

Abstract
Geometric isomerization can expand the scope of biological activities of natural products. The observed chemical diversity among the pseurotin-type fungal secondary metabolites is part generated by a trans-to-cis isomerization of an olefin. In vitro characterizations of pseurotin biosynthetic enzymes revealed that the glutathione S-transferase PsoE required a participation of the bifunctional C-methyltransferase–epoxidase PsoF to complete the trans-to-cis isomerization of a pathway intermediate, presynerazol. The PsoE–glutathione–presynerazol complex crystal structure indicated stereospecific glutathione–presynerazol conjugate formation is the principal function of PsoE. Moreover, PsoF was identified to have an additional, unexpected oxidative isomerase activity, making it a trifunctional enzyme that is key to the complexity generation in pseurotin biosynthesis. Through the study, we identified a novel mechanism of accomplishing a seemingly simple trans-to-cis isomerization reaction.
Keywords: isomerization, glutathione S-transferase, multifunctional enzyme, biosynthesis, fungal natural product
Graphical Abstract
In vitro study revealed that the glutathione S-transferase PsoE required the bifunctional PsoF to complete the isomerization of a pseurotin biosynthetic pathway intermediate. The PsoE crystal structure indicated that the intermediate–glutathione conjugation as the sole function of PsoE. PsoF was identified with an additional oxidative isomerase activity, making it a trifunctional enzyme key to complexity generation in pseurotin biosynthesis.
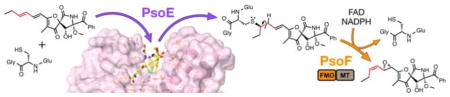
Geometric isomerization can significantly alter the physical and biological properties of natural products.[1] Pseurotin-type complex fungal natural products are a prominent example of geometric isomerization yielding a collection of related but distinct compounds. Identifying and understanding the mechanism of geometric isomerization is of great interest not only for understanding natural product biosynthesis but also for developing effective methods for preparing geometric isomers. The structurally interesting and biologically active secondary metabolites synerazol 1[2] and azaspirene 2[3] are representative members of the pseurotin family of compounds that exhibit various biological activities of medicinal importance[4] (Scheme 1). The chemical diversity of the pseurotin-type compounds is thought to be generated during the post-polyketide synthase–nonribosomal peptide synthetase modification steps.[5] Recently, we discovered a bifunctional enzyme PsoF that catalyzes a C-methylation and an epoxidation of various intermediates to generate multiple products, including pseurotin A 3 and D 4.[6] However, involvement of multiple enzymes and complex intermediates in the pseurotin biosynthetic pathway prevented determination of the exact activity of PsoE, a predicted glutathione S-transferase (GST). Here, we present successful biochemical characterization and crystallographic analysis of the biosynthetic enzymes that revealed the unique mechanism involved in the trans-to-cis isomerization of an olefin in the azaspirene-type intermediates to form the 12,13Z-configured products.
Scheme 1.

Proposed mechanism of the transformation of azaspirene 2 into synerazol 1 catalyzed by PsoE and PsoF from the pseurotin biosynthetic pathway.
Our previous gene knockout study in the A. fumigatus ΔpyrG/Δku70 strain AfKW1[6] allowed us to determine the involvement PsoE in the C12,C13 cis olefin formation. Thus, we decided to undertake in vitro analyses of this GST-type enzyme to establish its function clearly. When recombinantly produced PsoE was mixed with glutathione (GSH) and its substrate presynerazol 5, no reaction took place (Figure 1i, ii). However, when PsoF was added to the reaction mixture, 12,13E-configured 5 was completely converted into 12,13Z-configured 1 and 3 (Figure 1iii, iv). The chemical structures of the compounds discussed above were characterized with electrospray ionization LC–HRMS (liquid chromatography–high-resolution mass spectrometry), 1H NMR and 13C NMR showed in our previous report.[6] We chose to include PsoF in the reaction, because it was proposed to be responsible for the epoxidation step immediately following the isomerization of the 12,13E olefin, which was previously speculated to be catalyzed by PsoE in the pseurotin biosynthetic pathway.[6] The results from the in vitro assays carried out with PsoE and PsoF established clearly that PsoE indeed played a role in the trans-to-cis isomerization of the C12–C13 olefin for the ultimate formation of the 12,13Z-configured product 1 from the 12,13E-configured substrate 5. The lack of isomerization of the C12–C13 olefin in the absence of PsoF also revealed that PsoF, a previously established bifunctional methyltransferase–epoxidase, is also indispensable to the trans-to-cis isomerization step.
