

Abstract
A rhodium(II) Dibenzotetramethylaza[14]annulene dimer ([(tmtaa)Rh]2) undergoes metathesis reactions with [CpCr-(CO)3]2, [CpMo(CO)3]2, [CpFe(CO)2]2, [Co(CO)4]2, and [Mn-(CO)5]2 to form (tmtaa)Rh-M complexes (M = CrCp(CO)3, MoCp(CO)3, FeCp(CO)2, Co(CO)4, or Mn(CO)5). Molecular structures were determined for (tmtaa)Rh-FeCp(CO)2, (tmtaa)Rh-Co(μ-CO)(CO)3, and (tmtaa)Rh-Mn(CO)5 by X-ray diffraction. Equilibrium constants measured for the metathesis reactions permit the estimation of several (tmtaa)Rh-M bond dissociation enthalpies (Rh—Cr = 19 kcal mol−1, Rh—Mo = 25 kcal mol−1, and Rh—Fe = 27). Reactivities of the bimetallic complexes with synthesis gas to form (tmtaa)Rh-C(O)H and M-H are surveyed.
Graphical abstract
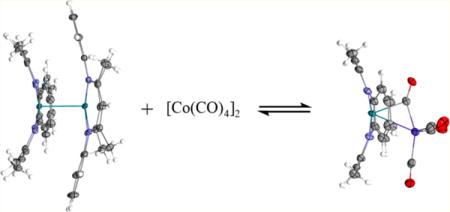
INTRODUCTION
The easily prepared, low cost N42− macrocyclic ligand Dibenzotetramethylaza[14]annulene dianion (tmtaa) has been suggested as a substitute for porphyrins.1–3 A rhodium(II) tmtaa dimer ([(tmtaa)Rh]2, 1) has recently been reported to react with synthesis gas (H2/CO) in toluene and pyridine to form large equilibrium concentrations of a metallo-formyl complex ((tmtaa)Rh-C(O)H) (Figure 1).3 Rhodium(II) porphyrins and related chelates and macrocycles provide precedents for reactions of H2/CO that produce η1-carbon-bonded formyl complexes.4–17 Several approaches attempting to advance the utilization of this type of metal-formyl fragment in the production of organic oxygenates invoke reactions that occur at the formyl group, such as hydrogenation using a second catalyst.18,19 Bimetal catalyst systems for alkene hydroformylation that involve intermolecular processes illustrate the efficacy of this type of strategy.20–22 Heterobimetallic complexes can be anticipated to occur in bimetal catalyst systems as nonproductive traps for active metal catalyst species. Reactivity studies of bimetallic compounds are important to determine whether or not these intermetallic intermediates are capable of returning the active catalyst units back into the productive cycle.
Figure 1.
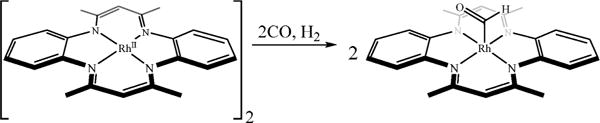
Reaction of [(tmtaa)Rh]2 with CO and H2.
This Article reports on the formation, structural features, and dissociation energetics for a series of bimetallic complexes ((tmtaa)Rh-M) produced by metathesis reactions of [(tmtaa)-Rh]2 (1) with [CpCr(CO)3]2 (2), [CpMo(CO)3]2 (3), [CpFe(CO)2]2 (4), [Co(CO)4]2 (5), and [Mn(CO)5]2 (6). Minimum energy structure optimized density functional theory (DFT) computations reproduce the primary structural features of the bimetal complexes, including an unexpected semi-bridging carbonyl in the molecular structure of (tmtaa)Rh-Co(μ-CO)(CO)3.
RESULTS AND DISCUSSION
The closed shell dimers [CpCr(CO)3]2, [CpMo(CO)3]2, and [Mn(CO)5]2, which have only terminal CO ligands, are unambiguously described as having M–M bonds, but the presence of bridging CO units complicates the description of the bonding in [CpFe(CO)2]2 and [Co(CO)4]223,24 Each of these dimers can homolyze to a metal-centered radical,25 and for the purpose of unifying and simplifying the presentation in this Article, all of the dimers (2–6) will be described as if the M–M bond were present.
Toluene solutions of the RhII–RhII bonded dimer (1) undergo metathesis reactions with 2, 3, 4, 5, and 6 to produce equilibrium distributions with heterobimetallic complexes of the general form (tmtaa)Rh-M, where M = CpCr(CO)3 (7), CpMo(CO)3 (8), CpFe(CO)2 (9), Co(CO)4 (10), and Mn(CO)5 (11), respectively (eq 1).
| (1) |
Reactions of dimers 2–6 usually involve thermal or photo homolysis to form monomeric d5, d7, and d9 metal-centered radicals. Thermal homolysis of compounds 1–6 into monomers occurs with reported dissociation enthalpies ranging from 15.5 to 33 kcal mol−1.2,26–28 Thermal reactions of the more strongly bonded dimer species that pass through metallo-radicals are slow because the dissociation enthalpies directly contribute to the activation parameters. Kinetics of the dimer reactions are often greatly increased by light-induced dimer homolysis. Both the π to σ* and the σ to σ* electronic transitions associated with M–M single-bonded complexes place an electron in the antibonding M–M σ molecular orbital and give photoinduced dimer homolysis.29
The lowest energy pathway for metathesis reactions of the form in reaction 1 is most likely a radical chain reaction that proceeds through an associative radical interchange (eqs 2, 3).30,31 The Rh(II) center in [(tmtaa)Rh]2 is a 5-coordinate, 16-electron site that can bind to M● and facilitate radical interchange (eqs 2, 3).
