

Abstract
The Gram-negative organism Campylobacter jejuni is the major cause of food poisoning. Unlike Escherichia coli, which has two major porins, OmpC and OmpF, C. jejuni has one, termed major outer membrane protein (MOMP) through which nutrients and antibiotics transit. We report the 2.1-Å crystal structure of C. jejuni MOMP expressed in E. coli and a lower resolution but otherwise identical structure purified directly from C. jejuni. The 2.1-Å resolution structure of recombinant MOMP showed that although the protein has timeric arrangement similar to OmpC, it is an 18-stranded, not 16-stranded, β-barrel. The structure has identified a Ca2 + bound at the constriction zone, which is functionally significant as suggested by molecular dynamics and single-channel experiments. The water-filled channel of MOMP has a narrow constriction zone, and single-molecule studies show a monomeric conductivity of 0.7 ± 0.2 nS and a trimeric conductance of 2.2 ± 0.2 nS. The ion neutralizes negative charges at the constriction zone, reducing the transverse electric field and reversing ion selectivity. Modeling of the transit of ciprofloxacin, an antibiotic of choice for treating Campylobacter infection, through the pore of MOMP reveals a trajectory that is dependent upon the presence metal ion.
Abbreviations: MOMP, major outer membrane protein; SeMet, selenomethionine; rMOMP, high-resolution structure of MOMP purified from an E. coli overexpression system; nMOMP, high-resolution structure of MOMP purified from Campylobacter; PEG, polyethylene glycol; RMSF, root mean square fluctuations; MD, molecular dynamics; Octyl-POE, n-octylpolyoxyethylene; CV, column volume
Keywords: Campylobacter, outer membrane proteins, antibiotic resistance, β-barrel, porins
Graphical Abstract
Highlights
-
•
The crystal structure of MOMP, the general diffusion porin of Campylobacter, has been determined.
-
•
The protein is an 18-stranded β-barrel that is different than the 16-stranded OmpC and OmpF proteins from E. coli, but like them, MOMP is trimeric.
-
•
The protein has a central pore size and conductivity intermediate between OmpC and OmpF.
-
•
A Ca2 + ion bound at the constriction zone influences the biophysical properties of porin.
-
•
The trajectory of the transit of the antibiotic ciprofloxacin through the pore is dependent on the presence of a metal ion.
Introduction
Campylobacter are Gram-negative bacteria belonging to the ε-proteobacteria, which are commensal in livestock, notably in poultry, and are thought to be the main natural reservoir. Human infection most commonly occurs via consumption of undercooked animal products or contaminated water and through contact with animals [1]. The pathogen Campylobacter jejuni is the major cause of human food poisoning and represents an economic burden [2]. In general, C. jejuni infection resolves without antibiotic treatment, but more serious cases (usually in infants) can be treated straightforwardly with antibiotics. Untreated campylobacteriosis can lead to a cycle of recurrent infections, and this in turn progresses to irritable bowel syndrome [3]. In rare but serious cases, infected individuals can develop non-trauma-related paralysis through Guillain–Barré syndrome [4].
In the recent years, multi-antibiotic-resistant Campylobacter species have emerged, and although not common, this trend has alarmed health care professionals [5]. Bacteria resist antibiotics by the expression of drug-modifying enzymes (e.g., β-lactamase), mutation of target (ribosome), drug efflux pumps (e.g., AcrAB-TolC in Enterobacteria, Mex family in Pseudomonads, and CmeABC in Campylobacterales), and reduced uptake (e.g., decreased expression or mutations of porins in Gram-negative bacteria) [6], [7]. Porins are responsible for the uptake of nutrients through the outer membrane of the Gram-negative bacteria by passive diffusion along the concentration gradients. Their substrate uptake can be specific or non-specific, depending on the nature of the protein [7].
Most antibiotics cannot pass through the outer membrane itself so instead must enter the cell via non-specific porins. In Escherichia coli, two outer membrane proteins, OmpC and OmpF, are predominant. A shift in expression profile from OmpF (higher conductance) to OmpC (lower conductance and less permeable to antibiotics) has been commonly observed in drug-resistant clinical isolates [8]. Combined structural, physical, and computational studies of a series of clinical OmpC mutations revealed a molecular basis for altered antibiotic uptake [9], [10]. In contrast to E. coli, C. jejuni has only one major outer membrane protein (MOMP), which is thus far present in all isolates and is highly (but not absolutely) conserved in other Campylobacteria. MOMP is a 44-kDa protein, which has the sequence signature typical of β-barrel porin, and CD spectroscopy [11] confirmed the predominance of β-strand typical of porins. MOMP has been reported to be critical to the stability and integrity of the outer membrane of C. jejuni [12], while other studies have shown MOMP to have a role in adhesion to epithelial cells [13]. As might be expected, given its location in the bacterial cell, antibodies are frequently raised against it by the human immune system [14], [15], [16], and this has led to ideas for vaccine development [17]. Structural and biophysical data are thus highly desired for this protein.
Here, we report the successful recombinant expression, extraction, purification, and structure determination of C. jejuni 85H strain MOMP from an E. coli overexpression system. Campylobacter are ε-proteobacteria, while E. coli belongs to the γ-proteobacteria, meaning there are large differences in lipid composition in the outer membrane. To allay concerns about the “nativeness” of this recombinant structure arising from the difference in the lipid environment during expression, we also determined the structure of MOMP purified directly from Campylobacter and we found it to be essentially identical. Crystals, but no structure of MOMP from C. jejuni, have been previously reported [18]. The lower purity of the MOMP obtained directly from Campylobacter highlighted the key advantage of E. coli expression. MOMP exhibits conductivity similar to OmpC; however, its constriction zone has a number of important differences that may relate to antibiotic permeability. Using this structure, accurate models can be generated for MOMPs from other strains and other species of Campylobacter.
Results
Structure of MOMP
Codon-optimized MOMP-coding gene from strain 85H was cloned in pTAMAHisTEV vector, which inserts a tobacco etch virus and a cleavable hexa-histidine tag between the protein and the TamA signal peptide. After the overexpression in E. coli and purification, a final yield of 12 mg of MOMP per 50 g of E. coli cell paste was obtained. In a different approach, native MOMP was purified from Campylobacter as described previously [11] and yielded 1 mg protein per 3 g of C. jejuni cell paste. Both proteins gave crystals, although in different conditions with different crystal symmetry.
The structure of the overexpressed selenomethionine (SeMet) variant MOMP was solved using single-wavelength anomalous diffraction to provide initial phases, which were improved by density modification. This structure was then used to solve the high-resolution structure of MOMP purified from an E. coli overexpression system (rMOMP). The protein purified from Campylobacter (nMOMP) was solved via molecular replacement using the recombinant MOMP structure as a search model (Table 1). Beyond small differences due to crystal packing and resolution, we have not detected any meaningful structural difference between the two proteins (RMSD of 0.45 Å for 398 Cα position using SSM [19]; Supplementary Fig. 1). Our discussion focuses on the higher-resolution rMOMP structure (2.1 Å). We use rMOMP and nMOMP, where we discuss the evaluation of each preparation method (to establish they are essentially identical), and MOMP to refer to the generic protein.
Table 1.
Data collection, refinement, and validation statistic
| MOMP Native | MOMP Recombinant | Recombinant MOMPSeMet | |
|---|---|---|---|
| Space group | P212121 | P63 | P63 |
| Cell dimensions a, b, c (Å) | 94.4, 99.4172.2 | 90.6, 90.6, 104.4 | 94.3, 94.3, 102.0 |
| Cell dimensions α, β, γ (°) | 90, 90, 90 | 90, 90, 120 | 90, 90, 120 |
| Resolution (Å) | 47.75–2.9(3.06–2.9) | 39.2–2.1(2.17–2.1) | 2.82 |
| Rmerge | 0.14 (0.86) | 0.06 (0.72) | 0.076 (0.90) |
| Completeness | 99.9 (99.8) | 99.5 (99.5) | 99.7 (99.7) |
| Multiplicity | 7.3 (7.2) | 13.5 (13.6) | 14.6 (15.0) |
| I/σ(I) | 12.9 (3.0) | 29.89 (3.92) | 23.1 (4.3) |
| Refinement | |||
| Rfactor/Rfree (%) | 23.9/27.1 | 19/22.27 | |
| No. of unique reflections | 36,693 | 28,313 | |
| No. of residues | 405 | 403 | |
| Water | 5 | 127 | |
| Bonds length (Å) | 0.01 | 0.01 | |
| Bonds angles (°) | 1.9 | 1.41 | |
| MolProbity score | 1.70 | 1.03 | |
Values in parentheses are for the highest-resolution shell.
