywords: Atherosclerosis, Berberine, Peroxisome proliferator‐activated receptor‐γ
Short abstract
An elevated level of plasma homocysteine has long been suspected as a metabolic risk factor for the development of atherosclerotic vascular diseases in diabetes. We reported that berberine, as a natural production, increase atherosclerotic plaque stability in hyperhomocysteinaemia mice, which is related to the activation of PPARγ and subsequent suppression of oxidative stress in endothelial cells.
Introduction
Atherosclerotic coronary artery disease, the underlying basis for ischemic heart disease, is the leading cause of death and disability in diabetes patients1, 2. Moderate hyperhomocysteinemia (HHCY) has been observed in some studies of diabetic patients with atherosclerosis. The homocysteine levels reflect the nature of the patients studied, including patients with poor glycemic control, variable duration of diabetes, and a variety of microvascular and macrovascular complications3. HHCY might play a role in the pathogenesis of vascular disorders, and is considered as an independent risk factor for atherosclerosis4.
Homocysteine occurs in human blood plasma in several forms, including the most reactive one, which is homocysteine thiolactone (HTL) of a cyclic trimester, and represents up to 0.29% of total plasma homocysteine5. HTL reacts with proteins by acylation of free basic amino groups. In particular, the epsilon‐amino group of lysine residues forms adducts, and induces structural and functional changes in plasma proteins. High levels of homocysteine impair endothelial function and cause endothelial damage in humans as well as in animal models6, 7, showing that the endothelial monolayer is very sensitive to changes in plasma homocysteine levels.
Berberine (BBR), an isoquinoline alkaloid originally isolated from the Chinese herb Coptis chinensis, is an antimicrobial drug routinely prescribed for the treatment of diarrhea in many Asian countries8. In this form it is reported to exert antifungal, antibacterial/viral and anti‐oncogenic effects, as well as a beneficial effect on diabetes, atherosclerosis and hyperlipidemia9, 10. Although protective effects of BBR have been observed in different animal models11, the effects of BBR on HHCY‐induced atherosclerotic plaque instability and the underlying mechanism are poorly understood. Here we reported that pharmacological activation of peroxisome proliferator‐activated receptor‐γ (PPARγ) by BBR increases atherosclerotic plaque stability in Apoe −/− mice with HHCY.
Materials and Methods
Materials
BBR, GW9662, pyrollidine dithiocarbamate (PDTC), dihydroethidium (DHE), apocynin, acetylcholine (Ach), sodium nitroprusside (SNP) and phenylephrine (PE) were purchased from Sigma Company (St. Louis, MO, USA). Commercial kits for determinations of malondialdehyde (MDA) and superoxide dismutase (SOD) activity were from Cayman Company (Ann Arbor, MI, USA).
Animals and collar placement around carotid artery
Apoe −/− mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). Mice were housed in temperature‐controlled cages with a 12‐h light–dark cycle. The animal protocol was reviewed and approved by Jilin University Animal Care and Use Committee. As described previously12, 13, a constrictive silastic tube (0.30‐mm inner diameter, 0.50‐mm outer diameter and 2.5‐mm long; Shandong Key Laboratory of Medical Polymer Materials, Jinan, China) was placed around the left common carotid artery near its bifurcation in male Apoe −/− mice at the age of 8–12 weeks.
Determination of plaque vulnerable index
The plaque vulnerable index was determined by using the ratio of CD68‐positive (%) plus Oil Red (%) to α‐actin (%) plus collagen (%) as described previously14. Two different lesion areas were chosen to account for each segment, and the mean was used in statistical analysis.
Cell culture
As described previously15, human umbilical vascular endothelial cells (HUVECs) from ATCC were grown in endothelial cells basal medium (Clonetics Inc., Walkersville, MD, USA) supplemented with 2% fetal bovine serum, penicillin (100 U/mL) and streptomycin (100 μg/mL). In all experiments, cells were between passages 3 and 8. All cells were incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Cells were grown to 70–80% confluency before being treated with different agents.
Organ chamber
An organ chamber study was carried out as described previously16. Mice were killed under anesthesia by intravenous injection with pentobarbital sodium (30 mg/kg). The descending aorta was isolated by removing the adhering perivascular tissue carefully, and was cut into rings (3–4 mm in length). Aortic rings were suspended and mounted to the organ chamber by using two stainless hooks. The rings were placed in organ baths filled with Krebs' buffer of the following compositions (in mmol/L): NaCl, 118.3; KCl, 4.7; MgSO4, 0.6; KH2PO4, 1.2; CaCl2, 2.5; NaHCO3, 25.0; ethylenediaminetetraacetic acid, 0.026; and pH 7.4 at 37°C, and gassed with 95% O2 plus 5% CO2, under a tension of 1.0 g, for a 90‐min equilibration period. During this period, the Krebs' solution was changed every 15 min. After the equilibration, aortic rings were challenged with 60 mmol/L KCl. After washing and another 30‐min equilibration period, contractile response was elicited by PE (1 μmol/L). At the plateau of contraction, accumulative Ach (0.01, 0.03, 0.1, 0.3, 1, 3 μmol/L) or SNP (0.01, 0.03, 0.1, 0.3, 1, 3, 10 μmol/L) was added into the organ bath to induce endothelium‐dependent or ‐independent relaxation.