Figure 1.
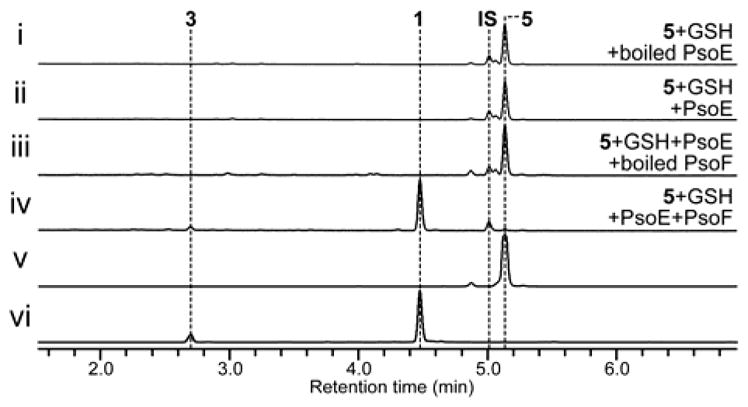
In vitro analyses of the activity of PsoE alone or with PsoF against 5 in the presence of glutathione (GSH). See Supporting Information for details. HPLC profiles of a reaction mixture containing (i) the heat-inactivated (boiled) PsoE with 5 and GSH, (ii) PsoE with 5 and GSH, (iii) PsoE and heat-inactivated PsoF with 5 and GSH,(iv) PsoE and PsoF with 5 and GSH. Traces are standardized to the height of the internal standard (IS) peak. All traces were monitored at 260 nm. The authentic reference of (v) 5 and (vi) 1 are also given.
Next, we examined the reaction conditions in detail to better understand how the task of isomerization is allocated between PsoE and PsoF. When the concentration of GSH was lowered in the reaction mixture containing PsoE and PsoF, we observed a substantially reduced formation of the Z-configured 1 and an increased production of the E-configured synerazol, 6 (Figure S2 i–iii). Similar product yield, which is higher than the enzyme concentration (20 μM), in the presence of 1 mM or 10 μM GSH suggests that the isomerization is enzyme-dependent and GSH is required. In addition, omission of NADPH from the reaction mixture prevented the formation of both 1 and 6 nor non-epoxidized products (Figure S2 iv), indicating the requirement for a reducing agent in this transformation. Also, concomitant analysis of the in vitro reactions for the presence of GSH-conjugated intermediates identified peaks having the mass/charge ratio corresponding to the 5–GSH conjugate 8 and its monoxide, presumably a sulfoxide intermediate 9, as determined by LC–HRMS (Figure S3). These results suggest that PsoE forms 8 that is isomerized and then epoxidized by PsoF to form 1. Low yield of 8 at low concentration of GSH leads to the formation of a shunt product 6.
To gain a better understanding of the role of PsoE, we determined the crystal structure of PsoE in complex with its native substrates 5 and GSH at 2.5 Å resolution using the single-wavelength anomalous dispersion method (see Supporting Information and Table S2, S3). Diffraction-grade crystals were obtained only for the PsoE–5–GSH ternary complex. The overall fold of PsoE (Figure 2A) is characteristic of the GST family of proteins.[7] Like other fungal GSTs, PsoE also shows low sequence similarity to other GSTs,[8] but the Theta-class human GST T2-2[9] is found to be most similar to PsoE (Figure S4). The main structural difference between PsoE and T2-2 is the absence of C-terminal α-helix α9 in PsoE. In GST T2-2, α8–α9 loop and α9 helix cover up the active site to bury the bound ligand deep inside the active site pocket (Figure S5 iii). However, the C-terminal residues of PsoE assume a random coil that is positioned away from the active site, leaving the ligand-binding site (H-site) without a lid (Figure S5 i and ii vs. iii). While lack of α9 helix also occurs in other classes of GSTs, including Zeta GSTs like the Ralstonia sp. strain U2 maleyl pyruvate isomerase[10] (Figure S5 iv), and Sigma GSTs like human prostaglandin synthases[11] (Figure S5 v and vi), shortened β2–α2 loop and shift in the positioning of α4–α4a loop and α4a helix reduce the depth of the H-site pocket in PsoE. Electron density for the bound ligand permitted modeling of 8, a true reaction intermediate with the GSH and 5 covalently linked in an S-configuration (Figure 2A). We designated the S-configured 8 as 8a and the R-configured counterpart as 8b (Scheme 1). PsoE forms hydrogen bonds with the GSH moiety using residues Arg 37, Lys 49, Val 50 (backbone amide and carbonyl groups), Glu 63 and Cys 64 (Figure 2B) from the N-terminal domain that form the G-site that is conserved among cytosolic or canonical GSTs (cGSTs)[8] (Figure S4, in bold red and purple letters). Only one residue from the C-terminal domain, Asn 99, forms a hydrogen bond with the GSH moiety. The formation of 8a likely involves Gln13, Cys 14 and Arg15 in PsoE, which structurally corresponds to Ser 13, Ser 14 and Cys15 in human GST Z1-1 that was shown to be important in catalysis and substrate binding.[12] Those residues in PsoE are thought to participate in catalysis by promoting the formation of the thiolate form of the bound GST. While the GSH sulfur and the side chain sulfur of Cys 14 are separated by 4.97 Å in our crystal structure, a conformational change in PsoE and an altered binding conformation of 5 can allow Cys 14 to interact with GSH prior to the formation of 8a. A similar shift in the GSH–catalytic residue interaction under different conditions was observed in the Pi-class GST P1-1.[13]
Figure 2.

