

Abstract
During development, hematopoietic stem cells (HSCs) emerge from aortic endothelial cells (ECs) through an intermediate stage called hemogenic endothelium by a process known as endothelial‐to‐hematopoietic transition (EHT). While Notch signaling, including its upstream regulator Vegf, is known to regulate this process, the precise molecular control and temporal specificity of Notch activity remain unclear. Here, we identify the zebrafish transcriptional regulator evi1 as critically required for Notch‐mediated EHT. In vivo live imaging studies indicate that evi1 suppression impairs EC progression to hematopoietic fate and therefore HSC emergence. evi1 is expressed in ECs and induces these effects cell autonomously by activating Notch via pAKT. Global or endothelial‐specific induction of notch, vegf, or pAKT can restore endothelial Notch and HSC formations in evi1 morphants. Significantly, evi1 overexpression induces Notch independently of Vegf and rescues HSC numbers in embryos treated with a Vegf inhibitor. In sum, our results unravel evi1–pAKT as a novel molecular pathway that, in conjunction with the shh–vegf axis, is essential for activation of Notch signaling in VDA endothelial cells and their subsequent conversion to HSCs.
Keywords: AKT, endothelial‐to‐hematopoietic transition, EVI1, hematopoietic stem cells, Notch, VEGF
Subject Categories: Development & Differentiation, Signal Transduction, Stem Cells
Introduction
The zebrafish (Danio rerio) model is increasingly used to study vertebrate hematopoiesis. Its rapid development, external fertilization, and embryonic transparency facilitate in vivo imaging of early embryonic processes and make it amenable for genetic and small molecule screens (Palis & Yoder, 2001; Davidson & Zon, 2004; Bertrand & Traver, 2009). Moreover, molecular pathways governing blood development appear largely conserved between fish and mammals, indicating that knowledge obtained in this model is likely transferrable to mammalian systems. As in other vertebrates, zebrafish hematopoiesis develops in sequential waves (Davidson & Zon, 2004). The first hematopoietic cells appear as the primitive wave at 12 h post‐fertilization (hpf) in the intermediate cell mass, an intra‐embryonic tissue derived from the ventral mesoderm (Detrich et al, 1995; Palis & Yoder, 2001). A second transient hematopoietic wave occurs in the caudal hematopoietic tissue (CHT), where multipotent erythromyeloid progenitors (EMPs) are generated as early as 24 hpf (Bertrand et al, 2007, 2010b). Shortly afterward, these cells are replaced by definitive hematopoietic stem/progenitor cells (HSPCs) that in fish start emerging approximately at 26 hpf in the ventral dorsal aorta (VDA)—the equivalent of the aorta–gonad–mesonephros (AGM) region in mammals—and then migrate to the CHT to expand in number (Bertrand et al, 2010a; Boisset et al, 2010). HSPCs subsequently colonize kidney marrow and thymus, the adult hematopoietic organs, to maintain the hematopoietic pool throughout the zebrafish life span (Jin et al, 2007; Chen & Zon, 2009).
The regulation of HSPC development in the VDA and AGM, respectively, is subject of intense research. On the cellular level, HSPCs arise from specialized VDA endothelial cells (ECs) referred to as hemogenic endothelium (HE), in a process known as endothelial‐to‐hematopoietic transition (EHT). Molecularly, several signaling pathways and genes have been demonstrated to regulate both artery and HSPC development (North et al, 1999; Burns et al, 2009; Chen et al, 2009). For example, the Notch–gridlock pathway controls the assembly of the first embryonic arteries, regulates arterial endothelial fate specification, and is critical for definitive hematopoietic stem cell (HSC) formation in both mammals and fish (Zhong et al, 2001; Kumano et al, 2003; Burns et al, 2005; Robert‐Moreno et al, 2005). Interestingly, Notch signaling is dispensable for the specification of primitive hematopoietic cells and EMPs (Bertrand et al, 2010b). Although arterial identity has been described as a prerequisite for HSCs, it is still controversial whether full arterial specification is required for HSC generation (de Bruijn et al, 2000). Indeed, most of the mouse and fish mutants that do not generate arteries show defective HSC development (Kumano et al, 2003; Burns, 2005; Robert‐Moreno et al, 2008; North et al, 2009). However, hematopoiesis is not necessarily affected in mutants with perturbed artery–vein boundaries, if intact Notch signaling is provided (Burns et al, 2005; You et al, 2005). Together, these data suggest that active Notch signaling is a key factor for ECs to generate HSCs, as well as needed for full arterial specification. Precise Notch activity levels, as achieved in ECs of the VDA at this developmental time point, might in fact distinguish cells capable of differentiating into blood from those that do not have this property and will remain part of the arteries (Gama‐Norton et al, 2015). Supporting this notion, in vitro cultured ECs derived from murine AGM, human embryonic stem (hES), or Macaca nemestrina‐induced pluripotent stem (iPS) cells require Notch induction (e.g., via genetic manipulation or co‐culture with ECs expressing the Notch ligands Jag1 and Dll4) for generating definitive HSPCs (Gori et al, 2015; Hadland et al, 2015). Similarly, the Notch ligand Dll4 was implicated as a hemato‐endothelial progenitor marker in hESCs and shown to regulate their hematopoietic differentiation (Ayllón et al, 2015).
The upstream regulatory mechanisms that control Notch levels in ECs are not fully understood. A major inductive role has been attributed to vascular endothelial growth factor (Vegf) and its upstream regulator sonic hedgehog (Ssh), which both have an essential role in regulating zebrafish HSC development in a Notch‐dependent manner (Lawson et al, 2002; Gering & Patient, 2005). However, in contrast to direct exposure to Notch ligands (Gori et al, 2015), standard addition of Vegf to cultured ES cells cannot by itself induce HSCs (Lengerke & Daley, 2005, 2010; Lengerke et al, 2008; Müller & Lengerke, 2009), indicating that additional factors may be involved in Notch regulation. Here, we identify the zebrafish homologue of the zinc finger transcription factor ecotropic viral integration site‐1 (Evi1, also termed Mecom) as a critical regulator of Notch pathway induction and HSC emergence in the zebrafish VDA endothelium.
Evi1 is a member of the SET/PR domain protein family and contains ten zinc finger motifs organized into two domains, each with distinct DNA‐binding specificities (Fig EV1A; Perkins et al, 1991; Delwel et al, 1993). Given its complex molecular structure, Evi1 can interact with different molecular partners, exerting inductive and/or repressive effects on several genes in a tissue‐dependent manner (Wieser, 2007). Identified several years ago as a common locus of retroviral integration in murine myeloid tumors (Mucenski et al, 1988), Evi1 has been mostly studied as an oncogene and poor risk factor in leukemia (Suzukawa et al, 1994; Ogawa et al, 1996; Konantz et al, 2013). However, Evi1 is also expressed in several embryonic and adult tissues (e.g., heart, somites, cranial ganglia, peripheral nervous system) and regulates proliferation and/or differentiation of various cell types (Hirai, 1999; Wieser, 2007). Evi1 −/− mice die early, between 10.5 (Hoyt et al, 1997; Yuasa et al, 2005) and 16.5 (Goyama et al, 2008) days post‐coitum, depending on the model, and show multiple abnormalities including paleness and reduced numbers of proliferating HSCs (Hoyt et al, 1997; Yuasa et al, 2005; Goyama et al, 2008). Here, we use the zebrafish model to investigate the mechanistic role of evi1 in developmental hematopoiesis. We find that evi1 regulates EHT and is required for the generation of HSPCs in the VDA. Molecularly, evi1 regulates this process by inducing pAKT‐Notch independently of Vegf activation.
Figure EV1. Validation of the evi1 morpholino oligonucleotides.
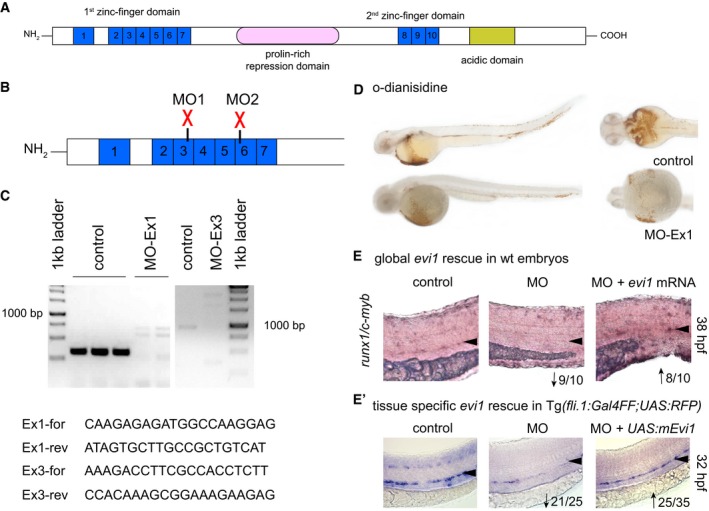
-
ASchematic representation of protein domains of the transcription factor Evi1. Blue boxes represent zinc finger motifs in the Evi1 protein. Other functional domains are indicated.
-
BThe evi1 splice donor MO, depicted by red crosses, target either the 3rd (MO1) or the 6th (MO2) zinc finger in the first zinc finger domain.
-
CRT–PCR of evi1 in embryos injected with evi1 MO1 or MO2 indicates splice modification. Corresponding primer pairs are shown. Expected wt bands for MO1 389 bp, in evi1 morphants 2,526 bp. For MO2: expected wt bands 1,100 bp, 5,695 bp for evi1 morphants.
-
DNo pooling was observed by o‐dianisidine staining in evi1 morphants at 38 hpf.
-
E, E′Co‐injection of capped evi1 mRNA (E) or UAS:mEvi1 (in Tg(fli.1:Gal4FF;UAS:RFP) embryos, E') together with the evi1 MO rescues the HSC phenotype, shown by restored runx1/c‐myb expression in the VDA, marked with black arrowheads. Numbers indicate the amount of embryos with the respective phenotype/total number of embryos analyzed in each experiment. Arrows indicate up‐ or downregulation of runx1/c‐myb in each condition. Lateral views are shown, anterior to the left, dorsal up.
Results
Zebrafish evi1 expression is detectable in the VDA and required for HSC development
Whole‐mount in situ hybridization (WISH) analyses were performed to document expression of the zebrafish homologue of the evi1 gene during early zebrafish development. Consistent with data collected in mice (Hoyt et al, 1997), evi1 expression is detectable in select cell types in the brain, the branchial arches, and the posterior pronephric duct (Fig 1A left and middle). Moreover, evi1 is expressed in the zebrafish VDA at the time point of HSC emergence and co‐localizes with the endothelial marker flk1 (Fig 1A right) and the HSC marker c‐myb (Appendix Fig S1), as shown by double WISH (Davidson & Zon, 2004). These results suggest that evi1 is present in hematopoietic cells as they emerge from the aortic endothelium and may regulate definitive hematopoiesis in a cell‐autonomous manner.
Figure 1. evi1 is expressed in emerging HSPCs and critically regulates definitive hematopoiesis.