For ex vivo experiments, the rings were contracted by PE (1 μmol/L) and then dilated with cumulative concentrations of Ach (0.01–3 μmol/L) to assess the integrity of the endothelium. The ring, which the maximal relaxation induced by Ach (3 μmol/L) was over 80%, was considered to have an intact endothelium and was used in the following study. Then, the rings were pretreated with BBR (10, 50, 100 μmol/L) for 30 min followed by the addition of HTL (1 mmol/L) for 90 min. After washing, Ach‐induced endothelium‐dependent relaxation and SNP‐induced endothelium‐independent relaxation were assayed, respectively. At the end of the experiments, the aortic rings were collected in liquid nitrogen for measurements of nitric oxide (NO) and MDA after being homogenized.
Measurements of MDA content, SOD activity and NO level
The contents of MDA content, SOD activity and NO level in aortic tissues or blood were assayed by using commercial kits following the recommend protocol as described previously17.
Evaluation of cell viability
Cell viability was assayed by using methylthiazolyldiphenyl‐tetrazolium bromide (MTT) as described previously18. Cells were seeded onto a 96‐well plate at the density of 10,000 cells/mL, and incubated for 24 h. After treatment, 10 μL MTT (5 mg/mL) was added into cultured medium in each well for 2–4 h until purple precipitate was visible. After removal of culture medium, 75 μL dimethylsulfoxide was added to each well and the cells were left at room temperature in the dark for 2 h. Absorbance at 570 nm was recorded.
Detection of reactive oxygen species
Reactive oxygen species (ROS) production in cultured cells was assayed by measuring the DHE fluorescence as described previously15, 19. Briefly, before the end of treatment, 10 μmol/L DHE was added to the medium and incubated for 30 min at 37°C, then washed with phosphate‐buffered saline twice. The image was taken by fluorescent microscope. The DHE fluorescent intensity was recorded by a fluorescent reader at the wave of excitation (485 nm) and emission (545 nm). Control was set up as 100%.
Statistical analysis
Data are reported as mean ± SE of the mean. All data were analyzed with the use of one‐ or two‐way anova followed by multiple t‐tests, and P < 0.05 were considered statistically significant.
Results
HHCY reduces atherosclerotic plaque stability in Apoe −/− mice
HHCY, as an independent risk factor of atherosclerosis, we first examined the effects of HHCY on the formation of prone‐to‐rupture plaque in advanced atherosclerosis, which is characterized by a thin fibrous cap, a large cholesterol deposition, rich in inflammatory cells and a few smooth muscle cells20. The HHCY model was induced by feeding Apoe −/− mice with HTL as described previously17. We fed Apoe −/− mice with HTL (50 mg/kg/day) for 8 weeks to mimic the model of HHCY. Treatment of mice with HTL dramatically increased plasma HTL levels, compared with control mice (7.38 ± 1.94 vs 2.91 ± 1.08 μmol/L, P < 0.01). The plasma level of HCY (7.28 ± 2.12 vs 5.64 ± 1.73 μmol/L, P < 0.05) was also slightly increased by treatment of HTL. These indexes showed that supplementation of HTL in the diet induced high blood levels of the HCY and HTL model in mice. The surgery of collar placement around the left common carotid artery was carried out to induce the formation of vulnerable plaque. As shown in Figure 1a–e, morphological and immunohistochemistry analysis showed that Oil Red+ lipid area and CD68+ area were significantly increased in mice fed with HTL, whereas Sirius Red+ collagen structures in atherosclerotic plaques were significantly decreased. The plaque vulnerable index was calculated according to the ratio of area I (Oil Red++CD68+) to area II [α‐smooth muscle actin (SMA)++collagen+] as described previously21. Statistically, HTL dramatically increased the plaque vulnerable index in mice (Figure 1f).
Figure 1.