| (2) |
| (3) |
Formation and Reactions of (tmtaa)Rh-CrCp(CO)3
Reaction of [(tmtaa)Rh]2 (1) in toluene with a three-fold excess of [CpCr(CO)3]2 (2) produces a 1H NMR-measurable equilibrium with the metathesis product CpCr(CO)3 (7) (K4 = 0.19 (0.05), ΔG4° = 0.98 kcal mol−1) (eq 4) in a period of hours at 298 K in the absence of light.
| (4) |
Reaction 4 is relatively fast for this type of process when light is absent. The dissociation free enthalpy for 2 is small (15.5 kcal mol−1),27 which makes 2 a facile source of Cp(CO)3Cr●, but reaction 4 is observed to be further accelerated by visible and UV irradiation.
The temperature dependence of Δν1/2 (Hz) for the η5-cyclopentadienyl ligand hydrogens in (tmtaa)Rh-CrCp(CO)3 is shown in Figure 2. The observed increase in the η5-Cp 1H NMR full line width at half height for (tmtaa)Rh-CrCp(CO)3 as the temperature is elevated is ascribed to lifetime broadening that results from dissociation of the diamagnetic (tmtaa)Rh-CrCp(CO)3 complex into paramagnetic (S = 1/2) metal-centered radicals (tmtaa)Rh● and ●CrCp(CO)3.
Figure 2.
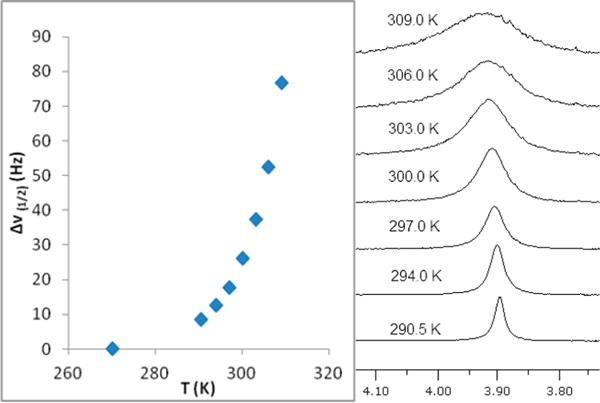
Temperature dependence of the 1H NMR line width for the cyclopentadienyl ligand in (tmtaa)Rh-CrCp(CO)3.
The observed 1H NMR line width (Δν1/2(obs)) is given as the sum of the natural line width and the line width resulting from dissociation into paramagnetic components (Δν1/2(obs) = Δν1/2(nat) + Δν1/2(ex)). Equation 5 gives the general relationship of the contribution of this exchange to the NMR line width (Δν1/2(ex)) with the electron–proton coupling constant (AH) and the diamagnetic (τd) and paramagnetic lifetimes (τp). Equation 5 simplifies to eq 6 (T2−1 = kex) for the case in which each exchange event results in nuclear spin relaxation. This limiting case occurs when the electron–nuclear coupling constant (A) is large enough such that (AτP/2)2≫ 1.
| (5) |
| (6) |
The mean lifetime for the diamagnetic species (τd) that results from the observed T2−1 yields the apparent rate constant (τd−1 = kex) for bond homolysis events that produce paramagnetic species with efficient nuclear relaxation ((Aτp/2)2 ≫ 1). Activation parameters for homolytic dissociation of (tmtaa)Rh-CrCp(CO)3 were obtained by application of transition state theory (K⧧ = kex(h/kT); −RT ln K⧧ex = ΔG⧧ = ΔH⧧ − TΔS⧧).6,32–36 Using a Δν1/2(nat) of 1.0 Hz for the Cp hydrogens of 7 yields the apparent activation parameters of ΔH⧧ = 21 (1) kcal mol−1 and ΔS⧧ex,4 = 19.6 (1.2) cal K−1 mol−1 for homolytic dissociation of (tmtaa)Rh-CrCp(CO)3 (Figure 3). Activation enthalpies for homolytic bond dissociation (ΔH⧧app) in a low viscosity medium like toluene are about 2 kcal mol−1 larger than the bond dissociation enthalpies (BDE; ΔH°),34,35,37,38 which places the Rh-Cr BDE in (tmtaa)Rh-CrCp(CO)3 at ∼19 kcal mol−1.
Figure 3.
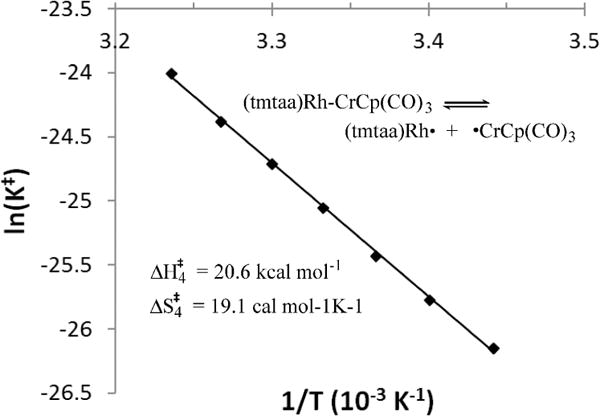
Determination of activation parameters for homolytic dissociation of (tmtaa)Rh-CrCp(CO)3 into (tmtaa)Rh● and (CO)3CpCr● in d8-toluene by 1H NMR line width measurements of the cyclopentadienide hydrogens (ΔH‡4 = 21 (1) kcal mol−1, ΔS‡ex,4 = 19.6 (1.2) cal K−1 mol−1).