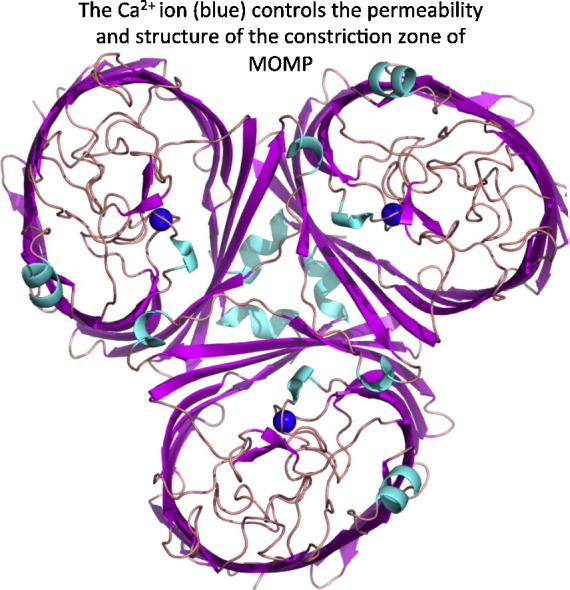
MOMP is an 18-stranded antiparallel β-barrel porin with an elliptical shape common to other 18-stranded porins. The signal peptide of nMOMP from C. jejuni strain 85H (uniprot entry number: Q659I5) was predicted to cleave between Ala22 and Thr23. Henceforth, Thr23 being the first residue in the tertiary structure was defined as the first residue in our residue count and was renamed to Thr1. In rMOMP, however, due to cloning artifacts, we have three additional non-native residues after signal peptide cleavage (Gly-Ala-Met), followed by a single point mutation of Thr1 to Gly. Typical of porins, the axis of the strands is offset, relative to the membrane normal. The long axis of the ellipse is approximately 37 Å (the distance from Gly179 to Glu280) and the minor axis is 31 Å (Arg45 to Ala234). The vertical height of the barrel varies between 19 Å (distance between Val10 and Phe86) and 37 Å (distance between Thr221 and Val334, on the opposite side; Fig. 1a–b). The N terminus on the periplasmic face has a strand, followed by an N-terminal α-helix that points away from the barrel wall (Fig. 1c). In the protein purified from Campylobacter, the helix is also present and adopts the same organization but lacks the short strand seen in the overexpressed recombinant protein. In keeping with convention for porins, extracellular loops connecting the strands are numbered sequentially and prefixed by L, whereas periplasmic loop connections are prefixed by T (also numbered sequentially). Four extracellular loops L1, L3, L4, and L6 fold inside of the barrel, with L3, L4, and L6 forming the constriction zone. Loop 4 contains Phe173-Lys174, the site of the proteinase K cleavage [20]; the structure shows that the site to be exposed is consistent with its cleavage. MOMP from another strain (79AH) is not cleaved at this position as it lacks the protease site. We observed additional electron density adjacent to L3 and L4 that we identify as a Ca2 + (rather than Zn2 + or Mg2+, or K+ or Na+) based on electron density and coordination (distance and geometry). The proteins expressed in E. coli have not had any Ca2 + ion added and we conclude that the ion was bound during expression in E. coli and has remained attached in throughput purification. The ion has octahedral coordination, with one oxygen atom from the side chain of Asp120, Gln152, Asp155, and Glu288, and has the carbonyl of Asp120 with a water molecule filling the final sixth position. The water is in turn coordinated by the amino acid Asp116 and Asp289 (Fig. 1d–e). The metal ion creates a cross-link among L3, L4, and L6. Additionally, the difference electron density map showed a residual elongated density that was modeled as an ethylene glycol like the molecule [from polyethylene glycol (PEG) 400 or C8E4] in the rMOMP. This molecule sits below the constriction zone underneath L3. The protein purified from Campylobacter was crystallized in the presence of Ca2 + ions and has an ion in an identical position but also has a second ion located outside the barrel at the interface between subunits. This ion is coordinated by Asp145 (main chain and side chain), Asn180 (side chain), and the main of Gly72 from the neighboring subunit. The recombinant structure shows a different side-chain conformation, implying that in the recombinant protein, this site does not exist. It had been hypothesized, according to molecular modeling, that in the 79AH strain, there is a metal-ion-binding site at Ser81 and Asp82 (found as Glu82 and Lys83 in 85H) that is important for the trimerization of 79AH [20]. These residues are located within a regular strand (β4); consequently, the side chains point in opposite directions and we suggest that there is no such metal-ion-binding site.
Fig. 1.

The structure of the MOMP monomer. (a) Viewed from the side, parallel to the membrane. (b) The structure has been rotated 90° so that it is viewed from the outside of the cell (looking in), perpendicular to the membrane. (c) The trimer viewed as in (b), the periplasmic N-terminal α-helix is colored magenta and the calcium in purple. An XYZ axis is shown to orientation. (d) The calcium is depicted as a purple sphere and the water molecule as a red sphere. Residues involved in the calcium coordination are shown as sticks. An XYZ axis shows the view that has been rotated by 90° around the X axis when compared to (c). (e) A detailed view of the amino acids involved in calcium-binding site; same orientation as in (d). The Fo-Fc and 2Fo-Fc electron density maps at 5σ and 2σ, respectively, have shown the final refined coordinates. The phases for the calculation of the map were based on a model that had never included the metal ion.
Analysis of the molecular surface using the program CCP4MG [21] shows that the outside wall of the barrel is uncharged as expected for proteins embedded within a hydrophobic lipid membrane bilayer. The extracellular surface of MOMP has regions of both positive and negative charge with the funnel-shaped region leading to the constriction zone, termed the eyelet, which is strongly negatively charged. The periplasmic face of the protein, including the inside surface of the barrel below the constriction, is predominantly negatively charged (Fig. 2). The pore axis is slightly offset, relative to the membrane normal. When viewed from the extracellular face, L7 sits above the entrance. Analysis with MolAxis [22] reveals that the constriction zone narrows to a radius of 2.6 Å. The constriction zone, like the protein, has an elliptical, not circular, shape. On one surface of the constriction zone, Tyr304 sits among amino acids Asp116, Asp120, Asp155, Asp289, and Glu300, which form a strongly negatively charged region of the pore; significantly, these residues also coordinate the Ca2 + ion. On the opposite side of the constriction zone, the surface is formed by residues Arg17, Arg19, Arg45, and Arg398, which create a strongly positively charged region. This arrangement gives rise to a profound dipole across the short axis of the elliptical constriction zone, termed the transverse electric field. Lys43 and Lys404 are not in the constriction zone but point toward it from the extracellular and periplasmic sides, respectively. In addition to the metal ion, several contacts appear crucial to maintain the structure of the constriction zone; Arg362 makes salt bridges with Asp337 and Glu109; Arg45 with Asp65, Asp120, and Glu151.
Fig. 2.

The pore of MOMP. (a) The surface of MOMP is colored by electrostatic charge calculated in CCP4MG [21]. The structure has the same orientation as in Fig. 1a. (b) The structure is colored as in (a), with the constriction zone circled. The structure on the left is the same orientation as in Fig. 1b; the structure on the right has been rotated 180° (in effect, looking from the periplasm through the membrane to outside the cell). (c) MOMP trimer viewed from outside the cell; the structure has the same orientation as in Fig. 1c. (d) A detailed view of the charged amino acids at the constriction zone; the view is from outside the cell and the same as in Fig. 1b. The Ca2 + ion, which has been omitted for clarity, is bound to Glu155.
Although only one monomer is present in the asymmetric unit, analysis of the crystal packing reveals a trimeric arrangement reminiscent of that seen in OmpC [23] and OmpF [24]. The identical trimeric arrangement is found in the protein purified from Campylobacter; in this case, the trimer is found in the crystallographic asymmetric unit. Analysis of the contacts between monomers using the PISA server [25] (which assesses the likely stability of multimers) identifies this trimer as the stable unit of MOMP. Gel-filtration data point to a trimeric arrangement consistent with electron microscopy of MOMP reconstituted into lipid bilayers [26]. The N-terminal Thr1 (Gly1 in recombinant MOMP) makes hydrogen bonds to strand 1 of the neighboring monomer; consequently, the three N-terminal helices in the trimer form a triangle. The main points of contact that stabilize the trimer arise from interactions with L2 from one monomer with L2, strand 5, strand 6, and L3 from the other monomer. Notably, residues Asn80 to Lys84 in this loop make main chain and, in some cases, side-chain hydrogen bonds across this interface. There is also a smaller area of contact on the periplasmic face between T8 (Lys370 to Phe376) on one monomer and T1 (Asp56 to Phe58) from the other. Each monomer–monomer interface buries over 2400 Å2 of surface area; the trimer buries 11,600 Å2 of surface area.
