

Abstract
The Hippo signalling pathway regulates cellular proliferation, apoptosis and differentiation, thus exerting profound effects on cellular homeostasis. Inhibition of Hippo signalling has been frequently implicated in human cancers, indicating a well-known tumour suppressor function of the Hippo pathway. However, it is less certain whether and how hyperactivation of the Hippo pathway affects biological outcome in living cells. This review describes current knowledge of the regulatory mechanisms of the Hippo pathway, mainly focusing on hyperactivation of the Hippo signalling nexus. The disease implications of hyperactivated Hippo signalling have also been discussed, including arrhythmogenic cardiomyopathy, Sveinsson's chorioretinal atrophy, Alzheimer's disease, amyotrophic lateral sclerosis and diabetes. By highlighting the significance of disease-relevant Hippo signalling activation, this review can offer exciting prospects to address the onset and potential reversal of Hippo-related disorders.
Keywords: Hippo pathway, arrhythmogenic cardiomyopathy, Sveinsson's chorioretinal atrophy, Alzheimer's disease, amyotrophic lateral sclerosis, diabetes
1. Introduction: an overview of Hippo pathway regulation
The tumour suppressor Hippo pathway has emerged as a major regulator of organ size control, stem cell pluripotency and regeneration (figure 1). Deregulation of Hippo signalling leads to deleterious consequences including cancers [1–6]. Deregulation of the Hippo pathway often refers to inhibition of Hippo signal transduction and over-proliferative effects. Nevertheless, the Hippo pathway can also be deregulated in an opposite way, which causes hyperactivation of Hippo signalling and survival defects (table 1). In contrast with the inactive Hippo pathway, the biological significance of hyperactive Hippo signalling has been largely underestimated.
Figure 1.

Models of Hippo pathway in fly and mammals. A simplified version of Hippo pathway regulation is shown here. In both Drosophila and mammals, when Yki/YAP/TAZ is relieved from inhibition through phosphorylation-dependent or independent mechanisms, its nuclear translocation then drives target gene expression in regulation of cellular proliferation, apoptosis and differentiation. The phosphorylation mechanism relies on the core kinase cascade including Hpo/MST, Wts/LATS, Sav/SAV1 and Mats/MOB1. In Drosophila, the FERM domain protein Ex has been shown to physically associate with Yki and block its nuclear translocation. Similarly, in mammals, the adherens protein AMOT and CRB3 complex inhibit target gene expression via sequestering YAP/TAZ in cytoplasm.
Table 1.
Hyperactive Hippo pathway and diseases.
| disease | Hippo components | affected organ or tissue | evidence | references |
|---|---|---|---|---|
| arrhythmogenic cardiomyopathy | NF2, MST1/2, LATS1/2 and YAP | heart | gene expression | [7] |
| Sveinsson's chorioretinal atrophy | TEAD1 | eye | human, mouse mutation | [8,9] |
| retinal detachment | MST2 | eye | gene expression | [10] |
| Alzheimer's disease | MST1/2, YAP | brain, nerves | gene expression | [11,12] |
| skeletal muscle atrophy | MST1, YAP? | muscles, nerves | gene expression | [13] |
| amyotrophic lateral sclerosis | MST1, YAP | nerves | gene expression | [14,15] |
| diabetes | MST1 | multiple | gene expression | [16] |
The Hippo pathway was initially discovered in the fruit-fly Drosophila melanogaster by genetic screens for identifying genes required for growth and proliferation. The warts (wts) gene was first identified from the genetic mosaic screens, whose mutations caused dramatic overgrowth of the mutant tissues [17,18]. Subsequently, salvador (sav) and hippo (hpo) were discovered and, together with wts, defined a pathway controlling growth/proliferation and survival [19–24]. Hence, the Hippo pathway is also known as the Salvador–Warts–Hippo pathway.
Most components of the Hippo pathway are conserved from flies to mammals, although some differences may exist (details will be described below). Regulation of the Hippo pathway largely relies on kinase cascade of the core components. Inhibition of Hippo signalling (i.e. hypo-Hippo) means that the kinase cascade of the Hippo pathway is inactive, whereas hyperactivation of the Hippo pathway (i.e. hyper-Hippo) means that the kinase signalling cascade is active. The Hpo kinase (Ste20 family kinases, MST1/2 in vertebrates) forms a complex with the adaptor proteins Sav (SAV or WW45 in vertebrates) and Mats (Mob as tumour suppressor; MOB1A/B in vertebrates), which enhances Hpo/MST kinase activity and facilitates the interaction between Hpo/MST and the serine threonine kinase Wts (NDR family kinases, LATS1/2 in vertebrates). Hpo/MST phosphorylates and activates Wts/LATS via a sequential phosphorylation process [23,25–28]. Recent structural data have further proved the critical roles of MOB1 in this kinase activation loop through direct interactions with MST and LATS [26]. The activated Wts/LATS subsequently phosphorylates the final effector of the Hippo pathway, the transcriptional co-activator Yorkie (Yki; Yes associated protein (YAP) and TAZ in vertebrates) [29–37]. Phosphorylation of Yki/YAP/TAZ leads to cytoplasmic retention and subsequent protein degradation through β-TRCP (β-transducin repeat-containing E3 ubiquitin protein ligase)-dependent proteasomal degradation, thereby inhibiting their transcriptional activity [29,32,34,36,38–41]. Therefore, Yki/YAP/TAZ phosphorylation caused by active kinase cascade of the Hippo pathway indicates that Hippo signalling is hyperactivated. Recently, emerging evidence has uncovered additional kinases, which share similar roles with Hpo/MST and Wts/LATS in Hippo pathway regulation. MAP4K (mitogen-activated protein kinase kinase kinase kinase) kinases can activate LATS through a direct phosphorylation event [42,43]. Other members of the NDR family kinases, STK38 (NDR1) and STK38L (NDR2), also function as YAP kinases and inhibit YAP activity in certain cell types [44]. Similar to the LATS kinases, STK38 is activated by the binding of MOB1 as well as by MST-dependent phosphorylation [45–47]. Further studies will be needed to unravel the physiological roles of MAP4K and STK kinases in Hippo pathway regulation.
Phosphorylation-independent regulations also exist. Yki can directly bind to the FERM-domain-containing adaptor protein Expanded (Ex) and form a complex with Hpo/MST and Wts/LATS, thereby triggering the cytoplasmic retention of Yki [48,49]. Prior to these studies, Ex and Merlin (neurofibromin 2 (NF2) in vertebrates) were considered to function upstream of the core Hippo signalling cassette [50]. As an apical membrane-localized protein, Ex not only intrinsically modulates Yki activity but also transduces signal from outside of the cell through binding to the transmembrane protein Crumbs (Crb; CRB3 in vertebrates) [51–54]. The binding of Ex with Crb stabilizes its apical localization and then promotes the Hippo signalling pathway. Interestingly, however, vertebrate CRB3 regulates Hippo signalling through different mechanisms. CRB3 forms a complex with YAP/TAZ to sequester YAP/TAZ in the cytoplasm, thereby inhibiting their transcriptional activities. The prevention of YAP/TAZ nuclear localization by CRB3 may require components of junction-associated protein i.e. angiomotin family members (AMOT, AMOTL1 and AMOTL2), although the AMOT family has no homologue in Drosophila. AMOT proteins retain YAP/TAZ in the cytoplasm through direct (binding to YAP/TAZ) or indirect (stimulating LATS kinase activity) mechanisms [55–61]. Hence, in addition to kinase cascade regulation, the discovery of multiple protein–protein interactions indicates Hippo signalling is a regulatory network.
Conversely, when the Hippo pathway is inactivated, unphosphorylated Yki/YAP/TAZ translocates to the nucleus. Nuclear Yki/YAP/TAZ activates transcription through binding to Scalloped (Sd, the TEA domain family members 1–4 (TEAD1–4) in vertebrates) [62–66] or other transcription factors because Yki/YAP/TAZ lack their own DNA-binding domains. Binding to Sd/TEAD allows Yki/YAP/TAZ to activate expression of target genes that are involved in controlling cell growth, proliferation and survival [62–66]. Hence, the pathological role of the Hippo pathway in tumourigenesis is primarily caused by the aberrant activation of Yki/YAP/TAZ. On the contrary, little is known about how Yki/YAP/TAZ is inactivated in living cells (i.e. hyperactivation of the Hippo signalling pathway). In order to gain further understanding and consider potential disease implications of Hippo pathway activation, this review discusses the current knowledge of how and where the Hippo pathway is hyperactivated.
2. Hyperactivation of the Hippo pathway
Both intrinsic and extrinsic mechanisms modulate the Hippo pathway to maintain tissue homeostasis. This section will focus on recent advances in regulatory mechanisms that contribute to hyperactivation of the Hippo pathway.