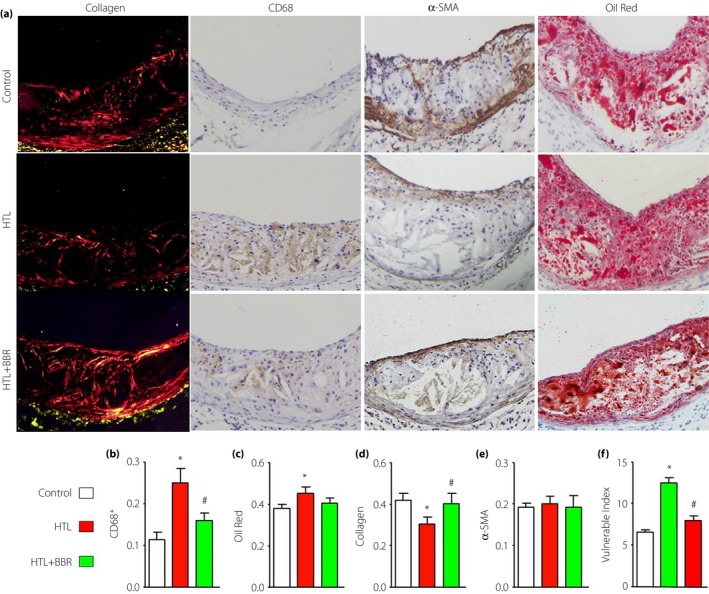
Administration of berberine (BBR) increases carotid atherosclerotic plaque stability in Apoe −/− mice. Male Apoe −/− mice at the age of 8–12 weeks fed with homocysteine thiolactone (HTL; 50 mg/kg/day) and having received administration of BBR (1.0 g/kg/day) for 4 weeks. Then a model of vulnerable atherosclerotic plaque was established by collar placement around the left common carotid near its bifurcation. Then, 8 weeks later, mice were killed under anesthesia. (a) Morphology of the left common carotid arteries by staining Oil Red for lipid content, PicroSirius Red for collagen, α‐smooth muscle actin (α‐SMA) for vascular smooth muscle cells and CD68 for macrophages, respectively. (b) Quantitative data for macrophage numbers. (c) Quantitative data for lipid content. (d) Quantitative data for collagen. (e) Quantitative data for vascular smooth muscle cells numbers. (f) Vulnerability index was determined by using the ratio of CD68+ (%) plus Oil Red (%) to α‐SMA (%) plus collagen (%). Each group n = 10–15. *P < 0.05 vs control mice. # P < 0.05 vs mice fed with HTL.
Administration of BBR increases plaque stability in HHCY mice
We then determined the effects of BBR on the stability of advanced atherosclerotic plaque. As shown in Figure 1a–e, compared with HTL‐fed mice, BBR significantly decreased the Oil Red+ lipid area and CD68+ macrophage area, and increased the Sirius Red+ collagen area, accompanied with the reduction of plaque vulnerable index (Figure 1f). Taking these data together, it suggests that BBR increases plaque stability in advanced vulnerable atherosclerosis in HHCY mice.
Administration of BBR improves endothelial function in mice wed with HTL
To investigate the mechanism of BBR on suppressing the formation of vulnerable atherosclerosis, we tested whether BBR improves vascular endothelial functions in mice with HHCY. Endothelial function was determined by measuring the vascular dilution induced by Ach. As shown in Figure 2a, HTL significantly inhibited Ach‐induced vascular relaxation. Importantly, the reduction of Ach‐induced vascular relaxation was reversed by administration of BBR. Both HTL and BBR did not change SNP‐induced vessel relaxation (Figure 2b), suggesting that the improvement of BBR on vascular bioactivity in HTL‐fed mice is due to maintaining endothelial function.
Figure 2.
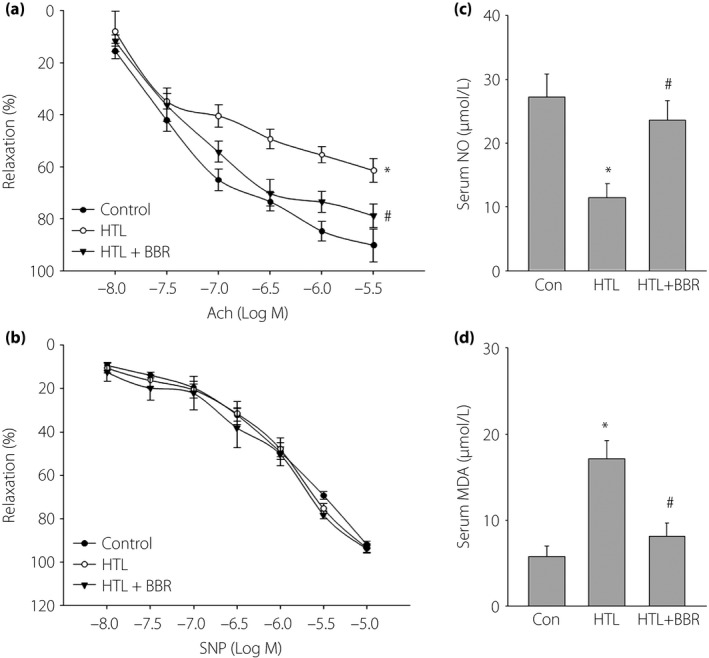
Berberine (BBR) suppresses oxidative stress and improves endothelial functions in mice fed with homocysteine thiolactone (HTL) in vivo. The mice were intragastrically gavaged HTL (50 mg/kg/day) and received BBR (1.0 g/kg/day) for 8 weeks. At the end of the experiments, the mice were killed under anesthesia. The artery from the descending aorta was subjected to (a) endothelium‐dependent relaxation induced by acetylcholine (Ach) assay and (b) endothelium‐independent relaxation induced by sodium nitroprusside (SNP) in the organ chamber assay. Blood was collected to assay the serum level of (c) nitric oxide (NO) by the Griess method and (d) malondialdehyde (MDA) by the thiobarbituric acid method. All data are expressed as mean ± SE of the mean. Each group n = 5–10. *P < 0.05 vs control (Con), # P < 0.05 vs HTL alone.