Single-channel conductance measurements
The pore-forming activities of both the rMOMP (Fig. 3) and nMOMP (Supplementary Fig. 3) were measured by single-channel ion-conductance measurements. Figure 3a and b shows the typical ion-current electrical signature of single monomeric and trimeric rMOMP, respectively. Figure 3a shows the monomers at negative (− 100 mV) and at positive (+ 100 mV) applied transmembrane voltages. It is interesting to note that at positive voltages, we observe only downward but never upward spikes, suggesting the presence of a monomer channel as opposed to the partially blocked oligomers. At negative voltages, the MOMP monomer has upward spikes leading to the closure of the pore (Fig. 3a and Supplementary Fig. 3b). After the single-channel insertion, we flush the cuvette intensively with fresh buffer to avoid further insertion. Figure 3b shows the typical traces of trimers. At positive voltages, the trimeric channel gives a smooth response, whereas at negative voltages, flickering noise occurs. The analysis of repeated single rMOMP channel reconstitution in 1 M KCl revealed two main conductance levels, a lower level of 0.7 ± 0.2 and a higher level of 2.2 ± 0.2 nS, suggesting a monomeric and trimeric state. The same analysis for nMOMP yields 0.7 ± 0.2 and 2.3 ± 0.3 nS, respectively. Like other trimeric porins, both monomer and trimer follow an ohmic conductance pattern between ± 200 mV (Supplementary Fig. 3a). No meaningful differences between the nMOMP and rMOMP proteins were observed. To investigate the homogeneity in the insertion of proteins, we followed multichannel measurements, and the distribution between monomers and trimers was random (Supplementary Fig. 3f). Control measurements with 1 M NaCl instead of KCl and 10 mM Mes (pH 6.0) revealed similar flickering at negative voltages (Supplementary Fig. 4a).
Fig. 3.

Representative ion-current traces of rMOMP in 1 M KCl and 10 mM Mes (pH 6.0). (a) The ion-current corresponding to one open monomer at negative (top panel, left) and positive (top panel, right) transmembrane potential of 100 mV. (b) The ion-current corresponding to the trimer at negative (top panel, left) and positive (top panel, right) transmembrane potential of 100 mV. (c) The ion-current corresponding to a trimer following the addition of 10 mM CaCl2 (1 M KCl at pH 6.0) at negative (bottom panel, left) and positive (bottom panel, right) transmembrane potential of 100 mV. (d) The ion-current corresponding to a trimer following the addition of 10 mM MgCl2 (1 M KCl at pH 6.0) at negative (bottom panel, left) and positive (bottom panel, right) transmembrane potential of 100 mV. The monomer and the trimer were produced from different bilayer measurements.
To elucidate the effect of divalent cation, we performed conductance measurements with 10 mM calcium chloride in the presence of 1 M KCl at pH 6.0. The presence of calcium chloride (Fig. 3c) eliminated the noise with a slight increase in conductance of 2.4 ± 0.2 nS at positive transmembrane potential of + 100 mV and a somewhat higher conductance of 2.8 ± 0.2 nS at negative transmembrane potential of − 100 mV. It is interesting to note that the measurements performed in either 10 mM MgCl2 (Fig. 3d) or 10 mM ZnCl2 (Supplementary Fig. 4b) caused flickering as observed in a calcium-free solution (Fig. 3a and b).
Conductance measurements were performed in the presence of the chelating agent EGTA. After the overnight incubation of the protein samples with 10 mM of EGTA, we observed in 1 M KCl, 10 mM Mes, and 10 mM EGTA at pH 6.0 a conductance of 2.4 ± 0.1 nS and, at negative potential, a conductance of 2.5 ± 0.1 nS (Supplementary Fig. 4c). As with native protein, noisy traces were observed with upward spikes seen at negative voltages. In a control measurement, we removed the EGTA by the addition of 10 mM CaCl2 in 1 M KCl buffer and observed the previous CaCl2 behavior (Supplementary Fig. 4d).
Ion selectivity
The ion selectivity was obtained from zero-current membrane potential measurements in the presence of up to 8-fold KCl concentration ratio (0.1 M KCl versus 0.8 M KCl). Approximately, 300–500 protein channels were reconstituted under a voltage of + 20 mV. After the saturation of channel insertion, we measured the zero-current membrane potential under a wide range of concentration gradient ratios ranging from 1.5 to 7.5. At a concentration gradient of 500 mM (0.1 M versus 0.5 M), the zero-current membrane potential was 28 ± 4 mV at the diluted side, revealing the preference for cations. Goldman–Hodgkin–Katz equation [27] determines the ratio of cation to anion (PK+/PCl-) to be 7 ± 2 for nMOMP and within the error identical for rMOMP (6 ± 1). Similarly, measurements were performed in the presence of 8-fold calcium chloride (0.1 M CaCl2 versus 0.8 M CaCl2) and no KCl. The zero-current membrane potential was measured as − 19 ± 1 mV, which implies that the cationic selectivity exhibited by the channel in monovalent cation (KCl) turns into anionic in the presence of divalent cation (CaCl2). This is consistent with the structure that shows that the narrow region of the constriction zone is lined with five negatively charged amino acid residues, which would favor the transport of cationic molecules over anionic ones. However, in the presence of calcium chloride, Ca2 + binds to the negatively charged residues reversing the ion selectivity (Supplementary Fig. 3g).
Interaction of ciprofloxacin with MOMP
A single trimeric channel of MOMP was reconstituted into artificial lipid bilayers containing 1 M KCl (without CaCl2). Addition of 1 mM ciprofloxacin to the trans side of the lipid bilayer decreased the conductance by 0.2 nS to 2.2 nS, along with an increase in the current noise (Supplementary Fig. 5a and b). The stronger flickering does not allow individual blocking events to be distinguished (Supplementary Fig. 5a). Increasing the concentration of ciprofloxacin to 2 mM reduces the conductance to ~ 2.1 nS at + 150 mV (Supplementary Fig. 5a and c). In the presence 1 M KCl, 10 mM Mes, and 10 mM CaCl2 addition of 0.5 mM ciprofloxacin to trimeric MOMP resulted in distinct blockages, which were concentration dependent (Fig. 4a and b). The blockage events were fitted into a kinetic binding model with an on rate, kon, of ʋ/3[c] (ʋ = number of blockages per second; [c] is concentration) and an off rate, koff, of 1/τ (τ = dwell time) [28]. The average dwell time did not depend on the concentration of the ciprofloxacin and was found to be in the range of 60 μs for the voltages applied between 125 and 199 mV (Supplementary Fig. 5d and e). The values of kon and koff obtained are shown in Table 2. Twofold increase in the blocking events on the addition of ciprofloxacin on the trans side when compared to the addition to the cis side was observed. This suggests that ciprofloxacin accessed the binding site more readily from the trans side. Addition of ciprofloxacin in the presence of magnesium chloride resulted in fast flickering events (Supplementary Fig. 6), consistent with a specific modulation in function by Ca2 +. In fact, in the presence of CaCl2, we saw mainly a trimeric protein, suggesting that the MOMP trimer was itself stabilized by CaCl2. The interaction of ciprofloxacin with MOMP trimers in the presence of calcium chloride resulted in clean blockage events. With monomeric protein, the blocking events were replaced with flickering.
Fig. 4.

Ciprofloxacin binds to and blocks channel conductance. (a) Transient blockages are observed in the ion-current trace of single trimeric rMOMP channel in the presence of 1 M KCl, 10 mM Mes, and 10 mM CaCl2 (pH 6.0) when 0.5 mM ciprofloxacin is added to trans side. The applied transmembrane potential was + 150 mV. (b) The number of binding events increases with the increase in the concentration of the ciprofloxacin from 0.25 to 1 mM measured at 150 mV.
Table 2.
Kinetics of ciprofloxacin entry into MOMP channel
| Rate constants | 0.5 mM ciprofloxacin |
|---|---|
| kon cis (× 103 M− 1 s− 1) | 47 ± 10 |
| kon trans (× 103 M− 1 s− 1) | 73 ± 7 |
| koff (× 103 s− 1) | 16 ± 1 |
Experimental conditions: 1 M KCl, 10 mM Mes, 10 mM CaCl2, and 0.5 mM ciprofloxacin (pH 6.0); T = 20 °C. The applied transmembrane voltage was 150 mV.
In silico modeling
Two rMOMP structures, one with calcium and one without, were equilibrated in silico, and the RMSD and root mean square fluctuations (RMSF) of the protein's backbone were calculated. With calcium, the structure is considerably more stable (RMSD of 1.45 Å versus 1.75 Å and RMSF halved). The instability was focused on the loops involved in calcium coordination (Fig. 5a). Although the empty site was occupied transiently by one of the sodium ions used to neutralize the system, its presence was not sufficient for the stabilization of the structure. Figure 5b shows that the pore size slightly increased in the absence of Ca2 +.
Fig. 5.