2.1. Extracellular cues to regulate the Hippo pathway
The Hippo pathway can be regulated by various upstream stimuli, including G protein-coupled receptor (GPCR) signalling, adhesion cues through cell–cell contact, polarity, mechanical signals and cellular stress. Studies have built on observations that soluble hormones or growth factors act through GPCRs to activate or inactivate the Hippo pathway. Epinephrine, glucagon or dopamine receptor agonist can induce Hippo pathway activation through binding to Gαs and increase YAP phosphorylation [67]. However, in most cases, soluble factors have been shown to inhibit Hippo signalling activity. For instance, EGF and IGF can inhibit LATS and stimulate nuclear accumulation of YAP through phosphoinositide 3-kinase (PI3K) and pyruvate dehydrogenase kinase 1 (PDK1) signalling [68–70]. Lysophosphatidic acid (LPA), sphingosine 1-phosphate (S1P) and thrombin signal have been reported to act through G12/13 and Gq/11 to inactivate the Hippo pathway via stimulating Rho GTPases [67,71,72]. Moreover, Wnt ligands, such as Wnt5a/b and Wnt 3a, are found to mediate Gα12/13 and Rho to inhibit LATS activity, and then activate YAP/TAZ-dependent transcription. Many secreted Wnt inhibitors, including DKK1, BMP4, IGFBP4 and WNT5a/b, are YAP/TAZ target genes of this Wnt–Gα12/13-Rho–LATS–YAP/TAZ signalling (also called alternative Wnt-YAP/TAZ signalling) [73]. Therefore, alternative Wnt–YAP/TAZ signalling acts as a negative regulation of canonical Wnt signalling. Recently, studies have revealed several aspects regarding integrations between the Hippo pathway and Wnt pathway. As both signalling pathways are known to play essential roles in numerous cellular functions, understanding the crosstalk of Hippo and Wnt pathways may provide potential therapeutic targets.
Several upstream regulators established by cell–cell contact are known to promote the Hippo pathway by linking YAP to both adherens junctions (AJ) and tight junctions (TJ) proteins and reveal additional mechanisms of YAP inactivation. For example, homophilic binding of E-cadherin at AJ suppresses transcriptional activity of YAP by modulating MST activity [74]. An AJ component, α-catenin, has been shown to sequester YAP in the cytoplasm through physical interactions in mouse keratinocytes. Depletion of α-catenin leads to nuclear accumulation of YAP, thus triggering over-proliferative effects [75,76]. Recruitment of AMOT to AJ protein complexes (E-cadherin–catenin) also leads to cytoplasmic retention of YAP and stimulation of Hippo signalling, which is required to maintain pluripotent embryonic stem cells (ESCs) of inner cell mass in early blastocyst. Conversely, in outer cells of trophectoderm, the cell polarity restricts AMOT localization in the apical domain of outer cells, thereby inactivating the Hippo pathway [55]. In addition, fibronectin (an extracellular matrix protein) mediated cell–cell adhesion has been reported to act through focal adhesion kinase (FAK)–Src signalling to promote nuclear accumulation of YAP in a LATS-dependent manner. Inhibition of FAK–Src signalling is able to activate the Hippo pathway through cytoplasmic retention of YAP [77]. These studies indicate that differential inputs (e.g. adhesion cues or cell polarity) may regulate diverse cellular functions through modulating the Hippo pathway in distinct ways. The physical properties of cells, such as cell shape and cytoskeletal tension, have also been found to regulate the Hippo pathway [78,79]. GPCR-mediated regulation on YAP/TAZ activity is likely to act through modulating the actin cytoskeleton [67]. Disruption of the actin cytoskeleton activates Hippo signalling in a LATS-dependent manner [79–81]. By contrast, other studies proposed that this mechanical regulation controls YAP/TAZ activity independently of the LATS kinases, but through the Rho–Rock pathway [78,82–84]. This discrepancy could be due to different cell contexts or methodology, or the actual mechanism may involve LATS-dependent and -independent regulation of YAP/TAZ activity. Taken together, these findings illustrate how Hippo signal transduction is tightly regulated by the presence of neighbouring cells.
2.2. Intrinsic mechanisms for Hippo pathway activation
Intracellular mechanisms are also important for the activation of Hippo signalling. Although Wnt signal is initiated by secreted ligand binding, studies have revealed crosstalk between the Hippo pathway and Wnt pathway in various intracellular axes: (i) the cytoplasmic retention of phosphorylated YAP/TAZ interacts with Disheveled (DVL), thereby inhibiting Wnt target gene expression [85,86]; (ii) phosphorylated YAP/TAZ sequesters β-catenin in the cytoplasm [87], then β-TRCP is in turn recruited to this complex and triggers the proteasomal degradation of β-catenin [88,89]; and (iii) the tyrosine phosphatase SHP2 is restricted by phosphorylated YAP/TAZ in the cytoplasm of high density cells. Conversely, unphosphorylated YAP/TAZ facilitates nuclear translocation of β-catenin and SHP2, which in turn promotes Wnt/β-catenin target gene expression [90]. Therefore, phosphorylation of YAP/TAZ plays a critical role in controlling the integrated Hippo/Wnt signalling. Recent study has reported that protein kinase C zeta (PKCζ, belongs to atypical PKCs (aPKC)) can inhibit YAP and β-catenin via phosphorylation, indicating that PKCζ is a positive regulator to activate the Hippo pathway. This regulation is essential to maintain intestinal epithelial homeostasis [91]. Conversely, overexpression of aPKC stimulates Yki activity to promote cell proliferation and survival in Drosophila epithelial cells [52,54]. The discrepancy is also found in breast cancer cells where PKC acts downstream of an oestrogen-stimulated GPCR (G protein-coupled oestrogen receptor, GPER) to inhibit LATS, thereby activating YAP/TAZ [92]. In addition, different PKC isoforms can oppositely regulate the Hippo pathway in different cell types [93]. Conventional PKC (cPKC, including α, β and γ) inhibits LATS kinase activity while novel PKC (nPKC, including δ, θ, ε and η) promotes YAP/TAZ phosphorylation in HEK293A cells, HeLa cells and U251MG glioma cells. On the contrary, the effects of LATS-dependent YAP/TAZ phosphorylation in response to cPKC or nPKC are completely different in Swiss3T3 cells, MEF cells and A549 lung cancer cells [93]. It is likely that LATS-dependent phosphorylation is differentially regulated by different PKC isoforms in a cell type-specific manner.
The Hippo pathway has been shown to be regulated by the important transcriptional regulators of organogenesis, E proteins and ID proteins [94,95]. The widely expressed E proteins belong to the basic helix-loop-helix (bHLH) family, which heterodimerize with tissue-specific bHLH proteins to regulate cell growth, commitment and differentiation in many tissues [96,97]. The heterodimeric bHLH proteins bind to the E-box sequence (CANNTG) and drive gene expression. ID proteins lack a basic DNA-binding motif, so that the heterodimers of bHLH proteins with ID proteins inhibit their regulatory activities. Recent study in Drosophila has demonstrated that high levels of Drosophila E protein homologue (or loss of Drosophila ID protein) can activate ex transcription and promote Hippo signalling independent of any responses to cell–cell interactions. The hyperactivation of the Hippo pathway inhibits cell survival, which prevents progenitor cells from undergoing misspecified differentiation, thereby functioning as an intrinsic surveillance mechanism [94]. Previously, the binding with transcription factors (e.g. Sd/TEAD, Hth, RUNX, PAX, TBX5 and SMAD) is thought to be required for the transcriptional activity of Yki/YAP/TAZ and results in the inhibition of Hippo signalling [63–66,98–104]. Discovery of this regulation by which the Hippo pathway is hyperactivated by bHLH transcription factors may provide novel insights into the pathological involvement of hyperactive Hippo signalling.
In response to intracellular oxidative stress or DNA damage, nuclear translocated YAP binds to p73, a transcription factor that belongs to the tumour suppressor p53 family, and induces transactivation of proapoptotic genes such as PUMA and BAX, thereby triggering apoptosis [105–107]. A tumour suppressor, Ras association domain family 1A (RASSF1A), has been shown to mediate YAP phosphorylation and facilitate YAP–p73-mediated cell death [106]. The role of RASSF1A in YAP phosphorylation is thought to scaffold the interaction between MST and LATS and enables activation of their kinase activities [106,108]. In contrast with the common model that phosphorylated YAP is restricted in the cytoplasm, RASSF1A-dependent YAP phosphorylation induces nuclear translocation of YAP [106]. As decreased expression of RASSF1A has been reported in various cancers, which accounts for the tumour suppressor function of RASSF1A, these findings provide one possible mechanism by which elevated RASSF1A levels may contribute to apoptosis through enhancing MST–LATS–YAP phosphorylation (i.e. hyperactivation of the Hippo pathway). Future studies will be needed to address whether and how RASSF1A is induced in response to cellular stimuli. In addition, YAP can be phosphorylated by AKT (also known as protein kinase B), a downstream effector of PI3K signalling. Intriguingly, AKT-dependent YAP phosphorylation plays an opposing role in regulating YAP–p73 mediated apoptosis. AKT-dependent YAP phosphorylation results in a cytoplasmic retention of YAP, thereby attenuating YAP–p73 mediated apoptosis [105]. Collectively, these findings indicate that different sites of YAP phosphorylation may result in different subcellular localizations of phospho-YAP and cause distinct consequences of Hippo signalling [109], although the underlying mechanisms remain to be addressed in detail.
3. Prospective disease model of hyper-Hippo signalling
Over-proliferative effects caused by inactivation of Hippo signal transduction have been extensively studied in various cancers [1–6]. Conversely, the hyperactivation of the Hippo pathway is implicated in some human diseases. Cells with hyperactive Hippo signalling have been shown to undergo apoptosis and be eliminated in vivo. Excess cell death is often associated with neurodegenerative disease, ischaemia, autoimmune disease and metabolic disease [110]. This section includes current evidence, that has linked the hyperactivation of the Hippo pathway to human disease or disease models (table 1).