An estimate for the Rh-Cr BDE in (tmtaa)Rh-CrCp(CO)3 is alternately obtained from the equilibrium studies in toluene for reaction 4. Using ΔG4° (298 K) of 0.98 kcal mol−1, Rh(II)-Rh(II) BDE for [(tmtaa)Rh]2 of 22 kcal mol−1, and [CpCr(CO)3]2 Cr–Cr BDE of 15.5 kcal mol−1,27 and assuming that ΔS4° is approximately zero, results in an estimate of 18 kcal mol−1 for the Rh-Cr BDE compared to 19 kcal mol−1 estimated by bond homolysis kinetics (Figure 3).
Exhaustive efforts to obtain single crystals of (tmtaa)Rh-CrCp(CO)3 for X-ray diffraction were unsuccessful. The small equilibrium constant for reaction 4 precluded obtaining (tmtaa)Rh-CrCp(CO)3 in the absence of [(tmtaa)Rh]2 and [CpCr(CO)3]2, which reduced the probability of obtaining a crystal of 7. Fourier transform infrared spectroscopy (FTIR) for a KBr pellet of the reaction mixture containing (tmtaa)Rh-CrCp(CO)3 shows a new CO stretching band at 1766 cm−1 that is indicative of an intermetal-bridging CO unit. DFT calculations39 were carried out using the B3LYP functional and a mixed basis set (6-31G* for C, H, N, O, and Cr; 3-21G for Rh) for (tmtaa)Rh-CrCp(CO)3. DFT computations for 7 starting from an all-terminal CO structure reached a computed minimum energy structure that contains a bridging CO unit consistent with the CO stretching frequency of 1766 cm−1 (Figure 4). Calculated Mulliken atomic charges from DFT show a smaller positive charge on the carbon of the semibridging CO (C = 0.197, O = −0.317) compared to that of the terminal CO units (Cavg = 0.257, Oavg = −0.320).
Figure 4.
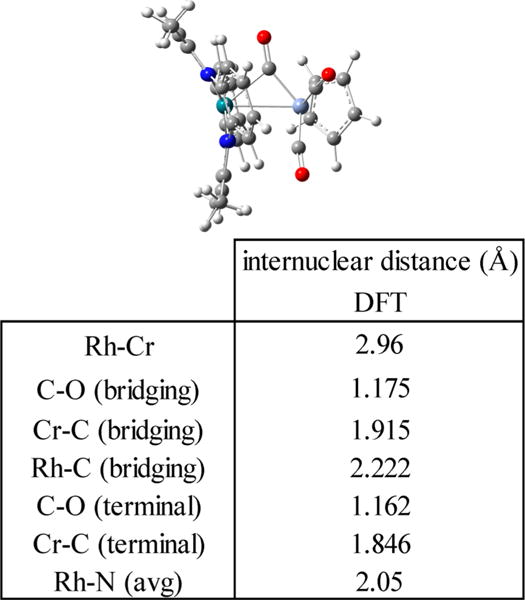
Minimum energy structural optimization of (tmtaa)Rh-CrCp(CO) by DFT (B3LYP, 3-21G/6-31G*) and a table of selected internuclear distances.
Toluene solutions of 7 with [(tmtaa)Rh]2 and [CpCr-(CO)3]2 in a vacuum-adapted NMR tube were charged with 1.0 atm of a 1:1 molar mixture of H2 and CO, and changes in the solution composition were followed by 1H NMR. At these conditions, 7 is fully consumed to form (tmtaa)Rh-C(O)H and [CpCr(CO)3]2 (eq 7). There are mutiple pathways to accomplish this transformation, but the distribution of species is defined by equilibria described by eqs 7–9.
| (7) |
| (8) |
| (9) |
The equilibrium constant for the reaction of (tmtaa)Rh-H with CO (eq 8) to produce a rhodium formyl complex (tmtaa)Rh-C(O)H in toluene (K8(298 K) = (10.8 (0.3)) × 103, ΔG8° (298 K) = −5.5 kcal mol−1) is sufficiently large such that at a pressure of 0.5 atm of CO the conversion of (tmtaa)Rh-H to the formyl complex (tmtaa)Rh-C(O)H is effectively complete, and the (tmtaa)Rh-H concentration is too small for observation by 1H NMR.3 Favorable thermodynamics for reaction 9 also give high conversion of [CpCr(CO)3]2 to Cp(CO)3Cr-H at a pressure of 0.5 atm of H2 at 298 K.40
Formation and Reactions of (tmtaa)Rh-MoCp(CO)3
Reaction of [(tmtaa)Rh]2 (1) with [CpMo(CO)3]2 (3) in the absence of light slowly produces a 1H NMR observable equilibrium with (tmtaa)Rh-MoCp(CO)3 (8) over a period of days (eq 10). Reaction 10 is greatly accelerated by light and reaches completion in a period of minutes when carried out in a Rayonet RMR-400 photochemical reactor with a 300 nm light source.