Crystal structure of PsoE in complex with the GSH conjugate of presynerazol 8a. (A) Overall structure of PsoE (PDB ID: 5F8B) as a dimer. In the first PsoE molecule (left), α-helices and β-strands are colored in pink and blue, respectively, whereas the second molecule (right) is colored only in purple. Carbon atoms in the stick model of the bound ligand 8a are in yellow, whereas oxygen, nitrogen and sulfur atoms are in red, blue and green, respectively. Electron density for 8a in the first PsoE molecule (2Fo–Fc map contoured at 1.2 σ, green mesh) is shown. (B) The active site of PsoE, showing the interactions between 8a and PsoE. The protein side chain carbon atoms are in orange. Green dashed lines represent hydrogen bonds.
As to substrate recognition, the H-site pocket is normally enclosed by residues from α1, α4, α4a and α6 helices and β1–α1 and β2–α2 loops in cGSTs[8] (Figure S4, bold green and purple letters). However, PsoE employs only the exterior surface of α4a helix at the dimer interface to interact with the 5 moiety (Figures 2B, S5 i), making the substrate-binding mode of PsoE highly unique among other GSTs. PsoE–5 interactions are primarily hydrophobic, but hydrogen bonds between 5 and Lys 49 and Gln 108 side chains may help align the C12–C13 olefin of 5 and the GSH thiol for the ensuing C–S bond formation. However, due to the exposed nature of the 5-binding site, PsoE does not seem to bind 5 rigidly. Loose binding of 5 was also observed experimentally in the PsoE–GSH–5 complex crystalized in the absence of cobalt ion (Table S3), indicated by sparse electron density for the bound ligand, particularly for the 5 moiety (Figure S6A). Inclusion of cobalt in the crystallization condition altered the packing of PsoE dimers such that the α2-containing loop from the symmetry-related molecule covers the open active site, limiting the wiggle space for the bound 5 moiety (Figure S6B). Nevertheless, very limited specific interactions with 5 in the loosely packing substrate binding site of PsoE (Figures 2 and S5 i, ii) suggests that PsoE may be predisposed to releasing 8a relatively easily. These observations suggest a possible reaction mechanism of the trans-to-cis isomerization reaction involved in the biosynthesis of synerazol.
Cis–trans isomerization reactions are known to be catalyzed by several different GSTs, including the Zeta-class GSTs that perform isomerization of maleyl (cis) substrates to fumaryl (trans) products.[14] However, our earlier study has established that PsoE alone is unable to perform the isomerization. Examination of the PsoE–8a complex structure also identifies no residue in the active site that is suitably positioned for acid–base catalysis of the isomerization reaction. Thus, it appears that PsoE is designed only to perform the stereospecific conjugation of GSH with 5 to form 8a and pass the product to PsoF for isomerization and epoxidation to form 1. The transfer of the substrate from PsoE to PsoF may be facilitated by the formation of a transient complex between those two enzymes. Such an interaction may be promoted by the C-terminal segment of PsoE, which is about 30 residues long and does not assume an ordered conformation in our crystal structure.