3.1. Hippo activation leads to adipogenesis in arrhythmogenic cardiomyopathy
Activation of the Hippo pathway has been linked to adipogenesis in the heart disease model. Increased levels of phosphorylated NF2, MST1/2, LATS1/2 and YAP have been detected in the myocardial samples with arrhythmogenic cardiomyopathy (AC) from human patients and mouse models. β-Catenin activity has been shown to be affected in AC, suggesting that the involvement of the Hippo pathway may also require the Wnt pathway [7]. AC, also known as arrhythmogenic right ventricular cardiomyopathy because it predominantly affects the right ventricular walls, is a hereditary cardiomyopathy that accounts for 15–25% of sudden cardiac deaths in patients younger than 35 years. The pathological hallmark of AC is the replacement of myocardium by fibroadipocytes, ventricular enlargement and dysfunction and lethal ventricular arrythmias. AC is a disease of the desmosomes, intercellular junctional complexes that join the ends of cardiomyocytes [111]. Desmosome disruption results in changes of mechanical control of upstream regulators of the Hippo pathway, leading to alteration of YAP/TAZ activity and localization [7]. Additionally, deregulated desmosome proteins have been considered to contribute to AC pathogenesis by enhancing adipogenesis driven by adipogenic transcription factor PPARγ [112]. The adipogenic effects can be antagonized by downregulation of Hippo kinases, indicating that hyperactive Hippo signalling could be a potential pathogenesis for AC [7]. This is consistent with the previous finding that adipogenesis can be induced by low YAP/TAZ activity [78,99]. It is likely that loss of mechanical integrity causes aberrant activation of the Hippo signalling pathway and promotes adipogenesis, thus resulting in AC. However, it is unclear whether desmosome mutation is exclusively required for Hippo-mediated AC pathology. Several questions remain to be addressed. For instance, is there Hippo pathway mutation(s) existing in AC patients? If so, is the Hippo pathway mutation(s) sufficient to trigger AC? It will be noteworthy to study in depth the underlying mechanisms in the future.
3.2. Degenerative disease
Sd/TEAD is the best-characterized DNA-binding partner of Yki/YAP/TAZ. The binding of Sd/TEAD with Yki/YAP/TAZ ensures its transcriptional activity [62–66]. Recent progress proves the interaction between YAP/TAZ and TEAD is essential for the maintenance and differentiation of retinal pigment epithelium in zebrafish [113]. Interestingly, a missense mutation in TEAD1 has been identified that leads to Sveinsson's chorioretinal atrophy, a rare genetic disease in which choroid and retina are gradually degenerated [8,9]. This missense mutation disrupts the interaction of TEAD with YAP/TAZ and therefore blocks its transcriptional activity [9,62–66]. These findings imply that activation of the Hippo pathway (i.e. disruption of YAP/TAZ–TEAD function) may contribute to the pathogenesis of Sveinsson's chorioretinal atrophy and other ocular diseases. The Tondu-domain-containing proteins, Drosophila Vestigial (Vg) and Tgi (Vestigial-like proteins 1–4 (VGLL1–4) in vertebrates), also regulate Sd/TEAD-dependent transcription through physical interactions. Binding of Vg/VGLL1–3 with Sd/TEAD stimulates its transcriptional activity [114–117], whereas Tgi/VGLL4 acts as a repressor when interacting with Sd/TEAD and may compete for Yki/YAP/TAZ binding to Sd/TEAD [118,119]. This raises the possibility that disease-associated TEAD mutations might disrupt its binding to VGLL proteins, although this warrants further investigation.
In addition to the final effectors of Hippo signalling, Matsumoto et al. [4] demonstrate that MST2 kinase acts as a regulator to trigger photoreceptor apoptosis in a mouse model of retinal detachment [10]. Retinal detachment can cause permanent vision loss due to photoreceptor cell death. Apoptotic indications are reduced in MST2 homozygous null mice after retinal detachment [10]. It will be interesting to further study whether (i) the effect of MST2 in retinal detachment requires YAP/TAZ activity and (ii) this regulation is involved in other retinal degenerative disorders.
Moreover, activation of MST1/2 has been connected to multiple neurodegenerative diseases. Alzheimer's disease (AD), the most common progressive neurodegenerative disease, is defined by the formation of amyloid plaques and neurofibrillary tangles in the brain and apoptotic cell death that causes synapse and neuron loss. AD pathogenesis is considered to be the accumulation and oligomerization of amyloid β (Aβ) peptide produced by defective proteolytic processing of the precursor of Aβ (AβPP) [120,121]. Recent study has reported that AβPP can promote nuclear translocation of a Forkhead transcription factor FOXO3a by inducing MST1-dependent phosphorylation of FOXO3a [11]. The nuclear FOXO proteins (e.g. FOXO1 and FOXO3) activate a pro-apoptotic member of Bcl-2 family and trigger an intrinsic apoptotic pathway, thus resulting in neuron death [11,122]. The MST–FOXO-mediated neuron death could be considered as a branch of the Hippo pathway. Activation of MST kinase is also induced by oxidative stress, which has been associated with various diseases including neurodegeneration [122–124]. Knockdown of FOXO can rescue MST1 overexpression- or oxidative stress-induced neuron death [125], supporting that FOXO acts as a downstream effector of MST1. However, it remains unclear whether depletion of MST1 is sufficient to rescue the AβPP-mediated neuron death. AKT has also been reported to phosphorylate FOXO, whereas AKT-dependent FOXO phosphorylation blocks the kinase activity of MST1 towards FOXO [126]. In contrast with MST1 phosphorylation, FOXO phosphorylation by AKT promotes its cytoplasmic retention, thereby preventing FOXO-mediated apoptosis [127–131]. In addition, activation of AKT can prevent the toxic effect of AβPP [132–134]. Collectively, these studies suggest a protective role of AKT in AβPP-mediated neuron death. Notably, AKT activation is inhibited by MST phosphorylation, indicating a mutual inhibition between MST and AKT kinases in FOXO regulation [126]. Intriguingly, YAP/TAZ has been proposed to act as downstream mediators of amyloid precursor protein signalling through physically interacting with the amyloid precursor protein and forming a transcriptionally active protein complex [12]. These findings may link the core components of the Hippo pathway to AD pathogenesis, although the detailed mechanisms await further investigation.
MST1 kinase has also been reported as an important regulator in skeletal muscle atrophy caused by denervation, ageing and metabolic diseases. Upregulation of MST1 induces muscle atrophy through phosphorylating and inducing nuclear accumulation of FOXO3a, thereby activating multiple autophagy genes. Furthermore, the neurogenic atrophy can be attenuated in Mst1 homozygous null mice [13]. Amyotrophic lateral sclerosis (ALS) is a severe progressive neurodegenerative disorder that involves the death of motor neurons. When the motor neurons die, patients progressively lose the ability to control muscle movement, thereby disrupting speech, eating, moving and breathing. The elevation of MST1 activity has been reported in motor neurons from a mouse model of ALS [14]. Mutations of Cu/Zn superoxide dismutase type-1 (SOD1), a crucial enzyme for cellular antioxidant defence mechanisms, have been linked to a hereditary form of ALS [135]. ALS-associated SOD1 (G93A) mutant induces MST1 activation in neurons in an oxidative stress-dependent manner. Increased MST1 causes autophagosome accumulation and death of motor neurons through activation of p38 and caspases. ALS phenotypes can be attenuated when MST1 is depleted in an ALS mouse model [14], indicating that MST1 activity plays a critical role for ALS pathogenesis. In this regard, future studies on the pathogenic roles of MST1 in ALS will inspire a potential route for targeting therapies. Moreover, YAP–p73-mediated neuron death has been reported in an ALS mouse model [15]. These findings indicate that the pro-apoptotic signalling mediated by MST1–YAP–p73 not only causes multiple types of neurodegenerative disorders but also suppresses tumourigenesis in certain tumour types [106,107,136,137].
3.3. Metabolic disorder
Recent study has revealed that hyperactivation of MST1 kinase plays an essential role in triggering the initiation of β-cell death and disruption of insulin secretion, thereby resulting in diabetes [16]. The destruction of insulin-producing β-cells caused by apoptotic cell death is a hallmark of both type 1 and type 2 diabetes. In diabetic human and mouse β-cells, MST1 is highly activated and directly phosphorylates the critical β-cell transcription factor, pancreatic and duodenal homeobox 1 (PDX1), leading to the subsequent degradation of PDX1 and insulin secretion failure. Notably, loss of MST1 is sufficient to rescue survival and insulin tolerance of pancreatic β-cells through preservation of PDX1 [16]. Mst1−/− mice are also protected from diabetogenic stimulation, suggesting MST1 could be a potential therapeutic target for treating diabetes. TEAD–YAP has been shown to activate key pancreatic transcription factors such as PDX1 [138]. However, it is currently unknown whether YAP/TAZ activity is inhibited in the MST1-mediated β-cell death. Hence, it will be attractive to elucidate the cellular mechanisms by which MST1 is hyperactivated under diabetic stimuli.
3.4. Infertility disease
Polycystic ovarian syndrome (PCOS) is the most common endocrine disorder among reproductive-age women, which results in infertility, menstrual disorders, metabolic symptoms and endometrial cancer. Although PCOS has been strongly suggested as a genetic disease, the pathology and cause of PCOS is largely uncertain. In patients with PCOS or primary ovarian insufficiency (POI), infertility treatment has been proposed to promote ovarian follicle growth. Facilitating ovarian follicle growth by promoting actin polymerization is sufficient to induce the nuclear translocation of YAP and subsequent activation of the downstream target genes BIRC (baculoviral inhibitors of apoptosis repeat containing) and CTGF (connective tissue growth factor) in a PCOS mouse model and POI patients [139,140]. Hence, inhibition of Hippo signal transduction provides a treatment for ovarian disorders, although the underlying mechanism remains elusive. It will be noteworthy to further address: (i) whether the Hippo pathway is hyperactivated in defective ovaries, (ii) which component(s) of the Hippo pathway is deregulated in animal models or human patients with defective ovaries and (iii) whether the pathogenesis of PCOS or POI is caused by decreased cellular proliferation and/or increased apoptosis upon Hippo pathway activation.