| (10) |
A BDE for Rh-Mo of 25 kcal mol−1 is estimated for 8 from the equilibrium constant for reaction 10 at 298 K in toluene (K10(298 K) = 7.8 × 10−2, ΔG10° (298 K) = 1.5 kcal mol−1) along with the Rh–Rh and Mo–Mo BDEs for [(tmtaa)Rh]2 (22 kcal mol−1)2 and [CpMo(CO)3]2 (30 ± 3 kcal mol−1),26 respectively. The Rh-Mo BDE in (tmtaa)Rh-MoCp(CO)3 is estimated at 25 ± 3 kcal mol−1 assuming that ΔS10° is close to zero. The Rh-Mo BDE in 8 is about 7 kcal mol−1 larger than the Rh-Cr BDE in 7, which is in the range expected for second transition series metal complexes compared to analogous first transition series metal species. Minimum energy structural optimization by DFT (B3LYP, 3-21G)39 anticipates a semi-bridging carbonyl in complex 8 (Figure 5), and calculated Mulliken atomic charges for this unit show a smaller positive charge on the semibridging CO (C = 0.162, O = −0.312) compared to that of the terminal CO units (Cavg = 0.201, Oavg = −0.305).
Figure 5.
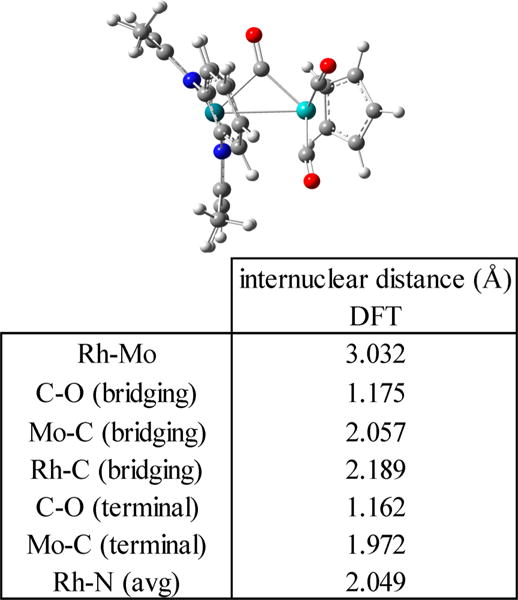
Minimum energy structural optimization of (tmtaa)Rh-MoCp(CO)3 by DFT (B3LYP, 3-21G/6-31G*) and a table of selected internuclear distances.
Solutions of 3 in equilibrium with 1 and 8 in toluene were charged with 706 Torr of a 50:50 mixture of CO/H2, and the subsequent slow reactions were followed by 1H NMR for a period of weeks. When the sample reached equilibrium through reactions 8, 11, and 12, compounds 3, (tmtaa)Rh-C(O)H, and Cp(CO)3Mo-H were observed in the 1H NMR spectrum.
| (11) |
| (12) |
The equilibrium constant for the reaction of [CpMo(CO)3]2 with H2 to form Cp(CO)2Mo-H (eq 12) was evaluated by integration of the 1H NMR (K12(298 K) ≅ 1.7; ΔG12° (298 K) ≅ −0.3 kcal mol−1). An estimate of 67 (3) kcal mol−1 for the Mo-H BDE in Cp(CO)2Mo-H is obtained by using a Mo–Mo BDE of 30 (3) kcal mol−1 for [CpMo(CO)3]2 along with an H–H BDE of 104 kcal mol−1 and using ΔH12° ≅ ΔG12° (298 K).26 This estimate of 67 (3) kcal mol−1 for the Mo-H BDE in Cp(CO)2Mo-H compares favorably with prior published estimates in the range of 66–69 kcal mol−1.41,42 The Mo-H BDE is ∼7 kcal mol−1 larger than the Cr-H BDE in Cp(CO)2M-H, which is in the range expected for second versus first transition series metal hydrides.
The rhodium formyl complex ((tmtaa)Rh-C(O)H) is the only observed product that contains the (tmtaa)Rh unit because K8(298 K) is very large. Removing H2/CO gases from solution by three freeze–pump–thaw cycles results in the formyl complex completely reverting to a mixture of 1 and 8 during a period of one week, but Cp(CO)3Mo-H did not decrease at an observable rate over this time. Reaction of [MoCp(CO)3]2 with 700 Torr of a 50:50 mole fraction ratio of CO/H2 in the absence of 1 did not produce 1H NMR-observable concentrations of Cp(CO)3Mo-H over a period of one week. Thermal reaction of [MoCp(CO)3]2 with H2/CO must be catalyzed by the presence of a (tmtaa)Rh species such as (tmtaa)Rh-H.