Administration of BBR suppresses oxidative stress in mice fed with HTL
We also determined whether BBR preserves the normal redox state in HTL‐fed mice. As expected, HTL induced the alternations, such as decreased serum NO level (Figure 2c) and increased serum levels of MAD (Figure 2d). All these defects induced by HTL were normalized by administration of BBR. This evidence indicate that the in vivo protective effects of BBR might be related to suppression of oxidative stress.
BBR through PPARγ preserves endothelium‐dependent relaxation impaired by HTL in mice isolated aortic rings
We then examined how BBR suppressed oxidative stress and protected vascular endothelial functions. Exposure of the aortic ring to HTL dramatically impaired Ach‐induced endothelium‐dependent relaxation, which was reversed by BBR dose‐dependently in aortic rings (Figure 3a). By using GW9662 of PPARγ antagonist22, we observed that GW9662 significantly abolished the protective effects of BBR on Ach‐induced endothelium‐dependent relaxation (Figure 3b), indicating that BBR through PPARγ activation protects endothelial function. In addition, SNP‐induced endothelium‐independent relaxation was not altered in all groups (Figure 3c), suggesting that the protective effects produced by BBR on vascular function are limited to endothelium, but not vascular smooth muscle cells.
Figure 3.
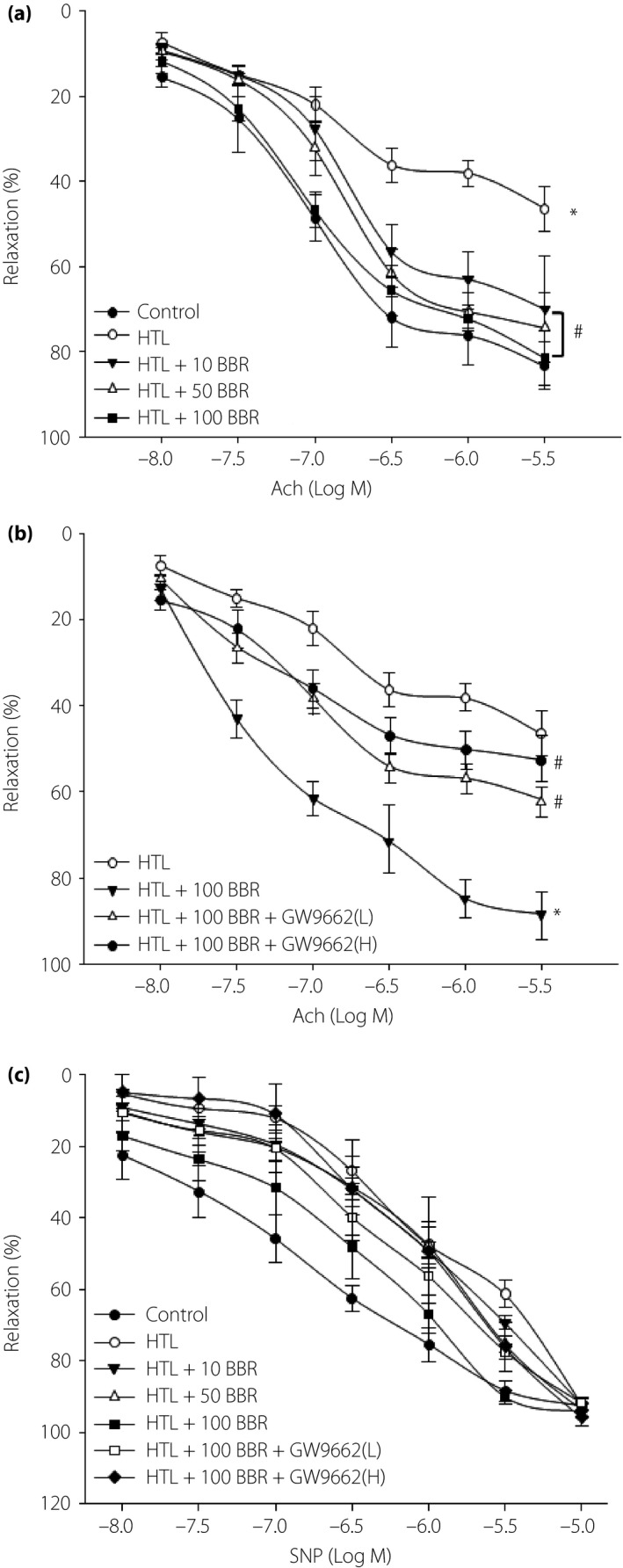
Berberine (BBR) through peroxisome proliferator‐activated receptor‐γ (PPARγ) prevents the impairment of endothelium‐dependent relaxation induced by homocysteine thiolactone (HTL) in isolated mouse aortas. (a) The isolated mouse aortic rings were exposed to HTL (1 mmol/L) for 1 h after pre‐incubation with BBR (10, 50, 100 μmol/L) for 30 min. The endothelium‐dependent relaxation induced by acetylcholine (Ach) was assayed by organ chamber. All data were expressed as mean ± SE of the mean. Each group n = 5. *P < 0.05 vs control, # P < 0.05 vs HTL alone. (b) The isolated mouse aortic rings were exposed to HTL (1 mmol/L) for 1 h after pre‐incubation with BBR (100 μmol/L) for 30 min with GW9662 (0.01, 0.1 mmol/L). The endothelium‐dependent relaxation induced by Ach was assayed by organ chamber. All data are expressed as mean ± SE of the mean. Each group n = 5. *P < 0.05 vs HTL alone, # P < 0.05 vs HTL+BBR. (c) Endothelium‐independent relaxation was assayed in all groups by using sodium nitroprusside (SNP).