3.5. Disease implications of helix-loop-helix proteins and the Hippo pathway
Deregulated E proteins and ID proteins are known to contribute to a variety of diseases [95,141–143]. Therefore, the discovery of the regulatory mechanism between E/ID proteins and the Hippo pathway [94] pinpoints an important insight for the physiological control to maintain organ integrity. To date, there has been no direct evidence linking the hyperactivated Hippo pathway, HLH proteins and disease, but there are some intriguing clues for the potential involvement in diseases. For instance, Id4 homozygous null (Id4−/−) mice have been shown to enhance adipogenesis and reduce osteogenenic differentiation [144]. Similarly, depletion of TAZ has been reported to promote adipogenesis [99]. The promising phenotypes may motivate further investigations to study whether the aforementioned intrinsic regulation involving the HLH proteins–Hippo pathway axis can promote adipogenesis. Future studies may also extend the current view that the involvement of YAP/TAZ in mesenchymal stem cell differentiation (adipogenesis and osteogenesis) is governed by mechanical cues.
A role for ID proteins has been suggested in circadian rhythm. ID proteins have been reported to regulate circadian rhythm through sequestering the circadian bHLH transcription factors CLOCK and BMAL, thus reducing expression of the Period (Per) clock protein [145,146]. By modulating the activity of CLOCK, BMAL and Per, casein kinase 1 (CK1) has been thought to be an important regulator for circadian rhythms. Intriguingly, CK1 promotes YAP/TAZ degradation through β-TRCP E3 ligase [38,41], suggesting YAP/TAZ may be implicated in circadian regulation. Deregulation of β-TRCP E3 ligase-dependent protein degradation has been reported to contribute to autosomal dominant polycystic kidney disease (PKD), which is frequently caused by inactivating mutations in the PKD1 and PKD2 genes [147]. TAZ homozygous null mutant mice display symptoms of PKD due to accumulation of product of the PKD2 gene, polycystin-2. TAZ binds and targets polycystin-2 for degradation through the β-TRCP E3 ligase pathway [148]. Polycystin-2 can sequester ID proteins in the cytoplasm through direct interaction. PKD phenotypes have been observed when the interaction between polycystin-2 and ID2 is disrupted [149]. Taken together, these findings suggest that the crosstalk between E/ID proteins and the Hippo pathway may be involved in circadian rhythm and PKD. However, the connections between these lines of evidence are only correlations without clear mechanistic support. Further studies are needed to characterize the connection and involvement of YAP/TAZ and ID proteins in circadian cycles and PKD pathogenesis.
Id3 homozygous null mice have been reported to induce features of Sjogren's syndrome [150], a chronic autoimmune disease, which disrupts salivary and lachrymal glands. Unexpectedly, the nuclear accumulation of TAZ has been reported to be sufficient to cause Sjogren's syndrome [151]. Although this is in opposition to the current knowledge of the HLH–Hippo regulatory aspect (figure 2), it remains interesting to determine the potential connection between E/ID proteins and Hippo pathway in Sjogren's syndrome. For instance, it will be useful to understand whether depletion of TAZ in Id3−/− mice relieves the symptoms of Sjogren's syndrome.
Figure 2.
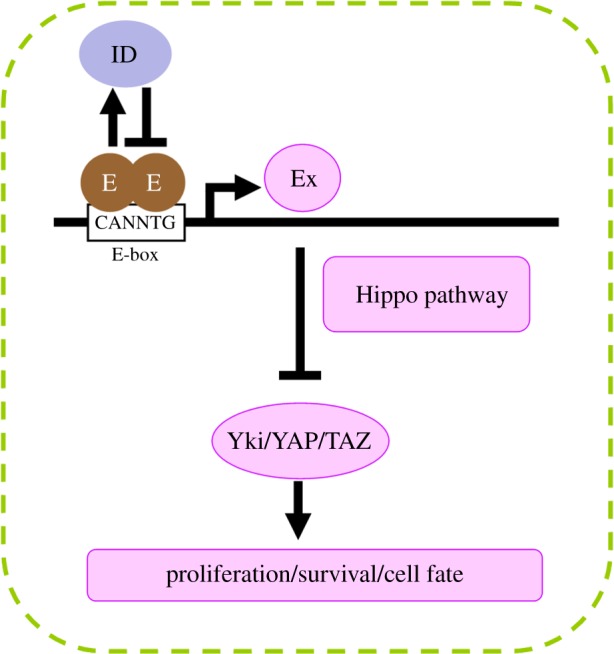
Intrinsic Hippo activation by Drosophila HLH proteins. Depletion of Drosophila ID protein results in elevated Drosophila E protein. The high levels of E protein activate ex transcription through binding to the E-box sites in the cis-regulatory element, thereby activating the Hippo pathway. The hyperactivated Hippo pathway prevents cellular proliferation and survival, leading to the elimination of misspecified progenitor cells.
4. Conclusion
At present, most dysfunctions of upstream regulators, kinases and downstream effectors of the Hippo pathway often lead to inhibition of Hippo signal transduction and are associated with overproliferative disorders such as cancer. However, aspects regarding activation of the Hippo pathway are also fundamental. By describing the recent advances in disease implications of Hippo activation, this review attempts to inspire future research of the underlying mechanisms and potential therapeutics for these diseases. Accumulating evidence has strongly linked TEAD1 disruption or MST kinases activation to many diseases, such as Sveinsson's chorioretinal atrophy, AC, AD, skeletal muscle atrophy, ALS and diabetes. Nevertheless, how these diseases are related to the roles of the Hippo pathway in normal development is still unclear and remains to be investigated. Small molecule inhibitors targeting Hippo pathway components have been reported to suppress tumourigenesis in various cancer cell lines [1]. However, some molecules may function oppositely. For instance, compound 9E1 has been shown to inhibit MST1 kinase activity and LPA, S1P or thrombin can target GPCR signalling to inhibit LATS kinase activity, thus promoting transcriptional activity of YAP/TAZ [67,71,72,152]. Therefore, these kinase inhibitors could be considered as potential therapeutic strategies to treat disorders caused by hyperactive Hippo signalling.
Interestingly, aberrant activation of MST kinases appears to cause a broad range of diseases occurring in multiple organs. For instance, MST activation can result in AD and diabetes. A possible mechanistic link between diabetes and AD is suggested by studies indicating that type 2 diabetes patients have a higher risk of developing AD [153]. It is interesting to speculate that hyperactive Hippo signalling is a common cause of diverse diseases in different organs. Future studies will be required to determine whether and how the Hippo pathway becomes globally hyperactive in vivo.
Apoptosis caused by hyperactive MST has been shown to act through FOXO (FOXO3) or AKT in some cases, although it remains to be tested if, and how, YAP/TAZ is involved in MST–FOXO- or MST–AKT-mediated apoptotic signalling. Interestingly, it has been reported that YAP can act as a transcriptional co-activator of FOXO1 in regulating the transcription of antioxidant genes in cardiomyocytes. Thus, hyperactive Hippo signalling stimulates cell death by inhibiting YAP–FOXO1-mediated gene expression in response to oxidative stress [154]. Recent work illustrates that dysregulated HLH transcription factors are sufficient to activate the Hippo signalling pathway. Results from this work demonstrate that the hyperactive Hippo pathway indeed contributes to apoptosis dependent upon the elimination of Yki/YAP/TAZ activity [94]. So far, connections between HLH proteins and core components of the Hippo pathway have not been proposed as direct causes of particular diseases or defects. However, independent evidence has implicated the roles of HLH proteins or Hippo components in adipogenesis, circadian regulation and Sjogren's syndrome. It would be informative to substantiate the association of HLH proteins with Hippo components in adipogenesis, circadian regulation, Sjogren's syndrome and other potential diseases. Clear challenges remain to clarify the pathogenesis at the molecular and cellular level.
Acknowledgements
The author thanks N. Baker (Albert Einstein College of Medicine, USA), Y. H. Sun (Academia Sinica, Taiwan), and the anonymous reviewers for comments on the manuscript. The author also apologizes to co-workers whose work could not been cited because of space constraints.
Authors' contributions
L.-H.W. conceived the idea for the review, wrote the article and gave final approval for publication. S.-P.W. contributed to the final version.
Competing interests
I declare I have no competing interests.
Funding
This paper is supported by the Ministry of Science and Technology of Taiwan (MOST 105-2311-B-016-001-MY2) to L.-H.W. S.-P.W was supported by a long-term fellowship from the Human Frontier Science Program Organization (HFSPO).