Based on these results, we theorize that PsoE catalyzes the stereospecific conjugation of GSH and 5 to form 8a, which converts the C12–C13 olefin into an sp3-hybridized sigma bond. The cis olefin can be formed by rotating 8a around the C12–C13 sigma bond and then eliminating the GSH group to regenerate the C12–C13 olefin in the Z-configuration. The crystal structure of PsoE has revealed that PsoE is not equipped to carry out the isomerization step. However, the uniquely open active site of PsoE would allow 8a to be released from PsoE and passed to PsoF. The flavin-containing monooxygenase domain of PsoF can oxidize the sulfur atom of 8a with its flavin hydroperoxide to generate 9, setting up the molecule for a subsequent pericyclic syn-elimination that leads to the release of the oxidized GSH and the formation of the 12,13Z-configured stereoisomer of 5. Lastly, the isomerized product remaining in the PsoF active site undergoes a subsequent epoxidation[15] of the C10–C11 olefin to form 1. For PsoF to be able to perform the trans-to-cis isomerization of 8a, its active site needs to have sufficient room to allow binding of the relatively bulky GSH moiety in the vicinity of the C12–C13 olefin. The fact that PsoF can accept substrates having either E- or Z-configuration at the C12–C13 olefin for the epoxidation of the C10–C11 olefin suggests that the section of the active site pocket that accommodates the terminal portion of the synerazol diene side chain is likely somewhat spacious. The bulky GSH group of 8a can force it to be bound in the least hindered, extended conformation, which is in fact the pro-cis conformation, inside the PsoF active site, to promote the formation of the cis olefin at C12–C13 upon release of the GSH molecule. There was a report of another GST that was shown to catalyze a cis–trans isomerization reaction involved in the biosynthesis of hypothemycin. This GST, Hpm2, was shown to isomerize 7′,8′-trans-containing aigialomycin A and 7′,8′-cis-containing hypothemycin to an equilibrated mixture comprised of 85% trans and 15% cis products.[16] While Hpm2 catalyzes the isomerization reaction alone, it is unable to shift the equilibrium toward the formation of the higher-energy cis product. In the pseurotin biosynthesis, employment of an additional enzyme, namely PsoF, to perform the isomerization reaction at the expense of FAD and NADPH seems to be driving the conversion of trans-containing 5 into cis-containing 1.
In conclusion, our study has shown that PsoE is a unique GST that works with another enzyme PsoF to accomplish the trans-to-cis isomerization of an olefin in the azaspirene-type compounds, giving rise to a family of geometric isomers. This unexpected finding adds yet another function to PsoF, a previously established bifunctional enzyme,[6] making it a trifunctional enzyme capable of performing C-methylation, isomerization and epoxidation reactions on a range of substrates. Through the study, we have successfully uncovered the activities of key enzymes that play a critical role in the biosynthetic strategy employed for generating chemical diversity of pseurotin family of natural products.
Supplementary Material
Footnotes
We wish to thank the financial support from the Japan Society for the Promotion of Science (JSPS) Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers (No. G2604) (K.W.). This work was also supported in part by Amano Enzyme Foundation (K.W.), Japan Antibiotics Research Association (K.W.), Mochida Memorial Foundation for Medical and Pharmaceutical Research (K.W.), the Institution of Fermentation at Osaka (K.W.), Nagase Science and Technology Foundation Japan (K.W.), Tokyo Biochemical Research Foundation (K.W.) and JSPS Fellowship for Research in Japan (K. Hotta). We acknowledge the beamline staff at Photon Factory for their kind support on data collection
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201xxxxxx.
