

Abstract
Aneuploidy leads to severe developmental defects in mammals and is also a hallmark of cancer. However, whether aneuploidy is a driving cause or a consequence of tumor formation remains controversial. Paradoxically, existing studies based on aneuploid yeast and mouse fibroblasts have shown that aneuploidy is usually detrimental to cellular fitness. Here, we examined the effects of aneuploidy on mouse embryonic stem (ES) cells by generating a series of cell lines that each carries an extra copy of single chromosomes, including trisomy 6, 8, 11, 12, or 15. Most of these aneuploid cell lines had rapid proliferation rates and enhanced colony formation efficiencies. They were less dependent on growth factors for self‐renewal and showed a reduced capacity to differentiate in vitro. Moreover, trisomic stem cells formed teratomas more efficiently, from which undifferentiated cells can be recovered. Further investigations demonstrated that co‐culture of wild‐type and aneuploid ES cells or supplementation with extracellular BMP4 rescues the differentiation defects of aneuploid ES cells.
Keywords: aneuploidy, differentiation, ES cells, extracellular factors, tumorigenesis
Subject Categories: Cancer, Development & Differentiation, Stem Cells
Introduction
Aneuploidy, a state of abnormal chromosomal number that is not a multiple of the haploid complement, is the leading cause of miscarriages and developmental defects in humans (Hassold & Jacobs, 1984). Aneuploidy is also one of the fundamental differences between normal and tumor cells and has been observed in more than 90% of human solid tumors with rapid proliferation (Weaver & Cleveland, 2006). More than 100 years ago, Theodor Boveri initially proposed that aneuploidy was the potential cause of cancer (Boveri, 1914). Recent studies showed that gains of single chromosomes in yeast (Torres et al, 2007), mouse embryonic fibroblasts (MEFs) (Williams et al, 2008) and human Down syndrome (DS) fibroblasts (Kimura et al, 2005) impaired cell proliferation in vitro, which implied that aneuploidy may be a barrier to tumorigenesis. This is the so‐called “aneuploidy paradox” (Weaver & Cleveland, 2008). Moreover, aneuploidy also endows organisms with adaptive potentials under stress conditions (Rancati et al, 2008; Chen et al, 2012; Selmecki et al, 2015; Sunshine et al, 2015). Thus far, the role of aneuploidy in cancer initiation and progression remains largely unknown.
Pluripotent stem cells (PSCs) share the features of rapid proliferation, accelerated metabolism and some gene expression profiles with malignant cancer cells (Zhang et al, 2012). Recent studies have linked an embryonic stem (ES) cell‐like gene signature with poorly differentiated breast, brain, and bladder cancers (Ben‐Porath et al, 2008). Moreover, PSCs can acquire chromosomal aberrations during prolonged cell culture and exhibit some features of malignant progression (Werbowetski‐Ogilvie et al, 2009). Therefore, PSCs could be valuable tools for studying the role of aneuploidy in cancer, but it is still limited by the availability of experimental models. To systematically investigate the effects of aneuploidy on the proliferation and differentiation of ES cells, we generated several aneuploid cell lines in which each line carries an additional chromosome, including trisomy 6, 8, 11, 12, or 15. Our results indicate that aneuploidy affects differentiation of ES cells and promotes teratoma formation by impeding exit from self‐renewal of stem cells. We found that dysregulation of secreted extracellular factors contributes to the differentiation defects of aneuploid ES cells, and the addition of exogenous factors can rescue the defects. Our study may provide new insights for cancer therapy.
Results
Generation and characterization of trisomic ES cell lines
Previously, we found that aneuploid mouse ES cell clones could be isolated by applying a novel genetic selection method in Blm‐deficient mouse ES cells designed to identify homozygous mutants (Huang et al, 2011). In this study, the same strategy was used to generate “newborn” aneuploid cell lines, each carrying an additional chromosome from wild‐type ES cells (AB1 and JM8 cell lines) that have been widely used in mouse genetic studies. Briefly, the piggyBac (PB) transposon vector (DHSV) carrying the conditional drug selection cassettes (Bsd and PuroΔTK) was inserted into the wild‐type mouse ES cell genome randomly. The clones containing a single PB insertion in the intergenic region were expanded to allow copy number increase, which might contribute to the double‐resistant population by the action of Cre recombinase (Fig 1A). Three alternative pathways were responsible for the increase in copy number: trisomy, tetraploidy, and loss of heterozygosity (LOH) (Fig 1B). We successfully obtained mouse ES cell lines that were trisomic for chromosome 6, 8, 11, or 15 (Ts6, Ts8, Ts11, or Ts15 respectively), from AB1 cells. Cell lines trisomic for chromosome 8 or 12 were also obtained from JM8 cells (Ts8‐JM8 or Ts12‐JM8) (Table 1). ES cells in which the marker became homozygous but retained a normal diploid karyotype could be used as isogenic diploid control cell lines (Homo‐).
Figure 1. Generation of trisomic ES cell lines.
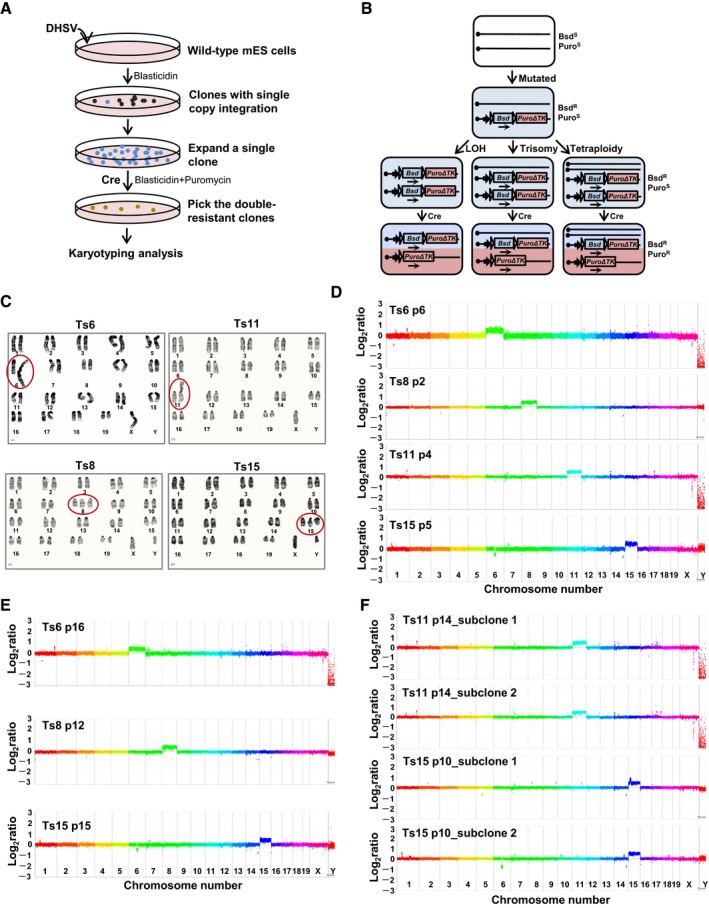
- The experimental flowchart to isolate trisomic ES cell lines.
- The molecular events during isolation of trisomic ES cell lines. BsdS: sensitive to blasticidin, BsdR: resistant to blasticidin, PuroS: sensitive to puromycin, PuroR: resistant to puromycin.
- Karyograms of trisomic ES cells show that these cell lines have extra copies of chromosomes 6, 8, 11, or 15. The Ts6 and Ts11 ES cells harbored isochromosomes of the two identical trisomic chromosomes. Ts, trisomy.
- CGH analysis of trisomic ES cell lines. The relative gene copy number is plotted using a log2 scale.
- CGH analysis of the genome stability of trisomic ES cells. The relative gene copy number is plotted using a log2 scale.
- CGH analysis of the genome integrity of trisomic ES cell subclones. The relative gene copy number is plotted using a log2 scale.
Table 1.
Summary of isolated trisomic ES cell lines
| Trisomic cell lines | Karyotype | Centromeric fusiona | No. of ES cell clones | Extra chromosomes | ||
|---|---|---|---|---|---|---|
| Trisomic ES cell clones | Homozygous ES cell clonesb | Chromosome sizec (kb) | No. of coding genesc | |||
| AB1‐derived trisomic ES cell lines | ||||||
| Ts6 | 40, XO, +6 | Yes | 2 | 0 | 149,736 | 1,309 |
| Ts8 | 41, XY, +8 | No | 24 | 1 | 129,401 | 1,072 |
| Ts11 | 40, XO, +11 | Yes | 4 | 2 | 122,082 | 1,634 |
| Ts15 | 41, XY, +15 | No | 5 | 2 | 104,043 | 797 |
| JM8‐derived trisomic ES cell lines | ||||||
| Ts8‐JM8 | 40, XO, +8 | Yes | 8 | 0 | 129,401 | 1,072 |
| Ts12‐JM8 | 41, XY, +12 | No | 2 | 0 | 120,129 | 836 |
Centromeric fusion was often correlated with aneuploidy.
The homozygous ES cell clones were isolated when obtaining trisomic ES cell lines.
The chromosome and gene information is from GRCm38.
Chromosome counting, karyotype analysis, and chromosome painting were used to determine the existence of three copies of a particular chromosome in these trisomic ES cell lines (Fig 1C and Appendix Fig S1A–D). We noticed two kinds of trisomies existed. The cell lines of Ts8, Ts15, and Ts12‐JM8 carry the freely segregating extra chromosomes, while Ts6, Ts11, and Ts8‐JM8 have fusion chromosomes of the two identical trisomic chromosomes. Array comparative genomic hybridization (CGH) validated the gene copy number increase in the chromosomes that were selected. A small number of other minor interstitial genomic changes and loss of the Y chromosome were detected (Fig 1D and Appendix Fig S1E), which are typical of the variation normally seen in ES cells (Liang et al, 2008). To test the genomic stability of the trisomic ES cells, we performed array CGH analysis after serial passaging in cell culture. Ten more passages later, the copy number variants observed in trisomic ES cell lines resembled those in earlier cultures (Fig 1E). To explore possible genetic heterogeneity within the cultures, single‐cell clones were isolated. There was no additional numerical aneuploidy, but multiple variable clonal small rearrangements existed (Fig 1F).
Cultured trisomic ES cells formed alkaline phosphatase (AP)‐positive dome‐shaped colonies after plating at low density (Appendix Fig S2A and B) and expressed markers indicative of pluripotency, as detected by immunofluorescence, reverse transcription (RT)‐qPCR, and Western blot analyses (Appendix Fig S2C–F). We examined the proliferation abilities of AB1‐derived trisomic ES cells in culture and found most cell lines showed rapid proliferation except for the slightly slower growth of Ts15 lines as compared to their non‐trisomic controls (Fig 2A). The Ts8‐JM8 and Ts12‐JM8 cells also proliferated rapidly and reached higher saturation densities (Appendix Fig S3A). Flow cytometry cell cycle analysis revealed that aneuploidy mildly affected cell cycle progression, but the effects were not uniform among different trisomies (Fig 2B and Appendix Fig S3B). The expression of apoptosis marker Annexin V was similar in wild‐type and trisomic ES cells as detected by flow cytometry (Fig 2C and Appendix Fig S3C).