References
- 1.Johnson R, Halder G. 2014. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 13, 63–79. (doi:10.1038/nrd4161) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pan D. 2010. The Hippo signaling pathway in development and cancer. Dev. Cell 19, 491–505. (doi:10.1016/j.devcel.2010.09.011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irvine KD, Harvey KF. 2015. Control of organ growth by patterning and hippo signaling in Drosophila. Cold Spring Harb. Perspect. Biol. 7, a019224 (doi:10.1101/cshperspect.a019224) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meng Z, Moroishi T, Guan KL. 2016. Mechanisms of Hippo pathway regulation. Genes Dev. 30, 1–17. (doi:10.1101/gad.274027.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grusche FA, Richardson HE, Harvey KF. 2010. Upstream regulation of the hippo size control pathway. Curr. Biol. 20, R574–R582. (doi:10.1016/j.cub.2010.05.023) [DOI] [PubMed] [Google Scholar]
- 6.Genevet A, Tapon N. 2011. The Hippo pathway and apico-basal cell polarity. Biochem. J. 436, 213–224. (doi:10.1042/BJ20110217) [DOI] [PubMed] [Google Scholar]
- 7.Chen SN, Gurha P, Lombardi R, Ruggiero A, Willerson JT, Marian AJ. 2014. The Hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 114, 454–468. (doi:10.1161/CIRCRESAHA.114.302810) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fossdal R, Jonasson F, Kristjansdottir GT, Kong A, Stefansson H, Gosh S, Gulcher JR, Stefansson K. 2004. A novel TEAD1 mutation is the causative allele in Sveinsson's chorioretinal atrophy (helicoid peripapillary chorioretinal degeneration). Hum. Mol. Genet. 13, 975–981. (doi:10.1093/hmg/ddh106) [DOI] [PubMed] [Google Scholar]
- 9.Kitagawa M. 2007. A Sveinsson's chorioretinal atrophy-associated missense mutation in mouse Tead1 affects its interaction with the co-factors YAP and TAZ. Biochem. Biophys. Res. Commun. 361, 1022–1026. (doi:10.1016/j.bbrc.2007.07.129) [DOI] [PubMed] [Google Scholar]
- 10.Matsumoto H, Murakami Y, Kataoka K, Lin H, Connor KM, Miller JW, Zhou D, Avruch J, Vavvas DG. 2014. Mammalian STE20-like kinase 2, not kinase 1, mediates photoreceptor cell death during retinal detachment. Cell Death Dis. 5, e1269 (doi:10.1038/cddis.2014.218) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanphui P, Biswas SC. 2013. FoxO3a is activated and executes neuron death via Bim in response to beta-amyloid. Cell Death Dis. 4, e625 (doi:10.1038/cddis.2013.148) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swistowski A, Zhang Q, Orcholski ME, Crippen D, Vitelli C, Kurakin A, Bredesen DE. 2009. Novel mediators of amyloid precursor protein signaling. J. Neurosci 29, 15 703–15 712. (doi:10.1523/JNEUROSCI.4351-09.2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei B, Dui W, Liu D, Xing Y, Yuan Z, Ji G. 2013. MST1, a key player, in enhancing fast skeletal muscle atrophy. BMC. Biol. 11, 12 (doi:10.1186/1741-7007-11-12) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JK, et al. 2013. MST1 functions as a key modulator of neurodegeneration in a mouse model of ALS. Proc. Natl Acad. Sci. USA 110, 12 066–12 071. (doi:10.1073/pnas.1300894110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morimoto N, Nagai M, Miyazaki K, Kurata T, Takehisa Y, Ikeda Y, Kamiya T, Okazawa H, Abe K. 2009. Progressive decrease in the level of YAPdeltaCs, prosurvival isoforms of YAP, in the spinal cord of transgenic mouse carrying a mutant SOD1 gene. J. Neurosci. Res. 87, 928–936. (doi:10.1002/jnr.21902) [DOI] [PubMed] [Google Scholar]
- 16.Ardestani A, et al. 2014. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat. Med. 20, 385–397. (doi:10.1038/nm.3482) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Justice RW, Zilian O, Woods DF, Noll M, Bryant PJ. 1995. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev 9, 534–546. (doi:10.1101/gad.9.5.534) [DOI] [PubMed] [Google Scholar]
- 18.Xu T, Wang W, Zhang S, Stewart RA, Yu W. 1995. Identifying tumor suppressors in genetic mosaics: the Drosophila lats gene encodes a putative protein kinase. Development 121, 1053–1063. [DOI] [PubMed] [Google Scholar]
- 19.Harvey KF, Pfleger CM, Hariharan IK. 2003. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 114, 457–467. (doi:10.1016/S0092-8674(03)00557-9) [DOI] [PubMed] [Google Scholar]
- 20.Pantalacci S, Tapon N, Leopold P. 2003. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat. Cell Biol. 5, 921–927. (doi:10.1038/ncb1051) [DOI] [PubMed] [Google Scholar]
- 21.Tapon N, Harvey KF, Bell DW, Wahrer DC, Schiripo TA, Haber D, Hariharan IK. 2002. Salvador promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 110, 467–478. (doi:10.1016/S0092-8674(02)00824-3) [DOI] [PubMed] [Google Scholar]
- 22.Udan RS, Kango-Singh M, Nolo R, Tao C, Halder G. 2003. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 5, 914–920. (doi:10.1038/ncb1050) [DOI] [PubMed] [Google Scholar]
- 23.Wu S, Huang J, Dong J, Pan D. 2003. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 114, 445–456. (doi:10.1016/S0092-8674(03)00549-X) [DOI] [PubMed] [Google Scholar]
- 24.Jia J, Zhang W, Wang B, Trinko R, Jiang J. 2003. The Drosophila Ste20 family kinase dMST functions as a tumor suppressor by restricting cell proliferation and promoting apoptosis. Genes Dev. 17, 2514–2519. (doi:10.1101/gad.1134003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoa L, Kulaberoglu Y, Gundogdu R, Cook D, Mavis M, Gomez M, Gomez V, Hergovich A. 2016. The characterisation of LATS2 kinase regulation in Hippo-YAP signaling. Cell Signal. 28, 488–497. (doi:10.1016/j.cellsig.2016.02.012) [DOI] [PubMed] [Google Scholar]
- 26.Ni L, Zheng Y, Hara M, Pan D, Luo X. 2015. Structural basis for Mob1-dependent activation of the core Mst-Lats kinase cascade in Hippo signaling. Genes Dev. 29, 1416–1431. (doi:10.1101/gad.264929.115) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan EH, Nousiainen M, Chalamalasetty RB, Schafer A, Nigg EA, Sillje HH. 2005. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene 24, 2076–2086. (doi:10.1038/sj.onc.1208445) [DOI] [PubMed] [Google Scholar]
- 28.Callus BA, Verhagen AM, Vaux DL. 2006. Association of mammalian sterile twenty kinases, Mst1 and Mst2, with hSalvador via C-terminal coiled-coil domains, leads to its stabilization and phosphorylation. FEBS J. 273, 4264–4276. (doi:10.1111/j.1742-4658.2006.05427.x) [DOI] [PubMed] [Google Scholar]
- 29.Dong J et al. . 2007. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 130, 1120–1133. (doi:10.1016/j.cell.2007.07.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao Y, Chun A, Cheung K, Rashidi B, Yang X. 2008. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 283, 5496–5509. (doi:10.1074/jbc.M709037200) [DOI] [PubMed] [Google Scholar]
- 31.Huang J, Wu S, Barrera J, Matthews K, Pan D. 2005. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila homolog of YAP. Cell 122, 421–434. (doi:10.1016/j.cell.2005.06.007) [DOI] [PubMed] [Google Scholar]
- 32.Lei QY, Zhang H, Zhao B, Zha ZY, Bai F, Pei XH, Zhao S, Xiong Y, Guan KL. 2008. TAZ promotes cell proliferation and epithelial–mesenchymal transition and is inhibited by the Hippo pathway. Mol. Cell Biol. 28, 2426–2436. (doi:10.1128/MCB.01874-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Praskova M, Xia F, Avruch J. 2008. MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Curr. Biol. 18, 311–321. (doi:10.1016/j.cub.2008.02.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao B, et al. 2007. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747–2761. (doi:10.1101/gad.1602907) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei X, Shimizu T, Lai ZC. 2007. Mob as tumor suppressor is activated by Hippo kinase for growth inhibition in Drosophila. EMBO J. 26, 1772–1781. (doi:10.1038/sj.emboj.7601630) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oh H, Irvine KD. 2008. In vivo regulation of Yorkie phosphorylation and localization. Development 135, 1081–1088. (doi:10.1242/dev.015255) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oka T, Mazack V, Sudol M. 2008. Mst2 and Lats kinases regulate apoptotic function of Yes kinase-associated protein (YAP). J. Biol. Chem. 283, 27 534–27 546. (doi:10.1074/jbc.M804380200) [DOI] [PubMed] [Google Scholar]