MOMP permeability. (a) RMSF of protein backbone for the two simulated structures: not Ca2 + bound and Ca2 + bound. The loops presenting bigger changes in fluctuation values are highlighted in orange. (b) The pore radius of MOMP monomer moving from the extracellular side to the periplasm without bound calcium (gray) and with bound calcium (green). (c) The macroscopic intrinsic electric field of MOMP with and without calcium. A reduction of the transversal components from ~ 18 mV/Å in the structure without calcium to ~ 7 mV/Å when calcium is present. (d) The putative pathway for water diffusion shifts toward the calcium ion when it is present (ice blue), while in the structure without calcium ion (purple), it avoids the cluster of negative residues that coordinate Ca2 +. MOMP monomer is shown in cartoon representation. (e) Free energy surface for the translocation of ciprofloxacin through MOMP without and with calcium. Antibiotic dipole moment orientation is depicted onto a licorice representation of ciprofloxacin in red. Each isocontour corresponds to a free energy difference of 2 kcal mol− 1. Free energy values were rescaled for each surface in order to have the absolute minimum equal to zero. The most relevant minima for ciprofloxacin translocation have been labeled for both scenarios: one in the extracellular region (EX) and two inside the constriction region (CR1, CR2).
In the absence of the calcium ion (Fig. 5c), the transverse internal electric field [29] of ~ 18 mV/Å was comparable to OmpC (~ 20 mV/Å) but lower than OmpF (36 mV/Å). When calcium was present, the internal electric field was reduced to almost a third of its value due to charge screening, which in turn reduced the transversal electric field in the constriction region to ~ 7 mV/Å. The divalent ion induced a rotation of the transversal electric field and a change in the conformation of the internal loops, which led to a shift in the putative path that water was predicted to diffuse along. When calcium was not present, water avoided passing near the cluster of negative residues that bind the divalent ion located in the region (Fig. 5d), whereas when calcium was present and the transversal electric field rotated, the water pathway was shifted toward the ion position.
In order to understand the effects of the presence of the cation in the permeation of polar antibiotics, the free energy profiles were calculated for ciprofloxacin translocation for both calcium-bound and calcium-free protein. The free energy maps show that the main barrier to translocation was, as expected, located at the constriction zone (Fig. 5e). Both free energy maps share common features but differed at the calcium-binding site. In the absence of calcium, ciprofloxacin approaches the protein constriction region by entering with its positive group, which is attracted toward the negatively charged calcium-binding site (Supplementary Fig. 7). This is similar to what happens in OmpF from E. coli, where the negative patch on L3 modulated the translocation of antibiotics and traps a divalent cation [30].
In the presence of calcium ion (Supplementary Fig. 7), ciprofloxacin entered with the opposite orientation, pointing its carboxylic group toward the ion. Translocation of ciprofloxacin starting from the “cis” side required us to apply a voltage above + 100 mV to achieve this favorable orientation. Critically, molecular dynamics (MD) predicts that the antibiotic follows a different path and has a higher barrier to translocation in the presence of bound Ca2 + ion.
Discussion
We detected no difference in structure or function between the MOMP purified from C. jejuni and the MOMP heterologously expressed in E. coli. Unusually for an 18-stranded β-barrel, MOMP adopts the same trimeric arrangement as seen for OmpF and OmpC, which are 16-stranded β-barrels. Similar to these trimers, a loop from one monomer reaches across and partly into the central pore of another monomer. Whether the trimer has functional significance, other than stability, is unclear. A distinctive structural feature of MOMP is the α-helix at the N terminus. The other known outer membrane proteins with an N-terminal helix are OprB from Pseudomonas aeruginosa and the palmitoyl transferase PagP from E. coli; both proteins are monomers [31], [32]. The trimeric sucrose-specific porin (ScrY) from Salmonella typhimurium comprises a coiled-coil domain at the N-terminal periplasmic domain [33]. Although the secondary structure of the N terminus was not determined from its crystal structure [33], spectroscopic and biophysical analyses suggested that it forms α-helical coiled-coil complexes that might be involved in the supramolecular stabilization and low-affinity sugar binding of ScrY [34]. The role of the N-terminal α-helix in MOMP has not been experimentally investigated, but structural analysis suggests that it favors trimerization. The most striking difference between MOMP and OmpF/OmpC from E. coli is the presence of a Ca2 +-binding site in the constriction zone of MOMP. The site clearly has a high affinity for calcium, since the overexpressed protein had selectively bound the ion from the culture and held onto it during purification. The ion is bound to a number of key loops that form the constriction zone, suggesting its structural role. Addition of EDTA to isolated protein, which would remove any calcium, has been previously shown to destabilize the protein [20]. We see that EGTA addition results in noisy conductance (Supplementary Fig. 4) comparable to native protein, whereas the addition of 10 mM CaCl2 containing 1 M KCl gives stable conductance traces (Fig. 3c). Addition of Mg2 + (Fig. 3d) or Zn2 + (Supplementary Fig. 4b) did not result in a stable ion-current, suggesting that this is a specific feature of Ca2 +. MD analysis showed that in the absence of calcium, the loops become unstable and disrupted the order of the constriction zone. We suggest that the binding of Ca2 + anchors the loops, thus ordering the eyelet, preventing them from “flopping” into the pore, and hence eliminating the spikes seen in ion-current in the absence of calcium (Fig. 3a and b).
Comparison of MOMP sequences (one each) from C. jejuni, Campylobacter coli, Campylobacter fetus, Campylobacter lari, and Campylobacter upsaliensis shows that three (Asp116, Glu288, and Asp289) of the five amino acids involved in metal binding are absolutely conserved. Asp120, which coordinates the metal ion in C. jejuni is conserved in C. coli and C. upsaliensis, but not in C. fetus or C. lari where there is a deletion of 4 aa. Gln152, which coordinates the metal ion with its side-chain oxygen, is fully conserved in C. jejuni, C. coli, and C. upsaliensis but is replaced by an asparagine in C. fetus and an aspartic acid in C. lari (Supplementary Fig. 2). MOMP, in the absence of Ca2 +, is a cation-selective intermediate between OmpC (most) and OmpF (least). The calculated radius at the narrowest point in the constriction zone of MOMP is 2.6 Å, somewhat larger than OmpC (2.2 Å) and smaller than OmpF (3.3 Å). Liposome swelling studies [35] have shown that MOMP is capable of transporting larger neutral molecules (such as arabinose, glucose, mannose, α-ketoglutarate, etc.) than OmpC, consistent with a pore that is larger than that of OmpC.
The single-channel conductance for rMOMP and nMOMP from the 85H strain of C. jejuni was 0.7 ± 0.2 for the monomeric and 2.2 ± 0.2 nS for the trimeric state in 1 M KCl. Previously, Dé and colleagues reported lower conductances for the monomer (0.51 ± 0.04 nS) and trimer (1.5 ± 0.06 nS) [36]. Given that the different buffer condition (here at pH 6.0, rather than pH 7.4, as reported by Dé et al.) may reflect differences in experimental conditions, both the rMOMP and nMOMP channels from C. jejuni exhibit two distinct channel conductance values related to the trimer and the monomer. We are unable to determine if the monomer species represents a trimer with two closed channels or a genuine monomer arising from dissociation of the trimer. The trimer exhibits a large single-channel conductance of 2.2 nS, similar to the conductance of cyanobacterium species Synechocystis sp. PCC 6714 [37] and comparable to the E. coli OmpC (2.5 nS) [38]. The conductance of the monomer is similar to the unitary conductance of cation-selective monomeric channel P. aeruginosa OccD3 (around 0.7 nS) [39] and E. coli OmpF (1 nS; all in 1 M KCl) [40], [41]. The monomeric value is also comparable to the triton/EDTA-solubilized protein of C. jejuni UA580 strain [42] and C. coli UA30 [35], whose single-channel conductances are 0.82 and 0.53 nS, respectively. In KCl solution, the channel was selective for cations, but after the addition of CaCl2, the channel switched its selectivity for anions. This inversion of selectivity mirrors that seen for OmpF in the presence of MgCl2 [30].
The presence of Ca2 + (but once again not Mg2 +) also altered the behavior of MOMP with ciprofloxacin in single-channel experiments (Fig. 4). The antibiotic blocked the channel with frequent binding events of short residence time (60 μs). When Ca2 + ions were not present, ciprofloxacin reduced the current flow in a concentration-dependent manner (Supplementary Fig. 5a), but individual blocking events were not seen. We conclude that in the absence of the Ca2 +, antibiotic translocation occurs too rapidly to see individual events under our experimental conditions. Metal-ion-dependent blockages were observed with imipenem added to OmpPst1 channel from Providencia stuartii [28] in the presence of La3 + and with enrofloxacin added to OmpF channel in the presence of Mg2 + [30]. In both these cases, the ion does not co-purify with the protein or seem to have any role in stability.
The presence of a calcium ion at the constriction zone partly neutralizes the negative charges and thus in turn profoundly weakens the transverse electric field at the eyelet. The transverse field is known to play a key role in the translocation of polar molecules [43]. MD shows that the presence of the Ca2 + increased the barrier to transport compared to an artificial model without the ion; a prediction entirely consistent with the experimental single-channel data. A review of the antibiotic sensitivity of C. jejuni showed that the organism is less susceptible to singly or doubly anionic antibiotics and to larger, dipolar ionic molecules [35]. The increased resistance to the charged antibiotics is consistent with a reduced transverse field, which would increase the barrier to translocation.