Figure 2. Proliferation characteristics of the trisomic ES cell lines.
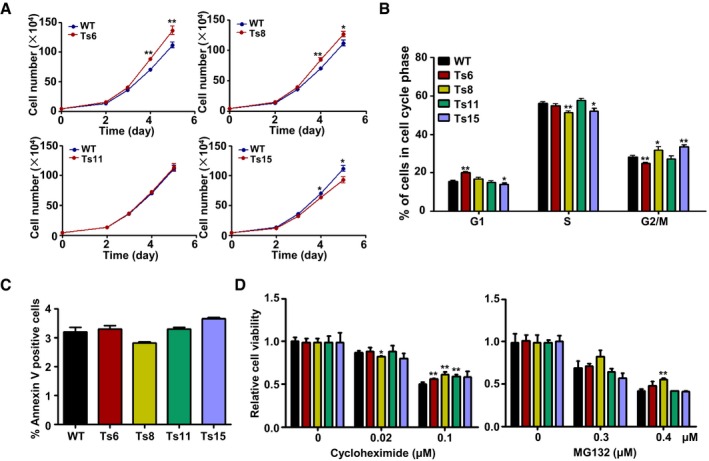
- Cumulative growth curves of trisomic and wild‐type ES cells. The number of ES cells was counted at the indicated days in culture. The cell lines were tested in triplicates. Error bars, ± SD. *P < 0.05, **P < 0.01.
- Cell cycle distribution of trisomic and wild‐type ES cell lines. The percentage of cells in G1, S, and G2/M phases was determined via flow cytometry. Error bars, ± SD. n = 3. *P < 0.05, **P < 0.01.
- The percentage of Annexin V+ apoptotic cells was quantified. Error bars, ± SD. n = 3.
- Trisomic and wild‐type ES cells were treated with cycloheximide or MG132 for 48 h at the indicated doses. Error bars, ± SD. n = 3. *P < 0.05, **P < 0.01.
Source data are available online for this figure.
Because the previously reported aneuploid yeasts and MEFs showed increased sensitivity to compounds causing protein or energy stress (Torres et al, 2007; Tang et al, 2011), we wanted to test whether there is proteotoxic stress in trisomic ES cells. The proliferation viabilities were tested in the presence of the compounds that cause proteotoxic stress (cycloheximide, an inhibitor of protein synthesis, and MG132, an inhibitor of proteasome). Nearly all cell lines could not be selectively inhibited by these compounds (Fig 2D). Thus, under ES cell culture conditions, trisomic cells did not show the same alterations in protein synthesis and degradation observed in aneuploid yeasts and MEFs.
Aneuploidy impeded the differentiation of ES cells
We examined the status of spontaneous differentiation of trisomic ES cells under ES cell culture conditions by the colony‐forming assay. The results showed that more AP‐positive colonies were observed in all trisomic cell lines compared with wild‐type ES cells (Fig 3A and Appendix Fig S3D), indicating the reduced spontaneous differentiation of the trisomic ES cells in culture. To further determine whether aneuploidy affects the differentiation capacities of ES cells under differentiation conditions, we first tested the dependency of the trisomic ES cells on the cytokine leukemia inhibitory factor (LIF). The colonies were scored as undifferentiated, partially differentiated or totally differentiated based on AP staining. Most of the wild‐type ES cells rapidly progressed to partial or total differentiation upon LIF withdrawal, while the trisomic ES cells formed more AP‐positive colonies after LIF withdrawal for 5 days (Figs 3B and C, and EV1A and B).
Figure 3. Limited differentiation abilities of trisomic ES cells in vitro .
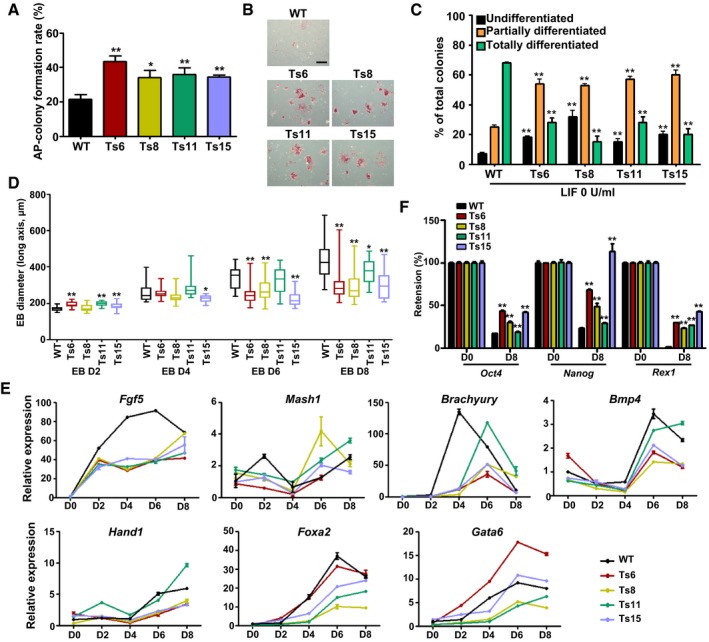
- The colony formation rates of the trisomic and wild‐type ES cells. Error bars, ± SD. n = 3. *P < 0.05, **P < 0.01.
- Alkaline phosphatase (AP) staining of trisomic and wild‐type ES cells after LIF withdrawal (LIF 0 U/ml) for 5 days. Scale bar, 500 μm.
- Evaluation of AP‐positive and AP‐negative colonies after LIF withdrawal. Black bars, undifferentiated colonies; orange bars, partially differentiated colonies; green bars, totally differentiated colonies. Error bars, ± SD. n = 3. **P < 0.01.
- Box plots of EB diameter at the indicated days of EB formation. Each box plot was plotted for more than 20 EBs. Boxes indicate the 25th to 75th percentiles and the central bars represent the median. The ends of the whiskers represent the maximum and minimum values. *P < 0.05, **P < 0.01.
- Time course analysis of marker gene expression during EB formation by RT‐qPCR. Error bars, ± SD. n = 3.
- Quantification of Oct4, Nanog, and Rex1 expression in trisomic and wild‐type ES cell‐derived EBs at day 8 of differentiation. Error bars, ± SD. n = 3. **P < 0.01.
Source data are available online for this figure.
Figure EV1. Decreased differentiation abilities of trisomic ES cells in vitro .

- AP staining for the differentiated ES cells. The LIF concentration is 1 U/ml (upper panels) and 10 U/ml (lower panels), respectively. Scale bar, 200 μm.
- Evaluation of AP‐positive and AP‐negative colonies under decreased LIF concentration (1 and 10 U/ml). Black bars, undifferentiated colonies; orange bars, partially differentiated colonies; green bars, totally differentiated colonies. Error bars, ± SD. n = 3. *P < 0.05, **P < 0.01.
- Morphology of wild‐type and trisomic EB formation at the indicated time points. Black arrows indicate the cystic structures. Scale bar, 200 μm.
We cultured the trisomic and wild‐type ES cells in suspension and found that all the trisomic ES cells could aggregate and form embryoid body (EB)‐like structures for 8 or more days. Wild‐type EBs grew very rapidly from day 4 to day 6 of differentiation and were efficiently cavitated and formed large cystic structures at day 8. However, the trisomic ES cell‐derived EBs were more compact and smaller (Fig EV1C). The average EB size of trisomic EBs at different differentiation time points are shown in Fig 3D. This result was further evidenced by the expression of germ layer‐specific marker genes. We found that all the trisomic ES cells aberrantly expressed the ectoderm marker genes (Fgf5, Mash1), mesoderm marker genes (Brachyury, Bmp4, Hand1) and endoderm marker genes (Foxa2, Gata6). The expression magnitude of Fgf5, Bmp4, Hand1, Foxa2, and Gata6 was decreased in most trisomic ES cell lines during EB formation, and the peak expression of Brachyury was delayed, and also the transient increase in Mash1 expression at day 2 of EB formation cannot be observed in most trisomic cells (Fig 3E). These results indicated that the differentiation timing of trisomic ES cells to multiple lineages had been delayed. Moreover, higher expression of the pluripotency‐associated genes, including Oct4 (also known as Pou5f1), Nanog, and Rex1, was observed in trisomic EBs as compared to wild‐type EBs (Fig 3F). It indicated that the “exit from pluripotency” was abnormal in trisomic ES cells.
We also tested the lineage commitment potential of the trisomic ES cells. Neuronal axons extended from the wild‐type ES cell‐derived neurospheres and formed network structures, while few visible neuronal axons were observed in trisomic ES cell‐derived neurospheres (Fig EV2A and B). When differentiated into the cardiac lineage, almost 100% of the cardiac lineage‐committed embryoid bodies (cEBs) from wild‐type ES cells exhibited prominent beating from day 11. However, about 75% of the cEBs from Ts8 and Ts11 ES cells showed beating contraction, and the proportion of the beating cEBs was just 37.5% for Ts6 ES cells and nearly 0% for Ts15 ES cells. Additionally, all trisomic ES cell cEBs exhibited beating contractions later than the wild‐type cells (Fig EV2C).
Figure EV2. Neural and cardiac differentiation of trisomic ES cells.

- Neurosphere differentiation of trisomic and wild‐type ES cells. Scale bar, 100 μm.
- Immunofluorescence staining of the neuronal markers. The cells were stained with βIII‐tubulin (green, top), MAP2 (green, bottom), and NeuN (red, bottom). The nuclei were stained with DAPI. Scale bars, 100 μm.
- A time course of wild‐type and trisomic cEBs that showed heart‐like spontaneous contraction among all EBs. Eight EBs were continuously observed for each cell line. Error bars, ± SD. *P < 0.05, **P < 0.01.