BBR through PPARγ reserves redox state in aortas, which is disturbed by HTL
Decreased NO bioavailability, which is as a result of decreased NO production or aberrant conversion of NO to ONOO− by ROS, contributes to impairment of Ach‐induced endothelium‐dependent relaxation in the cardiovascular system23. We then examined whether HTL maintains a normal redox state in mouse isolated aortic rings. We found that HTL dramatically decreased SOD activity (Figure 4a) and increased the content of MDA (Figure 4b), which is formed when ROS reacts with polyunsaturated fatty acid chain in membrane lipids24. However, pretreatment of cells with GW9662 significantly abolished BBR‐rescued abnormalities in HTL‐incubated aortas, suggesting that BBR through PPARγ/SOD‐MDA reserves the normal balance of the anti‐oxidative system.
Figure 4.
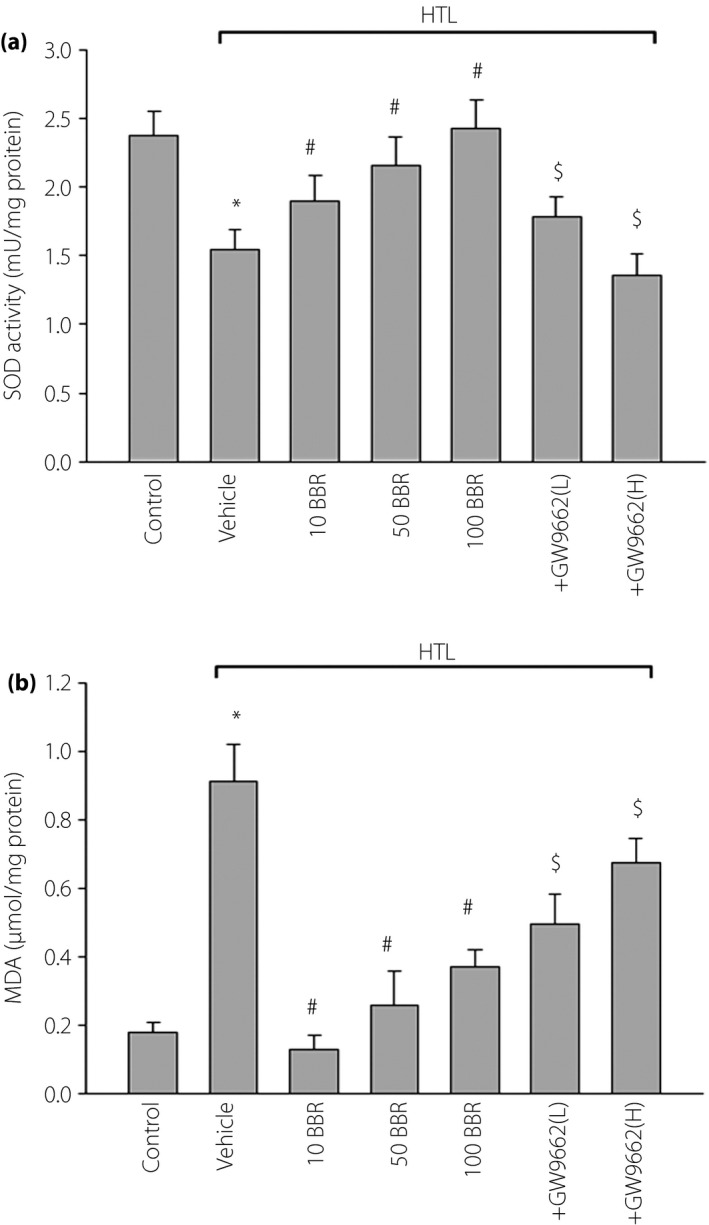
Berberine (BBR) through activation of peroxisome proliferator‐activated receptor‐γ (PPARγ) suppresses oxidative stress in isolated mouse aortas. The isolated mouse aortic rings were exposed to homocysteine thiolactone (HTL; 1 mmol/L) for 1 h after pre‐incubation with BBR (10, 50, 100 μmol/L) for 30 min. Aortas treated with 100 μmol/L BBR were in the presence of GW9662 (0.01, 0.1 mmol/L). Homogenates of aortic tissues were subjected to assay for (a) superoxide dismutase activity and (b) malondialdehyde (MDA) content by commercial kits. All data are expressed as mean ± SE of the mean. Each group n = 5. *P < 0.05 vs control, # P < 0.05 vs HTL alone, $ P < 0.05 vs HTL+100BBR.