Food poisoning by ingestion of Campylobacter is common and, as resistant strains have emerged, is causing concern. Unlike E. coli, Campylobacter have evolved to possess only one MOMP; thus, it cannot switch between OmpC and OmpF expression as E. coli does to alter antibiotic entry to the cell. In structural terms, MOMP is distinct from both OmpC and OmpF; however, in terms of pore size, it is intermediate. Uniquely, MOMP may rely on a permanently bound Ca2 + ion to modulate translocation across the outer membrane.
Materials and Methods
Molecular biology
A gene encoding MOMP from C. jejuni strain 85H (Uniprot entry number: Q659I5) was synthesized with codon optimization for E. coli by Eurofins MWG (Germany). The gene carried an NcoI enzymatic cleavage site at the 5′-end and a stop codon followed by HindIII site at the 3′-end. The momp gene was cloned into a pTAMAHisTEV expression vector. The pTAMAHisTEV vector was created by placing a TamA signal peptide into the T7 promoter ampicillin resistance pHisTEV vector [44]. Consequently, the pTAMAHisTEV plasmid expressed a protein with a TAMA signal peptide at the N terminus, followed by a histidine tag, then the TEV recognition sequence ENLYFQG, and finally the N-terminus of the target protein (here MOMP).
Protein expression
The plasmid pTAMAHisTEV harboring the MOMP gene was transformed into C43(DE3) competent cells. A 250-ml LB startup culture containing 100 μg ml− 1 ampicillin was incubated at 37 °C and at 200 RPM. Then, 20 ml of startup culture was transferred to 10 × 1 L LB containing the same amount of ampicillin and was grown at 37 °C until an OD600 of ~ 0.6. At this stage, cells were induced with 0.4 mM IPTG, the temperature was dropped to 25 °C, and growth continued for 16 h. Cells were harvested by centrifugation at 6200g (JLA8.1000 rotor, Beckman Coulter).
Cell pellets were resuspended in lysis buffer containing 20 mM Tris–HCl (pH 8.0), 300 mM NaCl, 10% glycerol, 20 μg ml− 1 DNAse, 100 μg ml− 1 lysozyme (both Sigma-Aldrich), and EDTA-free protease inhibitor cocktail (Roche). Cells were lysed by two passes through a chilled cell disruptor at 30 kpsi. Cellular debris was removed by centrifugation at 10,000g (JA 25.50 rotor, Beckman Coulter). The membrane fraction was retained by ultracentrifugation at 100,000g (50.2 Ti rotor, Beckman Coulter) at 4 °C for 1 h. A two-step process failed in our hands, in which the inner membrane was with N-lauroyl sarcosine and then discarded, followed by solubilization of the outer membrane with SB3.14, N,N-dimethyldodecylamine N-oxide (LDAO), n-octylpolyoxyethylene (Octyl-POE), or other detergents; the MOMP remained insoluble. Direct solubilization of total cell membrane with either 1% SB3.14 or 5% Elugent™ gave much improved extraction. As TEV protease was inactive in the presence of SB3.14, extraction was carried out in 20 mM Tris–HCl (pH 8.0), 150 mM NaCl, 5% (vol/vol) Elugent™ (Merck Millipore), and EDTA-free protease inhibitor cocktail using a tissue grinder and subsequent incubation at 4 °C overnight with gentle rotation. Insoluble material was removed by centrifugation (100,000g for 1 h). Elugent concentration was reduced by dilution to a final concentration of 1.25% (vol/vol). This protein solution was cycled over Ni-NTA column at 4 °C overnight using a peristaltic pump to ensure binding. The column was washed extensively with a buffer containing 20 mM Tris–HCl (pH 8.0), 30 mM imidazole, 150 mM NaCl, and 0.25% (vol/vol) Elugent™. MOMP was eluted with 2 column volume (CV) of buffer containing 250 mM imidazole. The eluent was supplemented with 1 mg His6-tagged TEV protease and dialyzed in SnakeSkin tubing (Thermo Scientific) against a buffer containing 10 mM imidazole at room temperature overnight. The dialyzed sample was passed through a 0.45-μm syringe filter to remove insoluble particles before the sample was applied to 2-ml Ni-NTA resin. Cleaved MOMP was collected from the flow-through fraction. MOMP was further purified by gel filtration (16/60 Superdex 200 pg; GE Healthcare) on an Äkta Express purifier during which Elugent™ was exchanged for 0.45% (wt/vol) C8E4. MOMP was concentrated to 10 mg ml− 1 for crystallization. Purity and integrity were monitored on SDS-PAGE (NuPAGE, Invitrogen) and mass spectrometry. SeMet-labeled MOMP was produced according to the metabolic inhibition method [45] using SeMet media from Molecular Dimensions and was purified as described above.
C. jejuni 85H strain [46] was grown according to Bolla et al. [11]. C. jejuni strain was spread onto a blood agar plate and incubated for 24 h at 42 °C. Bacteria were recovered with 1 ml of 2YT medium. Four Columbia agar plates supplemented with the appropriate amount of Campylobacter selective antibiotics supplement (Oxoid) were inoculated with 150 μl of the recovered bacteria solution and incubated for 48 h at 42 °C. 2YT medium was used to recover the bacteria, which were subsequently inoculated on 40 plates of Columbia agar and incubated for 48 h at 42 °C. Subsequently, each plate was rinsed with 5 ml of 10 mM Tris–EDTA buffer (pH 7.4) and agitated for 15 min at room temperature; then, bacterial suspension was recovered and the OD600 checked. From this point, all the following steps were performed at 4 °C. The bacteria were pelleted by centrifugation at 10,000g for 30 min. The pellet was resuspended in 200 ml of 200 mM Glycine–HCl (pH 2.2) and agitated for 15 min. Bacteria were harvested by centrifugation at 10,000g for 30 min and washed in 100 mM Tris–HCl (pH 7.4). Cells resuspended in Tris–HCl 10 mM (pH 7.4) were then lysed by using two passes at 30 Kpsi through a high-pressure cell disruption for micro volumes (Constant System Ltd). Unbroken cells were removed by spinning the cell lysate at 10,000g for 30 min. The supernatant was kept and spun down at 100,000g for 1 h. The pellet was then homogenized in 10 mM Tris–HCl (pH 7.4) and 0.1% (wt/vol) of sodium lauryl sarcosinate (Sigma) and was left rocking for 30 min. The outer membrane was recovered by ultracentrifugation at 100,000g for 1 h. The supernatant containing the inner membrane protein fraction was discarded, and the pellet was homogenized with 20 mM sodium phosphate buffer (pH 7.4) and 1% of Octyl-POE (Bachem AG) and was left rocking at 4 °C for 30 min. Solubilized proteins were recovered by ultracentrifugation at 100,000g for 1 h. Protein was loaded onto a MonoQ HR ion exchange column (GE Healthcare) and was equilibrated with 5 CV of buffer A (30 mM Na2HPO4, 10 mM NaCl, and 0.6% Octyl-POE). The bounded proteins were eluted stepwise with 5, 12, 20, 70, and 100% of buffer A supplemented with 1 M NaCl. The chromatography was performed on ÄKTA Explorer 10 system (GE Healthcare). Each fraction was examined with SDS-PAGE and Western blot with specific antibodies. Eluted fractions containing MOMP were collected and concentrated to 5 ml and injected onto a Superdex 200 16/60 GL (GE Healthcare) column equilibrated with 2 CV of 20 mM Tris–HCl (pH 8.0), 150 mM NaCl, and 0.45% (wt/vol) C8E4. Fractions containing MOMP were combined and concentrated to 10 mg ml− 1.
Crystallization
Recombinantly expressed MOMP crystallized in 50 mM sodium citrate (pH 4.25), 35% (vol/vol) PEG 400, and 70 mM KCl in a hanging-drop vapor-diffusion experiment at 20 °C with 500 μl reservoir solution and a crystallization droplet of 2 μl protein and 1 μl precipitant. SeMet MOMP crystallized under identical conditions. Crystals grew to full size after 1 week and were hexagonal in shape.
MOMP purified from Campylobacter crystallized in 0.05 M calcium chloride, 0.05 M barium chloride, 0.1 M Tris (pH 7.5), and 30% (vol/vol) PEG 400. Crystals were optimized using hanging-drop vapor-diffusion technique. Crystals appeared after 3 days in a drop made of 1 μl protein and 1 μl reservoir made of 0.05 M calcium chloride, 0.05 M barium chloride, 0.1 M Tris (pH 8), and 32% (vol/vol) PEG 400. In all cases, crystals were flash cooled before data collection, but no cryoprotectant was added.