Aneuploidy promoted teratoma formation
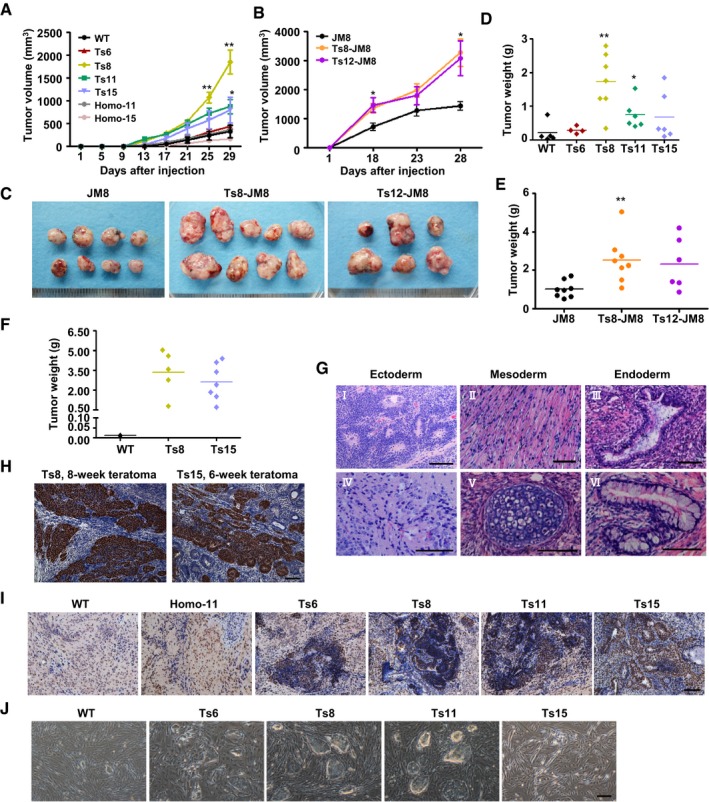
The teratoma assay has come to be considered a quantitative index of the frequency of teratoma‐initiating cells (TICs) in ES cell cultures (Baker et al, 2007; Werbowetski‐Ogilvie et al, 2009). To understand the effects of aneuploidy on teratoma formation, we conducted limiting‐dilution teratoma assays in severe combined immunodeficiency (SCID) mice. The trisomic ES cells formed palpable tumors on day 7–9 after cell injection, which was 3–6 days earlier than that of the wild‐type ES cells. Trisomic ES cell‐derived teratomas grew rapidly and were much larger and heavier than the diploid teratomas within 4 weeks of cell injection (Figs 4A and B, EV3A–F and Appendix Table S1). Moreover, when fewer cells were injected (2 × 105 cells/site), Ts8 and Ts15 ES cells still formed teratomas at high efficiencies (62–87%), compared with 12.5% for wild‐type ES cells (Appendix Table S1). Histological analysis showed that the teratomas produced by the trisomic ES cells contained cells of three germ layers, indicating pluripotency in vivo (Fig EV3G). However, large proportions of more primitive undifferentiated or low differentiated regions were detected in the trisomic ES cell‐derived teratomas (Fig 4C). Further immunohistochemical staining for Oct4 confirmed the existence of undifferentiated stem cells in the trisomic ES cell‐derived teratomas (Figs 4D and EV3H). This finding was further evidenced by the widespread expression of Ki67 in 4‐week trisomic ES cell‐derived teratomas (Fig EV3I). We conclude that aneuploidy limited the differentiation of PSCs during teratoma formation and hence promoted tumor formation.
Figure 4. Enhanced teratoma formation capacities of trisomic ES cells.
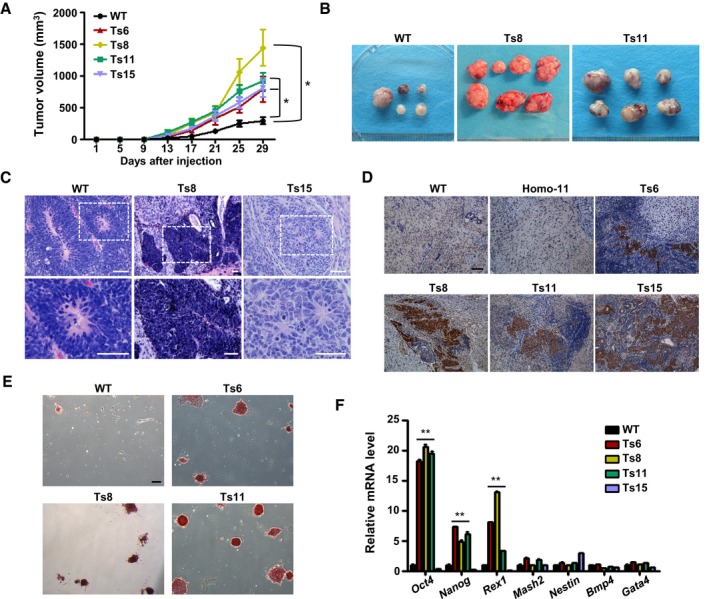
- Teratoma growth curves of trisomic and wild‐type ES cells after subcutaneous injection into SCID mice. A total of 1 × 106 cells were injected per site (6 sites per group). Error bars, ± SD. *P < 0.05.
- Four‐week teratomas derived from wild‐type and trisomic (Ts8 or Ts11) ES cell lines. A total of 5 × 105 cells were injected per site (8 sites per group).
- Representative images of undifferentiated regions in Ts8 ES cell‐derived teratomas (middle, top, and bottom) and the primitive neural rosettes in the teratomas derived from Ts15 ES cells (right, top, and bottom). The images of mature neuroectoderm from wild‐type ES cell‐formed teratomas (left, top, and bottom) are shown. The bottom panels are the enlarged views of the white dotted boxes of the top panels. Hematoxylin and eosin staining. Scale bars, 50 μm.
- Immunohistochemical analysis of Oct4 expression in wild‐type, Homo‐11, Ts6, Ts8, Ts11, or Ts15 ES cell teratomas produced via the injection of 5 × 105 ES cells. Scale bar, 100 μm.
- AP staining of the primary cultured teratoma cells derived from trisomic and wild‐type ES cells. Ts6, Ts8, or Ts11 ES cell‐derived teratoma cells grew ES‐like colonies that stained positive for AP. Scale bar, 200 μm.
- The expression of marker genes in cultured teratoma cells (p3). Error bars, ± SD. n = 3. **P < 0.01.
Source data are available online for this figure.
Figure EV3. Evaluation of wild‐type and trisomic ES cell‐derived teratomas.
- Growth curves of teratomas produced by injection of 5 × 105 AB1‐derived ES cells (8 injection sites/group). Error bars, ± SD. *P < 0.05, **P < 0.01.
- Growth curves of teratomas produced by injection of 6 × 105 JM8‐derived ES cells (8 injection sites/group). Error bars, ± SD. *P < 0.05.
- Images of 4‐week teratomas derived from JM8 and trisomic (Ts8‐JM8 or Ts12‐JM8) ES cells. The injected cell number was 6 × 105 per site.
- Scatter plot of 4‐week teratoma weight (AB1‐derived). Wild‐type, Ts6, Ts8, Ts11, or Ts15 ES cells were injected at the dose of 5 × 105 cells/site. *P < 0.05, **P < 0.01.
- Scatter plot of 4‐week teratoma weight (JM8‐derived). JM8, Ts8‐JM8, and Ts12‐JM8 ES cells were injected at the dose of 6 × 105 cells/site. **P < 0.01.
- Scatter plot of 6‐week teratoma weight (AB1‐derived). Wild‐type, Ts8, or Ts15 ES cells were injected at the dose of 2 × 105 cells/site.
- Histopathological analysis of the 4‐week teratomas. Hematoxylin and eosin staining of the teratoma sections detected all three germ layer‐derived tissues in teratomas derived from trisomic ES cells. Ectoderm‐derived tissues: neuroectoderm (I) and neuronal cells (IV); mesoderm‐derived tissues: muscle (II) and cartilage (V); endoderm‐derived tissues: respiratory (III) and intestine‐like epithelium (VI). Scale bars, 100 μm.
- Immunohistochemical analysis of Oct4 expression in Ts8 and Ts15 ES cell teratomas produced by injection of 2 × 105 ES cells. Scale bar, 100 μm.
- Immunohistochemical analysis of Ki67 expression in the teratoma sections. Scale bar, 100 μm.
- Four‐week teratomas derived from wild‐type or trisomic ES cells were digested with collagenase IV and then cultured under ES cell culture conditions for two passages. Scale bar, 200 μm.
We next tested whether the undifferentiated cells could be recovered from the trisomic teratomas. Teratomas were explanted, and cells derived from teratomas were cultured under normal ES cell culture conditions. Obviously, the Ts6, Ts8, and Ts11 ES cell‐derived teratoma cells grew out with the typical ES cell morphology after two passages (Fig EV3J). Few colonies were observed from the Ts15 ES cell‐derived teratomas, which may be attributable to the chromosome‐specific effects of this trisomy. The ES‐like colonies stained positive for AP (Fig 4E) and the Ts6, Ts8, and Ts11 ES cell‐derived teratoma cells expressed higher levels of PSC markers (Oct4, Nanog, and Rex1) compared with the diploid teratoma cells, whereas the germ layer markers of Mash2, Nestin, Bmp4, or Gata4 were not changed so much in these trisomic cell‐derived teratoma cells (Fig 4F). These results confirmed the existence of undifferentiated stem cells in the trisomic ES cell‐derived teratomas.
Trisomy correction rescued the differentiation defects of trisomic ES cells in vitro and in vivo
To test whether aneuploidy is the direct cause of differentiation defects of these trisomic ES cells, we investigated whether trisomy correction could rescue the defects. A negative selection strategy was designed to select for ES cells with spontaneous trisomic chromosome loss (named corrected diploid ES cells) (Fig 5A). Only cells which had lost the puroΔTK active cassette survived after FIAU selection. A similar strategy has been used to remove the extra copy of chromosome 21 in Down syndrome induced pluripotent stem cells (iPSCs) previously (Li et al, 2012). We have derived the corrected diploid ES cells from Ts8, Ts11, and Ts15 lines (Di8, Di11, and Di15) and validated the correction by chromosome counting and painting (Appendix Fig S4A and B). There were no additional chromosomal changes in these corrected cell lines (Fig 5B). Upon LIF withdrawal, the corrected diploid ES cells formed much less AP‐positive colonies than counterpart trisomic ES cells (Fig 5C). They formed EBs with large cystic structures, in sharp contrast to the compact EBs formed by trisomic ES cells (Appendix Fig S4C). We further examined the causality between aneuploidy and the enhanced teratoma formation. We injected Di8, Di11, and Di15 ES cells and their parental trisomic ES cells into SCID mice subcutaneously and found the teratomas derived from the corrected diploid ES cells were much smaller and lighter than their parental trisomic ES cell‐derived teratomas (Fig 5D and E, and Appendix Fig S4D, Appendix Table S1). The Oct4‐positive regions were nearly undetectable in the corrected diploid ES cell‐derived teratomas (Appendix Fig S4E). To test whether some possible epigenetic changes occurred in these corrected diploid clones, we analyzed the levels of histone modification and DNA methylation in these cell lines. The levels of H3K27me3 and H4K20me1 in the corrected ES cell lines were comparable to that of wild‐type ES cells (Appendix Fig S4F). These cells also maintained the hypomethylation status of the promoters of pluripotency‐associated genes Oct4 and Nanog (Appendix Fig S4G). We thus concluded that aneuploidy itself, instead of some elusive additional genetic and epigenetic variations generated during cloning and drug treatment, was sufficient to increase teratoma size.
Figure 5. Trisomy correction rescued the differentiation defects of trisomic ES cells.

- Schematic overview for correcting trisomic cells.
- Karyograms of Di8, Di11, and Di15 ES cell lines. Di8 is the corrected ES cell line from Ts8, showing diploid karyotype and Y chromosome loss. Di11 and Di15 were derived from Ts11 and Ts15 ES cells, respectively.