BBR normalizes cell viabilities and oxidative stress in HTL‐treated endothelial cells
We also investigated whether HTL affected cell viabilities in cultured HUVECs. As shown in Figure 5a, incubation of HUVECs with HTL (1 mmol/L) for 24 h dramatically reduced cell viabilities detected by MTT assay, consistent with previous reports17. BBR alone did not affect endothelial cell viabilities, but dose‐dependently reversed cell viabilities reduced by HTL.
Figure 5.
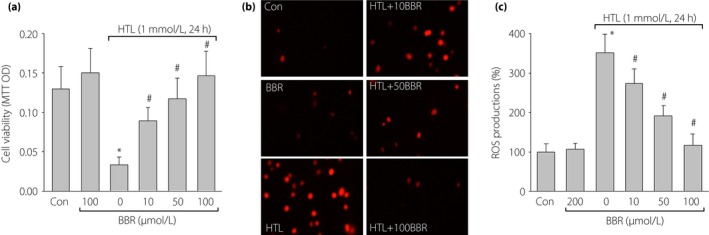
Berberine (BBR) dose‐dependently suppresses homocysteine thiolactone (HTL)‐induced oxidative stress and improves cell viabilities in cultured human umbilical vascular endothelial cells (HUVECs). Cultured HUVECs were pretreated with BBR (10, 50, 100 μmol/L) for 1 h and then incubated with HTL (1 mmol/L) for 24 h. (a) Cell viability was measured by methylthiazolyldiphenyl‐tetrazolium bromide. (b) Intracellular reactive oxygen species (ROS) production was detected by dihydroethidium fluorescence. (c) Quantitative analysis of ROS production. All data are expressed as mean ± SE of the mean. Each group n = 3. *P < 0.05 vs control (Con), # P < 0.05 vs HTL alone.
We determined whether BBR through suppression of oxidative stress maintains the normal phenotypes of vascular endothelial cells. In Figure 5b and c, incubation of HUVECs with HTL (1 mmol/L) for 24 h remarkably increased ROS production in cultured cells. However, pre‐incubation of these cells with BBR inhibited the enhancements of ROS production induced by HTL in a dose‐dependent manner. Taking these together, it shows that BBR protects cell viabilities reduced by HTL, which is possibly related to suppression of oxidative stress.
BBR through PPARγ protects HTL‐treated endothelial cells
Finally, we next determined whether BBR through activation of PPARγ provided beneficial effects in endothelial cells by using GW9662. As expected, inhibition of PPARγ by GW9662 significantly abolished the protective effects of BBR on the improvement of cell viabilities (Figure 6a), reductions of ROS production (Figure 6b,c) in a dose‐dependent manner. Taking these data together, it shows that BBR through PPARγ activation suppresses oxidative stress and protects cell viability.
Figure 6.
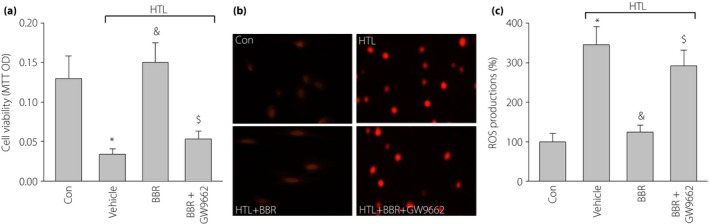
Inhibition of peroxisome proliferator‐activated receptor‐γ (PPARγ) abolished berberine (BBR)‐suppressed oxidative stress and maintains cell viability in homocysteine thiolactone (HTL)‐treated cells. Cultured human umbilical vascular endothelial cells (HUVECs) were pretreated with BBR (100 μmol/L) for 1 h and then incubated with HTL (1 mmol/L) for 24 h in the presence of GW9662 (0.01 mmol/L). Cells were subjected to assay; (a) cell viability by methylthiazolyldiphenyl‐tetrazolium bromide and (b) intracellular reactive oxygen species (ROS) production by dihydroethidium fluorescence. (c) Quantitative analysis of ROS production. All data are expressed as mean ± SE of the mean. Each group n = 3. *P < 0.05 vs control (Con), & P < 0.05 vs HTL alone, $ P < 0.05 vs HTL+BBR.
Discussion
The present study first shows that HTL in vitro or in vivo causes accelerated oxidative stress, endothelial dysfunction and formation of atherosclerotic vulnerable plaque, all of which are abrogated by BBR. Mechanistically, the protective effect of BBR on vascular function is attributable to PPARγ activation, resulting in suppression of oxidative stress.