Data collection, and structure determination and refinement
X-ray data of recombinant SeMet crystals were collected at beamline ID23-1 in ESRF, Grenoble, France and native data at Diamond. Data were processed with Xia2 [47]. Both belong to spacegroup P63 with one monomer per asymmetric unit. The SeMet structure was solved by single-wavelength anomalous dispersion to a 2.7-Å resolution with Autosolve program of Phenix [48]. Using this as search model, the 2.1-Å native structure was solved with the PHASER MR from Phenix [49]. The model was built using Arp/Warp [50]. X-ray data from a crystal of MOMP purified from Campylobacter crystals were collected in house using a Rigaku Micromax™-007HF Cu anode with VariMax optics and a Rigaku Saturn 944 + CCD detector and processed with Xia2 [47]. The structure was solved to a resolution of 2.9 Å by molecular replacement using the recombinant MOMP structure as a search model with the program Phaser [49]. Crystals belong to spacegroup P212121 with three molecules per asymmetric unit and a Matthew's coefficient of 45%.
The three models were manually completed in Coot [51] and refined with REFMAC [52]. Structures were validated with Molprobity [53]. Data collection and refinement statistic are listed in Table 1. The webserver “CheckmyMetal” validates the identification as calcium [54]. Additionally, the difference electron density map showed a residual elongated density that was modeled as an ethylene glycol like the molecule (from PEG 400 or C8E4) in the recombinant MOMP. This molecule sits below the constriction zone underneath loop 3.
Single-channel conductance measurements
A planar lipid bilayer was formed using solvent-free lipid bilayer technique [55]. In brief, the cuvettes used for our bilayer experiments consist of cis and trans chambers separated by a 25-μm thick teflon film (Goodfellow) carrying an aperture with a diameter of 40–70 μm. The spherical hole in the teflon film was made by a high-voltage cathode discharge (Electrotechnic Products). In order to form the lipid bilayer, the aperture is pre-painted with 1 μl of 1% hexadecane in hexane. Due to its high mechanical and chemical stability, 5% solution of diphytanoyl phosphatidylcholine (DPhPC, Avanti Polar Lipids) is commonly used [56]. The bilayer it forms allows for the insertion of transmembrane pores. The chambers are filled with electrolyte solution, which usually consist of 1 M KCl and 10 mM Mes (pH 6.0) with a total solution volume of 2.5 ml. MOMP was added to the cis side (which is the electrical ground or reference potential) of the chamber at a final concentration of 2 ng ml− 1, and the channel insertion was facilitated by the rapid mixing of the contents of the chamber while applying a transmembrane potential of − 199 mV. Electrical recordings were made through a pair of Ag/AgCl electrodes (World Precision Instruments), attached to an Axon Instruments 200B amplifier (Axon Instruments Inc.) in the voltage clamp mode. Data were filtered by a low-pass Bessel filter at 10 kHz and directly saved into the computer memory with a sampling frequency of 50 kHz. Data analyses were performed using Clampfit 10.0 software (Axon Instruments Inc). As observed previously [35], multiple channel events for MOMP were non-homogeneous and hence were not analyzed further (Supplementary Fig. 3h).
Ion-selectivity measurements
The ion selectivity of ~ 44-kDa MOMP was carried out by measuring the zero-current membrane potential as described elsewhere [27]. The Teflon cuvettes consist of two chambers, with a pre-designed spherical hole having a diameter of 0.5–0.5 mm2. In chloroform, 2% DPhPC serves as the pre-painting solution, and 1% solution of DPhPC in n-decane serves as the lipid for the formation of black lipid membranes. After the formation of a stable membrane, 50–100 ng/ml of MOMP protein was added to both sides of the Teflon chambers containing 0.1 M KCl solution. After the incorporation of 200–500 channels, a salt gradient was established by adding 3 M solution of KCl on one side of the membrane, and an equal volume of 0.1 M solution of KCl was added on the other side with uniform stirring of the contents. The resulting zero-current membrane potentials were measured using the high-impedance electrometer (Keithley 617).
MD
The high-resolution X-ray structure of trimeric MOMP was used as starting coordinates for MD simulations. Loop 2 from Ala77 to Glu81 was missing in the crystal structure and was modeled using MODELLER [57] that considered the optimal structure among 100 generated models. Amino acid residues were simulated in its ionization state at neutral pH. The entire trimer was embedded in a pre-equilibrated 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine bilayer of 471 lipids and the system was oriented in order to center the protein at the origin of the coordinate system and align the channel along the z-axis (positive z: extracellular side; negative z: periplasmic side). We added 53 sodium ions to neutralize the system total charge. The system was solvated with ~ 40,997 TIP3P water molecules (simulation box size: 13.72, 23.80, 10.71 nm; total number of atoms: ~ 204 k). Simulations were performed with and without the calcium ion to evaluate its effect on the structure and dynamics of loops.
After 1 ps of energy minimization (conjugate gradients), a slow heating from 10 to 300 K was carried out for 1 ns. During this stage, positional restraints were applied on the protein α-carbons (all three dimensions) and on the lipids' phosphorus atoms (along z only). After releasing the constraints on the 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, an equilibration stage follows for 4 ns in the isothermal-isobaric (NPT) ensemble at 1.0 bar and 300 K. Finally, 0.9-μs MD simulations were performed in the canonical (NVT) ensemble after the elimination of the protein restraints.
The NPT equilibration was performed with the program NAMD [58], with 1.0 fs time step, and by treating long-range electrostatics with the soft particle mesh Ewald method (64 grid points and order 4 with direct cutoff at 1.0 nm and 1.0 Å grid size). Pressure control was applied using the Nose–Hoover method (extended Lagrangian) with isotropic cell, integrated with the Langevin Dynamics (200 fs and 100 fs of piston period and decay, respectively). The latter was also applied for temperature control with 200-fs thermostat damping time. Production run in the NVT ensemble was performed with the ACEMD code [59] compiled for graphic processing units (GPUs) by rescaling the hydrogen mass to 4 au and increasing the time step up to 4.0 fs [60]. The Langevin thermostat was used with 1-ps damping time. Soft particle mesh Ewald was used to treat the electrostatics as for the equilibration stage. The Amber99SB-ILDN force field [61] was used for the protein and lipids, and the TIP3P [62] for waters.
The GAFF force field parameters [63] were used to describe ciprofloxacin (DrugBank [64] n. DB00537). Partial atomic charges were evaluated according to the restrained electrostatic potential (RESP) approach [65]: the molecule was first optimized at the HF/6-31G(d) level, up to a convergence in energy of 10− 5 au, using the Gaussian03 package [66]. Atomic RESP charges were derived from the electrostatic potential using the antechamber module of the AMBER package [67]. Parameters are freely available† [68].
Starting from the final configuration of the MOMP simulation without the calcium ion described above, the antibiotic was placed inside the lumen of the first monomer. The difference between the z-coordinate of the center of mass (com) of the antibiotic two-ring system and the z-coordinate of the com of the protein monomer was + 32.9 Å. A thousand steps of energy minimization were performed. The equilibration stage followed for 1 ns in the NVT ensemble at 300 K as described hereinbefore. Well-tempered metadynamics simulation (500 ns) was performed with the ACEMD code, until the first effective translocation through the protein constriction region was observed [69], [70]. Then, four configurations were randomly selected, two with the antibiotic located in the extracellular vestibule and two in the periplasmic vestibule. Correspondingly, four multiple walkers [71] were set to extend the metadynamics reconstruction of the free energy surface. Two biased collective variables were used, namely, the antibiotic position and the projection of the dipole moment of the antibiotic onto the x-axis of the channel. In practice, the “position” Δz was defined as the difference of the z-coordinate between the com of the antibiotic two-ring system and that of the porin first monomer. We run 4 × 1 μs, arriving to a total simulation time of 4.5 μs. During the metadynamics, energy biases were added every 1.25 ps to each collective variable (initial height: 1.0 kcal mol− 1; σ 0.3 kcal mol− 1 and 5.0 degree for position and dipole moment orientation, respectively). Well-tempered ΔT was 5000 K.
Accession numbers
Coordinates and structure factors have been deposited in the Protein Data Bank with the ID codes 5ldt and 5ldv.
Acknowledgments
L.G.M.F. and S.A.-G. are funded by EU FP7-PEOPLE-2013-ITN Translocation network Nr. 607694. The research leading to these results was conducted as part of the Translocation consortium (www.translocation.com) and has received support from the Innovative Medicines Initiatives Joint Undertaking under Grant Agreement no.115525, resources of which are composed of financial contribution from the European Union's seventh framework programme (FP7/2007-2013) and of the EFPIA companies in kind contribution. J.H.N. is Royal Society Wolfson Merit Award holder, Senior Investigator Wellcome Trust (WT100209MA), and Chinese Academy of Science 1000 Talent Scholar. M.C. thanks the PRACE consortium for the use of the Research Infrastructure CURIE based in France at TGCC through the project Tier-0 nr. RA2699. The use of beamlines at both Diamond and ESRF is acknowledged.
Edited by James Bowie
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jmb.2016.09.021.
Appendix A. Supplementary Data
Supplementary material 1.
Supplementary material 2.
Supplementary material 3.