- AP staining of trisomic and the corrected diploid ES cells after LIF withdrawal (LIF 0 U/ml) for 5 days. Scale bar, 500 μm.
- Teratoma growth curves of trisomic and the corrected diploid ES cells. The injected cell number was 5 × 105/site (8 sites per group). Error bars, ± SD. *P < 0.05, **P < 0.01.
- Scatter plot of 4‐week teratoma weight. The teratomas were formed by the corrected diploid ES cells and their paternal trisomic ES cells. *P < 0.05.
Gene expression profiling of aneuploid ES cells
To further understand the molecular basis of aneuploidy‐associated differentiation defects, we compared the global transcription profiles of the trisomic (Ts6, Ts8, Ts11, and Ts15) and wild‐type AB1 ES cell lines by microarray analysis. Overall, most of the genes on the additional chromosomes were transcribed at 117–128% of those in control cell lines, correlating with their increased copy number but not attaining the full 150% (Fig EV4A and Dataset EV1). The principal component analysis (PCA) showed that Ts6, Ts8, and Ts11 ES cell lines were more similar to each other based on the global gene expression profiles, apart from that of wild‐type and Ts15 ES cell lines (Fig 6A). We further performed hierarchical clustering of the whole gene expression data, which showed that the Ts6, Ts8, and Ts11 ES cell lines clustered together and the expression patterns were far different from wild‐type ES cells. Although the Ts15 line was more similar to wild‐type ES cells, thousands of genes exhibited changes in their expression (Figs 6B and EV4B). These data suggested that the additional chromosomes in ES cells were sufficient to induce the genomewide dysregulation of gene expression.
Figure EV4. Gene expression profiles of trisomic ES cells.
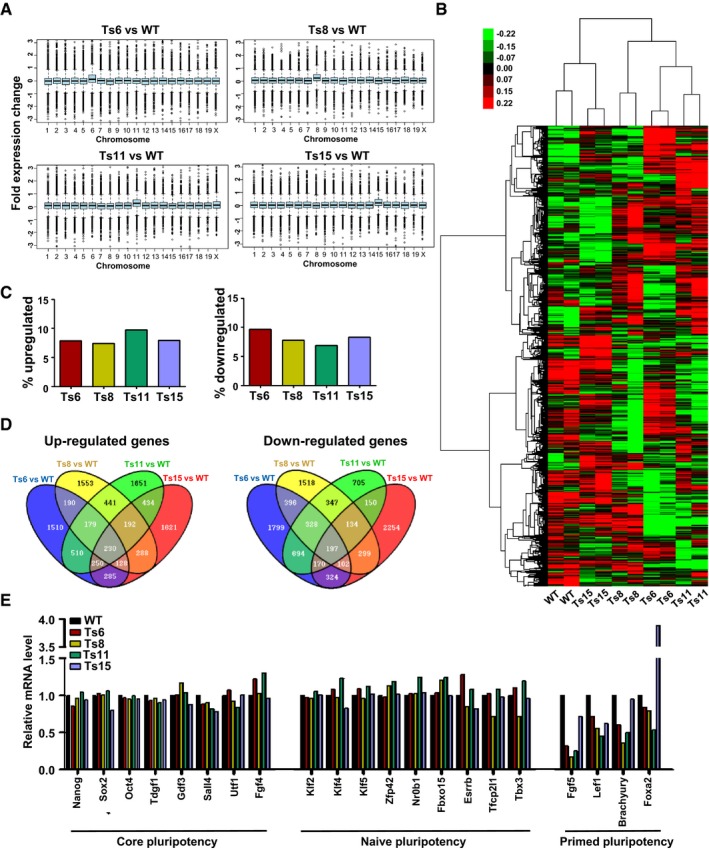
- The average gene expression changes for each chromosome in the trisomic ES cell lines. Average gene expression values were binned by chromosome. P < 1 × 10−49, Student's t‐test.
- Hierarchically clustered gene expression data. Clustering was performed to the coding genes except for those mapped on the aneuploid chromosomes and Y chromosome. The columns represent cell lines and the rows represent genes.
- The percentage of up‐ or downregulated genes in each trisomic cell line.
- The Venn diagrams depict the overlapping genes that were upregulated or downregulated among different trisomic ES cell lines.
- The relative mRNA level of genes related to core, naïve, and primed pluripotency from the graph represents a summary of the microarray data.
Figure 6. Gene expression of trisomic and wild‐type ES cells.
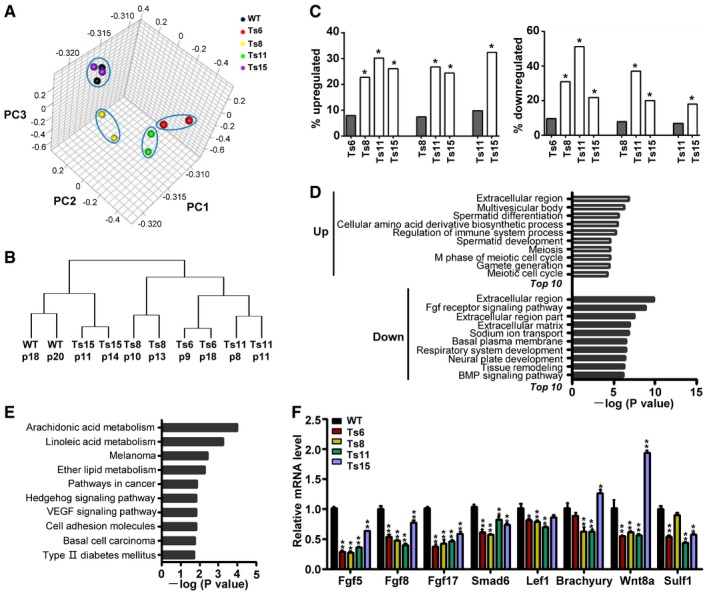
- Principal component analysis (PCA) of wild‐type, Ts6, Ts8, Ts11, and Ts15 ES cells, based on whole gene expression signatures except for those residing on aneuploid chromosomes and the Y chromosome.
- The phylogenetic tree of the two replicates of wild‐type, Ts6, Ts8, Ts11, and Ts15 ES cells based on the unsupervised hierarchial clustering of all protein‐coding genes excluding those encoded from aneuploid chromosomes and the Y chromosome.
- Different trisomic ES cells exhibited overlapping gene expression changes. Gray bars indicate the percentage of up‐ or downregulated genes among all genes in the indicated cell line. White bars indicate the percentage of the overlapping genes that were up‐ or downregulated among all the up‐ or downregulated genes in this cell line when compared with the cell line indicated by the gray bar. *P < 0.05, hypergeometric test.
- Enriched GO terms of up‐ and downregulated genes across the trisomic ES cell lines.
- Enriched pathways of up‐ and downregulated genes across the trisomic ES cell lines identified by KEGG pathway analysis.
- RT‐qPCR analysis of gene expression in trisomic and wild‐type ES cells. Error bars, ± SD. n = 3. *P < 0.05, **P < 0.01.
Source data are available online for this figure.
The percentage of up‐ and downregulated genes in each trisomic line was similar. Seven to ten percent of genes were upregulated in the four trisomic lines, and the percentage of downregulated genes varied between 6 and 10% (Fig EV4C). We then compared the up‐ and downregulated genes among all trisomic lines and found that the different trisomic lines exhibited overlapping gene expression changes (Fig EV4D). Genes that were up‐ or downregulated in one trisomic ES cell line were likely to have a similar change in another trisomic line (Fig 6C). Further analysis showed that 981 genes were upregulated and 942 genes were downregulated in at least three out of the four trisomic cell lines (Dataset EV2). Among these, 503 upregulated genes and 510 downregulated genes have Gene Ontology (GO) annotations. The population of upregulated genes showed relatively poor enrichment of GO categories, but were enriched with genes for extracellular region, cellular amino acid derivative biosynthetic process, regulation of immune system process, spermatid development, meiosis, gamete generation, etc. (Fig 6D and Dataset EV3). Although those were involved in more general processes, many terms were related to the reproductive processes. Since the gametes are error‐prone in segregating their chromosomes during meiosis and have an innate susceptibility to aneuploidy (Jones & Lane, 2013), some genes that are associated with the reproductive processes might participate in monitoring the occurrence of aneuploidy. The GO terms significantly enriched in the downregulated genes were specific to the development‐related processes, including fibroblast growth factor receptor signaling pathway, system development, neural plate development, and BMP signaling pathway. (Fig 6D and Dataset EV3). It suggested that the differentiation potential of trisomic ES cells might be impaired. In addition, we performed KEGG pathway analysis for differentially expressed genes shared by three or four trisomic ES cell lines and identified 18 significantly enriched pathways. Pathways related to metabolism and cancer were significantly enriched (Fig 6E and Appendix Table S2). Interestingly, 6 out of 18 pathways were related to cancer, including melanoma, pathways in cancer, Hedgehog signaling pathway, VEGF signaling pathway, cell adhesion molecules (CAMs), and basal cell carcinoma. The results provided the molecular basis for aneuploidy associated with alterations in the risk of cancers, which may give an explanation to the enhanced teratoma formation abilities of trisomic ES cells.
To test whether aneuploidy disturbs the expression of pluripotency‐associated genes, we evaluated the expression of genes related to core, naïve, and primed pluripotency with microarray data. In all trisomic lines, the expression level of core and naïve pluripotency‐associated genes were comparable to that in wild‐type ES cells, whereas several genes associated with primed pluripotency (differentiation‐related) were significantly dysregulated. Of them, the expression of Fgf5 and Lef1 was downregulated in all four trisomic lines, and the expression of Brachyury and Foxa2 was downregulated in three out of four lines (Fig EV4E). Further RT‐qPCR analysis confirmed that genes such as Fgf5, Fgf8, Fgf17, Smad6, Lef1, Brachyury, Wnt8a, and Sulf1 were downregulated in at least three out of four trisomic lines (Fig 6F). These data indicated that aneuploidy does not disturb the stem cell core circuity, but rather dysregulates the expression of some differentiation‐associated genes.
Rescue of differentiation defects in aneuploid cells by extracellular factors
To further investigate how aneuploidy leads to differentiation defects in ES cells, we analyzed some cell‐autonomous effects and the impacts of extracellular microenvironment. The high‐throughput gene expression analysis had prompted us to focus on the defective fibroblast growth factor (FGF) signaling in blocking proper differentiation of aneuploid ES cells. FGF/Erk signaling is essential for ES cells to exit from pluripotency and initiate differentiation (Kunath et al, 2007; Lanner & Rossant, 2010). Inhibition of FGF/Erk signaling is required to reset the mouse ES cell genome to the ground state (Ying et al, 2008) and has been reported to induce rapid global DNA demethylation (Ficz et al, 2013). However, no significant difference in the levels of 5‐methylcytosine (5mC) could be observed between serum‐cultured aneuploid and wild‐type ES cells (Appendix Fig S5A). We further evaluated the global DNA methylation levels of aneuploid ES cells cultured under feeder (high or low density) or feeder‐free conditions using luminometric methylation assay (LUMA) and found no major differences between aneuploid and wild‐type ES cells grown under serum conditions (Appendix Fig S5B). Also the trisomic ES cells maintained the hypomethylation status of the promoters of Oct4 and Nanog (Appendix Fig S5C). Moreover, the levels of two histone marks, H3K27me3 and H4K20me1, were similar among the trisomic ES cells and their diploid controls (Appendix Fig S5D). Therefore, downregulation of FGF family members was not sufficient to reset the epigenetic state in aneuploid ES cells.