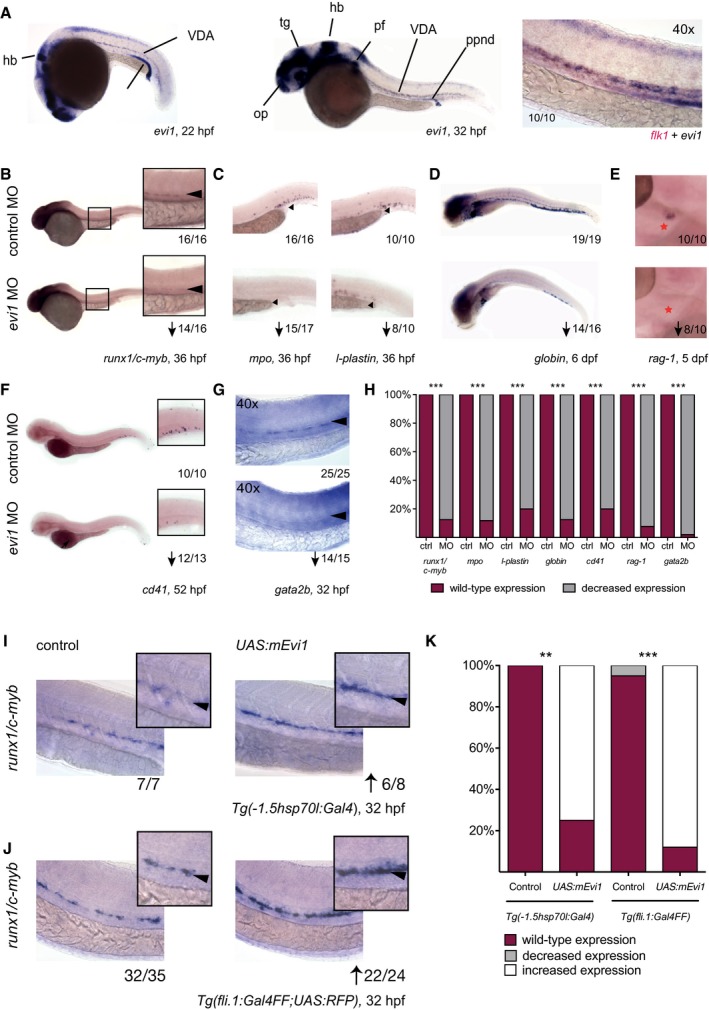
-
AWhole‐mount in situ hybridization (WISH) of evi1 at 20 (left) and 32 (middle) hpf. evi1 expression is visible in various structures of the brain, neuronal structures, the posterior pronephric duct (ppnd), and the branchial arches (ba), as well as in the VDA (ventral dorsal aorta) region. Additionally, evi1 co‐localizes with the endothelial marker flk1 (right).
-
B, CWISH of runx1/c‐myb in HSPCs (B), mpo in neutrophils (left), and l‐plastin (right) in monocytes/macrophages (C) at 36 hpf in control (upper)‐ and evi1 MO (lower)‐injected embryos.
-
D–GWISH of globin in erythrocytes of 6 dpf embryos (D), of rag‐1 in the thymus of 5 dpf embryos (E; red asterisk), of cd41 at 52 hpf (F), and gata2b at 32 hpf (G) for both control (upper)‐ and evi1 morpholino (lower)‐injected embryos.
-
HQuantitation of results is shown for each gene, displaying the percentages of embryos with normal vs. changed expression in each condition. A Fisher's exact test was applied to calculate statistical significance. ***P < 0.001.
-
IWISH of runx1/c‐myb in HSPCs of uninjected control and UAS:mEvi1 plasmid DNA‐injected Tg(‐1.5hsp70l:Gal4) embryos with heat‐shock induction performed at 14 hpf.
-
JWISH of runx1/c‐myb in HSPCs of uninjected control and UAS:mEvi1 mRNA in Tg(fli.1:Gal4FF;UAS:RFP) embryos, leading to endothelial‐specific evi1 overexpression.
-
KGraph displays quantitation of results from (I) and (J), displaying the percentages of embryos with normal vs. changed expression in each condition. A Fisher's exact test was applied to calculate statistical significance. **P < 0.01, ***P < 0.001.
To investigate the role of evi1 in zebrafish hematopoiesis, in vivo loss‐of‐function experiments were performed and embryos treated with two different antisense morpholino oligonucleotides (MO) to inhibit evi1 pre‐mRNA splicing. Both MOs result in intron preservation, thereby leading to a premature stop either in the 2nd (MO1) or the 6th (MO2) zinc finger of the first zinc finger domain (Fig EV1A and B). Transcripts in morphants were mis‐spliced, whereas uninjected and control‐injected embryos showed normal splicing (Fig EV1C). o‐Dianisidine staining was performed to exclude major defects in vascular integrity, including hemorrhaging and/or blood pooling, in MO‐injected fish (Fig EV1D). Control and evi1 MO‐injected fish were analyzed by WISH for the expression of hematopoietic genes and/or flow cytometric quantification using transgenic reporter lines (Fig 1B–H and Appendix Fig S2). Indeed, reduced numbers of runx1 +/c‐myb + HSPCs were observed in the VDA region of evi1 morphants vs. control embryos at 36 hpf (Figs 1B and H, and EV2A and C). Co‐injection with zebrafish evi1 mRNA in wild‐type fish (Fig EV1E) and, respectively, with UAS:mEvi1 plasmid DNA in Tg(fli.1:Gal4FF;UAS:RFP) fish (Fig EV1E'), was able to rescue runx1/c‐myb expression in the VDA of evi1 morphants, indicating that the observed effects on HSCs were specific to evi1 inhibition.
Figure EV2. evi1 MO2 injection leads to the same phenotype as seen by injection of evi1 MO1.
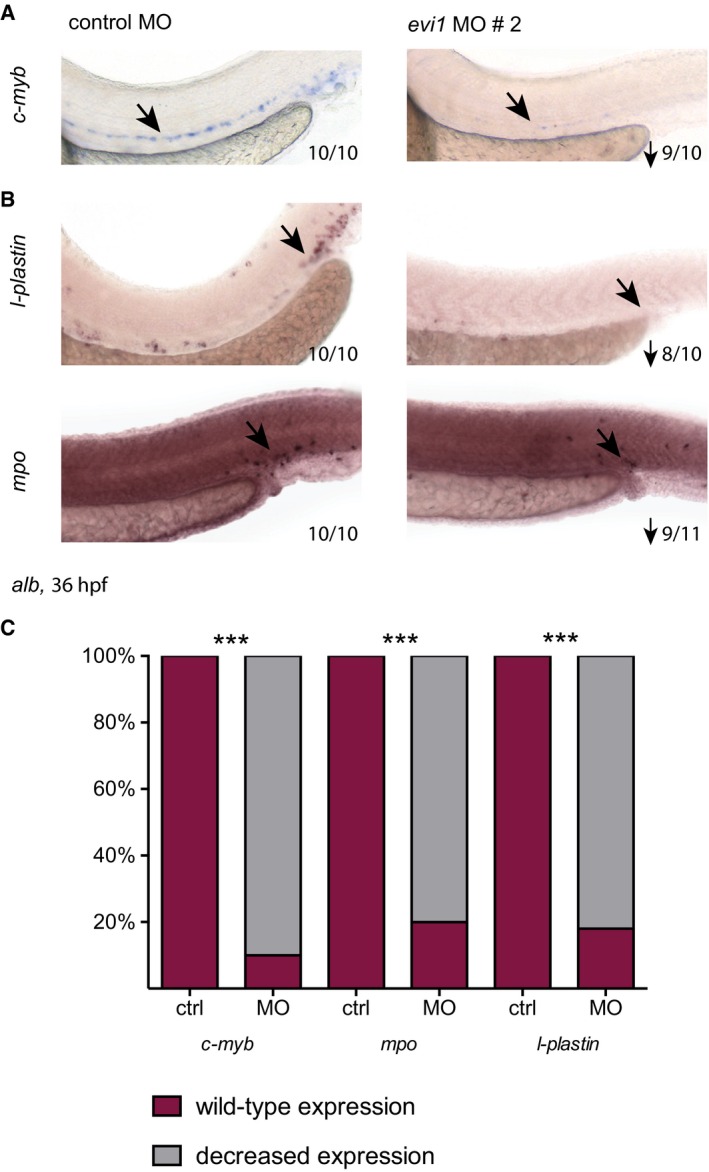
- WISH of c‐myb in control‐ and evi1 MO2‐injected albino (alb) embryos.
- WISH of l‐plastin (upper) and mpo (lower) in control‐ and evi1 MO2‐injected alb embryos.
- For each analyzed gene, quantitation of results is shown, displaying the percentages of embryos with normal vs. decreased gene expression for each condition. A Fisher's exact test was applied to calculate statistical significance (***P < 0.001).
Consistent with HSPC loss, lower numbers of mpo +, l‐plastin +, or lyz + myeloid cells (Fig 1C and Appendix Fig S2A, Tg(lyz:dsred): P = 0.0286) as well as hbae3 +, globin + erythroid cells (Fig 1D and Appendix Fig S2B, Tg(globin:GFP): P = 0.057), rag1 + T lymphocytes (Fig 1E, note that thymus epithelium is intact as shown in Fig EV3A), and cd41 + cells (Fig 1F and Appendix Fig S2C, Tg(cd41:eGFP): P = 0.0286) were documented by WISH and flow cytometry in evi morphants vs. control fish analyzed at 36 hpf to 6 dpf (Figs 1C–H and EV2B and C). Interestingly, we also observe downregulation of gata2b, a recently identified early regulator of the HE (Butko et al, 2015), in our evi1 morphants (Fig 1G). Together, these data suggest that suppression of evi1 expression severely affects VDA‐derived HSPCs, thereby strongly reducing blood cell counts of all definitive lineages (Figs 1H and EV2C). Further supporting the notion that evi1 expression regulates definitive hematopoiesis, increased numbers of HSPCs were observed at 32 hpf in the VDA of UAS:mEvi1‐injected Tg(‐1.5hsp70l:Gal4) or Tg(fli.1:Gal4FF;UAS:RFP) embryos, in which Evi1 expression was conditionally induced by heat shock or via the Gal4:UAS binary system specifically in endothelial cells, respectively (Fig 1I–K).
Figure EV3. Thymus epithelium, notochord, and vasculature are not affected upon evi1 knockdown.
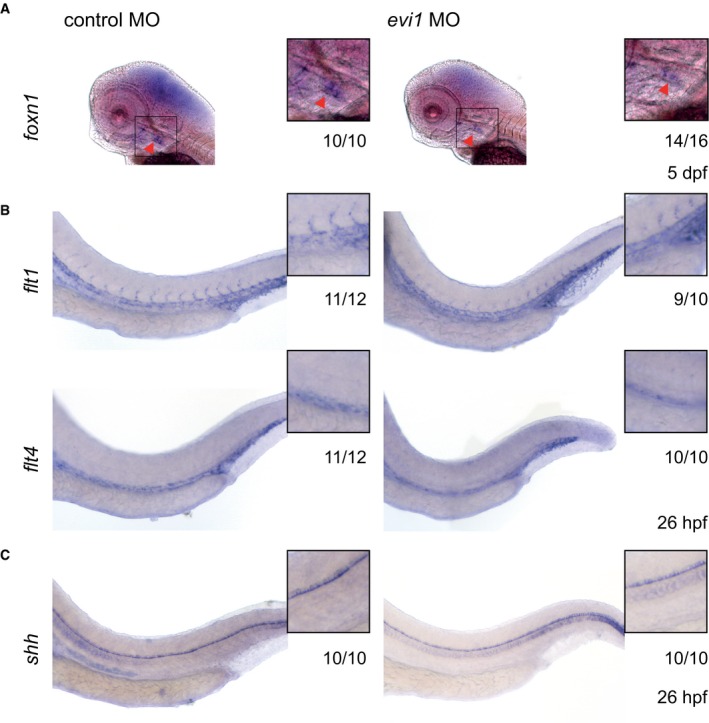
- WISH of foxn1 in the thymus epithelium of both control (left)‐ and evi1 MO‐injected (right) 5 dpf embryos.