Contributor Information
Dr. Tsuyoshi Yamamoto, Department of Pharmaceutical Sciences, University of Shizuoka, Shizuoka 422-8526 (Japan)
Dr. Yuta Tsunematsu, Department of Pharmaceutical Sciences, University of Shizuoka, Shizuoka 422-8526 (Japan)
Dr. Kodai Hara, Department of Pharmaceutical Sciences, University of Shizuoka, Shizuoka 422-8526 (Japan)
Dr. Tomohiro Suzuki, Research Institute of Green Science and Technology, Graduate School of Agriculture, Graduate School of Science and Technology Shizuoka University, Shizuoka 422-8529 (Japan)
Prof. Dr. Hirokazu Kawagishi, Research Institute of Green Science and Technology, Graduate School of Agriculture, Graduate School of Science and Technology Shizuoka University, Shizuoka 422-8529 (Japan)
Prof. Dr. Hiroshi Noguchi, Department of Pharmaceutical Sciences, University of Shizuoka, Shizuoka 422-8526 (Japan)
Prof. Dr. Hiroshi Hashimoto, Department of Pharmaceutical Sciences, University of Shizuoka, Shizuoka 422-8526 (Japan)
Prof. Dr. Yi Tang, Department of Chemical and Biomolecular Engineering, and Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095 (United States)
Prof. Dr. Kinya Hotta, School of Biosciences, The University of Nottingham Malaysia Campus, Selangor 43500 (Malaysia)
Prof. Dr. Kenji Watanabe, Email: kenji55@u-shizuoka-ken.ac.jp, Department of Pharmaceutical Sciences, University of Shizuoka, Shizuoka 422-8526 (Japan)
References
- 1.a) Koziol A, Stryjewska A, Librowski T, Salat K, Gawel M, Moniczewski A, Lochynski S. Mini Rev Med Chem. 2014;14:1156–1168. doi: 10.2174/1389557514666141127145820. [DOI] [PubMed] [Google Scholar]; b) Pariza MW, Park Y, Cook ME. Prog Lipid Res. 2001;40:283–298. doi: 10.1016/s0163-7827(01)00008-x. [DOI] [PubMed] [Google Scholar]
- 2.Ando O, Satake H, Nakajima M, Sato A, Nakamura T, Kinoshita T, Furuya K, Haneishi T. J Antibiot (Tokyo) 1991;44:382–389. doi: 10.7164/antibiotics.44.382. [DOI] [PubMed] [Google Scholar]
- 3.Asami Y, Kakeya H, Onose R, Yoshida A, Matsuzaki H, Osada H. Org Lett. 2002;4:2845–2848. doi: 10.1021/ol020104+. [DOI] [PubMed] [Google Scholar]
- 4.Maebayashi Y, Horie Y, Satoh Y, Yamazaki M. Mycotoxins. 1985;1985:33–34. [Google Scholar]
- 5.Wiemann P, Guo CJ, Palmer JM, Sekonyela R, Wang CC, Keller NP. Proc Natl Acad Sci USA. 2013;110:17065–17070. doi: 10.1073/pnas.1313258110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsunematsu Y, Fukutomi M, Saruwatari T, Noguchi H, Hotta K, Tang Y, Watanabe K. Angew Chem Int Ed Engl. 2014;53:8475–8479. doi: 10.1002/anie.201404804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheehan D, Meade G, Foley VM, Dowd CA. Biochem J. 2001;360:1–16. doi: 10.1042/0264-6021:3600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oakley AJ. Curr Opin Struct Biol. 2005;15:716–723. doi: 10.1016/j.sbi.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Rossjohn J, McKinstry WJ, Oakley AJ, Verger D, Flanagan J, Chelvanayagam G, Tan KL, Board PG, Parker MW. Structure. 1998;6:309–322. doi: 10.1016/s0969-2126(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 10.Marsh M, Shoemark DK, Jacob A, Robinson C, Cahill B, Zhou NY, Williams PA, Hadfield AT. J Mol Biol. 2008;384:165–177. doi: 10.1016/j.jmb.2008.09.028. [DOI] [PubMed] [Google Scholar]
- 11.Hohwy M, Spadola L, Lundquist B, Hawtin P, Dahmén J, Groth-Clausen I, Nilsson E, Persdotter S, von Wachenfeldt K, Folmer RH, Edman K. J Med Chem. 2008;51:2178–2186. doi: 10.1021/jm701509k. [DOI] [PubMed] [Google Scholar]
- 12.Board PG, Taylor MC, Coggan M, Parker MW, Lantum HB, Anders MW. Biochem J. 2003;374:731–737. doi: 10.1042/BJ20030625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oakley AJ, Lo Bello M, Battistoni A, Ricci G, Rossjohn J, Villar HO, Parker MW. J Mol Biol. 1997;274:84–100. doi: 10.1006/jmbi.1997.1364. [DOI] [PubMed] [Google Scholar]
- 14.Deponte M. Biochim Biophys Acta. 2013;1830:3217–3266. doi: 10.1016/j.bbagen.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 15.Mao XM, Zhan ZJ, Grayson MN, Tang MC, Xu W, Li YQ, Yin WB, Lin HC, Chooi YH, Houk KN, Tang Y. J Am Chem Soc. 2015;137:11904–11907. doi: 10.1021/jacs.5b07816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reeves CD, Hu Z, Reid R, Kealey JT. Appl Environ Microbiol. 2008;74:5121–5129. doi: 10.1128/AEM.00478-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.