The extracellular matrix is essential in cell proliferation, migration, and differentiation (Bonnans et al, 2014), and the extracellular differentiation factors can stimulate the signaling cascade in ES cells to trigger their differentiation (Ng & Surani, 2011; Young, 2011). A study had demonstrated that Fgfr2 mutant EBs failed to form basement membrane and thus did not cavitate. However, the incomplete differentiation can be rescued by secreted extracellular factors from the normal cells in mixed cultures (Li et al, 2001). We started to explore whether the incomplete differentiation of aneuploid ES cells could be rescued by wild‐type ES cells. Single‐cell suspensions of wild‐type and individual trisomic ES cell line were mixed and cultured to form EBs (Fig EV5A). When cultured for 8–10 days, most trisomic ES cells themselves failed to cavitate and formed solid EBs but the mixed ES cells formed the cystic EBs, comparable to wild‐type ES cell‐derived EBs (Figs 7A and EV5B). Trisomic cells were found to participate in the formation of cystic EBs in mixed cell cultures (Fig EV5C). However, in most cases, the proportion of trisomic cells decreased in mixed EBs (Ts6, Ts11, and Ts15), which suggested the diploids outgrew the trisomies in the differentiated state. The rapid proliferation of trisomic cells may be restricted to the undifferentiated state. We then analyzed the expression of pluripotency marker Nanog and found it declined more rapidly in mixed EBs at day 8 of differentiation than that in the trisomic ES cell‐formed solid EBs (Fig 7B). However, the extent of rescue by wild‐type ES cells varied among different trisomic ES cell lines. This suggests that the extracellular signals from the neighboring wild‐type ES cells could facilitate the complete differentiation of aneuploid ES cells in a dose‐dependent manner.
Figure EV5. Rescue of differentiation defects in trisomic cells by wild‐type ES cells.

- Experimental scheme. The EBs were formed by mixed wild‐type and trisomic ES cells.
- Morphology of EBs grown for 10 days derived from wild‐type, trisomic, or mixed ES cell lines. Wild‐type and trisomic ES cells were mixed in the proportion of 3:7. Black arrows indicate the cystic structures. Scale bar, 200 μm.
- The relative proportion of trisomic cells in mixed EBs cultured at day 8. Wild‐type and trisomic ES cells were mixed in the proportion of 3:7 at the start of EB formation. Error bars, ± SD.
Source data are available online for this figure.
Figure 7. Rescue of differentiation defects of trisomic ES cells by extracellular factors.

- Morphology of EBs grown for 8 days derived from individual or mixed ES cell lines. Hematoxylin and eosin staining. Scale bar, 100 μm.
- Quantification of Nanog expression in trisomic, wild‐type, and mixed ES cell‐derived EBs cultured for 8 days. Error bars, ± SD. n = 3. **P < 0.01.
- The morphology of EBs at day 10 of differentiation. M15 was the normal EB formation condition and was used as a control. Black arrows indicate the cystic structures. Scale bar, 200 μm.
- Box plots of EB diameter at day 10 of EB formation under M15, CM, and M15 supplemented with Bmp4 conditions. Each box plot was plotted for more than 10 EBs. Boxes indicate the 25th to 75th percentiles and the central bars represent the median. The ends of the whiskers represent the maximum and minimum values. EBs cultured under CM or Bmp4 conditions were compared to EBs of the same cell line cultured under M15 conditions. *P < 0.05, **P < 0.01.
Source data are available online for this figure.
We then investigated the effects of the extracellular factors on trisomic EB formation by using wild‐type EB conditioned media (CM). The CM were collected, filtered, and administrated to trisomic EBs every 24 h. In the cultures with CM, large cystic structures were observed in Ts8 or Ts15 EBs at day 10 of differentiation and trisomic EBs seemed not to be compact (Fig 7C). The average EB size was larger in Ts8, Ts11, or Ts15 EBs cultured with CM compared with those in M15 media (Fig 7D), which indicated that cells in CM‐cultured EBs might proliferate and differentiate properly. As the morphology and average size of trisomic EBs cultured with CM did not reach the level of wild‐type EBs, we inferred that the extracellular factors partially rescued the differentiation of trisomic EBs. To test what kinds of extracellular factors functioned in promoting trisomic ES cell differentiation, we investigated several differentiation factors whose expression was downregulated across most trisomic ES cell lines (Fgf5, Fgf8, Tgfβ2) or that were reported to promote EB cavitation (Bmp4) (Zarei Fard et al, 2015). After exposure of trisomic cells with these factors in a time‐dependent manner, the morphology and size of EBs were detected. Among them, the short‐time exposure of Bmp4 (+Bmp4) promoted cavitation of Ts8, Ts11 and T15 EBs (Fig 7C). The average size of Ts8, Ts11 and T15 EBs in +Bmp4 was larger than in M15 medium (Fig 7D). The Ts15 EBs also formed large cystic structures after long‐time exposure to Bmp4 (++Bmp4) (Appendix Fig S6A). There was significant increase in the size of Ts11 and Ts15 EBs cultured under ++Bmp4 conditions (Appendix Fig S6B). However, the short‐ and long‐time exposure of Tgfβ2 did not show positive effects on trisomic EB growth and cavitation (Appendix Fig S6A and B). Combined exposure of Fgf5 and Fgf8 made Ts15 EBs more even and their average size was slightly increased (Appendix Fig S6A and B). Thus, for the exogenous differentiation factors that were tested, Bmp4 could partially improve the differentiation defects of Ts8, Ts11, and T15 EBs in a time‐dependent manner.
Discussion
In this study, we used genetic manipulation to purposefully isolate ES cell lines trisomic for chromosome 6, 8, 11, 12, or 15. There are several advantages of this system. First, this method can be used to isolate any trisomic ES cell lines if the cells are viable and undifferentiated. We isolated trisomic ES cells covering a wide spectrum of mouse chromosomes, including cell lines trisomic for chromosome 8 and 11, which occur frequently during prolonged cell culture (Liu et al, 1997; Ben‐David & Benvenisty, 2012). Second, the trisomic ES cells that we isolated here were “newborn” trisomies that occur spontaneously during short‐term cell culture. These cells are different from the “culture‐adapted” aneuploid ES cells that have undergone prolonged cell culture and potentially harbor abundant genetic and epigenetic variations (Liang et al, 2008; Liang & Zhang, 2013). Third, our trisomic ES cells harbored a positive/negative selection cassette (PuroΔTK), which made it easy to obtain the corrected diploid ES cells and then establish the causality between aneuploidy and differentiation defects. A similar strategy has been successfully applied to correct trisomy 21 in human Down syndrome iPS cells (Li et al, 2012). Therefore, the trisomic ES cells that we isolated here are valuable resources for studying the roles of aneuploidy in cancer.
Previous studies have shown that aneuploidy impaired the proliferation of budding yeast (Torres et al, 2007), fission yeast (Pavelka et al, 2010), trisomic MEFs (Williams et al, 2008), primary foreskin fibroblasts of DS individuals (Segal & McCoy, 1974), and human trisomic and tetrasomic cell lines (Stingele et al, 2012). However, our results revealed that most of the trisomic ES cells exhibited a similar or increased proliferation rate compared with wild‐type ES cells, which showed some similarities with the culture‐adapted ES cells (Baker et al, 2007; Werbowetski‐Ogilvie et al, 2009; Ben‐David et al, 2014; Gaztelumendi & Nogues, 2014). According to our and the previous reported karyotypically abnormal PSCs, aneuploidy may not always be related to growth disadvantage. As we know, cancer cells are usually highly aneuploid and have uncontrolled proliferation. We infer that the decreased spontaneous differentiation of trisomic ES cells in culture may be responsible for maintaining the cell population, because trisomic ES cells have higher colony formation efficiencies than wild‐type cells. The transcriptional analysis of aneuploid ES cells indicates that aneuploidy can induce similar changes at the gene expression level that are independent of the specific identity of the extra chromosomes. These results are in agreement with some previous studies revealing transcriptional similarities among aneuploid somatic cells (Sheltzer et al, 2012) and human trisomic and tetrasomic cell lines (Durrbaum et al, 2014). However, contrary to previous studies, we did not observe any downregulation of genes associated with cell cycle and cell proliferation in aneuploid ES cells. Instead, genes involved in the development and differentiation processes were downregulated in these cells. Consistently, further differentiation assays showed that all trisomic ES cells had a reduced capacity to differentiate in vitro and in vivo. These results indicate that aneuploidy does not inevitably lead to an antiproliferative response, but rather, its effect depends on the cell type and the environmental conditions.
Several previous studies also reported differentiation capacity changes in aneuploid human cells, but nearly all of them are limited to trisomy 21. The impact of trisomy 21 on the early stages of embryonic hematopoiesis has been illustrated by the isolation of isogenic human trisomic 21 PSCs (MacLean et al, 2012). Blood progenitor cells generated from trisomic 21 PSCs proliferated normally but their differentiation capacities were altered with increased erythropoiesis and decreased myelopoiesis (Chou et al, 2012). In human primary fetal liver, trisomy 21 increased the frequency of hematopoietic stem cells (HSC) and megakaryocyte–erythroid progenitors and impaired B‐lymphoid development (Roy et al, 2012). The earliest stages of abnormal cardiogenesis were investigated by using the trisomy 21 sibling human ES cells (Bosman et al, 2015). More interestingly, mouse ES cells with an extra human chromosome 21 (HSA21) showed limited neuroectodermal differentiation potential in the teratoma model (Mensah et al, 2007). The above studies together with ours supported that aneuploidy impaired stem/progenitor cell differentiation.