- WISH of endothelial‐specific flt1 (upper) and flt4 (lower) in both control (left)‐ and evi1 MO‐injected (right) 26 hpf embryos.
- WISH of shh in the notochord of both control‐ and evi1 MO‐injected embryos.
evi1 regulates HSC development by inducing EHT
Next, we explored the mechanism by which evi1 expression regulates HSPCs. Genes marking the blood vessels (flt1 and flt4, Fig EV3B) and nearby tissues (e.g., shh in the notochord, see Fig EV3C) showed unaltered expression. Importantly, circulation was intact in evi1 morphants, suggesting that the HSPC defect was not due to gross alteration of vascular development or blood flow, previously shown to affect HSC formation (Adamo et al, 2009; North et al, 2009; Movies EV1, EV2, and EV3). However, evi1 morphants showed strong reduction in the expression of notch1b and dll4 (Fig 2A), while conditional induction of Evi1 leads to increased notch1b expression (Fig 2B), suggesting that evi1 might regulate HSCs by modulating Notch activity. Since evi1 co‐stained with the endothelial marker flk1 in the VDA (Fig 1B), but evi1 suppression did not overtly impair endothelial cells (Fig EV3B), we asked whether EHT, the next step in HSC development, was perturbed. Double‐transgenic Tg(kdrl:mKate‐CAAX;c‐myb:eGFP) embryos were examined by in vivo confocal time‐lapse microscopy between 28 and 42 hpf, to analyze the emergence of HSPCs (c‐myb:eGFP + (green)) from the VDA (kdrl:mKate + (red)). Consistent with the results above, microscopy did not reveal any obvious endothelial phenotype in evi1 morphants. In control‐injected embryos, c‐myb:eGFP + hematopoietic cells directly arose from kdrl:mKate + ECs along the VDA as previously described: ECs, normally flattened in appearance, transformed into double‐positive kdrl + c‐myb + cells of spherical shape before budding into the lumen of the DA (Fig 3A, control MO, upper panel) (Bertrand et al, 2010a; Kissa & Herbomel, 2010). However, after evi1 MO injection, kdrl + c‐myb + cells started transitioning to spherical shape but could not emerge from the DA (Fig 3A, evi1 MO, lower panel). In particular, endothelial kdrl + cells from the VDA of evi1 morphants began to turn yellow indicative of acquisition of the green signal due to c‐myb induction, but afterward failed to be released into the lumen of the venous system as seen in control embryos (see Movie EV1). To quantify these observations, kdrl + c‐myb + cells detectable at any time in the VDA, their proliferation capacity, as well as numbers of HSPCs directly emerging from kdrl + c‐myb + cells (as documented by live imaging) were counted in n = 10 movies of evi1 morphants and control embryos, each taken over 8 h. Interestingly, evi1 MO‐injected embryos showed a significantly lower amount of emerging HSPCs than control‐injected embryos (Fig 3B, P < 0.01), while no differences were detected in dividing or total numbers of kdrl + c‐myb + cells (“kdrl + c‐myb + cells in view”). In sum, these data indicate that evi1 expression is required for full transition to the hematopoietic fate and specifically regulates HSPCs emergence (Fig 3B) from the VDA.
Figure 2. Endothelial Notch signaling is dependent on evi1 expression levels.
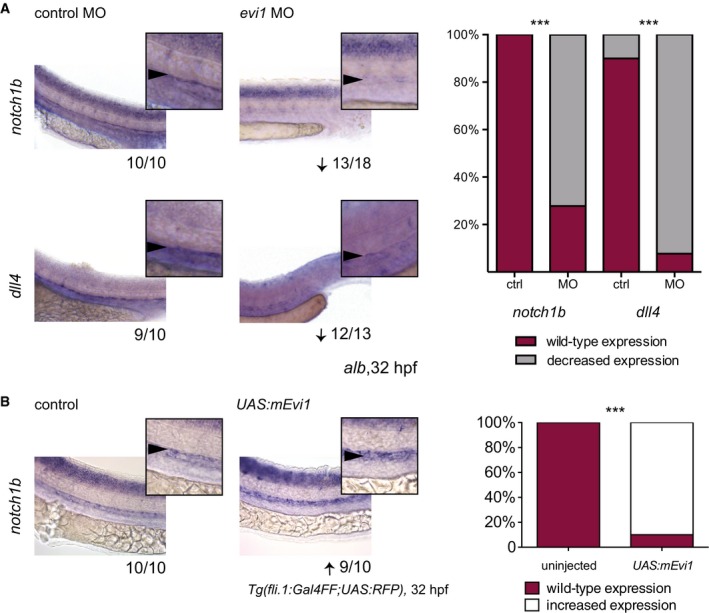
- WISH of notch1b (upper) and dll4 (lower) in both control (left)‐ and evi1 MO‐injected embryos (right).
- WISH of notch1b in uninjected (left) and UAS:mEvi1 plasmid DNA‐injected (right) Tg(fli.1:Gal4FF;UAS:RFP) embryos resulting in endothelial‐specific evi1 induction.
Figure 3. evi1 regulates HSC emergence from the VDA .
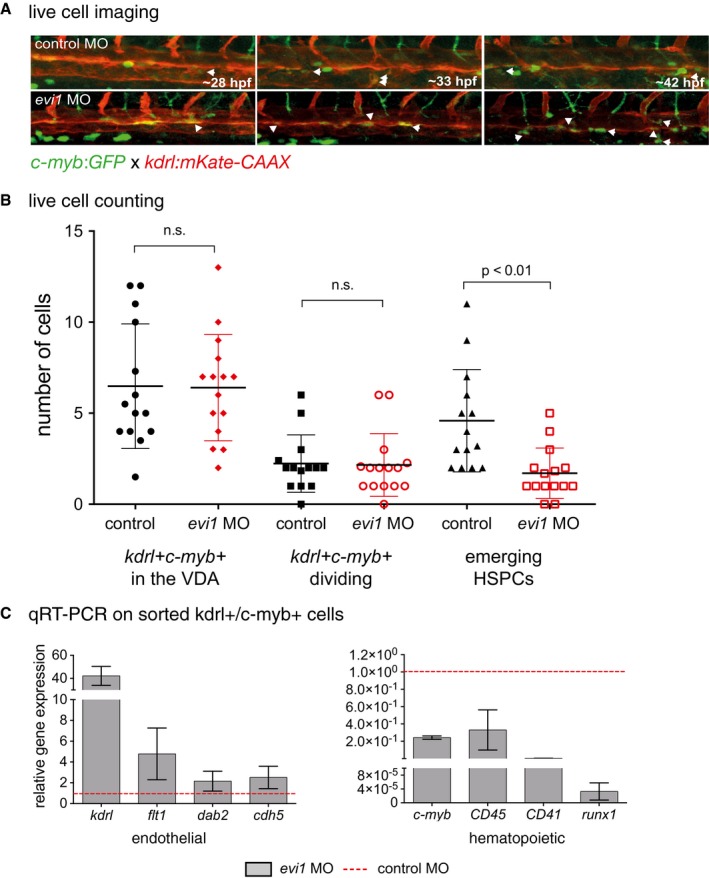
- Confocal time‐lapse live imaging was performed in Tg(c‐myb:GFP; kdrl:mKate‐CAAX) embryos from 28 to 42 hpf (Movie EV1). Shown are three representative time points in which the transformation from hemogenic endothelial cells to the hematopoietic fate is visible, indicated by the white arrowheads. For each time point, merged images are shown. White arrowheads denote double‐positive cells.
- Cell counts of total numbers of kdrl + cmyb1 + cells “in view”, dividing and, respectively, emerging HSPCs. n = 10 movies from n = 3 biological replicates were analyzed. A nonparametric Mann–Whitney U‐test was used to test for statistical significance, and error bars are shown as ± s.d.
- Gene expression analysis of endothelial (kdrl, flt1, dab2, cdh5) and blood‐specific genes (c‐myb, CD45, CD41, runx1) in sorted kdrl + c‐myb + cells from 34 hpf Tg(c‐myb:GFP; kdrl:mKate‐CAAX) embryos. Cells were isolated from 15 to 25 pooled embryos per sample. Three biological experiments were performed with one representative shown. Error bars indicate s.d. of three technical replicates for each representative experiment.
To further investigate the influence of evi1 during EHT, we isolated kdrl + c‐myb + cells from evi1 MO and control Tg(kdrl:mKate; c‐myb:eGFP) embryos, respectively, by FACS (Appendix Fig S3) and performed qRT–PCR expression analyses of hematopoietic and endothelial genes. evi1 knockdown quantitatively reduced the kdrl + c‐myb + cell population (2.59% vs. 1.4% of total gated live embryonic cells, Appendix Fig S3, P < 0.05), which comprises the kdrl + c‐myb + cells in the VDA as well as most of emerging HSPCs, and may retain some fluorescent signal indicative of an endothelial origin (due to the half‐life of the introduced fluorescent protein). Interestingly, the kdrl + c‐myb + cell population of evi1 morphants also showed severe upregulation of endothelial and downregulation of hematopoietic genes (Fig 3C), indicative of an arrest in the transition from endothelial‐to‐hematopoietic cell fate. Together, these data argue that evi1 expression is required for the transition of ECs to hematopoietic fate and thereby for HSC emergence.
HSPC emergence is determined by parallel evi1 and Vegf‐mediated Notch induction
One prominent regulator of early HSC development is the Notch–Runx1 pathway (Burns et al, 2005). Although morphologically normal, endothelial cells in the VDA of evi1 morphants showed strongly reduced expression of dll4 and notch1b (Fig 2A), suggesting that evi1 might impact HSPC emergence via Notch regulation. Treatment with DAPT, a γ‐secretase inhibitor that blocks the intracellular cleavage of Notch receptors required for signal propagation, strongly inhibited HSPC formation but did not influence evi1 expression, suggesting that evi1 is upstream of Notch signaling at this stage of development in the VDA (Appendix Fig S4). However, evi1 morphants showed unchanged expression of the Notch ligand delta C (dlc) and the Notch target gene efnb2a, a well‐established arterial marker commonly associated with HE capable of HSC generation (Fig EV4A and B) suggestive of a more complex regulatory interaction.
Figure EV4. Unchanged expression of genes involved during the somitic requirement of HSC emergence.