- 38.Liu CY, et al. 2010. The Hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCFβ-TrCP E3 ligase. J. Biol. Chem. 285, 37 159–37 169. (doi:10.1074/jbc.M110.152942) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ren F, Zhang L, Jiang J. 2010. Hippo signaling regulates Yorkie nuclear localization and activity through 14-3-3 dependent and independent mechanisms. Dev. Biol. 337, 303–312. (doi:10.1016/j.ydbio.2009.10.046) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kanai F, et al. 2000. TAZ: a novel transcriptional co-activator regulated by interactions with 14-3-3 and PDZ domain proteins. EMBO J. 19, 6778–6791. (doi:10.1093/emboj/19.24.6778) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao B, Li L, Tumaneng K, Wang CY, Guan KL. 2010. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev. 24, 72–85. (doi:10.1101/gad.1843810) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng Y, Wang W, Liu B, Deng H, Uster E, Pan D. 2015. Identification of Happyhour/MAP4K as alternative Hpo/Mst-like kinases in the Hippo kinase cascade. Dev. Cell 34, 642–655. (doi:10.1016/j.devcel.2015.08.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meng Z, et al. 2015. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat. Commun. 6, 8357 (doi:10.1038/ncomms9357) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang L, et al. 2015. NDR functions as a physiological YAP1 kinase in the intestinal epithelium. Curr. Biol. 25, 296–305. (doi:10.1016/j.cub.2014.11.054) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bichsel SJ, Tamaskovic R, Stegert MR, Hemmings BA. 2004. Mechanism of activation of NDR (nuclear Dbf2-related) protein kinase by the hMOB1 protein. J. Biol. Chem. 279, 35 228–35 235. (doi:10.1074/jbc.M404542200) [DOI] [PubMed] [Google Scholar]
- 46.Hergovich A, Kohler RS, Schmitz D, Vichalkovski A, Cornils H, Hemmings BA. 2009. The MST1 and hMOB1 tumor suppressors control human centrosome duplication by regulating NDR kinase phosphorylation. Curr. Biol. 19, 1692–1702. (doi:10.1016/j.cub.2009.09.020) [DOI] [PubMed] [Google Scholar]
- 47.Vichalkovski A, Gresko E, Cornils H, Hergovich A, Schmitz D, Hemmings BA. 2008. NDR kinase is activated by RASSF1A/MST1 in response to Fas receptor stimulation and promotes apoptosis. Curr. Biol. 18, 1889–1895. (doi:10.1016/j.cub.2008.10.060) [DOI] [PubMed] [Google Scholar]
- 48.Badouel C, Gardano L, Amin N, Garg A, Rosenfeld R, Le BT, McNeill H. 2009. The FERM-domain protein Expanded regulates Hippo pathway activity via direct interactions with the transcriptional activator Yorkie. Dev. Cell 16, 411–420. (doi:10.1016/j.devcel.2009.01.010) [DOI] [PubMed] [Google Scholar]
- 49.Oh H, Reddy BV, Irvine KD. 2009. Phosphorylation-independent repression of Yorkie in Fat-Hippo signaling. Dev. Biol. 335, 188–197. (doi:10.1016/j.ydbio.2009.08.026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hamaratoglu F, Willecke M, Kango-Singh M, Nolo R, Hyun E, Tao C, Jafar-Nejad H, Halder G. 2006. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat. Cell Biol. 8, 27–36. (doi:10.1038/ncb1339) [DOI] [PubMed] [Google Scholar]
- 51.Ling C, Zheng Y, Yin F, Yu J, Huang J, Hong Y, Wu S, Pan D. 2010. The apical transmembrane protein Crumbs functions as a tumor suppressor that regulates Hippo signaling by binding to Expanded. Proc. Natl Acad. Sci. USA 107, 10 532–10 537. (doi:10.1073/pnas.1004279107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robinson BS, Huang J, Hong Y, Moberg KH. 2010. Crumbs regulates Salvador/Warts/Hippo signaling in Drosophila via the FERM-domain protein Expanded. Curr. Biol. 20, 582–590. (doi:10.1016/j.cub.2010.03.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen CL, Gajewski KM, Hamaratoglu F, Bossuyt W, Sansores-Garcia L, Tao C, Halder G. 2010. The apical–basal cell polarity determinant Crumbs regulates Hippo signaling in Drosophila. Proc. Natl Acad. Sci. USA 107, 15 810–15 815. (doi:10.1073/pnas.1004060107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grzeschik NA, Parsons LM, Allott ML, Harvey KF, Richardson HE. 2010. Lgl, aPKC, and Crumbs regulate the Salvador/Warts/Hippo pathway through two distinct mechanisms. Curr. Biol. 20, 573–581. (doi:10.1016/j.cub.2010.01.055) [DOI] [PubMed] [Google Scholar]
- 55.Hirate Y, et al. 2013. Polarity-dependent distribution of angiomotin localizes Hippo signaling in preimplantation embryos. Curr. Biol. 23, 1181–1194. (doi:10.1016/j.cub.2013.05.014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paramasivam M, Sarkeshik A, Yates JR, Fernandes MJ, McCollum D. 2011. Angiomotin family proteins are novel activators of the LATS2 kinase tumor suppressor. Mol. Biol. Cell 22, 3725–3733. (doi:10.1091/mbc.E11-04-0300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Varelas X, Samavarchi-Tehrani P, Narimatsu M, Weiss A, Cockburn K, Larsen BG, Rossant J, Wrana JL. 2010. The Crumbs complex couples cell density sensing to Hippo-dependent control of the TGF-beta-SMAD pathway. Dev. Cell 19, 831–844. (doi:10.1016/j.devcel.2010.11.012) [DOI] [PubMed] [Google Scholar]
- 58.Zhao B, Li L, Lu Q, Wang LH, Liu CY, Lei Q, Guan KL. 2011. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 25, 51–63. (doi:10.1101/gad.2000111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yi C, et al. 2011. A tight junction-associated Merlin-angiomotin complex mediates Merlin's regulation of mitogenic signaling and tumor suppressive functions. Cancer Cell 19, 527–540. (doi:10.1016/j.ccr.2011.02.017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan SW, Lim CJ, Chong YF, Pobbati AV, Huang C, Hong W. 2011. Hippo pathway-independent restriction of TAZ and YAP by angiomotin. J. Biol. Chem. 286, 7018–7026. (doi:10.1074/jbc.C110.212621) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang W, Huang J, Chen J. 2011. Angiomotin-like proteins associate with and negatively regulate YAP1. J. Biol. Chem. 286, 4364–4370. (doi:10.1074/jbc.C110.205401) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. 2001. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 15, 1229–1241. (doi:10.1101/gad.888601) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang L, Ren F, Zhang Q, Chen Y, Wang B, Jiang J. 2008. The TEAD/TEF family of transcription factor Scalloped mediates Hippo signaling in organ size control. Dev. Cell 14, 377–387. (doi:10.1016/j.devcel.2008.01.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhao B, et al. 2008. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 22, 1962–1971. (doi:10.1101/gad.1664408) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Goulev Y, Fauny JD, Gonzalez-Marti B, Flagiello D, Silber J, Zider A. 2008. SCALLOPED interacts with YORKIE, the nuclear effector of the hippo tumor-suppressor pathway in Drosophila. Curr. Biol. 18, 435–441. (doi:10.1016/j.cub.2008.02.034) [DOI] [PubMed] [Google Scholar]
- 66.Wu S, Liu Y, Zheng Y, Dong J, Pan D. 2008. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev. Cell 14, 388–398. (doi:10.1016/j.devcel.2008.01.007) [DOI] [PubMed] [Google Scholar]
- 67.Yu FX, et al. 2012. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150, 780–791. (doi:10.1016/j.cell.2012.06.037) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fan R, Kim NG, Gumbiner BM. 2013. Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc. Natl Acad. Sci. USA 110, 2569–2574. (doi:10.1073/pnas.1216462110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reddy BV, Irvine KD. 2013. Regulation of Hippo signaling by EGFR-MAPK signaling through Ajuba family proteins. Dev. Cell 24, 459–471. (doi:10.1016/j.devcel.2013.01.020) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Strassburger K, Tiebe M, Pinna F, Breuhahn K, Teleman AA. 2012. Insulin/IGF signaling drives cell proliferation in part via Yorkie/YAP. Dev. Biol. 367, 187–196. (doi:10.1016/j.ydbio.2012.05.008) [DOI] [PubMed] [Google Scholar]
- 71.Miller E, Yang J, DeRan M, Wu C, Su AI, Bonamy GM, Liu J, Peters EC, Wu X. 2012. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem. Biol. 19, 955–962. (doi:10.1016/j.chembiol.2012.07.005) [DOI] [PubMed] [Google Scholar]
- 72.Mo JS, Yu FX, Gong R, Brown JH, Guan KL. 2012. Regulation of the Hippo-YAP pathway by protease-activated receptors (PARs). Genes Dev. 26, 2138–2143. (doi:10.1101/gad.197582.112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Park HW, et al. 2015. Alternative Wnt signaling activates YAP/TAZ. Cell 162, 780–794. (doi:10.1016/j.cell.2015.07.013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim NG, Koh E, Chen X, Gumbiner BM. 2011. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl Acad. Sci. USA 108, 11 930–11 935. (doi:10.1073/pnas.1103345108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Silvis MR, Kreger BT, Lien WH, Klezovitch O, Rudakova GM, Camargo FD, Lantz DM, Seykora JT, Vasioukhin V. 2011. α-Catenin is a tumor suppressor that controls cell accumulation by regulating the localization and activity of the transcriptional coactivator Yap1. Sci. Signal. 4, pra33. (doi:10.1126/scisignal.2001823) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schlegelmilch K, et al. 2011. Yap1 acts downstream of alpha-catenin to control epidermal proliferation. Cell 144, 782–795. (doi:10.1016/j.cell.2011.02.031) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim NG, Gumbiner BM. 2015. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J. Cell Biol. 210, 503–515. (doi:10.1083/jcb.201501025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dupont S, et al. 2011. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183. (doi:10.1038/nature10137) [DOI] [PubMed] [Google Scholar]