The major finding of the present study was that BBR through activation of PPARγ prevents HTL‐induced endothelial dysfunctions. Recent studies have found that the conversion of homocysteine into HTL plays a critical role in the progress of cardiovascular diseases in patients with HHCY6, 25, 26. In this present study, we used HTL to treat isolated aortic ring ex vivo or mice in vivo, by which both impaired Ach‐induced endothelium‐dependent relaxation, consistent with our previous study27. This observation strongly supports the finding that the detrimental effects of homocysteine are related to the high reactivity of HTL, though the level of plasma thiolactone is very low. Most importantly, HTL‐induced endothelial dysfunction both ex vivo and in vivo was reversed by BBR, which is in a dose‐dependent manner. Collectively, the present results suggest that BBR functions as a protector of endothelial cells. This discovery is also supported by several published studies carried out in cultured endothelial cells or animals, which have shown that BBR protects endothelial function28, 29, 30. However, a recent study on humans reported that BBR caused endothelial dysfunction in normal volunteers31. Of course, the reason for this discrepancy between healthy and HHCY requires further investigation.
Another finding of the present study is that BBR suppresses the formation of vulnerable plaque in advanced atherosclerosis. Previous studies have reported that BBR improved endothelial dysfunction and prevented atherosclerosis28, 32. In the present study, we further uncovered that BBR carried out its anti‐oxidative action to increase plaque stability by providing in vitro, ex vivo or in vivo experimental evidence.
In summary, these studies support a novel function of BBR that activates PPARγ to suppress oxidative stress. This, in turn, leads to the improvement of endothelial function and atherosclerotic plaque stability. The finding that BBR attenuates endothelial dysfunction induced by HTL through suppression of oxidative stress could have broad applications for cardiovascular diseases, as endothelial dysfunction is a common characteristic at the beginning and in the progress of a number of vascular diseases including atherosclerosis33, 34 and diabetes35. Thus, BBR might be a useful drug for more effective treatments of atherosclerosis and hypertension.
Disclosure
The authors declare no conflict of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81472030, 81472454, 31100930 and 81572082), Natural Science Foundation of Jilin Provincial Science and Technology Department, China (20150414015GH, 20110739 and 20150204001YY).
J Diabetes Investig 2016; 7: 824–832
References
- 1. Chan CP, Rainer TH. Pathophysiological roles and clinical importance of biomarkers in acute coronary syndrome. Adv Clin Chem 2013; 59: 23–63. [DOI] [PubMed] [Google Scholar]
- 2. Heeschen C. Biomarkers in acute coronary syndromes and their role in diabetic patients. Diab Vasc Dis Res 2005; 2: 122–127. [DOI] [PubMed] [Google Scholar]
- 3. Hadi HA, Suwaidi JA. Endothelial dysfunction in diabetes mellitus. Vasc Health Risk Manag 2007; 3: 853–876. [PMC free article] [PubMed] [Google Scholar]
- 4. McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. Am J Pathol 1969; 56: 111–128. [PMC free article] [PubMed] [Google Scholar]
- 5. Malinowska J, Kolodziejczyk J, Olas B. The disturbance of hemostasis induced by hyperhomocysteinemia; the role of antioxidants. Acta Biochim Pol 2012; 59: 185–194. [PubMed] [Google Scholar]
- 6. Kolodziejczyk‐Czepas J, Talar B, Nowak P, et al Homocysteine and its thiolactone impair plasmin activity induced by urokinase or streptokinase in vitro. Int J Biol Macromol 2012; 50: 754–758. [DOI] [PubMed] [Google Scholar]
- 7. Liu Y, Tian T, Zhang H, et al The effect of homocysteine‐lowering therapy with folic acid on flow‐mediated vasodilation in patients with coronary artery disease: a meta‐analysis of randomized controlled trials. Atherosclerosis 2014; 235: 31–35. [DOI] [PubMed] [Google Scholar]
- 8. Affuso F, Mercurio V, Fazio V, et al Cardiovascular and metabolic effects of Berberine. World J Cardiol 2010; 2: 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mokhber‐Dezfuli N, Saeidnia S, Gohari AR, et al Phytochemistry and pharmacology of berberis species. Pharmacogn Rev 2014; 8: 8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang Q, Zhang M, Liang B, et al Activation of AMP‐activated protein kinase is required for berberine‐induced reduction of atherosclerosis in mice: the role of uncoupling protein 2. PLoS ONE 2011; 6: e25436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lau CW, Yao XQ, Chen ZY, et al Cardiovascular actions of berberine. Cardiovasc Drug Rev 2001; 19: 234–244. [DOI] [PubMed] [Google Scholar]
- 12. von der Thusen JH, van Berkel TJ, Biessen EA. Induction of rapid atherogenesis by perivascular carotid collar placement in apolipoprotein E‐deficient and low‐density lipoprotein receptor‐deficient mice. Circulation 2001; 103: 1164–1170. [DOI] [PubMed] [Google Scholar]