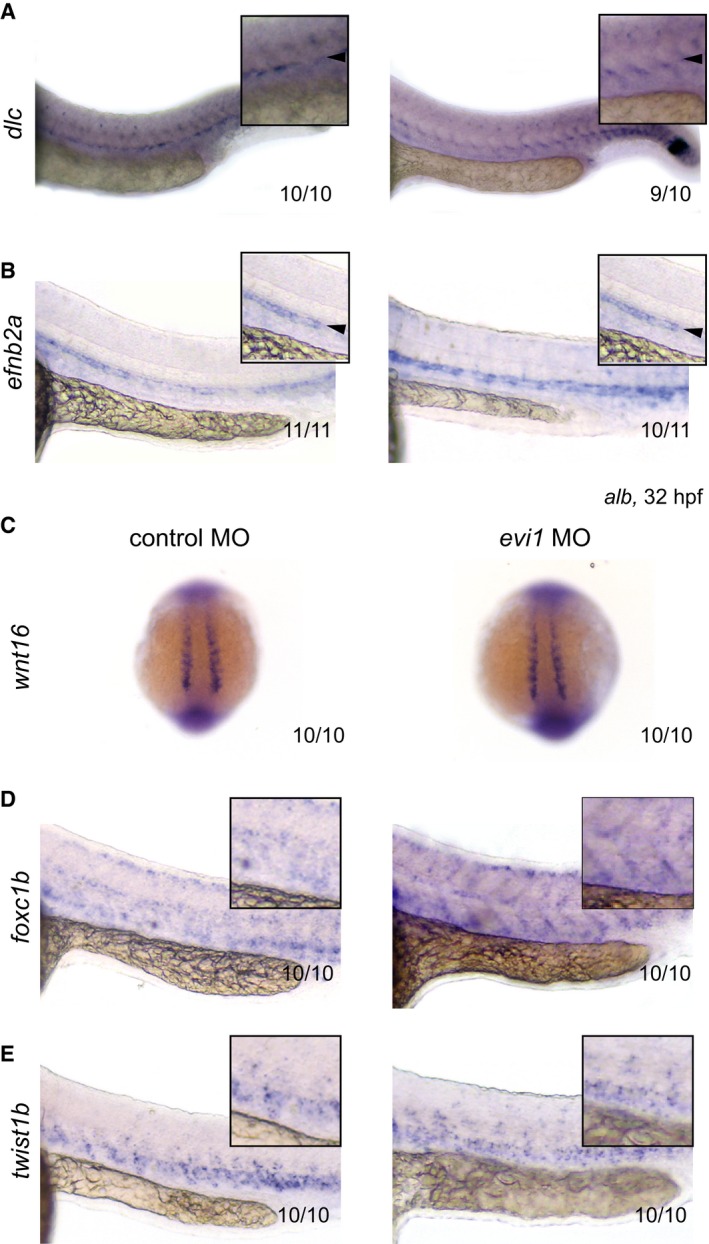
-
A–EWISH of dlc (A), efnb2a (B), wnt16 (C), foxc1b (D), and twist1b (E) in control‐injected (left) or evi1 MO‐injected alb embryos (right).
To further investigate the relationship between evi1 and the Notch pathway, we next performed rescue experiments using a heat‐shock‐inducible notch intracellular domain (NICD) transgenic zebrafish line. Indeed, defined hyper‐activation of NICD at both 14 and 20 hpf robustly rescued the loss of runx1/c‐myb expression in the VDA of evi1 morphants (Fig 4A and F; Appendix Fig S5A and C). In order to determine whether spatially restricted endothelial expression of NICD was sufficient to rescue HSC development in the VDA of evi1 suppressed fish, we used an endothelial‐specific Gal4 driver line (Tg(cdh5 BAC :gal4ff) mu101) to induce NICD (Tg(UAS:myc‐Notch1a‐intra)). Indeed, forced NICD expression restricted to the endothelium was equally able to restore runx1/c‐myb expression in the VDA of evi1 morphants (Fig 4B and F). Moreover, the activation of Notch signaling (at both 14 or 20 hpf) via global induction of its upstream activator vegfaa, using a heat‐shock inducible line (Carroll et al, 2014), also rescued HSPC development (Fig 4C and F, Appendix Figs S5B and C, and S6), while ectopic induction of the Notch target gene gata2a, via mRNA co‐injection, provided a partial rescue (Fig 4D and F). Together, these data suggest that evi1 induces EHT by regulating local Notch levels (see also Appendix Fig S7). To further explore this hypothesis, we performed live imaging analyses of evi1 morphant fish with spatial‐restricted endothelial expression of NICD (Fig 5A and B) by using an endothelial‐specific Gal4 driver line (Tg(fli.1:Gal4:RFP; c‐myb:GFF)) to induce NICD (Tg(UAS:myc‐Notch1a‐intra)). HSPCs directly emerging from fli.1 + c‐myb + cells in these fish were counted in n = 5 movies all from independent experiments taken over 8 h (Fig 5, Movies EV2 and EV3). Confirming our previous results, evi1 MO‐injected embryos showed significantly lower numbers of HSPCs emerging from fli.1 + c‐myb + cells of the endothelial niche, and enforced endothelial NICD expression could rescue these effects (Fig 5B, P < 0.05).
Figure 4. Global and tissue‐specific activation of Notch or upstream Vegf signaling is sufficient to rescue HSPCs in evi1 morphants.
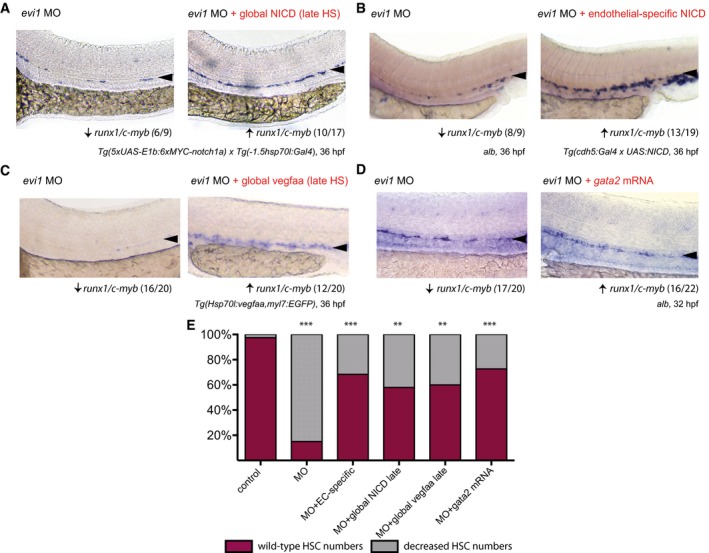
- WISH for runx1/c‐myb in 36 hpf evi1 morphant (left) and evi1 morphant transgenic Tg(5xUAS‐E1b:6xMYC‐notch1a;‐1.5hsp70l:Gal4) embryos (right) with global heat‐shock (HS) induction at 20 hpf.
- WISH for runx1/c‐myb in 36 hpf evi1 morphant (left) and evi1 morphant transgenic Tg(5xUAS‐E1b:6xMYC‐notch1a; cdh5:gal4ff) embryos with endothelial‐specific NICD induction (right).
- WISH for runx1/c‐myb in 36 hpf evi1 morphant (left) and evi1 morphant transgenic Tg(Hsp70l:vegfaa; myl7:eGFP) embryos (right) with global heat‐shock induction at 20 hpf.
- WISH for runx1/c‐myb in 32 hpf evi1 MO‐injected embryos (left) and embryos injected with both evi1 MO and capped gata2 mRNA (right).
- Quantitation of all rescue experiments. Shown are the percentages of embryos displaying normal or changed runx1/c‐myb expression. A Fisher's exact test was applied to calculate statistical significance. n.s., not significant; ***P < 0.001; **P < 0.01.
Figure 5. Endothelial NICD induction rescues HSC emergence from the VDA .
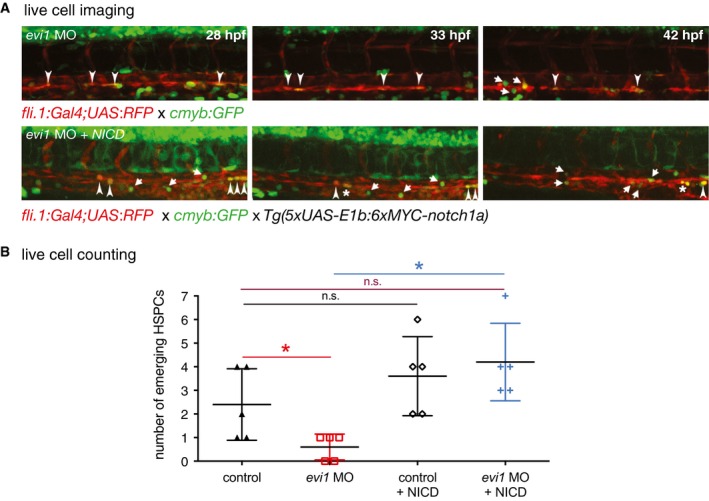
- Confocal time‐lapse live imaging was performed in Tg(c‐myb:GFP; fli.1:UAS;Gal4:RFP) evi1 morphant embryos (top, Movie EV2) or in Tg(c‐myb:GFP; fli.1:UAS;Gal4:RFP) crossed to Tg(5xUAS‐E1b:6xMYC‐notch1a) embryos prior to MO injection (bottom, Movie EV3) from 28 to 42 hpf. Shown are three representative time points, in which the transformation from hemogenic endothelial cells to the hematopoietic fate is visible. For each time point, merged images are shown. NICD induction controlled by endothelial fli.1 (lower panel) can rescue the evi1 morphant phenotype and increases the number of HSPCs cells emerging from the VDA (arrows). Arrowheads indicate double‐positive cells. Dividing cells are marked by an asterisk.
- Cell counts of emerging HSPCs in control and evi1 morphant fish with or without endothelial NICD induction. At least n = 5 movies from n = 3 biological replicates were analyzed. A nonparametric Mann–Whitney U‐test was used to test for statistical significance, and error bars are shown as ± s.d. n.s., not significant, *P < 0.05.
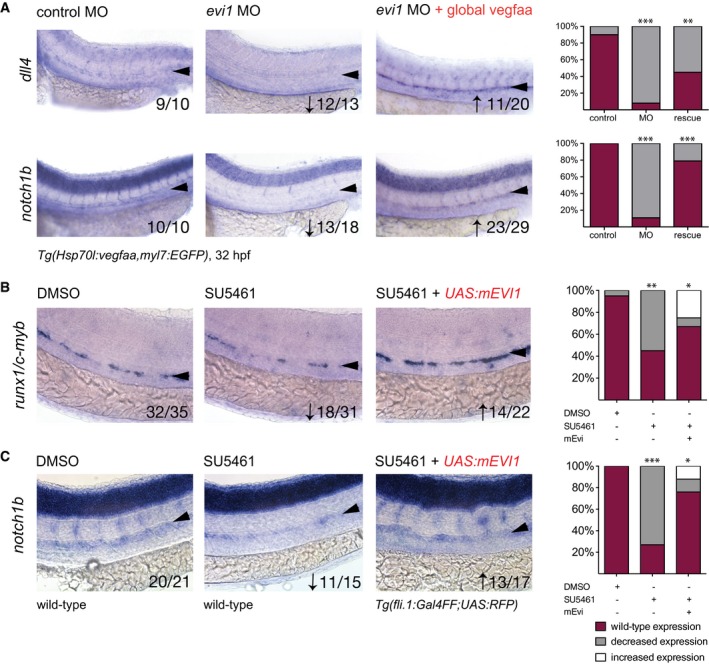
Since both ectopic NICD and its activator Vegf functionally rescued HSPC formation in evi1 morphants, we next asked whether evi1 controls Notch via Vegf. ISH for the Vegf receptor genes flt1 and flt4 as well as the known vegf upstream regulator shh showed unaltered expression upon evi1 inhibition at 26 hpf (Fig EV3B and C). Moreover, whereas Vegf is critical for vasculature development (Liang et al, 2001), evi1 morphants did not present overt vascular abnormalities, suggesting that vegf levels were not grossly influenced by evi1 expression. Of note, overexpression of vegfaa not only restored the expression of runx1/c‐myb (Fig 4C, Appendix Fig S5B and S6) but also of Notch pathway‐associated molecules in the VDA of evi1 morphants (dll4, notch1b, Fig 6A). In order to analyze whether evi1 acts in parallel or downstream of Vegf, we next treated embryos with the Vegf receptor inhibitor SU5461 from 24 to 32 hpf. As expected, SU5461 treatment reduced runx1/c‐myb and notch1b expression in the VDA in a dose‐dependent manner (Fig 6B and C, middle and Appendix Fig S8). However, overexpression of evi1 (achieved by co‐injection of UAS:mEvi1 in Tg(fli.1:Gal4FF;UAS:RFP embryos) was able to rescue runx1 + /c‐myb + cells and notch1b expression in evi1 MO‐injected fish (Fig 6B and C, left). Together, these data indicate that evi1 critically regulates EHT by activating Notch via a mechanism that is separate from and perhaps complementary to the classical shh–vegf axis.