- 79.Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. 2011. Hippo pathway regulation by cell morphology and stress fibers. Development 138, 3907–3914. (doi:10.1242/dev.070987) [DOI] [PubMed] [Google Scholar]
- 80.Zhao B, Li L, Wang L, Wang CY, Yu J, Guan KL. 2012. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 26, 54–68. (doi:10.1101/gad.173435.111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kim M, Kim M, Lee S, Kuninaka S, Saya H, Lee H, Lee S, Lim DS. 2013. cAMP/PKA signalling reinforces the LATS-YAP pathway to fully suppress YAP in response to actin cytoskeletal changes. EMBO J. 32, 1543–1555. (doi:10.1038/emboj.2013.102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Low BC, Pan CQ, Shivashankar GV, Bershadsky A, Sudol M, Sheetz M. 2014. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett. 588, 2663–2670. (doi:10.1016/j.febslet.2014.04.012) [DOI] [PubMed] [Google Scholar]
- 83.Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. 2013. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 154, 1047–1059. (doi:10.1016/j.cell.2013.07.042) [DOI] [PubMed] [Google Scholar]
- 84.Calvo F, et al. 2013. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 15, 637–646. (doi:10.1038/ncb2756) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barry ER, et al. 2013. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature 493, 106–110. (doi:10.1038/nature11693) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Varelas X, et al. 2010. The Hippo pathway regulates Wnt/beta-catenin signaling. Dev. Cell 18, 579–591. (doi:10.1016/j.devcel.2010.03.007) [DOI] [PubMed] [Google Scholar]
- 87.Imajo M, Miyatake K, Iimura A, Miyamoto A, Nishida E. 2012. A molecular mechanism that links Hippo signalling to the inhibition of Wnt/beta-catenin signaling. EMBO J. 31, 1109–1122. (doi:10.1038/emboj.2011.487) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Azzolin L, Zanconato F, Bresolin S, Forcato M, Basso G, Bicciato S, Cordenonsi M, Piccolo S. 2012. Role of TAZ as mediator of Wnt signaling. Cell 151, 1443–1456. (doi:10.1016/j.cell.2012.11.027) [DOI] [PubMed] [Google Scholar]
- 89.Azzolin L, et al. 2014. YAP/TAZ incorporation in the beta-catenin destruction complex orchestrates the Wnt response. Cell 158, 157–170. (doi:10.1016/j.cell.2014.06.013) [DOI] [PubMed] [Google Scholar]
- 90.Tsutsumi R, Masoudi M, Takahashi A, Fujii Y, Hayashi T, Kikuchi I, Satou Y, Taira M, Hatakeyama M. 2013. YAP and TAZ, Hippo signaling targets, act as a rheostat for nuclear SHP2 function. Dev. Cell 26, 658–665. (doi:10.1016/j.devcel.2013.08.013) [DOI] [PubMed] [Google Scholar]
- 91.Llado V, et al. 2015. Repression of intestinal stem cell function and tumorigenesis through direct phosphorylation of beta-catenin and Yap by PKCzeta. Cell Rep. 10, 740–754. (doi:10.1016/j.celrep.2015.01.007) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou X, et al. 2015. Estrogen regulates Hippo signaling via GPER in breast cancer. J. Clin. Invest. 125, 2123–2135. (doi:10.1172/JCI79573) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gong R, Hong AW, Plouffe SW, Zhao B, Liu G, Yu FX, Xu Y, Guan KL. 2015. Opposing roles of conventional and novel PKC isoforms in Hippo-YAP pathway regulation. Cell Res. 25, 985–988. (doi:10.1038/cr.2015.88) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang LH, Baker NE. 2015. Salvador-warts-hippo pathway in a developmental checkpoint monitoring helix-loop-helix proteins. Dev. Cell 32, 191–202. (doi:10.1016/j.devcel.2014.12.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang LH, Baker NE. 2015. E proteins and ID proteins: helix-loop-helix partners in development and disease. Dev. Cell 35, 269–280. (doi:10.1016/j.devcel.2015.10.019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Massari ME, Murre C. 2000. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol. Cell Biol. 20, 429–440. (doi:10.1128/MCB.20.2.429-440.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kee BL. 2009. E and ID proteins branch out. Nat. Rev. Immunol. 9, 175–184. (doi:10.1038/nri2507) [DOI] [PubMed] [Google Scholar]
- 98.Di Palma T, D'Andrea B, Liguori GL, Liguoro A, de Cristofaro T, Del Prete D, Pappalardo A, Mascia A, Zannini M. 2009. TAZ is a coactivator for Pax8 and TTF-1, two transcription factors involved in thyroid differentiation. Exp. Cell Res. 315, 162–175. (doi:10.1016/j.yexcr.2008.10.016) [DOI] [PubMed] [Google Scholar]
- 99.Hong JH, et al. 2005. TAZ, a transcriptional modulator of mesenchymal stem cell differentiation. Science 309, 1074–1078. (doi:10.1126/science.1110955) [DOI] [PubMed] [Google Scholar]
- 100.Varelas X, Sakuma R, Samavarchi-Tehrani P, Peerani R, Rao BM, Dembowy J, Yaffe MB, Zandstra PW, Wrana JL. 2008. TAZ controls Smad nucleocytoplasmic shuttling and regulates human embryonic stem-cell self-renewal. Nat. Cell Biol. 10, 837–848. (doi:10.1038/ncb1748) [DOI] [PubMed] [Google Scholar]
- 101.Alarcon C, et al. 2009. Nuclear CDKs drive Smad transcriptional activation and turnover in BMP and TGF-beta pathways. Cell 139, 757–769. (doi:10.1016/j.cell.2009.09.035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Murakami M, Nakagawa M, Olson EN, Nakagawa O. 2005. A WW domain protein TAZ is a critical coactivator for TBX5, a transcription factor implicated in Holt-Oram syndrome. Proc. Natl Acad. Sci. USA 102, 18 034–18 039. (doi:10.1073/pnas.0509109102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cui CB, Cooper LF, Yang X, Karsenty G, Aukhil I. 2003. Transcriptional coactivation of bone-specific transcription factor Cbfa1 by TAZ. Mol. Cell Biol. 23, 1004–1013. (doi:10.1128/MCB.23.3.1004-1013.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Peng HW, Slattery M, Mann RS. 2009. Transcription factor choice in the Hippo signaling pathway: homothorax and yorkie regulation of the microRNA bantam in the progenitor domain of the Drosophila eye imaginal disc. Genes Dev. 23, 2307–2319. (doi:10.1101/gad.1820009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Basu S, Totty NF, Irwin MS, Sudol M, Downward J. 2003. Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol. Cell 11, 11–23. (doi:10.1016/S1097-2765(02)00776-1) [DOI] [PubMed] [Google Scholar]
- 106.Matallanas D, et al. 2007. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol. Cell 27, 962–975. (doi:10.1016/j.molcel.2007.08.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Strano S, et al. 2005. The transcriptional coactivator Yes-associated protein drives p73 gene-target specificity in response to DNA damage. Mol. Cell 18, 447–459. (doi:10.1016/j.molcel.2005.04.008) [DOI] [PubMed] [Google Scholar]
- 108.Guo C, Tommasi S, Liu L, Yee JK, Dammann R, Pfeifer GP. 2007. RASSF1A is part of a complex similar to the Drosophila Hippo/Salvador/Lats tumor-suppressor network. Curr. Biol. 17, 700–705. (doi:10.1016/j.cub.2007.02.055) [DOI] [PubMed] [Google Scholar]
- 109.Oh H, Irvine KD. 2009. In vivo analysis of Yorkie phosphorylation sites. Oncogene 28, 1916–1927. (doi:10.1038/onc.2009.43) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Robertson JD, Fadeel B, Zhivotovsky B, Orrenius S. 2002. ‘Centennial’ nobel conference on apoptosis and human disease. Cell Death. Differ. 9, 468–475. (doi:10.1038/sj.cdd.4401014) [DOI] [PubMed] [Google Scholar]
- 111.Campuzano O, et al. 2013. Genetics of arrhythmogenic right ventricular cardiomyopathy. J. Med. Genet. 50, 280–289. (doi:10.1136/jmedgenet-2013-101523) [DOI] [PubMed] [Google Scholar]
- 112.Lombardi R, Marian AJ. 2011. Molecular genetics and pathogenesis of arrhythmogenic right ventricular cardiomyopathy: a disease of cardiac stem cells. Pediatr. Cardiol. 32, 360–365. (doi:10.1007/s00246-011-9890-2) [DOI] [PubMed] [Google Scholar]
- 113.Miesfeld JB, Gestri G, Clark BS, Flinn MA, Poole RJ, Bader JR, Besharse JC, Wilson SW, Link BA. 2015. Yap and Taz regulate retinal pigment epithelial cell fate. Development 142, 3021–3032. (doi:10.1242/dev.119008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Halder G, Polaczyk P, Kraus ME, Hudson A, Kim J, Laughon A, Carroll S. 1998. The Vestigial and Scalloped proteins act together to directly regulate wing-specific gene expression in Drosophila. Genes Dev. 12, 3900–3909. (doi:10.1101/gad.12.24.3900) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pobbati AV, Chan SW, Lee I, Song H, Hong W. 2012. Structural and functional similarity between the Vgll1-TEAD and the YAP-TEAD complexes. Structure 20, 1135–1140. (doi:10.1016/j.str.2012.04.004) [DOI] [PubMed] [Google Scholar]
- 116.Vaudin P, Delanoue R, Davidson I, Silber J, Zider A. 1999. TONDU (TDU), a novel human protein related to the product of vestigial (vg) gene of Drosophila melanogaster interacts with vertebrate TEF factors and substitutes for Vg function in wing formation. Development 126, 4807–4816. [DOI] [PubMed] [Google Scholar]