The extra chromosome in ES cells was sufficient to induce genomewide gene expression changes (Fig EV4B), which meant that the aneuploidy effects were not limited to the specific chromosome. A recent study revealed a genomewide flattening of gene expression levels (named gene expression dysregulation domains, GEDDs) in trisomy 21 individual by comparison of the transcriptome of fetal fibroblasts from the monozygotic twins discordant for trisomy 21 (Letourneau et al, 2014). It will be interesting to use our trisomic ES cell lines to test whether GEDDs exist in other trisomies. Although we cannot deeply elucidate why and how different trisomies induced some similar gene expression changes and the similar differentiation defects at present, we obviously observed that the extracellular signals secreted from the neighbor wild‐type ES cells can partially improve the differentiation defects of trisomic ES cells. More interestingly, most trisomic ES cell‐derived EBs grew relatively well and cavitated when cultured under wild‐type CM conditions or provided with the exogenous differentiation factors, such as Bmp4. It means that the differentiation defects of aneuploid ES cells are caused, at least partially, by the insufficient production of some critical extracellular differentiation factors. Considering the elusive timing of CM collection and administration, and the accurate dose of exogenous differentiation factors, the effects of rescue might be optimized in the future. Moreover, some extracellular factors were upregulated in trisomic ES cells, which might also function in maintaining the proper differentiation. In addition, cell–cell contact also plays critical role in stem cell differentiation (Tang et al, 2010; Pieters & van Roy, 2014); it should be taken into account when rescuing trisomic ES cell differentiation by CM and additional factors. The function of microenvironment in cell differentiation has been paid attention for a long time. Studies on members of fibroblast growth factors, transforming growth factors, and other cell‐secreted factors revealed the importance of extracellular matrix on the normal cell differentiation and the early development (Li et al, 2001; Singla & Sun, 2005; Przybyla & Voldman, 2012; Taylor‐Weiner et al, 2013; Ochiai et al, 2015). Based on these observations, we propose the following model to depict the mechanisms of aneuploidy‐induced differentiation defects (Fig 8): Aneuploidy disturbs the genomewide gene expression, and it further dysregulates the secretion of extracellular factors. The dysregulation of the extracellular factors impedes the proper differentiation of ES cells. However, the differentiation defects can be rescued by extracellular factors secreted by wild‐type ES cells. Future studies should be designed to test which components of the extracellular factors are important for the proper differentiation of aneuploid ES cells.
Figure 8. Working model.
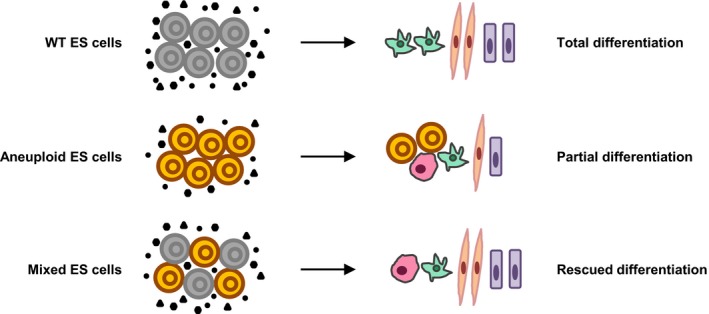
Proposed mechanism of aneuploidy impeding the differentiation of ES cells. The black circles, triangles, and hexagons represent the extracellular factors.
ES cells share proliferation and metabolism characteristics, as well as gene expression signatures, with cancer cells. ES cells are more likely to become malignant once the balance of self‐renewal and differentiation is disturbed, which is usually caused by genetic and epigenetic variation (Ben‐David & Benvenisty, 2011). Consistently, the culture‐adapted PSCs with chromosome aberrations showed a reduced capacity for differentiation and reflected tumorigenic events (Baker et al, 2007; Werbowetski‐Ogilvie et al, 2009; Ben‐David et al, 2014). The teratoma assay of trisomic ES cells revealed that teratomas from trisomic ES cells differ from diploid teratomas in that large parts of undifferentiated regions (reflected by Oct4‐positive cells) were observed. Moreover, undifferentiated cells could be recovered from trisomic ESC‐derived teratomas, suggesting these tumors have the teratocarcinoma‐like features. This finding also resembled the one obtained from culture‐adapted PSCs, although the latter reflected the effects of a specific chromosome. Our data suggest that aneuploidy can promote tumorigenesis in a chromosome‐independent manner. Interestingly, the failure of X‐inactivation of female mouse ES cells and human iPSCs correlated with the poorer differentiation potential and the upregulation of cancer‐related genes (Anguera et al, 2012; Schulz et al, 2014), which supports our findings.
Aneuploidy can be regarded as large‐scale gene copy number variations (CNV) (Iafrate et al, 2004; Sebat et al, 2004; Tang & Amon, 2013). The relationship between aneuploidy and cancer is a hotly debated issue (Gordon et al, 2012). A mouse model of the mitotic checkpoint protein BubR1 overexpression protected against aneuploidization and attenuated spontaneous, carcinogen, and genetically induced tumorigenesis (Baker et al, 2013). This finding evidently supports the hypothesis of a causal relationship between aneuploidy and tumorigenesis. Moreover, based on computational mining, large cancer genome databases, or new single‐cell analysis of circulating tumor cells (CTCs), several studies indicate that CNVs, including aneuploidy, are possibly driving genetic events instead of passenger ones for tumorigenesis and metastasis (Davoli et al, 2013; Ni et al, 2013). Here, we systemically analyzed the traits of different aneuploid ES cells and demonstrated that aneuploidy could impede the differentiation of ES cells and promote teratoma formation. It is tempting to speculate that in multiform cancer stem cells (CSCs), aneuploidy could limit their differentiation and enhance their self‐renewal and proliferation. This process will shed light on understanding the roles of aneuploidy in cancer initiation and progression and provide support for cancer differentiation therapy (Sell et al, 2016). It also prompts us to revisit Boveri's theory one century later (Boveri, 1914).
Materials and Methods
Trisomic ES cell derivation and trisomy correction
The method to isolate trisomic ES cell lines has been described previously (Huang et al, 2011). Firstly, we co‐transfected 10 μg CAG‐PBase and 200 ng PB transposon into the wild‐type male ES cell (AB1 or JM8) genome randomly. Blasticidin S (8 μg/ml, Invitrogen) selection was initiated 36 h later and continued for 6 days to allow the individual colony growth. The clones with transposon single insertions in non‐coding genomic region of chromosome 6, 8, 11, 12, or 15 were then expanded to allow trisomy occurrence. Subsequently, 20 μg CAGGS‐Cre plasmid was electroporated into 1.0 × 107 ES cells. The selection of puromycin (3 μg/ml, Sigma) and blasticidin S (8 μg/ml) was performed 36 h later and continued for 7 days until individual ES cell clones were visible. For trisomy correction, trisomic ES cells (p10–p20) were plated at a low density and selected by FIAU. FIAU was used to select for ΔTK inactive cells. Trisomic ES cells were resistant to blasticidin and puromycin and sensitive to FIAU. During consecutive cell culture, a very small number of cells might undergo spontaneous chromosome loss. Once the chromosome with active puroΔTK was lost, the cells would be resistant to FIAU and sensitive to puromycin. The positive colonies were picked 7 days after selection.
Karyotype analysis
ES cells were cultured on six‐well plates until 60% confluent. Exponentially growing cells were treated with colcemid (Sigma, D1925) at 0.2 μg/ml for 2–3 h at 37°C. After trypsinization, the cells were collected via centrifugation. Collected cells were incubated in 0.075 M KCl hypotonic solution for 15 min at 37°C. Hypotonic solution‐treated ES cells were fixed in 10 ml fresh ice‐cold 3:1 methanol/acetic acid at room temperature for 20 min three times and resuspended in 200 μl fix solution. Fixed cell suspensions were dropped onto precleaned slides and were dispersed via vapor for 5 s. The slides were allowed to age at room temperature overnight. Dried slides were digested by 0.005% trypsin and stained with Giemsa solution (10% v/v Giemsa to pH 7.0 phosphate buffer) for 5 min and washed in ddH2O. More than 30 metaphase spreads were analyzed for each cell line. Metaphase spreads were imaged by microscope on a Zeiss Imager M2 using SmartCapture 3 software (Digital Scientific UK) and analyzed using SmartType software (Digital Scientific UK).
Alkaline phosphatase staining
At day 5 after plating, the cells were fixed with 4% paraformaldehyde for 2 min, washed in a rinse buffer, and stained with Alkaline Phosphatase Kit 86R (Sigma, 86R‐1KT) according to the manufacturer's instructions. The positive colonies were counted using an inverted microscope (Olympus X41). For AP staining after LIF withdrawal, “undifferentiated” means AP‐positive cells only, “partially differentiated” indicates the mixture of AP‐positive and AP‐negative cells, and “totally differentiated” represents AP‐negative cells only. This criteria have been used previously (Caillier et al, 2010; Yamamizu et al, 2012).
Array comparative genomic hybridization
The genomic DNA of the trisomic and wild‐type ES cells were extracted using the Wizard Genomic DNA Purification Kit (Promega, A1120) and sent to the CapitalBio Corporation (Changping District, Beijing) for CGH analysis using NimbleGen 3 × 720 K mouse whole‐genome tiling arrays with an average probe spacing of 3.5 kb. The wild‐type AB1 or JM8 ES cell genomic DNA was used as a reference. All aCGH data are available at GEO accession GSE55500.
Global gene expression analysis
Two biological replicates were used for each cell line. The samples were: WT (P18 and P20), Ts6 (P9 and P18), Ts8 (P10 and P13), Ts11 (P8 and P11), and Ts15 (P11 and P14). Total RNA was extracted from diploid and aneuploid ES cells by Trizol (Invitrogen). It was purified by NucleoSpin RNA clean‐up kit (Macherey‐Nagel) according to the manufacturer's instructions. RNA was then transcribed and labeled with a cRNA amplification kit (CapitalBio Corporation) and hybridized to Agilent SurePrint G3 Mouse Gene Expression 8 × 60 K microarrays (Agilent). Arrays were then scanned on the Agilent G2565CA Microarray Scanner. The images were processed using Agilent Feature Extraction software and raw expression data were normalized with quantile and 75th percentile shift methods by the GeneSpring GX Software (Agilent). Probe sets that map to genes on aneuploid chromosomes and chromosome Y were excluded when identifying the values of 75th percentile. The genes expressed at significantly different levels between the trisomies and wild‐type were identified using a ± 1.5‐FC cutoff.
The box plots summarized the average fold change data by chromosomes that were created in R package. The average fold change data of each chromosome was calculated by subtracting the average wild‐type value from the trisomy value. Expression of the mRNA data sets in the two replicates of each cell line was analyzed using a principal component analysis (PCA). Hierarchical clustering was performed using Cluster 3.0 and the data was displayed using TreeView software. The detected protein‐coding genes across trisomies and wild‐type ES cells were input for PCA or clustering. The probes that mapped to aneuploid chromosomes and Y chromosome and unmapped to any chromosomes were excluded. To test whether there are overlapping up‐ and downregulated genes among different trisomic ES cell lines, we performed 12 out of 12 pairwise comparisons across the trisomies and used the hypergeometric test at a P < 0.05 significance threshold. To determine whether the same genes were affected in three or four trisomic cell lines, we applied a permutation test, in which the expression values were randomized within each compared trisomy. We identified the same up‐ and downregulated genes in different trisomies among 10,000 random permutations. The genes on aneuploid chromosomes were excluded in each trisomic ES cell line to avoid a dosage effect. The GO term analysis was performed using agriGO (a web‐based database, http://bioinfo.cau.edu.cn/agriGO/index.php) with a Hochberg (FDR)‐corrected P‐value of 0.05. The minimum number of mapping entries is 5. The KEGG pathways were used to perform pathway annotation. The P‐values were calculated using hypergeometric distribution.