Figure 6. evi1 regulates Notch independently of the Vegf pathway and can compensate for Vegf inhibition.
-
AWISH of dll4 (upper) and notch1b (lower) in control‐injected embryos (left), evi1 morphants (middle), and evi1 morphant transgenic Tg(Hsp70l:vegfaa; myl7:eGFP) embryos at 32 hpf after heat‐shock induction at 20 hpf (right).
-
B, CWISH of runx1/c‐myb (B) or notch1b (C) after treatment with DMSO (left), the VEGF receptor inhibitor SU5461 (middle), and, respectively, SU5461 after injection of UAS:mEvi1 (inducing endothelial evi1 expression) in Tg(fli.1:Gal4FF;UAS:RFP) embryos (right).
evi1 activates endothelial Notch via pAKT
PI3K/AKT/mTOR signaling has been implicated in Notch regulation during development and is also known to be molecularly linked to Evi1 in intestinal epithelial (Liu et al, 2006) and adult murine bone marrow cells (Yoshimi et al, 2011). Therefore, we sought to investigate the relationship between evi1, Notch, and AKT during HSC development. As shown in Fig 7A, embryos treated with the PI3Kγ inhibitor wortmannin (WM) from 14 to 36 hpf were analyzed for effects of pAKT suppression on runx1/c‐myb expression in the VDA (Li et al, 2015). As observed in evi1 morphants, vessel development remained intact, but expression of runx1/c‐myb and notch1b was strongly suppressed in the VDA region upon treatment with WM (Fig 7A and B), reflective of efficiently suppressing pAKT (Appendix Fig S9A). Importantly, enforced, heat‐shock induced NICD expression was able to restore runx1/c‐myb expression in WM‐treated fish (Fig 7A, right), as previously documented in evi1 MO‐injected embryos (Fig 4A), suggesting these factors may work in conjunction to regulate Notch.
Figure 7. evi1 regulates Notch signaling via pAKT .
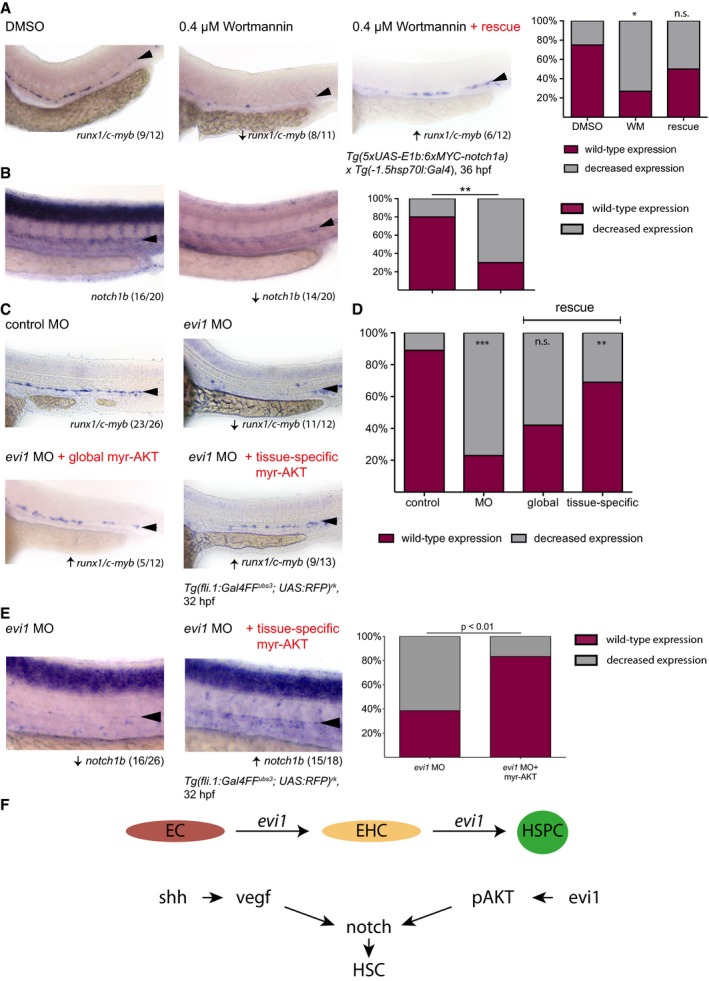
- Expression of runx1/c‐myb in the VDA after treatment with DMSO vehicle control (left), the PI3K/AKT inhibitor wortmannin (WM, middle), and after joined treatment with WM and enforced NICD expression via HS at 14 hpf in Tg(5xUAS‐E1b:6xMYC‐notch1a;‐1.5hsp70l:Gal4) embryos (right). Graph displays quantitation of results. Shown are the percentages of embryos with normal or decreased runx1/c‐myb expression for each condition.
- Expression of notch1b after treatment with DMSO vehicle control (left) or wortmannin (WM, middle). Graph displays quantitation of results. Shown are the percentages of embryos with normal or decreased notch1b expression for each condition.
- WISH for runx1/c‐myb in control‐injected embryos (upper left), evi1 morphants (upper right), or, respectively, evi1 MO with myr‐AKT‐injected embryos with HS at 14 hpf (lower right) and evi1 MO with UAS:myr‐AKT‐injected transgenic Tg(fli.1:Gal4FF ubs3 ; UAS:RFP) rk embryos.
- Quantitation of results from (C). Shown are the percentages of embryos with normal or decreased runx1/c‐myb expression for each condition.
- WISH for notch1b in evi1 MO‐injected (left) and evi1 plus UAS:myr‐AKT‐injected transgenic Tg(fli.1:Gal4FF ubs3 ; UAS:RFP) rk embryos (right). Graph displays quantitation of results. Shown are the percentages of embryos with normal or decreased notch1b expression for each condition.
- Schematic illustration of the role of Evi1 during HSPC emergence and the dual regulation of Notch by the shh–vegf and the evi1–pAKT axes.
In adult hematopoietic cells, Evi1 has been shown to positively regulate AKT phosphorylation via PTEN suppression (Yoshimi et al, 2011). Consistently, evi1 morphants displayed suppressed pAKT but mainly unaltered AKT levels (Appendix Fig S9B, left and middle). However, no induction of PTEN protein levels were observed in evi1 morphant vs. control‐injected embryos (Appendix Fig S9B, right), suggesting that alternative regulatory mechanisms may be active. To further explore pAKT as a downstream target of evi1 in VDA cells, rescue experiments were performed using a myristoylated AKT (myr‐AKT) construct to induce pAKT in evi1 morphants after heat‐shock induction (Appendix Fig S9C and D). Indeed, mosaic expression of myr‐AKT (Appendix Fig S9C and D) was sufficient to partially restore runx1/c‐myb expression in the VDA of evi1 MO‐injected embryos (Fig 7C and D). Finally, forced endothelial pAKT expression, using a UAS:myr‐AKT construct injected into Tg(fli.1:Gal4FF ubs3; UAS:RFP) rk embryos, rescued both runx1/c‐myb and notch1b expressions (Fig 7C–E). In sum, these data indicate an endothelial evi1–pAKT molecular axis that acts in parallel to VEGF to control the precise induction of Notch expression levels required for HSPC emergence (Fig 7F).
Discussion
Evi1 has been primarily studied as an oncogene in myeloid and more recently lymphoid leukemia, as well as a marker of healthy and malignant hematopoietic stem cells (Suzukawa et al, 1994; Ogawa et al, 1996; Konantz et al, 2013; Sato et al, 2014; Saito & Morishita, 2015). Evi1 and its target genes are preferentially expressed in long‐term repopulating (LT)‐HSCs, and Evi1 has been functionally associated with stemness and self‐renewal (Forsberg et al, 2010). Moreover, Evi1 was shown to induce HSC proliferation via Gata2 and apoptotic resistance as a target of Survivin (Yuasa et al, 2005; Goyama et al, 2008; Kataoka et al, 2011; Bard‐Chapeau et al, 2014; Fukuda et al, 2015). This manuscript investigates the contribution of evi1 to HSPC development during embryogenesis. In vivo loss‐of‐function studies were performed in zebrafish embryos, which, in contrast to mammalian embryos, develop ex utero, allowing direct visualization of HSC emergence. evi1 suppression was achieved via the injection of inhibiting morpholino oligonucleotides. Notably, embryos injected with multiple gRNAs died, suggesting that introduction of mutations using the CRISPR‐Cas9 system might be lethal even at mosaic levels. Consistent with murine Evi1 −/− AGM explant culture studies (Goyama et al, 2008) showing reduced HSC numbers, evi1 zebrafish morphants displayed a profound suppression of VDA HSCs and consequently of definitive hematopoietic cells of all lineages in vivo (Fig 1). To explore the mechanistic role of evi1 specifically on this cell population, we performed live imaging analysis on double‐transgenic zebrafish embryos of the Tg(kdrl:mKate‐CAAX) and Tg(c‐myb:eGFP) lines, two marker previously used to visualize the generation of HSCs from the HE in the zebrafish VDA (Bertrand et al, 2010a; Kissa & Herbomel, 2010). Surprisingly, a yet unknown and very striking defect in HSPC specification from ECs was noted in detailed live imaging and molecular analyses of sorted cell populations, which all reinforced that indeed evi1 promotes HSC development by controlling the completion of EHT and HSC emergence (Fig 3).