- 117.Simmonds AJ, Liu X, Soanes KH, Krause HM, Irvine KD, Bell JB. 1998. Molecular interactions between Vestigial and Scalloped promote wing formation in Drosophila. Genes Dev. 12, 3815–3820. (doi:10.1101/gad.12.24.3815) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhang W, et al. 2014. VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex. Cell Res. 24, 331–343. (doi:10.1038/cr.2014.10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Koontz LM, Liu-Chittenden Y, Yin F, Zheng Y, Yu J, Huang B, Chen Q, Wu S, Pan D. 2013. The Hippo effector Yorkie controls normal tissue growth by antagonizing scalloped-mediated default repression. Dev. Cell 25, 388–401. (doi:10.1016/j.devcel.2013.04.021) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Huang Y, Mucke L. 2012. Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222. (doi:10.1016/j.cell.2012.02.040) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tomiyama T. 2012. Involvement of beta-amyloid in the etiology of Alzheimer's disease. Brain Nerve 62, 691–699. [PubMed] [Google Scholar]
- 122.Yuan Z, Lehtinen MK, Merlo P, Villen J, Gygi S, Bonni A. 2009. Regulation of neuronal cell death by MST1-FOXO1 signaling. J. Biol. Chem. 284, 11 285–11 292. (doi:10.1074/jbc.M900461200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Graves JD, Gotoh Y, Draves KE, Ambrose D, Han DK, Wright M, Chernoff J, Clark EA, Krebs EG. 1998. Caspase-mediated activation and induction of apoptosis by the mammalian Ste20-like kinase Mst1. EMBO J. 17, 2224–2234. (doi:10.1093/emboj/17.8.2224) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu W, Wu J, Xiao L, Bai Y, Qu A, Zheng Z, Yuan Z. 2012. Regulation of neuronal cell death by c-Abl-Hippo/MST2 signaling pathway. PLoS ONE 7, e36562 (doi:10.1371/journal.pone.0036562) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lehtinen MK, et al. 2006. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell 125, 987–1001. (doi:10.1016/j.cell.2006.03.046) [DOI] [PubMed] [Google Scholar]
- 126.Jang SW, Yang SJ, Srinivasan S, Ye K. 2007. Akt phosphorylates MstI and prevents its proteolytic activation, blocking FOXO3 phosphorylation and nuclear translocation. J. Biol. Chem. 282, 30 836–30 844. (doi:10.1074/jbc.M704542200) [DOI] [PubMed] [Google Scholar]
- 127.Brunet A, et al. 1999. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868. (doi:10.1016/S0092-8674(00)80595-4) [DOI] [PubMed] [Google Scholar]
- 128.Biggs WH, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. 1999. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc. Natl Acad. Sci. USA 96, 7421–7426. (doi:10.1073/pnas.96.13.7421) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. 1999. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem. 274, 17 179–17 183. (doi:10.1074/jbc.274.24.17179) [DOI] [PubMed] [Google Scholar]
- 130.Tang ED, Nunez G, Barr FG, Guan KL. 1999. Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem. 274, 16 741–16 746. (doi:10.1074/jbc.274.24.16741) [DOI] [PubMed] [Google Scholar]
- 131.Kops GJ, de Ruiter ND, De Vries-Smits AM, Powell DR, Bos JL, Burgering BM. 1999. Direct control of the Forkhead transcription factor AFX by protein kinase B. Nature 398, 630–634. (doi:10.1038/19328) [DOI] [PubMed] [Google Scholar]
- 132.Li L, Xu B, Zhu Y, Chen L, Sokabe M, Chen L. 2010. DHEA prevents Abeta25-35-impaired survival of newborn neurons in the dentate gyrus through a modulation of PI3K-Akt-mTOR signaling. Neuropharmacology 59, 323–333. (doi:10.1016/j.neuropharm.2010.02.009) [DOI] [PubMed] [Google Scholar]
- 133.Zeng KW, Wang XM, Ko H, Kwon HC, Cha JW, Yang HO. 2011. Hyperoside protects primary rat cortical neurons from neurotoxicity induced by amyloid beta-protein via the PI3K/Akt/Bad/Bcl(XL)-regulated mitochondrial apoptotic pathway. Eur. J. Pharmacol. 672, 45–55. (doi:10.1016/j.ejphar.2011.09.177) [DOI] [PubMed] [Google Scholar]
- 134.Shang YC, Chong ZZ, Wang S, Maiese K. 2012. Prevention of beta-amyloid degeneration of microglia by erythropoietin depends on Wnt1, the PI3-K/mTOR pathway, Bad, and Bcl-xL. Aging (Albany. NY) 4, 187–201. (doi:10.18632/aging.100440) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bendotti C, Carri MT. 2004. Lessons from models of SOD1-linked familial ALS. Trends Mol. Med. 10, 393–400. (doi:10.1016/j.molmed.2004.06.009) [DOI] [PubMed] [Google Scholar]
- 136.Lapi E, et al. 2008. PML, YAP, and p73 are components of a proapoptotic autoregulatory feedback loop. Mol. Cell 32, 803–814. (doi:10.1016/j.molcel.2008.11.019) [DOI] [PubMed] [Google Scholar]
- 137.Tomlinson V, Gudmundsdottoir K, Luong P, Leung KY, Knebel A, Basu S. 2010. JNK phosphorylates Yes-associated protein (YAP) to regulate apoptosis. Cell Death Dis. 1, e29 (doi:10.1038/cddis.2010.7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cebola I, et al. 2015. TEAD and YAP regulate the enhancer network of human embryonic pancreatic progenitors. Nat. Cell Biol. 17, 615–626. (doi:10.1038/ncb3160) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cheng Y, Feng Y, Jansson L, Sato Y, Deguchi M, Kawamura K, Hsueh AJ. 2015. Actin polymerization-enhancing drugs promote ovarian follicle growth mediated by the Hippo signaling effector YAP. FASEB J. 29, 2423–2430. (doi:10.1096/fj.14-267856) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kawamura K, et al. 2013. Hippo signaling disruption and Akt stimulation of ovarian follicles for infertility treatment. Proc. Natl Acad. Sci. USA 110, 17 474–17 479. (doi:10.1073/pnas.1312830110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Belle I, Zhuang Y. 2014. E proteins in lymphocyte development and lymphoid diseases. Curr. Top. Dev. Biol. 110, 153–187. (doi:10.1016/B978-0-12-405943-6.00004-X) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lasorella A, Benezra R, Iavarone A. 2014. The ID proteins: master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer 14, 77–91. (doi:10.1038/nrc3638) [DOI] [PubMed] [Google Scholar]
- 143.Ling F, Kang B, Sun XH. 2014. Id proteins: small molecules, mighty regulators. Curr. Top. Dev. Biol. 110, 189–216. (doi:10.1016/B978-0-12-405943-6.00005-1) [DOI] [PubMed] [Google Scholar]
- 144.Tokuzawa Y, et al. 2010. Id4, a new candidate gene for senile osteoporosis, acts as a molecular switch promoting osteoblast differentiation. PLoS. Genet. 6, e1001019 (doi:10.1371/journal.pgen.1001019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Duffield GE, Watson NP, Mantani A, Peirson SN, Robles-Murguia M, Loros JJ, Israel MA, Dunlap JC. 2009. A role for Id2 in regulating photic entrainment of the mammalian circadian system. Curr. Biol. 19, 297–304. (doi:10.1016/j.cub.2008.12.052) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ward SM, Fernando SJ, Hou TY, Duffield GE. 2010. The transcriptional repressor ID2 can interact with the canonical clock components CLOCK and BMAL1 and mediate inhibitory effects on mPer1 expression. J. Biol. Chem. 285, 38 987–39 000. (doi:10.1074/jbc.M110.175182) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Peters DJ, Sandkuijl LA. 1992. Genetic heterogeneity of polycystic kidney disease in Europe. Contrib. Nephrol. 97, 128–139. (doi:10.1159/000421651) [DOI] [PubMed] [Google Scholar]
- 148.Tian Y et al. . 2007. TAZ promotes PC2 degradation through a SCFβ-Trcp E3 ligase complex. Mol. Cell Biol. 27, 6383–6395. (doi:10.1128/MCB.00254-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Li X, Luo Y, Starremans PG, McNamara CA, Pei Y, Zhou J. 2005. Polycystin-1 and polycystin-2 regulate the cell cycle through the helix-loop-helix inhibitor Id2. Nat. Cell Biol. 7, 1202–1212. (doi:10.1038/ncb1326) [DOI] [PubMed] [Google Scholar]
- 150.Li H, Dai M, Zhuang Y. 2004. A T cell intrinsic role of Id3 in a mouse model for primary Sjogren's syndrome. Immunity 21, 551–560. (doi:10.1016/j.immuni.2004.08.013) [DOI] [PubMed] [Google Scholar]
- 151.Enger TB, et al. 2013. The Hippo signaling pathway is required for salivary gland development and its dysregulation is associated with Sjogren's syndrome. Lab. Invest. 93, 1203–1218. (doi:10.1038/labinvest.2013.114) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Anand R, Maksimoska J, Pagano N, Wong EY, Gimotty PA, Diamond SL, Meggers E, Marmorstein R. 2009. Toward the development of a potent and selective organoruthenium mammalian sterile 20 kinase inhibitor. J. Med. Chem. 52, 1602–1611. (doi:10.1021/jm8005806) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kimura N. 2016. Diabetes mellitus induces Alzheimer's disease pathology: histopathological evidence from animal models. Int. J. Mol. Sci. 17, 503 (doi:10.3390/ijms17040503) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Shao D, Zhai P, Del Re DP, Sciarretta S, Yabuta N, Nojima H, Lim DS, Pan D, Sadoshima J. 2014. A functional interaction between Hippo-YAP signalling and FoxO1 mediates the oxidative stress response. Nat. Commun. 5, 3315–3324. (doi:10.1038/ncomms4315) [DOI] [PMC free article] [PubMed] [Google Scholar]