EB formation, neuronal and cardiac differentiation
Embryoid bodies (EBs) were formed from diploid and trisomic ES cells as described previously (Jackson et al, 2010). Briefly, cultured ES cells were dissociated with trypsin on the day of passage and sedimentated for 30 min at 37°C. A total of 1.5 × 106 cells were transferred to low attachment 90‐mm‐diameter bacteriological grade Petri dishes in EB differentiating medium (M15) containing high‐glucose DMEM (GIBCO), 15% fetal bovine serum (FBS), 2 mM GlutaMax, 1% non‐essential amino acids (NEAA), and 100 μM β‐mercaptoethanol. The cultures were replaced with fresh differentiation medium every other day. The EBs were cultured for 10 more days. Usually, EBs formed by this method had irregular sizes and shapes. To make a direct comparison between different EBs, we also cultured EBs using the hanging drop method under M15 conditions without LIF for 10 days. The trypsinized ES cells were plated as 20 μl drops (3 × 104 cells/ml) onto the lids of 150‐mm dishes and cultured for 2 days. EBs were then collected and cultured in suspension. EB diameters were measured with Image‐Pro Express software. Diameters of all EBs in the visual filed were measured and averaged. If the EBs were not round, the diameter along the longest dimension was measured.
For neuronal differentiation, the EBs cultured in differentiation medium for 4 days were supplemented with 5 μM all‐trans retinoic acid (ATRA). At day 8, the cultured EBs were transferred to poly‐L‐lysine/laminin‐coated coverslips on 24‐well plates in DMEM/F12 with N2 supplements. The cells were maintained in this medium for 2 days; the medium was then changed to Neurobasal medium supplemented with B27. Seven to fourteen days after plating, the cells were fixed for immunofluorescence staining.
Cardiac differentiation of the ES cells was performed as previously described (Fuegemann et al, 2010). Briefly, EBs were formed in hanging drops on the lids of 150‐mm Petri dishes for 2 days. The aggregated EBs were flushed and cultured in bacteriological low attachment dishes for 5 days, and the medium was changed every other day. At day 7, the EBs were transferred to 0.1% gelatin‐coated 24‐well plates. One day later, spontaneous beating areas could be detected.
Mixed culture of wild‐type and trisomic EBs
For the mixed culture of EBs, wild‐type and trisomic ES cells were mixed at the start of differentiation (typically at 3:7) and were cultured by hanging drop method. EBs at day 8 of differentiation were collected by centrifuge and fixed in ethanol and acetic acid solution (ethanol: acetic acid = 95:5) at room temperature overnight. After dehydration in ethanol, EBs were embedded in paraffin and sectioned in 3‐μm intervals. The samples were stained with hematoxylin and eosin and imaged using a standard light microscope (Zeiss, AIXO) at 100× and 200× magnification. Genomic DNA of wild‐type, trisomic, and mixed EBs was extracted at day 8 of EB formation. A pair of primers were designed to match the sequence of β‐geo cassette (from DHSV, two copies for each cell) in trisomic cells. A real‐time PCR was performed in triplicate. The primer sets used are listed in Appendix Table S3. The amount of ACTB expression was used to normalize all values.
EBs treated with CM or exogenous differentiation factors
For the conditioned medium (CM) experiment, wild‐type EBs were prepared 1 day earlier by using hanging drop method. Wild‐type EB‐derived CM was collected every 24 h during EB formation and filtered with a 0.22‐μm syringe membrane filter (Corning, 431229). The filtered CM was added directly to the growing trisomic and wild‐type EBs. The medium was replaced every 24 h. The amount of EBs for producing CM was equal to those in each tested cell line.
To test the effects of extracellular differentiation factors, wild‐type and trisomic EBs were cultured with 100 ng/ml Bmp4 (R&D, 5020‐BP‐010) from the start of EB formation (++Bmp4) or after 48 h (+Bmp4) of EB culture. EBs were then cultured with Bmp4 for an additional 3 days. At day 6, the medium was changed to M15 (−Bmp4). EBs were cultured until day 10. The administration of TGFβ2 (5 ng/ml, R&D, 7346‐B2‐005) was similar to Bmp4, with the long‐time exposure for 5 days (++TGFβ2) and the short‐time exposure for 3 days (+TGFβ2). EBs were also treated with 100 ng/ml Fgf5 (R&D, 237‐F5‐050) and 100 ng/ml Fgf8b (R&D, 423‐F8‐025) at the start of culture for 2 days.
Teratoma formation and histological analysis
For the generation of teratomas, 6‐week‐old SCID‐Beige male mice were used. The mice were purchased from Charles River. All mouse experimental protocols were approved by the Institutional Animal Care and Use Committee at Peking Union Medical College & Chinese Academy of Medical Sciences. All animal care and experimental methods were carried out in accordance with the ARRIVE guidelines for animal experiments. Three or four mice were randomly allocated into experimental groups according to their body weight. ES cells were injected into both dorsal flanks of male SCID‐Beige mice subcutaneously. Tumors were measured with digital calipers, and the tumor volume was calculated as the length × width2/2. Four to eight weeks after injection, teratomas were dissected, fixed in 10% buffered formalin phosphate, embedded in paraffin, and sectioned in 3‐μm intervals. The samples were stained with hematoxylin and eosin and imaged using a standard light microscope (Zeiss) at 100× and 200× magnification.
Statistical analysis
The data are shown as the means ± SD. A two‐tailed Student's t‐test was used to analyze significant differences. Welch's correction was used for variance heterogeneity. The hypergeometric test was used to estimate the overlapping gene expression changes of trisomic ES cell lines. P < 0.05 was considered significant (*P < 0.05, **P < 0.01). The data analyses were performed using Prism GraphPad software.
Accession numbers
Gene expression and CGH data sets can be accessed as the GEO reference series GSE55501. This series includes the GSE55499 (gene expression in mouse embryonic stem cells trisomic for chromosome 6, 8, 11, and 15) and GSE55500 (CGH analysis of mouse embryonic stem cells trisomic for chromosome 6, 8, 11, 12, and 15) data sets.
Author contributions
YH conceived the project; YH and MZ designed the experiments; MZ and LC carried out the majority of the experiments; YJ performed the FISH experiment; GL helped to conduct the cell culture; SS and CL conducted the bioinformatics analysis; all authors discussed the results; AB provided valuable reagents and advice; YH, MZ and AB wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank YF. Liu, N. Yao, B. Chen, and X. Wang for technical assistance. We thank FT. Yang (Sanger Institute) for assistance with karyotype analysis, J. Su (EMBL‐EBI) for assistance with bioinformatic analysis, YZ. Gao for assistance with histological analysis, FC. Tang (Peking University) for valuable comments of the manuscript, and GL. Xu (Shanghai Institutes for Biological Sciences, CAS) for providing 5mC antibody. This work was supported by grants from the National Key Basic Research Program of China (2013CB967002) to Y.H., the National Key Research & Development Program of China (2016YFA0100103) to Y.H., the National Natural Science Foundation of China (NSFC) (31021091, 91231111) to Y.H., and PhD Programs Foundation of Ministry of Education of China (20121106110022) to Y.H..
The EMBO Journal (2016) 35: 2285–2300
See also: Z Storchová (November 2016)
References
- Anguera MC, Sadreyev R, Zhang Z, Szanto A, Payer B, Sheridan SD, Kwok S, Haggarty SJ, Sur M, Alvarez J, Gimelbrant A, Mitalipova M, Kirby JE, Lee JT (2012) Molecular signatures of human induced pluripotent stem cells highlight sex differences and cancer genes. Cell Stem Cell 11: 75–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DE, Harrison NJ, Maltby E, Smith K, Moore HD, Shaw PJ, Heath PR, Holden H, Andrews PW (2007) Adaptation to culture of human embryonic stem cells and oncogenesis in vivo . Nat Biotechnol 25: 207–215 [DOI] [PubMed] [Google Scholar]
- Baker DJ, Dawlaty MM, Wijshake T, Jeganathan KB, Malureanu L, van Ree JH, Crespo‐Diaz R, Reyes S, Seaburg L, Shapiro V, Behfar A, Terzic A, van de Sluis B, van Deursen JM (2013) Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat Cell Biol 15: 96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐David U, Benvenisty N (2011) The tumorigenicity of human embryonic and induced pluripotent stem cells. Nat Rev Cancer 11: 268–277 [DOI] [PubMed] [Google Scholar]
- Ben‐David U, Benvenisty N (2012) High prevalence of evolutionarily conserved and species‐specific genomic aberrations in mouse pluripotent stem cells. Stem Cells 30: 612–622 [DOI] [PubMed] [Google Scholar]
- Ben‐David U, Arad G, Weissbein U, Mandefro B, Maimon A, Golan‐Lev T, Narwani K, Clark AT, Andrews PW, Benvenisty N, Carlos Biancotti J (2014) Aneuploidy induces profound changes in gene expression, proliferation and tumorigenicity of human pluripotent stem cells. Nat Commun 5: 4825 [DOI] [PubMed] [Google Scholar]
- Ben‐Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA (2008) An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet 40: 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnans C, Chou J, Werb Z (2014) Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol 15: 786–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosman A, Letourneau A, Sartiani L, Del Lungo M, Ronzoni F, Kuziakiv R, Tohonen V, Zucchelli M, Santoni F, Guipponi M, Dumevska B, Hovatta O, Antonarakis SE, Jaconi ME (2015) Perturbations of heart development and function in cardiomyocytes from human embryonic stem cells with trisomy 21. Stem Cells 33: 1434–1446 [DOI] [PubMed] [Google Scholar]
- Boveri T (1914) Zur Frage der Entstehung maligner Tumoren. Jena: Fischer; [Google Scholar]
- Caillier M, Thenot S, Tribollet V, Birot AM, Samarut J, Mey A (2010) Role of the epigenetic regulator HP1gamma in the control of embryonic stem cell properties. PLoS ONE 5: e15507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Bradford WD, Seidel CW, Li R (2012) Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature 482: 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou ST, Byrska‐Bishop M, Tober JM, Yao Y, Vandorn D, Opalinska JB, Mills JA, Choi JK, Speck NA, Gadue P, Hardison RC, Nemiroff RL, French DL, Weiss MJ (2012) Trisomy 21‐associated defects in human primitive hematopoiesis revealed through induced pluripotent stem cells. Proc Natl Acad Sci USA 109: 17573–17578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ (2013) Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155: 948–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrbaum M, Kuznetsova AY, Passerini V, Stingele S, Stoehr G, Storchova Z (2014) Unique features of the transcriptional response to model aneuploidy in human cells. BMC Genom 15: 139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficz G, Hore TA, Santos F, Lee HJ, Dean W, Arand J, Krueger F, Oxley D, Paul YL, Walter J, Cook SJ, Andrews S, Branco MR, Reik W (2013) FGF signaling inhibition in ESCs drives rapid genome‐wide demethylation to the epigenetic ground state of pluripotency. Cell Stem Cell 13: 351–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuegemann CJ, Samraj AK, Walsh S, Fleischmann BK, Jovinge S, Breitbach M (2010) Differentiation of mouse embryonic stem cells into cardiomyocytes via the hanging‐drop and mass culture methods. Curr Protoc Stem Cell Biol 15: 1F.11.1–1F.11.13 [DOI] [PubMed] [Google Scholar]