A major player required for HSPC formation is the Notch signaling pathway (Butko et al, 2016; Kanz et al, 2016), which is also important for the development of the VDA itself (Lawson et al, 2002; Quillien et al, 2014). As shown by multiple groups, Notch activity is mandatory to initiate specification of hematopoietic fate within the DA by controlling runx1 expression (Kumano et al, 2003; Burns et al, 2005; Gering & Patient, 2005; Richard et al, 2013). gata2b expression within the hemogenic sub‐compartment of the VDA is likewise Notch‐dependent, and its deregulation reduces runx1 (Butko et al, 2015). Upon evi1 inhibition, downregulation of notch1b and dll4 expressions was observed in VDA cells, although expression of the Notch target gene efnb2a was unchanged, indicating that cells have successfully undergone arterial specification (Figs 2A and EV4B). Moreover, production of EHT and HSPC was restored in evi1 morphants by global or EC‐specific induction of Notch, its upstream regulator Vegf, or its downstream target gata2 (Fig 4). Together, these data indicate that evi1 expression in EC is necessary for proper Notch signaling inducing EHT (Fig 5). In support of this hypothesis, the Evi1 was recently found to be the second most upregulated transcription factor in murine AGM‐derived HE as compared to EC cells (Solaimani Kartalaei et al, 2015). Furthermore, the Notch ligand Dll4 has been lately demonstrated to mark human hemato‐endothelial progenitor cells and regulate their hematopoietic fate (Ayllón et al, 2015; Gama‐Norton et al, 2015). This signal is lacking in evi1 morphants, and therefore, we propose that EHT can initiate but then arrest in an intermediate state, with HE unable to generate emerging HSCs without Evi1 activity.
Recently, two separate waves of Notch signaling requirement have been identified for HSC development: an early somitic (~14 hpf), non‐cell‐autonomous and a late endothelial (~20 hpf), cell‐autonomous wave (Clements et al, 2011; Kim et al, 2014). The fact that late induction of Notch (at 20 hpf) is sufficient to restore runx1/c‐myb cell numbers and HSC emergence in our model strongly suggests that evi1 regulates Notch levels required during this latter phase. Early induction of Notch at 14 hpf (via NICD or vegfaa) also provides a rescue (Appendix Fig S5), but in this experimental setting, induction is sustained throughout the second wave (20 hpf). In further support of the notion that evi1 regulates the late Notch signaling requirement wave, evi1 morphants showed unchanged expression of molecules shown to be involved during the first wave such as wnt16, downstream dlc, or of the sclerotome‐specific markers foxc1b and twist1b (Fig EV4) (Clements et al, 2011; Kim et al, 2014). Finally, we show that evi1 activates endothelial Notch independent of its well‐known upstream regulator Vegf, since evi1 expression restored Notch and HSPC numbers in embryos treated with the VEGF inhibitor SU5461. Consistently, Vegf‐dependent endothelial development was unaffected in evi1 morphants. We therefore propose that evi1 induces activation of an alternative pathway that cooperates with Vegf to induce Notch levels permissive for EHT and HSC emergence.
A promising intermediate between Evi1 and Notch is the PI3K/AKT/mTOR signaling pathway. Mice with disrupted PI3K and Notch signaling show phenotypic similarities during blood development (Clayton et al, 2002), and co‐culture with AKT‐activated AGM‐derived ECs was previously shown to stimulate the hematopoietic activity of E10‐E11 P‐Sp/AGM tissues (Hadland et al, 2015). Although more data exist on the role of Notch as an activator of the PI3K/AKT/mTOR pathway (Meurette et al, 2009; Cornejo et al, 2011), there is also evidence that, vice versa, PI3K/AKT can act upstream of Notch. For example, PI3K/AKT pathway activity has been shown to determine the cellular responsiveness to Notch signals in various cell types (McKenzie et al, 2006). Likewise, in T acute lymphoblastic leukemia (T‐ALL) cells, AKT inhibitors could downregulate the expression of Notch pathway molecules (Calzavara et al, 2008). In our model, treatment with the PI3Kγ inhibitor wortmannin suppresses pAKT and strongly reduces both notch1b expression (Fig 7B) and runx1/c‐myb expression in the VDA in a similar manner to that of evi1 inhibition (Fig 7A, middle). Moreover, the evi1 morphant/pAKT epistasis experiment confirmed that indeed evi1 exerts its effects on Notch activity and subsequent EHT via modulation of endothelial pAKT (Fig 7C). In adult murine bone marrow, Evi1 was shown to activate AKT/mTOR signaling by repressing PTEN either directly or via interaction with Polycomb group (PcG) proteins (Yoshimi et al, 2011). In spite showing reduction in pAKT, evi1 morphants did not show convincing changes in PTEN levels, suggesting that evi1 may modulate AKT phosphorylation by additional mechanisms (see Appendix Fig S9B). Finally, the G‐coupled protein receptor 56 (Gpr56), a direct molecular target of Evi1 in adult murine hematopoietic stem cells (Saito et al, 2013), has recently been identified by whole transcriptome analysis as a key factor in EHT in mice (Solaimani Kartalaei et al, 2015). In fish, HSCs were absent in gpr56 morphants, and gpr56 overexpression induced ectopic HSPC formation in the axial vein (Solaimani Kartalaei et al, 2015). How gpr56 acts in the EHT and how it molecularly relates to the evi1–pAKT–notch molecular axis identified here is currently unknown and subject of ongoing studies. Recently, a related G protein‐coupled receptor, gpr183, has also been found to regulate EHT in a Notch‐dependent manner, indicating potential functional redundancies (Zhang et al, 2015).
Together, our study specifically addresses the function of Evi1 during HSC emergence from endothelial cells. Previous reports indicate that Evi1 can regulate proliferation of fetal liver and bone marrow HSCs, thereby providing alternative mechanisms by which inhibition of this gene can impair stem cell numbers. Indeed, similar mechanisms of action during development as well as in the biology of preformed HSCs have been reported for gata2, a molecular downstream target of evi1. By taking advantage of the strengths of the zebrafish model for live imaging and genetic studies, we identify a novel Evi1–pAKT–Notch molecular axis that is essential for regulation of VDA EC, enabling EHT and subsequent HSC emergence during embryogenesis (Fig 7F).
Materials and Methods
Zebrafish husbandry and genetic strains
Zebrafish were bred and maintained as described in Nüsslein‐Volhard & Dahm (2002) at 28°C. Staging was done by hours post‐fertilization (hpf) as described by Warga and Kimmel (Warga & Kimmel, 1990) and according to FELASA and Swiss federal law guidelines. The following lines were used in this study: wild‐type Tuebingen strains, alb, Tg(globin: eGFP), Tg(c‐myb:GFP) (both provided by L. I. Zon, Children's Hospital, Harvard Medical School, Boston, MA), Tg(lyz:dsred), Tg(cd41:eGFP), Tg(UAS:myc‐Notch1a‐intra) kca3 , Tg(hsp70l:gal4) 1.5kca4 , Tg(hsp:70l:vegfaa, myl7:eGFP), Tg(cdh5 BAC :gal4ff) mu101, Tg(BAC:kdrl:mKate2‐CAAX) UBS16 , and Tg(fli.1:Gal4FF ubs3 ; UAS:RFP) rk (Scheer & Campos‐Ortega, 1999; Scheer et al, 2001; Lin et al, 2005; Hall et al, 2007; Parsons et al, 2009; Bussmann et al, 2011; Herwig et al, 2011; Wiley et al, 2011; Lenard et al, 2013).
Morpholino design and validation
Two evi1 splice morpholino oligonucleotides (MO) and a standard control MO were synthesized by Gene Tools (Gene Tools LLC, OR, USA): (1: CCAAAATAGTGGTGTTCTCACCTCT, 2: TGAAGGGTCTGAGTGACTTACATAT), preventing either splicing of the first or the third exon–intron splice junction. Embryos were injected at the one‐cell stage. About 0.05% of phenol red (Sigma‐Aldrich, St. Louis, MI, USA) was added as an injection tracer. Embryos were raised to appropriate stages and fixed in 4% paraformaldehyde (PFA)/1× phosphate‐buffered saline (PBS) for gene expression analysis. For validation, control‐injected and MO‐injected embryos were collected and mRNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany). cDNA was synthesized using Transcriptor First Strand cDNA Synthesis Kit (Roche, Basel, Switzerland) according to the manufacturer's protocol. RT–PCR was performed using the following primers to verify splice modification on agarose gels: CAAGAGAGATGGCCAAGGAG, GAGCAGGTCTTTCCTGATGC, ATAGTGCTTGCCGCTGTCAT, TATGAAGGGCTTGACGGAAG.
Generation of expression constructs
For rescue experiments, evi1 and gata2 were PCR‐amplified using gene‐specific primers (see Appendix Table S1), cloned into the pCR®2‐TOPO® vector, and then subcloned into the pCS2+ expression vector. Capped RNA was synthesized using the mMessage mMachine SP6 Kit (Ambion Life Technologies, Carlsbad, CA, USA). About 25–45 picograms of RNA was injected into the yolk of one‐cell stage embryos together with the evi1 MO to rescue the MO phenotype. UAS:mEvi1 was generated by cloning a codon‐optimized version of the murine Evi1 gene (Konantz et al, 2013) into pENTRY using the pENTRYD‐TOPO cloning system (Invitrogen). pENTRYmEvi1 and 4nrUAS (Akitake et al, 2011) were cloned into a Tol2 vector (pDestTol2CG2 (Kwan et al, 2007)). The final pUAS:mEvi1 plasmid was co‐injected with tol2 mRNA into the Tg(fli.1:Gal4FF ubs3 ;UAS:RFP) rk and Tg(‐1.5hsp70l:Gal4) embryos.
pBSFI‐myr‐akt1 was a gift from Peter Vogt (Addgene plasmid # 49186) (Aoki et al, 1998). To create the hsp70:myr‐akt1 construct, gene‐specific primers (see Appendix Table S1) with a 5′ BamHI and a 3′ BstBI restriction site were designed. Full‐length myr‐akt1 without the stop codon was PCR‐amplified from the pBSFI‐myr‐akt1 plasmid as template and purified using the Wizard® SV Gel and PCR Clean‐Up System (Promega, Fitchburg, WI, USA). After BamHI and BstBI digestion, the construct was subcloned in frame in a pMini Tol2 vector hsp70:cdc123 p2a Cherry (kindly provided by Judith Konantz, CRTD Dresden, Germany), substituting the cdc123 insert. Plasmid DNA containing the myr‐Akt1 insert was injected into the zygote, and embryos were raised to the appropriate stage.
Whole‐mount in situ hybridization (WISH) of zebrafish embryos
Riboprobes for WISH were generated using the following plasmid templates, restriction enzymes for DNA linearization, and RNA enzymes for further antisense probe production: runx1: HindIII, T7; c‐myb: pBK‐CMV, EcoRI, T7 (gift from Caroline E. Burns); evi1: pSPORT1, KpnI, SP6 (gift from Rebecca Wingert); shh: HindIII, T7 (gift from Judith Konantz); rag1: PCRII, HindIII, T7; efnb2a: pBS, HindII, T7; foxn1: XbaI, T7; wnt16: pCR2.1, NotI, SP6; notch1b: pCR‐Script, BamHI, T3; notch3: pCR‐Script, XhoI, T3; dlc: pBS(SK), XbaI, T7, dll4: PCRII, NotI, SP6; twist1b: pCRII, NotI, SP6, foxc1b: pCRII, BamHI, T7 (gifts from David Traver and Julien Bertrand); flk1: EcoRI, T7; flt4: EcoRI, T7 (gifts from Markus Affolter and Elin Ellertsdottir). RT/PCR‐based approaches were used to generate riboprobes for hbae3, mpo, and l‐plastin using gene‐specific primers (Appendix Table S1). For the PCR, an antisense primer was designed, which has the T7‐promoter sequence tagged onto its 5′‐end. The resultant PCR product will then include the target sequence flanked by the T7‐promoter sequence. Riboprobes were labeled with digoxigenin or fluorescein‐labeled UTP as recommended by Roche, 100‐fold diluted in Hyb solution, and stored at −20°C. WISH was performed according to standard procedures. Stained embryos were kept in 50% glycerol/PBS at 4°C until they were mounted in 89% glycerol and photographed using a digital AxioCam HRm camera attached on a Zeiss Axioplan 2 microscope or a digital MC170 HD camera attached to a Leica DM 2000 LED.