- 13. de Nooijer R, von der Thusen JH, Verkleij CJ, et al Overexpression of IL‐18 decreases intimal collagen content and promotes a vulnerable plaque phenotype in apolipoprotein‐E‐deficient mice. Arterioscler Thromb Vasc Biol 2004; 24: 2313–2319. [DOI] [PubMed] [Google Scholar]
- 14. Dong M, Zhong L, Chen WQ, et al Doxycycline stabilizes vulnerable plaque via inhibiting matrix metalloproteinases and attenuating inflammation in rabbits. PLoS ONE 2012; 7: e39695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang S, Zhang M, Liang B, et al AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26S proteasomes. Circ Res 2010; 106: 1117–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang S, Peng Q, Zhang J, et al Na+/H+ exchanger is required for hyperglycaemia‐induced endothelial dysfunction via calcium‐dependent calpain. Cardiovasc Res 2008; 80: 255–262. [DOI] [PubMed] [Google Scholar]
- 17. Yang XH, Li P, Yin YL, et al Rosiglitazone via PPARgamma‐dependent suppression of oxidative stress attenuates endothelial dysfunction in rats fed homocysteine thiolactone. J Cell Mol Med 2015; 19: 826–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yi J, Zheng Y, Miao C, et al Desflurane preconditioning induces oscillation of NF‐kappaB in human umbilical vein endothelial cells. PLoS ONE 2013; 8: e66576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang S, Zhang C, Zhang M, et al Activation of AMP‐activated protein kinase alpha2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat Med 2012; 18: 902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Spagnoli LG, Bonanno E, Sangiorgi G, et al Role of inflammation in atherosclerosis. J Nucl Med 2007; 48: 1800–1815. [DOI] [PubMed] [Google Scholar]
- 21. Dong M, Yang X, Lim S, et al Cold exposure promotes atherosclerotic plaque growth and instability via UCP1‐dependent lipolysis. Cell Metab 2013; 18: 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sharma S, Barton J, Rafikov R, et al Chronic inhibition of PPAR‐gamma signaling induces endothelial dysfunction in the juvenile lamb. Pulm Pharmacol Ther 2013; 26: 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang S, Xu J, Song P, et al Acute inhibition of guanosine triphosphate cyclohydrolase 1 uncouples endothelial nitric oxide synthase and elevates blood pressure. Hypertension 2008; 52: 484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shuang‐Xi W, Li‐Ying L, Hu M, et al Na+/H+ exchanger inhibitor prevented endothelial dysfunction induced by high glucose. J Cardiovasc Pharmacol 2005; 45: 586–590. [DOI] [PubMed] [Google Scholar]
- 25. Ventura E, Durant R, Jaussent A, et al Homocysteine and inflammation as main determinants of oxidative stress in the elderly. Free Radic Biol Med 2009; 46: 737–744. [DOI] [PubMed] [Google Scholar]
- 26. Malinowska J, Oleszek W, Stochmal A, et al The polyphenol‐rich extracts from black chokeberry and grape seeds impair changes in the platelet adhesion and aggregation induced by a model of hyperhomocysteinemia. Eur J Nutr 2013; 52: 1049–1057. [DOI] [PubMed] [Google Scholar]
- 27. Liu YH, You Y, Song T, et al Impairment of endothelium‐dependent relaxation of rat aortas by homocysteine thiolactone and attenuation by captopril. J Cardiovasc Pharmacol 2007; 50: 155–161. [DOI] [PubMed] [Google Scholar]
- 28. Wang Y, Huang Y, Lam KS, et al Berberine prevents hyperglycemia‐induced endothelial injury and enhances vasodilatation via adenosine monophosphate‐activated protein kinase and endothelial nitric oxide synthase. Cardiovasc Res 2009; 82: 484–492. [DOI] [PubMed] [Google Scholar]
- 29. Wang JM, Yang Z, Xu MG, et al Berberine‐induced decline in circulating CD31+/CD42− microparticles is associated with improvement of endothelial function in humans. Eur J Pharmacol 2009; 614: 77–83. [DOI] [PubMed] [Google Scholar]
- 30. Cheng F, Wang Y, Li J, et al Berberine improves endothelial function by reducing endothelial microparticles‐mediated oxidative stress in humans. Int J Cardiol 2013; 167: 936–942. [DOI] [PubMed] [Google Scholar]
- 31. Spinozzi S, Colliva C, Camborata C, et al Berberine and its metabolites: relationship between physicochemical properties and plasma levels after administration to human subjects. J Nat Prod 2014; 77: 766–772. [DOI] [PubMed] [Google Scholar]
- 32. Han Y, Wang Q, Song P, et al Redox regulation of the AMP‐activated protein kinase. PLoS ONE 2010; 5: e15420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Antoniades C, Shirodaria C, Warrick N, et al 5‐methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation 2006; 114: 1193–1201. [DOI] [PubMed] [Google Scholar]
- 34. Kawashima S, Yokoyama M. Dysfunction of endothelial nitric oxide synthase and atherosclerosis. Arterioscler Thromb Vasc Biol 2004; 24: 998–1005. [DOI] [PubMed] [Google Scholar]
- 35. Alp NJ, Mussa S, Khoo J, et al Tetrahydrobiopterin‐dependent preservation of nitric oxide‐mediated endothelial function in diabetes by targeted transgenic GTP‐cyclohydrolase I overexpression. J Clin Invest 2003; 112: 725–735. [DOI] [PMC free article] [PubMed] [Google Scholar]