Formation and Reactions of (tmtaa)Rh-FeCp(CO)2
Toluene solutions of [(tmtaa)Rh]2 (1) in the absence of light react very slowly with excess [η5-CpFe(CO)2]2 (4) over 18 days to produce an equilibrium distribution with the heterobimetallic complex (tmtaa)Rh-FeCp(CO)2 (9) (eq 13). Reaction 13 is accelerated in ambient laboratory light, and the reaction is completed after irradiation for 10 min in a Rayonet RMR-400 photochemical reactor with a 300 nm source.
| (13) |
Crystals of heterobimetallic complex in solutions of 9 were grown from slow diffusion of pentane in toluene, and the molecular structure was determined by single crystal X-ray diffraction. An ORTEP diagram for 9 at the 50% probability level is shown in Figure 6 along with an energy-minimized structure from DFT computations (B3LYP, 3-21G/6-31G*). The presence of a d7–d7 RhII-FeI bond (263 nm), an η5-Cp, and all terminal CO ligands are the primary structural features observed in the molecular structure. The energy-minimized structure from DFT (B3LYP, 3-21G/6-31G*)39 computations also shows all of the most important structural features for 9 (Figure 6).
Figure 6.
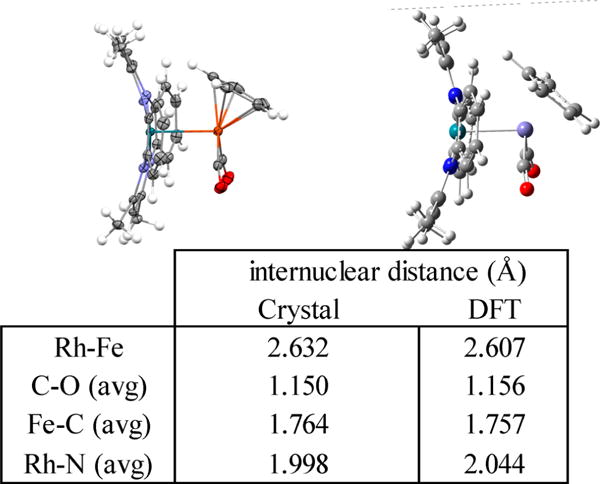
ORTEP representation of (tmtaa)Rh-FeCp(CO)2 (9) structure from single crystal X-ray diffraction with thermal ellipsoids shown on non-hydrogen atoms at 50% probability level, hydrogen atoms shown as open circles, and energy minimization by DFT computations (B3LYP, 3-21G/6-31G*). A table of selected internuclear distances observed in the molecular structure and calculated by DFT is also included.
Dihedral angles looking down the N-Cbenzene single bond of tmtaa in 9 show an inequivalency present in the ligand with larger dihedral angles (31.8°) in the half of tmtaa opposite the Cp ligand on iron and smaller dihedral angles (23.4°) on the alternate half. These dihedral angles define the tilting of the nonplanar saddle shape of the tmtaa macrocycle, and the inequivalency here shows that there is more pronounced tilting of the diiminato fragment on one-half of tmtaa to accommodate the steric demands of the η5-Cp ligand on iron.
The equilibrium constant for reaction 13 was directly evaluated by integration of the 1H NMR for each species in reaction 13 (K13(298 K) = 0.19; ΔG13° (298 K) = 0.98 kcal mol−1). An estimate of 27 (5) kcal mol−1 for the Rh-Fe BDE in (tmtaa)Rh-FeCp(CO)2 is obtained by using the Rh–Rh BDE of 22 kcal mol−1 for [(tmtaa)Rh]2 and the electrochemical estimate for the Fe–Fe BDE for [CpFe(CO)2]2 (33 ± 5 kcal mol−1)26 and assuming that ΔH13° ≅ ΔG13° = 0.98 kcal mol−1.
Formation of (tmtaa)Rh-Co(μ-CO)(CO)3
Reaction of [(tmtaa)Rh]2 (1) with an excess of [Co(CO)4]2 (5) in toluene occurs immediately and completely to form (tmtaa)Rh-Co(μ-CO)(CO)3 (10). Excess [Co(CO)4]2 was removed by evacuation of the toluene solution followed by vacuum sublimation; however, subsequent IR (KBr pellet) showed traces of dicobalt octacarbonyl remaining. The reverse reaction of 10 to form 1 and 5 was not observed after removing most of the excess [Co(CO)4]2, indicating a large equilibrium constant for the formation of 10. Crystals of 10 were obtained from slow diffusion of pentane into a toluene solution, and the molecular structure was determined by X-ray diffraction. An important structural feature of 10 is the presence of a semibridging carbonyl ligand supported by a Co-Rh single bond. The semibridging carbonyl results from the filled rhodium dπ donating into the π* of CO (Figure 7). The dπ to π* donation is reflected in the structure by an increased carbon-oxygen bond length for the semibridging CO (1.169 Å) compared to the average bond length of the terminal carbonyls (1.140 Å). DFT calculations (B3LYP, 3-21G/6-31G*)39 for 10 converge to an energy-minimized structure that mimics the observed bridging CO unit (Figure 8). Analysis of the Mulliken atomic charges from the DFT shows a smaller positive charge on the semibridging carbonyl (C = 0.206, O = −0.305) compared to that of the terminal carbonyls (Cavg = 0.308, Oavg = −0.281), which is consistent with the bonding model. The infrared spectrum of 10 in a KBr pellet has a peak at 1820 cm−1 that is attributed to the CO stretching vibration of the semibridging carbonyl. The analogous porphyrin complex (OEP)Rh-Co-(CO)4 shows only terminal CO stretches (νCO = 2054, 1996, and 1965 cm−1) in the infrared spectrum.
Figure 7.