Flow cytometric analysis
Embryos were dissociated into single cells by using Accumax dissociation reagent (Millipore, Billerica, MA, USA) at room temperature. From time to time, embryos were disrupted using a 20‐G needle. After dissociation was complete, cells were washed once in PBS and passed through a 35‐μM filter. The number of fluorescent‐labeled cells was them determined via flow cytometry on a FACSCanto II using FACSDiva™ software (BD Biosciences, Franklin Lakes, NJ, USA) and Sytox® blue for dead cell discrimination.
Isolation and time‐lapse imaging of kdrl + c‐myb + and, respectively, fli.1 + c‐myb + cells
Transgenic Tg(c‐myb:GFP) were crossed to either Tg(BAC:kdrl:mKate2‐CAAX) UBS16 or Tg(fli1:Gal4FF; UAS:RFP) fish. In order to isolate forming/emerging HSCs, embryos were screened for positive fluorescence, dissociated as described above, and sorted according to their fluorescence signal using a BD Influx™ Cell sorter. DAPI was used for live/dead cell discrimination. HSCs (kdrl:mKate + ;c‐myb:GFP +) were directly sorted into RNeasy lysis buffer (Qiagen), and RNA was isolated according to the manufacturer's protocol and amplified with QantiTect Whole Transcriptome Kit (Qiagen). Gene transcripts were detected by quantitative real‐time reverse transcription PCR (qRT–PCR) using an Applied Biosystems® Real‐Time PCR 7500 Fast System instrument and Fast Start Universal SYBR Green Master with Rox (Roche) and gene‐specific primers (Appendix Table S1). A β‐actin control for equal loading was used throughout the experiments to later normalize the real‐time PCR data. Fold change values of gene expression between cells isolated from morphants vs. control embryos represented by averages from triplicate measurements were calculated using the ∆∆CT method as described by Livak and Schmittgen (Livak & Schmittgen, 2001). For time‐lapse imaging, fish were screened for fluorescence, anaesthetized in tricaine, and embedded in 0.8% low‐melting agarose. Time‐lapse imaging was performed using double‐transgenic fish between 28 and 42 hpf on a Leica TCS SP5 confocal (Tg(BAC:kdrl:mKate2‐CAAX) UBS16 × Tg(c‐myb:GFP)) or a Visitron Systems CSU‐W1 spinning disk microscope (Tg(fli1:Gal4FF;UAS:RFP) × Tg(c‐myb:GFP)). Raw data were analyzed using Fiji software.
Rescue experiments
Tg(hsp70:gal4) were mated to Tg(UAS:notch1a‐intra) and Tg(hsp:70l:vegfaa,myl7:EGFP) to wild‐type Tuebingen stains, and (MO‐injected) embryos were then raised in E3 until 14 hpf for heat‐shock induction. Embryos were therefore collected in 5 ml of E3 and placed in a 38°C waterbath for 8 or 10 min, respectively. hsp70:myr‐Akt p2a Cherry plasmid DNA‐injected wild‐type Tuebingen fish were heat‐shocked in a 37°C water at the same age for 50 min. Subsequently, embryos were transferred to petri dishes and allowed to develop until the respective time points (26, 32, and 36 hpf) for further experiments. For tissue‐specific rescue experiments Tg(5xUAS‐E1b:6xMYC‐notch1a), fish were crossed to Tg(cdh5 BAC :gal4ff) mu101 animals prior to MO injection. For tissue‐specific AKT rescue, UAS‐AKT DNA (carrying cmlc:GFP as injection control; kind gift from M. Mione, Karlsruhe Institute of Technology) was injected together with Tol2 RNA into Tg(fli.1:Gal4FF ubs3 ; UAS:RFP) rk embryos. For tissue‐specific rescue in vivo live imaging experiments, Tg(5xUAS‐E1b:6xMYC‐notch1a) fish were crossed to Tg(fliGal4:UAS:RFP;cmyb:GFP) prior to MO injection. For rescue experiments with murine Evi1, the UAS‐construct was injected with or without evi1 MO in Tg(fli.1:Gal4FF;UAS:RFP) embryos.
Pharmacological inhibition of Notch, VEGF, and PI3K signaling
The γ‐secretase inhibitor DAPT (Sigma‐Aldrich) was dissolved in DMSO at 12.5 mM and applied at 100 μM as described before (Gering & Patient, 2005). The VEGF inhibitor SU5461 (Sigma‐Aldrich) was applied between 24 and 32 hpf at different concentrations. To attenuate PI3K/AKT signaling, 0.4 μM wortmannin (Sigma) was added to 14 hpf embryos until the appropriate stage (Li et al, 2015). For each experiment, DMSO was used as a vehicle control.
Immunoblot analysis
Total protein lysates were prepared at the indicated times and after treatments by homogenizing between 10 and 50 embryos with an equal amount of lysis buffer. Lysates were denatured in Laemmli buffer and then electrophoresed on 12% polyacrylamide gels. After electrophoresis, proteins were transferred to PVDF membranes (Amersham, Amersham, UK) in a semi‐dry blotting apparatus (Trans‐Blot Turbo, Bio‐Rad, Hercules, CA, USA) and then probed with the following antibodies: rabbit anti‐human phospho‐Akt (Ser473), anti‐human pan Akt, anti‐human PTEN, and anti β‐actin from Cell Signaling Technology (Danvers, MA, USA) and detected via ECL.
o‐Dianisidine staining
Whole embryo staining for heme expression was performed using o‐dianisidine staining according to previously described methods (Detrich et al, 1995).
Author contributions
MK and CL wrote the paper. MK, EA, JM, and AL performed experiments and analyzed the data. MK, AL, EA, TEN, and CL analyzed and interpreted the data. VE, KJC, and LK provided critical reagents and/or technical/administrative support. All authors contributed to editing of the manuscript. CL conceived and supervised the study.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Review Process File
Acknowledgements
We thank Loïc Sauteur, Judith Konantz, Christopher L. Antos, Caroline E. Burns, Leonard I. Zon, Julien Bertrand, David Traver, Rebecca Wingert, Marie Mayrhofer, Marina Mione, Markus Affolter, and Elin Ellertsdottir for critical reagents and Hui Wang, Tamara Pereboom, Sarah Hendel, the Imaging Core Facility of the Biozentrum Basel and the Department of Biomedicine (DBM), as well as the DBM Flow Cytometry Core Facility for technical support. This work was supported by grants from the Swiss National Science Foundation (SNF Project 149735), the Boehringer Ingelheim Fonds (Exploration Grant), and the Nora van Meeuwen Haefliger Stiftung to CL.
The EMBO Journal (2016) 35: 2315–2331
References
- Adamo L, Naveiras O, Wenzel PL, McKinney‐Freeman S, Mack PJ, Gracia‐Sancho J, Suchy‐Dicey A, Yoshimoto M, Lensch MW, Yoder MC, García‐Cardeña G, Daley GQ (2009) Biomechanical forces promote embryonic haematopoiesis. Nature 459: 1131–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akitake CM, Macurak M, Halpern ME, Goll MG (2011) Transgenerational analysis of transcriptional silencing in zebrafish. Dev Biol 352: 191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki M, Batista O, Bellacosa A, Tsichlis P, Vogt PK (1998) The akt kinase: molecular determinants of oncogenicity. Proc Natl Acad Sci USA 95: 14950–14955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayllón V, Bueno C, Ramos‐Mejía V, Navarro‐Montero O, Prieto C, Real PJ, Romero T, García‐León MJ, Toribio ML, Bigas A, Menendez P (2015) The Notch ligand DLL4 specifically marks human hematoendothelial progenitors and regulates their hematopoietic fate. Leukemia 29: 1741–1753 [DOI] [PubMed] [Google Scholar]
- Bard‐Chapeau EA, Szumska D, Jacob B, Chua BQ, Chatterjee GC, Zhang Y, Ward JM, Urun F, Kinameri E, Vincent SD, Ahmed S, Bhattacharya S, Osato M, Perkins AS, Moore AW, Jenkins NA, Copeland NG (2014) Mice carrying a hypomorphic Evi1 allele are embryonic viable but exhibit severe congenital heart defects. PLoS One 9: e89397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand JY, Chi NC, Santoso B, Teng S, Stainier DY, Traver D (2010a) Haematopoietic stem cells derive directly from aortic endothelium during development. Nature 464: 108–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand JY, Cisson JL, Stachura DL, Traver D (2010b) Notch signaling distinguishes 2 waves of definitive hematopoiesis in the zebrafish embryo. Blood 115: 2777–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand JY, Kim AD, Violette EP, Stachura DL, Cisson JL, Traver D (2007) Definitive hematopoiesis initiates through a committed erythromyeloid progenitor in the zebrafish embryo. Development 134: 4147–4156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand JY, Traver D (2009) Hematopoietic cell development in the zebrafish embryo. Curr Opin Hematol 16: 243–248 [DOI] [PubMed] [Google Scholar]
- Boisset JC, van Cappellen W, Andrieu‐Soler C, Galjart N, Dzierzak E, Robin C (2010) In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature 464: 116–120 [DOI] [PubMed] [Google Scholar]
- de Bruijn MF, Speck NA, Peeters MC, Dzierzak E (2000) Definitive hematopoietic stem cells first develop within the major arterial regions of the mouse embryo. EMBO J 19: 2465–2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CE (2005) Hematopoietic stem cell fate is established by the Notch‐Runx pathway. Genes Dev 19: 2331–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CE, Galloway JL, Smith AC, Keefe MD, Cashman TJ, Paik EJ, Mayhall EA, Amsterdam AH, Zon LI (2009) A genetic screen in zebrafish defines a hierarchical network of pathways required for hematopoietic stem cell emergence. Blood 113: 5776–5782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns CE, Traver D, Mayhall E, Shepard JL, Zon LI (2005) Hematopoietic stem cell fate is established by the Notch‐Runx pathway. Genes Dev 19: 2331–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussmann J, Wolfe SA, Siekmann AF (2011) Arterial‐venous network formation during brain vascularization involves hemodynamic regulation of chemokine signaling. Development 138: 1717–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butko E, Distel M, Pouget C, Weijts B, Kobayashi I, Ng K, Mosimann C, Poulain FE, McPherson A, Ni CW, Stachura DL, Del Cid N, Espín‐Palazón R, Lawson ND, Dorsky R, Clements WK, Traver D (2015) Gata2b is a restricted early regulator of hemogenic endothelium in the zebrafish embryo. Development 142: 1050–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butko E, Pouget C, Traver D (2016) Complex regulation of HSC emergence by the Notch signaling pathway. Dev Biol 409: 129–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzavara E, Chiaramonte R, Cesana D, Basile A, Sherbet GV, Comi P (2008) Reciprocal regulation of Notch and PI3K/Akt signalling in T‐ALL cells in vitro. J Cell Biochem 103: 1405–1412 [DOI] [PubMed] [Google Scholar]