References
- 1.Kaakoush N.O., Castaño-Rodríguez N., Mitchell H.M., Man S.M. Global epidemiology of Campylobacter infection. Clin. Microbiol. Rev. 2015;28:687–720. doi: 10.1128/CMR.00006-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ruiz-Palacios G.M. The health burden of Campylobacter infection and the impact of antimicrobial resistance: playing chicken. Clin. Infect. Dis. 2007;44:701–703. doi: 10.1086/509936. [DOI] [PubMed] [Google Scholar]
- 3.Kalischuk L.D., Buret A.G. A role for Campylobacter jejuni-induced enteritis in inflammatory bowel disease? Am. J. Physiol. Gastrointest. Liver Physiol. 2010;298:G1–G9. doi: 10.1152/ajpgi.00193.2009. [DOI] [PubMed] [Google Scholar]
- 4.Allos B.M. Association between Campylobacter infection and Guillain–Barré syndrome. J. Infect. Dis. 1997;176:S125–S128. doi: 10.1086/513783. [DOI] [PubMed] [Google Scholar]
- 5.Luangtongkum T., Jeon B., Han J., Plummer P., Logue C.M., Zhang Q. Antibiotic resistance in Campylobacter: emergence, transmission and persistence. Future Microbiol. 2010;4:189–200. doi: 10.2217/17460913.4.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X.-Z., Plésiat P., Nikaido H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 2015;28:337–418. doi: 10.1128/CMR.00117-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lavigne J.P., Sotto A., Nicolas-Chanoine M.H., Bouziges N., Pagès J.M., Davin-Regli A. An adaptive response of Enterobacter aerogenes to imipenem: regulation of porin balance in clinical isolates. Int. J. Antimicrob. Agents. 2013;41:130–136. doi: 10.1016/j.ijantimicag.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 8.Pratt L.A., Hsing W., Gibson K.E., Silhavy T.J. From acids to osmZ: multiple factors influence synthesis of the OmpF and OmpC porins in Escherichia coli. Mol. Microbiol. 1996;20:911–917. doi: 10.1111/j.1365-2958.1996.tb02532.x. [DOI] [PubMed] [Google Scholar]
- 9.Lou H., Chen M., Black S.S., Bushell S.R., Ceccarelli M., Mach T., Beis K., Low A.S., Bamford V.A., Booth I.R., Bayley H., Naismith J.H. Altered antibiotic transport in OmpC mutants isolated from a series of clinical strains of multi-drug resistant E. coli. PLoS One. 2011;6 doi: 10.1371/journal.pone.0025825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bajaj H., Scorciapino M.A., Moynié L., Page M.G.P., Naismith J.H., Ceccarelli M., Winterhalter M. Molecular basis of filtering carbapenems by porins from β-lactam-resistant clinical strains of Escherichia coli. J. Biol. Chem. 2015;291:2837–2847. doi: 10.1074/jbc.M115.690156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bolla J.M., Loret E., Zalewski M., Pages J.M. Conformational analysis of the Campylobacter jejuni porin. J. Bacteriol. 1995;177:4266–4271. doi: 10.1128/jb.177.15.4266-4271.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amako K., Wai S.N., Umeda A., Shigematsu M., Takade A. Electron microscopy of the major outer membrane protein of Campylobacter jejuni. Microbiol. Immunol. 1996;40:749–754. doi: 10.1111/j.1348-0421.1996.tb01136.x. [DOI] [PubMed] [Google Scholar]
- 13.Moser I., Schroeder W., Salnikow J. Campylobacter jejuni major outer membrane protein and a 59-kDa protein are involved in binding to fibronectin and INT 407 cell membranes. FEMS Microbiol. Lett. 1997;157:233–238. doi: 10.1111/j.1574-6968.1997.tb12778.x. [DOI] [PubMed] [Google Scholar]
- 14.Newell D.G., McBride H., Pearson A.D. The identification of outer membrane proteins and flagella of Campylobacter jejuni. J. Gen. Microbiol. 1984;130:1201–1208. doi: 10.1099/00221287-130-5-1201. [DOI] [PubMed] [Google Scholar]
- 15.Newell D.G., McBride H., Dolby J.M. Investigations on the role of flagella in the colonization of infant mice with Campylobacter jejuni and attachment of Campylobacter jejuni to human epithelial cell lines. J. Hyg. (Lond.) 1985;95:217–227. doi: 10.1017/s0022172400062653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wenman W.M., Chai J., Louie T.J., Goudreau C., Lior H., Newell D.G., Pearson A.D., Taylor D.E. Antigenic analysis of Campylobacter flagellar protein and other proteins. J. Clin. Microbiol. 1985;21:108–112. doi: 10.1128/jcm.21.1.108-112.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Islam A., Raghupathy R., Albert M.J. Recombinant PorA, the major outer membrane protein of Campylobacter jejuni, provides heterologous protection in an adult mouse intestinal colonization model. Clin. Vaccine Immunol. 2010;17:1666–1671. doi: 10.1128/CVI.00255-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolla J.M., Saint N., Labesse G., Pagès J.M., Dumas C. Crystallization and preliminary crystallographic studies of MOMP (major outer membrane protein) from Campylobacter jejuni. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:2349–2351. doi: 10.1107/S0907444904024795. [DOI] [PubMed] [Google Scholar]
- 19.Krissinel E., Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 20.Labesse G., Garnotel E., Bonnel S., Dumas C., Pages J.M., Bolla J.M. MOMP, a divergent porin from Campylobacter: cloning and primary structural characterization. Biochem. Biophys. Res. Commun. 2001;280:380–387. doi: 10.1006/bbrc.2000.4129. [DOI] [PubMed] [Google Scholar]
- 21.McNicholas S., Potterton E., Wilson K.S., Noble M.E.M. Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011;67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yaffe E., Fishelovitch D., Wolfson H.J., Halperin D., Nussinov R. MolAxis: a server for identification of channels in macromolecules. Nucleic Acids Res. 2008;36:W210–W215. doi: 10.1093/nar/gkn223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baslé A., Rummel G., Storici P., Rosenbusch J.P., Schirmer T. Crystal structure of osmoporin OmpC from E. coli at 2.0 Å. J. Mol. Biol. 2006;362:933–942. doi: 10.1016/j.jmb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Cowan S., Garavito R., Jansonius J., Jenkins J., Karlsson R., König N., Pai E., Pauptit R., Rizkallah P., Rosenbusch J., Rummel G., Schirmer T. The structure of OmpF porin in a tetragonal crystal form. Structure. 1995;3:1041–1050. doi: 10.1016/s0969-2126(01)00240-4. [DOI] [PubMed] [Google Scholar]
- 25.Krissinel E., Henrick K. Detection of protein assemblies in crystals. Comput. Life Sci. 2005;3695:163–174. [Google Scholar]
- 26.Zhuang J., Engel A., Pagés J.M., Bolla J.M. The Campylobacter jejuni porin trimers pack into different lattice types when reconstituted in the presence of lipid. Eur. J. Biochem. 1997;244:575–579. doi: 10.1111/j.1432-1033.1997.t01-1-00575.x. [DOI] [PubMed] [Google Scholar]
- 27.Benz R., Schmid A., Hancock R.E.W. Ion selectivity of gram-negative bacterial porins. J. Bacteriol. 1985;162:722–727. doi: 10.1128/jb.162.2.722-727.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bajaj H., Tran Q.T., Mahendran K.R., Nasrallah C., Colletier J.P., Davin-Regli A., Bolla J.M., Pagès J.M., Winterhalter M. Antibiotic uptake through membrane channels: role of Providencia stuartii OmpPst1 porin in carbapenem resistance. Biochemistry. 2014;51:10,244–10,249. doi: 10.1021/bi301398j. [DOI] [PubMed] [Google Scholar]
- 29.Gutiérrez S.A., Bodrenko I., Scorciapino M.A., Ceccarelli M. Macroscopic electric field inside water-filled biological nanopores. Phys. Chem. Chem. Phys. 2016;18:8855–8864. doi: 10.1039/c5cp07902k. [DOI] [PubMed] [Google Scholar]
- 30.Raj Singh P., Ceccarelli M., Lovelle M., Winterhalter M., Mahendran K.R. Antibiotic permeation across the OmpF channel: modulation of the affinity site in the presence of magnesium. J. Phys. Chem. B. 2012;116:4433–4438. doi: 10.1021/jp2123136. [DOI] [PubMed] [Google Scholar]
- 31.VaneDE Berg B. Structural basis for outer membrane sugar uptake in pseudo monads. J. Biol. Chem. 2012;287:41,044–41,052. doi: 10.1074/jbc.M112.408518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwang P.M., Choy W.-Y., Lo E.I., Chen L., Forman-Kay J.D., Raetz C.R.H., Privé G.G., Bishop R.E., Kay L.E. Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proc. Natl. Acad. Sci. U. S. A. 2002;99:13,560–13,565. doi: 10.1073/pnas.212344499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forst D., Welte W., Wacker T., Diederichs K. Structure of the sucrose-specific porin ScrY from Salmonella typhimurium and its complex with sucrose. Nat. Struct. Biol. 1998;5:37–46. doi: 10.1038/nsb0198-37. [DOI] [PubMed] [Google Scholar]