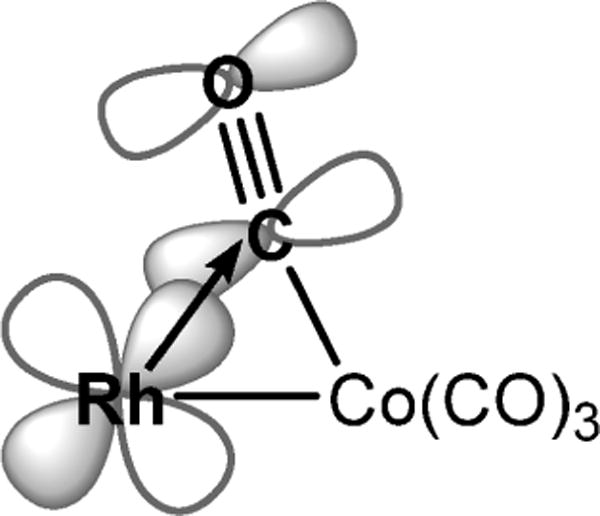
Drawing of the semibridging carbonyl in (tmtaa)Rh-Co(μ-CO)(CO)3 showing the donation of rhodium dπ into the C≡O π* orbital.
Figure 8.
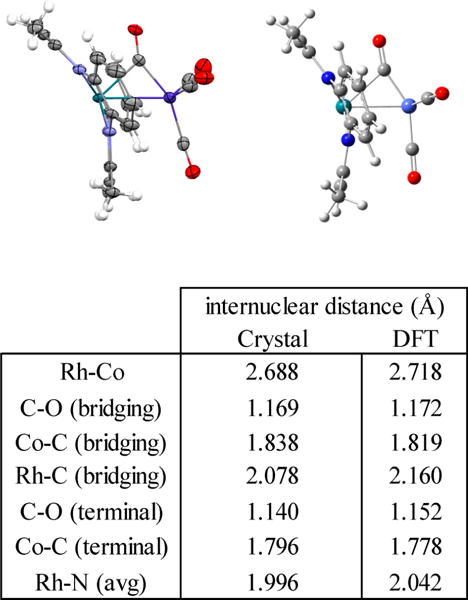
ORTEP representation of the (tmtaa)Rh-Co(CO)4 structure from single crystal X-ray diffraction with thermal ellipsoids shown on non-hydrogen atoms at 50% probability level, hydrogen atoms shown as open circles, and energy minimization by DFT computations (B3LYP, 3-21G/6-31G*). A table of selected internuclear distances observed in the molecular structure and calculated by DFT is also included.
Solutions of 10 in pyridine with a small excess of [Co(CO)4]2 immediately disproportionate into [(tmtaa)Rh-(pyr)2]+ and the cobalt tetracarbonyl anion (eq 14). Disproportionation is not observed by 1H NMR for [(tmtaa)-Rh]2 in pyridine, but 1 reacts via heterolytic pathways when treated with pyridinium chloride to form [(tmtaa)Rh-(pyr)2]+[Cl]− and (tmtaa)Rh-H. The cationic fragment [(tmtaa)Rh(pyr)2]+ shows an identical set of peaks with either cobalt tetracarbonyl or chloride anion, and the cobalt tetracarbonyl anion is observed by the FTIR (KBr pellet) T2 CO stretching mode centered at 1880 cm−1 diagnostic of Co(CO)4−.43,44
| (14) |
Solutions of 10 were charged with 200 Torr of ethene, resulting in the formation of an insoluble product that formed crystals in an NMR tube that were then used to obtain the molecular structure by X-ray diffraction (Figure 9). The product of the reaction of 10 with ethene is an ionic species with a cationic (tmtaa)Rh fragment, in which a CH2CH2 group bridges the rhodium and methine carbon centers, and a [Co(CO)4]− counteranion. The diiminato fragment bridged to rhodium lost the delocalized π system, which is demonstrated in the structure by shorter N-C double bonds and longer C–C single bonds as shown in Figure 9. The nucleophilicity of the methine position was previously reported by Cotton.45
Figure 9.
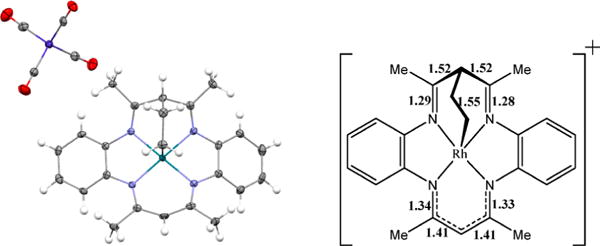
Crystal structure of ionic bridged ethyl complex and drawing of cationic (tmtaa)Rh fragment with bond distances labeled in angstroms.
Formation and Reactions of (tmtaa)Rh-Mn(CO)5
Reaction of [(tmtaa)Rh]2 (1) with an excess of [Mn(CO)5]2 (6) in toluene proceeds in the dark over several weeks to achieve equilibrium with (tmtaa)Rh-Mn(CO)5 (11) (eq 15), or 11 may be formed much more rapidly by photolysis (Rayonet RMR-400 photochemical reactor, 300 nm source).
| (15) |
The molecular structure of 11 determined by X-ray diffraction shows only terminal CO ligands (Figure 10). The carbon monoxide trans to rhodium has shorter Mn-C (0.06 Å) and longer C–O (0.01 Å) bond lengths compared to that of the cis-oriented carbonyl, which is ascribed to larger π backbonding for the Mn-CO unit trans to rhodium. Infrared spectroscopy also shows only terminal CO stretches for 11 (1950–2150 cm−1).