- Gaztelumendi N, Nogues C (2014) Chromosome instability in mouse embryonic stem cells. Sci Rep 4: 5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DJ, Resio B, Pellman D (2012) Causes and consequences of aneuploidy in cancer. Nat Rev Genet 13: 189–203 [DOI] [PubMed] [Google Scholar]
- Hassold TJ, Jacobs PA (1984) Trisomy in man. Annu Rev Genet 18: 69–97 [DOI] [PubMed] [Google Scholar]
- Huang Y, Pettitt SJ, Guo G, Liu G, Li MA, Yang F, Bradley A (2011) Isolation of homozygous mutant mouse embryonic stem cells using a dual selection system. Nucleic Acids Res 40: e21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C (2004) Detection of large‐scale variation in the human genome. Nat Genet 36: 949–951 [DOI] [PubMed] [Google Scholar]
- Jackson M, Taylor AH, Jones EA, Forrester LM (2010) The culture of mouse embryonic stem cells and formation of embryoid bodies. Methods Mol Biol 633: 1–18 [DOI] [PubMed] [Google Scholar]
- Jones KT, Lane SI (2013) Molecular causes of aneuploidy in mammalian eggs. Development 140: 3719–3730 [DOI] [PubMed] [Google Scholar]
- Kimura M, Cao X, Skurnick J, Cody M, Soteropoulos P, Aviv A (2005) Proliferation dynamics in cultured skin fibroblasts from Down syndrome subjects. Free Radic Biol Med 39: 374–380 [DOI] [PubMed] [Google Scholar]
- Kunath T, Saba‐El‐Leil MK, Almousailleakh M, Wray J, Meloche S, Smith A (2007) FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self‐renewal to lineage commitment. Development 134: 2895–2902 [DOI] [PubMed] [Google Scholar]
- Lanner F, Rossant J (2010) The role of FGF/Erk signaling in pluripotent cells. Development 137: 3351–3360 [DOI] [PubMed] [Google Scholar]
- Letourneau A, Santoni FA, Bonilla X, Sailani MR, Gonzalez D, Kind J, Chevalier C, Thurman R, Sandstrom RS, Hibaoui Y, Garieri M, Popadin K, Falconnet E, Gagnebin M, Gehrig C, Vannier A, Guipponi M, Farinelli L, Robyr D, Migliavacca E et al (2014) Domains of genome‐wide gene expression dysregulation in Down's syndrome. Nature 508: 345–350 [DOI] [PubMed] [Google Scholar]
- Li X, Chen Y, Scheele S, Arman E, Haffner‐Krausz R, Ekblom P, Lonai P (2001) Fibroblast growth factor signaling and basement membrane assembly are connected during epithelial morphogenesis of the embryoid body. J Cell Biol 153: 811–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LB, Chang KH, Wang PR, Hirata RK, Papayannopoulou T, Russell DW (2012) Trisomy correction in Down syndrome induced pluripotent stem cells. Cell Stem Cell 11: 615–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Conte N, Skarnes WC, Bradley A (2008) Extensive genomic copy number variation in embryonic stem cells. Proc Natl Acad Sci USA 105: 17453–17456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G, Zhang Y (2013) Genetic and epigenetic variations in iPSCs: potential causes and implications for application. Cell Stem Cell 13: 149–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu H, Loring J, Hormuzdi S, Disteche CM, Bornstein P, Jaenisch R (1997) Trisomy eight in ES cells is a common potential problem in gene targeting and interferes with germ line transmission. Dev Dyn 209: 85–91 [DOI] [PubMed] [Google Scholar]
- MacLean GA, Menne TF, Guo G, Sanchez DJ, Park IH, Daley GQ, Orkin SH (2012) Altered hematopoiesis in trisomy 21 as revealed through in vitro differentiation of isogenic human pluripotent cells. Proc Natl Acad Sci USA 109: 17567–17572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensah A, Mulligan C, Linehan J, Ruf S, O'Doherty A, Grygalewicz B, Shipley J, Groet J, Tybulewicz V, Fisher E, Brandner S, Nizetic D (2007) An additional human chromosome 21 causes suppression of neural fate of pluripotent mouse embryonic stem cells in a teratoma model. BMC Dev Biol 7: 131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Surani MA (2011) The transcriptional and signalling networks of pluripotency. Nat Cell Biol 13: 490–496 [DOI] [PubMed] [Google Scholar]
- Ni X, Zhuo M, Su Z, Duan J, Gao Y, Wang Z, Zong C, Bai H, Chapman AR, Zhao J, Xu L, An T, Ma Q, Wang Y, Wu M, Sun Y, Wang S, Li Z, Yang X, Yong J et al (2013) Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc Natl Acad Sci USA 110: 21083–21088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochiai H, Suga H, Yamada T, Sakakibara M, Kasai T, Ozone C, Ogawa K, Goto M, Banno R, Tsunekawa S, Sugimura Y, Arima H, Oiso Y (2015) BMP4 and FGF strongly induce differentiation of mouse ES cells into oral ectoderm. Stem Cell Res 15: 290–298 [DOI] [PubMed] [Google Scholar]
- Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R (2010) Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 468: 321–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieters T, van Roy F (2014) Role of cell‐cell adhesion complexes in embryonic stem cell biology. J Cell Sci 127: 2603–2613 [DOI] [PubMed] [Google Scholar]
- Przybyla LM, Voldman J (2012) Attenuation of extrinsic signaling reveals the importance of matrix remodeling on maintenance of embryonic stem cell self‐renewal. Proc Natl Acad Sci USA 109: 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancati G, Pavelka N, Fleharty B, Noll A, Trimble R, Walton K, Perera A, Staehling‐Hampton K, Seidel CW, Li R (2008) Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell 135: 879–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Cowan G, Mead AJ, Filippi S, Bohn G, Chaidos A, Tunstall O, Chan JK, Choolani M, Bennett P, Kumar S, Atkinson D, Wyatt‐Ashmead J, Hu M, Stumpf MP, Goudevenou K, O'Connor D, Chou ST, Weiss MJ, Karadimitris A et al (2012) Perturbation of fetal liver hematopoietic stem and progenitor cell development by trisomy 21. Proc Natl Acad Sci USA 109: 17579–17584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz EG, Meisig J, Nakamura T, Okamoto I, Sieber A, Picard C, Borensztein M, Saitou M, Bluthgen N, Heard E (2014) The two active X chromosomes in female ESCs block exit from the pluripotent state by modulating the ESC signaling network. Cell Stem Cell 14: 203–216 [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A et al (2004) Large‐scale copy number polymorphism in the human genome. Science 305: 525–528 [DOI] [PubMed] [Google Scholar]
- Segal DJ, McCoy EE (1974) Studies on Down's syndrome in tissue culture. I. Growth rates and protein contents of fibroblast cultures. J Cell Physiol 83: 85–90 [DOI] [PubMed] [Google Scholar]
- Sell S, Nicolini A, Ferrari P, Biava PM (2016) Cancer: a problem of developmental biology; scientific evidence for reprogramming and differentiation therapy. Curr Drug Targets 17: 1103–1110 [DOI] [PubMed] [Google Scholar]
- Selmecki AM, Maruvka YE, Richmond PA, Guillet M, Shoresh N, Sorenson AL, De S, Kishony R, Michor F, Dowell R, Pellman D (2015) Polyploidy can drive rapid adaptation in yeast. Nature 519: 349–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Torres EM, Dunham MJ, Amon A (2012) Transcriptional consequences of aneuploidy. Proc Natl Acad Sci USA 109: 12644–12649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla DK, Sun B (2005) Transforming growth factor‐beta2 enhances differentiation of cardiac myocytes from embryonic stem cells. Biochem Biophys Res Commun 332: 135–141 [DOI] [PubMed] [Google Scholar]
- Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z (2012) Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol Syst Biol 8: 608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunshine AB, Payen C, Ong GT, Liachko I, Tan KM, Dunham MJ (2015) The fitness consequences of aneuploidy are driven by condition‐dependent gene effects. PLoS Biol 13: e1002155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J, Peng R, Ding J (2010) The regulation of stem cell differentiation by cell‐cell contact on micropatterned material surfaces. Biomaterials 31: 2470–2476 [DOI] [PubMed] [Google Scholar]
- Tang YC, Williams BR, Siegel JJ, Amon A (2011) Identification of aneuploidy‐selective antiproliferation compounds. Cell 144: 499–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang YC, Amon A (2013) Gene copy‐number alterations: a cost‐benefit analysis. Cell 152: 394–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor‐Weiner H, Schwarzbauer JE, Engler AJ (2013) Defined extracellular matrix components are necessary for definitive endoderm induction. Stem Cells 31: 2084–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A (2007) Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317: 916–924 [DOI] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW (2006) Does aneuploidy cause cancer? Curr Opin Cell Biol 18: 658–667 [DOI] [PubMed] [Google Scholar]
- Weaver BA, Cleveland DW (2008) The aneuploidy paradox in cell growth and tumorigenesis. Cancer Cell 14: 431–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werbowetski‐Ogilvie TE, Bosse M, Stewart M, Schnerch A, Ramos‐Mejia V, Rouleau A, Wynder T, Smith MJ, Dingwall S, Carter T, Williams C, Harris C, Dolling J, Wynder C, Boreham D, Bhatia M (2009) Characterization of human embryonic stem cells with features of neoplastic progression. Nat Biotechnol 27: 91–97 [DOI] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A (2008) Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322: 703–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamizu K, Fujihara M, Tachibana M, Katayama S, Takahashi A, Hara E, Imai H, Shinkai Y, Yamashita JK (2012) Protein kinase A determines timing of early differentiation through epigenetic regulation with G9a. Cell Stem Cell 10: 759–770 [DOI] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle‐Morera L, Doble B, Woodgett J, Cohen P, Smith A (2008) The ground state of embryonic stem cell self‐renewal. Nature 453: 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RA (2011) Control of the embryonic stem cell state. Cell 144: 940–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarei Fard N, Talaei‐Khozani T, Bahmanpour S, Esmaeilpour T (2015) Comparison of cell viability and embryoid body size of two embryonic stem cell lines after different exposure times to bone morphogenetic protein 4. Iran J Med Sci 40: 110–117 [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Nuebel E, Daley GQ, Koehler CM, Teitell MA (2012) Metabolic regulation in pluripotent stem cells during reprogramming and self‐renewal. Cell Stem Cell 11: 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Source Data for Figure 7