- 34.Michels J., Geyer A., Mocanu V., Welte W., Burlingame A.L., Przybylski M. Structure and functional characterization of the periplasmic N-terminal polypeptide domain of the sugar-specific ion channel protein (ScrY porin) Protein Sci. 2002;11:1565–1574. doi: 10.1110/ps.2760102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Page W.J., Huyer G., Huyer M., Worobec E.A. Characterization of the porins of Campylobacter jejuni and Campylobacter coli and implications for antibiotic susceptibility. Antimicrob. Agents Chemother. 1989;33:297–303. doi: 10.1128/aac.33.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dé E., Jullien M., Labesse G., Pages J.M., Molle G., Bolla J.M. MOMP (major outer membrane protein) of Campylobacter jejuni; a versatile pore-forming protein. FEBS Lett. 2000;469:93–97. doi: 10.1016/s0014-5793(00)01244-8. [DOI] [PubMed] [Google Scholar]
- 37.Jürgens U.J., Benz R. Pore-forming activity of outer membrane extracts from the unicellular cyanobacterium Synechocystis sp. PCC 6714. Z. Naturforsch. 1989;44:65–169. [Google Scholar]
- 38.Mahendran K.R., Kreir M., Weingart H., Fertig N., Winterhalter M. Permeation of antibiotics through Escherichia coli OmpF and OmpC porins: screening for influx on a single-molecule level. J. Biomol. Screen. Off. J. Soc. Biomol. Screen. 2010;15:302–307. doi: 10.1177/1087057109357791. [DOI] [PubMed] [Google Scholar]
- 39.Liu J., Wolfe A.J., Eren E., Vijayaraghavan J., Indic M., Van Den Berg B., Movileanu L. Cation selectivity is a conserved feature in the OccD subfamily of Pseudomonas aeruginosa. Biochim. Biophys. Acta Biomembr. 2012;1818:2908–2916. doi: 10.1016/j.bbamem.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lout K.L., Saint N., Prilipov A., Rummel G., Benson S.A., Rosenbusch J.P., Schirmer T. Structural and functional characterization of OmpF porin mutants selected for larger pore size. I. Crystallographic analysis. J. Biol. Chem. 1996;271:20,669–20,675. [PubMed] [Google Scholar]
- 41.Chimerel C., Movileanu L., Pezeshki S., Winterhalter M., Kleinekathöfer U. Transport at the nanoscale: temperature dependence of ion conductance. Eur. Biophys. J. 2008;38:121–125. doi: 10.1007/s00249-008-0366-0. [DOI] [PubMed] [Google Scholar]
- 42.Huyer M., Jr Parr T.R., Hancock R.E.W., Page W.J. Outer membrane porin protein of Campylobacter jejuni. FEMS Microbiol. Lett. 1986;37:247–250. [Google Scholar]
- 43.Acosta-Gutierrez S., Scorciapino M.A., Bodrenko I., Ceccarelli M. Filtering with electric field: the case of E. coli porins. J. Phys. Chem. Lett. 2015;6:1807–1812. doi: 10.1021/acs.jpclett.5b00612. [DOI] [PubMed] [Google Scholar]
- 44.Liu H., Naismith J.H. A simple and efficient expression and purification system using two newly constructed vectors. Protein Expr. Purif. 2009;63:102–111. doi: 10.1016/j.pep.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Duyne G.D., Standaert R.F., Karplus P.A., Schreiber S.L., Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 46.Kervella M., Fauchere J.L., Fourel D., Pages J.M. Immunological cross-reactivity between outer-membrane pore proteins of Campylobacter jejuni and Escherichia coli. FEMS Microbiol. Lett. 1992;99:281–285. doi: 10.1016/0378-1097(92)90041-l. [DOI] [PubMed] [Google Scholar]
- 47.Winter G. Xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 2010;43:186–190. [Google Scholar]
- 48.Zwart P.H., Afonine P.V., Grosse-Kunstleve R.W., Hung L.-W., Ioerger T.R., McCoy A.J., McKee E., Moriarty N.W., Read R.J., Sacchettini J.C., Sauter N.K., Storoni L.C., Terwilliger T.C., Adams P.D. Automated structure solution with the PHENIX suite. Methods Mol. Biol. 2008;426:419–435. doi: 10.1007/978-1-60327-058-8_28. [DOI] [PubMed] [Google Scholar]
- 49.Terwilliger T.C., Grosse-Kunstleve R.W., Afonine P.V., Moriarty N.W., Zwart P.H., Hung L.W., Read R.J., Adams P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 2007:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perrakis A., Harkiolaki M., Wilson K.S., Lamzin V.S. ARP/wARP and molecular replacement. Acta Crystallogr. D Biol. Crystallogr. 2001;57:1445–1450. doi: 10.1107/s0907444901014007. [DOI] [PubMed] [Google Scholar]
- 51.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 52.Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 53.Davis I.W., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:615–619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zheng H., Chordia M.D., Cooper D.R., Chruszcz M., Müller P., Sheldrick G.M., Minor W. Validation of metal-binding sites in macromolecular structures with the CheckMyMetal web server. Nat. Protoc. 2014;9:156–170. doi: 10.1038/nprot.2013.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Montal M., Mueller P. Formation of bimolecular membranes from lipid monolayers and a study of their electrical properties. Proc. Natl. Acad. Sci. U. S. A. 1972;69:3561–3566. doi: 10.1073/pnas.69.12.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lamichhane U., Islam T., Prasad S., Weingart H., Mahendran K.R., Winterhalter M. Peptide translocation through the mesoscopic channel: binding kinetics at the single molecule level. Eur. Biophys. J. 2013;42:363–369. doi: 10.1007/s00249-012-0885-6. [DOI] [PubMed] [Google Scholar]
- 57.Sali A., Blundell T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 58.Phillips J.C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R.D., Kal L., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harvey M.J., Giupponi G., De Fabritiis G. ACEMD: accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 2009;5:1632–1639. doi: 10.1021/ct9000685. [DOI] [PubMed] [Google Scholar]
- 60.Buch I., Giorgino T., De Fabritiis G. Complete reconstruction of an enzyme-inhibitor binding process by molecular dynamics simulations. Proc. Natl. Acad. Sci. U. S. A. 2011;108:10,184–10,189. doi: 10.1073/pnas.1103547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J.L., Dror R.O., Shaw D.E. Improved side-chain torsion potentials for the amber ff99SB protein force field. Proteins Struct. Funct. Bioinf. 2010;78:1950–1958. doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926. [Google Scholar]
- 63.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 64.Law V., Knox C., Djoumbou Y., Jewison T., Guo A.C., Liu Y., MacIejewski A., Arndt D., Wilson M., Neveu V., Tang A., Gabriel G., Ly C., Adamjee S., Dame Z.T., Han B., Zhou Y., Wishart D.S. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014;42:1091–1097. doi: 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cornell W.D., Cieplak P., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M., Spellmeyer D.C., Fox T., Caldwell J.W., Kollman P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 66.Frisch M.J., Trucks G.W., Schlegel H.B., Scuseria G.E., Robb M.A., Cheeseman J.R., Scalmani G., Barone V., Mennucci B., Petersson G.A., Nakatsuji H., Caricato M., Li X., Hratchian H.P., Izmaylov A.F., Bloino J., Zheng G., Sonnenberg J.L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J.A., Jr., Peralta J.E., Ogliaro F., Bearpark M., Heyd J.J., Brothers E., Kudin K.N., Staroverov V.N., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J.C., Iyengar S.S., Tomasi J., Cossi M., Rega N., Millam J.M., Klene M., Knox J.E., Cross J.B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R.E., Yazyev O., Austin A.J., Cammi R., Pomelli C., Ochterski J.W., Martin R.L., Morokuma K., Zakrzewski V.G., Voth G.A., Salvador P., Dannenberg J.J., Dapprich S., Daniels A.D., Farkas Ö., Foresman J.B., Ortiz J.V., Cioslowski J., Fox D.J. Gaussian Inc; Wallingford CT: 2009. Gaussian 09, Revision A.02; p. 34. [Google Scholar]
- 67.Wang J., Wang W., Kollman D.A. Case, automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006;25:247–260. doi: 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 68.Malloci G., Vargiu A.V., Serra G., Bosin A., Ruggerone P., Ceccarelli M. A database of force-field parameters, dynamics, and properties of antimicrobial compounds. Molecules. 2015;20:13,997–14,021. doi: 10.3390/molecules200813997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barducci A., Bussi G., Parrinello M. Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008;100(2):100. doi: 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- 70.Laio A., Parrinello M. Escaping free-energy minima. Proc. Natl. Acad. Sci. U. S. A. 2002;99:12,562–12,566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Raiteri P., Laio A., Gervasio F.L., Micheletti C., Parrinello M. Efficient reconstruction of complex free energy landscapes by multiple walkers metadynamics. J. Phys. Chem. B. 2006;110:3533–3539. doi: 10.1021/jp054359r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 1.
Supplementary material 2.
Supplementary material 3.