Figure 10.
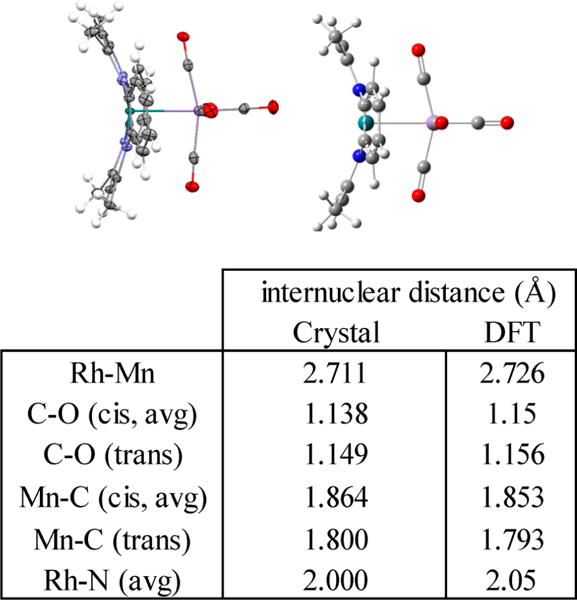
ORTEP representation of the (tmtaa)Rh-Mn(CO)5 structure from single crystal X-ray diffraction with thermal ellipsoids shown on non-hydrogen atoms at 50% probability level, hydrogen atoms shown as open circles, and energy minimization by DFT computations (B3LYP, 3-21G/6-31G*). A table of selected internuclear distances observed in the molecular structure and calculated by DFT is also included.
SUMMARY AND CONCLUSIONS
Transition metal complexes that are sources of d5, d7, and d9 17-electron metal-centered radicals, including [CpCr(CO)3]2, [CpMo(CO)3]2, [CpFe(CO)2]2, [Co(CO)4]2, and [Mn-(CO)5]2, react with rhodium(II) Dibenzotetramethylaza[14]-annulene dimers to form (tmtaa)Rh-M complexes (M = CrCp(CO)3, MoCp(CO)3, FeCp(CO)2, Co(CO)4, and Mn-(CO)5). The molecular structures for (tmtaa)Rh-FeCp(CO)2, (tmtaa)Rh-Co(μ-CO)(CO)3, and (tmtaa)Rh-Mn(CO)5 were determined by single crystal X-ray diffraction. The structure of (tmtaa)Rh-Co(μ-CO)(CO)3 has a semibridging carbon monoxide supported by a Rh-Co bond. DFT calculations correctly mimic the observed bridging CO in (tmtaa)Rh-Co(μ-CO)-(CO)3 and anticipate this type of bridging CO unit in (tmtaa)Rh-CrCp(CO)3 and (tmtaa)Rh-MoCp(CO)3, for which molecular structures could not be obtained. Equilibrium constants evaluated for the metathesis reactions show that the processes are nearly free energy neutral with ΔG° (298 K) varying in the range from +0.9 to +1.5 kcal mol−1. Measurements of reaction equilibria permit the estimation of several (tmtaa)Rh-M bond dissociation enthalpies (Rh—Cr = 19 kcal mol−1, Rh—Mo = 25 kcal mol−1, and Rh—Fe = 27 kcal mol−1). Several of the (tmtaa)Rh-M derivatives (M = CpCr(CO)3 (7), CpMo(CO)3 (8), and Mn(CO)5 (11)) react with synthesis gas (1:1 CO/H2) at 1 atm and 298 K to form (tmtaa)Rh-CHO and M-H derivatives, which makes these systems potential candidates for hydrogenation of the rhodium formyl unit under more forceful conditions.
Supplementary Material
Acknowledgments
This research was supported by the National Science Foundation through Grant CHE-1362016 and assisted by major research instrumentation Grant CNS-09-58854.
Footnotes
Supporting Information
Solution spectra, equilibrium measurements, full crystallographic tables, and crystallographic information files (cif). This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interests.
References
- 1.Imler GH, Bhagan S, Coffin VL, Wayland BB. Inorg Chem. 2012;51:3352. doi: 10.1021/ic3001224. [DOI] [PubMed] [Google Scholar]
- 2.Imler GH, Zdilla MJ, Wayland BB. Inorg Chem. 2013;52:11509. doi: 10.1021/ic401778c. [DOI] [PubMed] [Google Scholar]
- 3.Imler GH, Zdilla MJ, Wayland BB. J Am Chem Soc. 2014;136:5856. doi: 10.1021/ja501173d. [DOI] [PubMed] [Google Scholar]
- 4.Anderson DJ, Eisenberg R. Inorg Chem. 1994;33:5378. [Google Scholar]
- 5.Wayland BB, Sherry AE, Poszmik G, Bunn AG. J Am Chem Soc. 1992;114:1673. [Google Scholar]
- 6.Wayland BB. Polyhedron. 1988;7:1545. [Google Scholar]
- 7.Wayland BB, Sherry AE, Bunn AG. J Am Chem Soc. 1993;115:7675. [Google Scholar]
- 8.Wayland BB, Vanvoorhees SL, Wilker C. Inorg Chem. 1986;25:4039. [Google Scholar]
- 9.Farnos MD, Woods BA, Wayland BB. J Am Chem Soc. 1986;108:3659. [Google Scholar]
- 10.Wei ML, Wayland BB. Organometallics. 1996;15:4681. [Google Scholar]
- 11.Bunn AG, Wei ML, Wayland BB. Organometallics. 1994;13:3390. [Google Scholar]
- 12.Van Voorhees SL, Wayland BB. Organometallics. 1987;6:204. [Google Scholar]
- 13.Fu XF, Basickes L, Wayland BB. Chem Commun (Cambridge, UK) 2003:520. doi: 10.1039/b212027e. [DOI] [PubMed] [Google Scholar]
- 14.Bunn AG, Ni YP, Wei ML, Wayland BB. Inorg Chem. 2000;39:5576. doi: 10.1021/ic000647k. [DOI] [PubMed] [Google Scholar]
- 15.Fu X, Li S, Wayland BB. Inorg Chem. 2006;45:9884. doi: 10.1021/ic0615022. [DOI] [PubMed] [Google Scholar]
- 16.Fu XF, Wayland BB. J Am Chem Soc. 2005;127:16460. doi: 10.1021/ja054548n. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X-X, Parks GF, Wayland BB. J Am Chem Soc. 1997;119:7938. [Google Scholar]
- 18.West NM, Miller AJM, Labinger JA, Bercaw JE. Coord Chem Rev. 2011;255:881. [Google Scholar]
- 19.Elowe PR, West NM, Labinger JA, Bercaw JE. Organometallics. 2009;28:6218. [Google Scholar]
- 20.Hidai M, Fukuoka A, Koyasu Y, Uchida Y. J Mol Catal. 1986;35:29. [Google Scholar]
- 21.Li C, Widjaja E, Garland M. Organometallics. 2004;23:4131. [Google Scholar]
- 22.Rida MA, Smith AK. J Mol Catal A: Chem. 2003;202:87. [Google Scholar]
- 23.Green JC, Green MLH, Parkin G. Chem Commun (Cambridge, UK) 2012;48:11481. doi: 10.1039/c2cc35304k. [DOI] [PubMed] [Google Scholar]
- 24.Labinger JA. Inorg Chim Acta. 2015;424:14. [Google Scholar]
- 25.Baird MC. J Organomet Chem. 2014;751:50. [Google Scholar]
- 26.Pugh JR, Meyer TJ. J Am Chem Soc. 1992;114:3784. [Google Scholar]
- 27.Woska DC, Ni YP, Wayland BB. Inorg Chem. 1999;38:4135. [Google Scholar]
- 28.Klingler RJ, Rathke JW. J Am Chem Soc. 1994;116:4772. [Google Scholar]
- 29.Bitterwolf TE. Coord Chem Rev. 2000;206:419. [Google Scholar]
- 30.Stolzenberg AM, Cao Y. J Am Chem Soc. 2001;123:9078. doi: 10.1021/ja002954v. [DOI] [PubMed] [Google Scholar]
- 31.Li S, de Bruin B, Peng CH, Fryd M, Wayland BB. J Am Chem Soc. 2008;130:13373. doi: 10.1021/ja804010h. [DOI] [PubMed] [Google Scholar]
- 32.Coffin VL, Brennen W, Wayland BB. J Am Chem Soc. 1988;110:6063. doi: 10.1021/ja00226a022. [DOI] [PubMed] [Google Scholar]
- 33.Wayland BB, Coffin VL, Farnos MD. Inorg Chem. 1988;27:2745. [Google Scholar]
- 34.Koenig T, Finke RG. J Am Chem Soc. 1988;110:2657. [Google Scholar]
- 35.Koenig TW, Hay BP, Finke RG. Polyhedron. 1988;7:1499. [Google Scholar]
- 36.Koenig T, Scott TW, Franz James A. Bonding Energetics in Organometallic Compounds. Vol. 428. American Chemical Society; Washington, DC: 1990. p. 113. [Google Scholar]
- 37.Ng FTT, Rempel GL, Mancuso C, Halpern J. Organometallics. 1990;9:2762. [Google Scholar]
- 38.Halpern J. Polyhedron. 1988;7:1483. [Google Scholar]
- 39.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, revision B.1. Gaussian Inc; Wallingford, CT: 2009. [Google Scholar]
- 40.Kiss G, Zhang K, Mukerjee SL, Hoff CD, Roper GC. J Am Chem Soc. 1990;112:5657. [Google Scholar]
- 41.Tilset M, Parker VD. J Am Chem Soc. 1989;111:6711. [Google Scholar]
- 42.Tilset M, Parker VD. J Am Chem Soc. 1990;112:2843. [Google Scholar]
- 43.Friedel RA, Wender I, Shufler SL, Sternberg HW. J Am Chem Soc. 1955;77:3951. [Google Scholar]
- 44.Fumagalli A, Ulivieri P, Costa M, Crispu O, Della Pergola R, Fabrizi de Biani F, Laschi F, Zanello P, Macchi P, Sironi A. Inorg Chem. 2004;43:2125. doi: 10.1021/ic035133a. [DOI] [PubMed] [Google Scholar]
- 45.Cotton FA, Czuchajowska-Wiesinger J. Gazz Chim Ital. 1992;122:321. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.