- Carroll KJ, Esain V, Garnaas MK, Cortes M, Dovey MC, Nissim S, Frechette GM, Liu SY, Kwan W, Cutting CC, Harris JM, Gorelick DA, Halpern ME, Lawson ND, Goessling W, North TE (2014) Estrogen defines the dorsal‐ventral limit of VEGF regulation to specify the location of the hemogenic endothelial niche. Dev Cell 29: 437–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AT, Zon LI (2009) Zebrafish blood stem cells. J Cell Biochem 108: 35–42 [DOI] [PubMed] [Google Scholar]
- Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E, Speck NA (2009) Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 457: 887–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton E, Bardi G, Bell SE, Chantry D, Downes CP, Gray A, Humphries LA, Rawlings D, Reynolds H, Vigorito E, Turner M (2002) A crucial role for the p110delta subunit of phosphatidylinositol 3‐kinase in B cell development and activation. J Exp Med 196: 753–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements WK, Kim AD, Ong KG, Moore JC, Lawson ND, Traver D (2011) A somitic Wnt16/Notch pathway specifies haematopoietic stem cells. Nature 474: 220–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornejo MG, Mabialah V, Sykes SM, Khandan T, Lo Celso C, Lopez CK, Rivera‐Muñoz P, Rameau P, Tothova Z, Aster JC, DePinho RA, Scadden DT, Gilliland DG, Mercher T (2011) Crosstalk between NOTCH and AKT signaling during murine megakaryocyte lineage specification. Blood 118: 1264–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson AJ, Zon LI (2004) The ‘definitive’ (and ‘primitive’) guide to zebrafish hematopoiesis. Oncogene 23: 7233–7246 [DOI] [PubMed] [Google Scholar]
- Delwel R, Funabiki T, Kreider BL, Morishita K, Ihle JN (1993) Four of the seven zinc fingers of the Evi‐1 myeloid‐transforming gene are required for sequence‐specific binding to GA(C/T)AAGA(T/C)AAGATAA. Mol Cell Biol 13: 4291–4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detrich HW 3rd, Kieran MW, Chan FY, Barone LM, Yee K, Rundstadler JA, Pratt S, Ransom D, Zon LI (1995) Intraembryonic hematopoietic cell migration during vertebrate development. Proc Natl Acad Sci USA 92: 10713–10717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg EC, Passegue E, Prohaska SS, Wagers AJ, Koeva M, Stuart JM, Weissman IL (2010) Molecular signatures of quiescent, mobilized and leukemia‐initiating hematopoietic stem cells. PLoS One 5: e8785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda S, Hoggatt J, Singh P, Abe M, Speth JM, Hu P, Conway EM, Nucifora G, Yamaguchi S, Pelus LM (2015) Survivin modulates genes with divergent molecular functions and regulates proliferation of hematopoietic stem cells through Evi‐1. Leukemia 29: 433–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama‐Norton L, Ferrando E, Ruiz‐Herguido C, Liu Z, Guiu J, Islam AB, Lee SU, Yan M, Guidos CJ, López‐Bigas N, Maeda T, Espinosa L, Kopan R, Bigas A (2015) Notch signal strength controls cell fate in the haemogenic endothelium. Nat Commun 6: 8510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gering M, Patient R (2005) Hedgehog signaling is required for adult blood stem cell formation in zebrafish embryos. Dev Cell 8: 389–400 [DOI] [PubMed] [Google Scholar]
- Gori JL, Butler JM, Chan YY, Chandrasekaran D, Poulos MG, Ginsberg M, Nolan DJ, Elemento O, Wood BL, Adair JE, Rafii S, Kiem HP (2015) Vascular niche promotes hematopoietic multipotent progenitor formation from pluripotent stem cells. J Clin Invest 125: 1243–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyama S, Yamamoto G, Shimabe M, Sato T, Ichikawa M, Ogawa S, Chiba S, Kurokawa M (2008) Evi‐1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell 3: 207–220 [DOI] [PubMed] [Google Scholar]
- Hadland BK, Varnum‐Finney B, Poulos MG, Moon RT, Butler JM, Rafii S, Bernstein ID (2015) Endothelium and NOTCH specify and amplify aorta‐gonad‐mesonephros‐derived hematopoietic stem cells. J Clin Invest 125: 2032–2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall C, Flores MV, Storm T, Crosier K, Crosier P (2007) The zebrafish lysozyme C promoter drives myeloid‐specific expression in transgenic fish. BMC Dev Biol 7: 42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herwig L, Blum Y, Krudewig A, Ellertsdottir E, Lenard A, Belting HG, Affolter M (2011) Distinct cellular mechanisms of blood vessel fusion in the zebrafish embryo. Curr Biol 21: 1942–1948 [DOI] [PubMed] [Google Scholar]
- Hirai H (1999) The transcription factor Evi‐1. Int J Biochem Cell Biol 31: 1367–1371 [DOI] [PubMed] [Google Scholar]
- Hoyt PR, Bartholomew C, Davis AJ, Yutzey K, Gamer LW, Potter SS, Ihle JN, Mucenski ML (1997) The Evi1 proto‐oncogene is required at midgestation for neural, heart, and paraxial mesenchyme development. Mech Dev 65: 55–70 [DOI] [PubMed] [Google Scholar]
- Jin H, Xu J, Wen Z (2007) Migratory path of definitive hematopoietic stem/progenitor cells during zebrafish development. Blood 109: 5208–5214 [DOI] [PubMed] [Google Scholar]
- Kanz D, Konantz M, Alghisi E, North TE, Lengerke C (2016) Endothelial‐to‐hematopoietic transition: notch‐ing vessels into blood. Ann N Y Acad Sci 1370: 97–108 [DOI] [PubMed] [Google Scholar]
- Kataoka K, Sato T, Yoshimi A, Goyama S, Tsuruta T, Kobayashi H, Shimabe M, Arai S, Nakagawa M, Imai Y, Kumano K, Kumagai K, Kubota N, Kadowaki T, Kurokawa M (2011) Evi1 is essential for hematopoietic stem cell self‐renewal, and its expression marks hematopoietic cells with long‐term multilineage repopulating activity. J Exp Med 208: 2403–2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AD, Melick CH, Clements WK, Stachura DL, Distel M, Panáková D, MacRae C, Mork LA, Crump JG, Traver D (2014) Discrete Notch signaling requirements in the specification of hematopoietic stem cells. EMBO J 33: 2363–2373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissa K, Herbomel P (2010) Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature 464: 112–115 [DOI] [PubMed] [Google Scholar]
- Konantz M, Andre MC, Ebinger M, Grauer M, Wang H, Grzywna S, Rothfuss OC, Lehle S, Kustikova OS, Salih HR, Handgretinger R, Fend F, Baum C, Kanz L, Quintanilla‐Martinez L, Schulze‐Osthoff K, Essmann F, Lengerke C (2013) EVI‐1 modulates leukemogenic potential and apoptosis sensitivity in human acute lymphoblastic leukemia. Leukemia 27: 56–65 [DOI] [PubMed] [Google Scholar]
- Kumano K, Chiba S, Kunisato A, Sata M, Saito T, Nakagami‐Yamaguchi E, Yamaguchi T, Masuda S, Shimizu K, Takahashi T, Ogawa S, Hamada Y, Hirai H (2003) Notch1 but not Notch2 is essential for generating hematopoietic stem cells from endothelial cells. Immunity 18: 699–711 [DOI] [PubMed] [Google Scholar]
- Kwan KM, Fujimoto E, Grabher C, Mangum BD, Hardy ME, Campbell DS, Parant JM, Yost HJ, Kanki JP, Chien CB (2007) The Tol2kit: a multisite gateway‐based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn 236: 3088–3099 [DOI] [PubMed] [Google Scholar]
- Lawson ND, Vogel AM, Weinstein BM (2002) Sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev Cell 3: 127–136 [DOI] [PubMed] [Google Scholar]
- Lenard A, Ellertsdottir E, Herwig L, Krudewig A, Sauteur L, Belting HG, Affolter M (2013) In vivo analysis reveals a highly stereotypic morphogenetic pathway of vascular anastomosis. Dev Cell 25: 492–506 [DOI] [PubMed] [Google Scholar]
- Lengerke C, Daley GQ (2005) Patterning definitive hematopoietic stem cells from embryonic stem cells. Exp Hematol 33: 971–979 [DOI] [PubMed] [Google Scholar]
- Lengerke C, Schmitt S, Bowman TV, Jang IH, Maouche‐Chretien L, McKinney‐Freeman S, Davidson AJ, Hammerschmidt M, Rentzsch F, Green JB, Zon LI, Daley GQ (2008) BMP and Wnt specify hematopoietic fate by activation of the Cdx‐Hox pathway. Cell Stem Cell 2: 72–82 [DOI] [PubMed] [Google Scholar]
- Lengerke C, Daley GQ (2010) Autologous blood cell therapies from pluripotent stem cells. Blood Rev 24: 27–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Lahvic JL, Binder V, Pugach EK, Riley EB, Tamplin OJ, Panigrahy D, Bowman TV, Barrett FG, Heffner GC, McKinney‐Freeman S, Schlaeger TM, Daley GQ, Zeldin DC, Zon LI (2015) Epoxyeicosatrienoic acids enhance embryonic haematopoiesis and adult marrow engraftment. Nature 523: 468–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Chang JR, Chin AJ, Smith A, Kelly C, Weinberg ES, Ge R (2001) The role of vascular endothelial growth factor (VEGF) in vasculogenesis, angiogenesis, and hematopoiesis in zebrafish development. Mech Dev 108: 29–43 [DOI] [PubMed] [Google Scholar]
- Lin HF, Traver D, Zhu H, Dooley K, Paw BH, Zon LI, Handin RI (2005) Analysis of thrombocyte development in CD41‐GFP transgenic zebrafish. Blood 106: 3803–3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chen L, Ko TC, Fields AP, Thompson EA (2006) Evi1 is a survival factor which conveys resistance to both TGFbeta‐ and taxol‐mediated cell death via PI3K/AKT. Oncogene 25: 3565–3575 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- McKenzie G, Ward G, Stallwood Y, Briend E, Papadia S, Lennard A, Turner M, Champion B, Hardingham GE (2006) Cellular Notch responsiveness is defined by phosphoinositide 3‐kinase‐dependent signals. BMC Cell Biol 7: 10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurette O, Stylianou S, Rock R, Collu GM, Gilmore AP, Brennan K (2009) Notch activation induces Akt signaling via an autocrine loop to prevent apoptosis in breast epithelial cells. Cancer Res 69: 5015–5022 [DOI] [PubMed] [Google Scholar]
- Mucenski ML, Taylor BA, Ihle JN, Hartley JW, Morse HC 3rd, Jenkins NA, Copeland NG (1988) Identification of a common ecotropic viral integration site, Evi‐1, in the DNA of AKXD murine myeloid tumors. Mol Cell Biol 8: 301–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller R, Lengerke C (2009) Patient‐specific pluripotent stem cells: promises and challenges. Nat Rev Endocrinol 5: 195–203 [DOI] [PubMed] [Google Scholar]
- North T, Gu TL, Stacy T, Wang Q, Howard L, Binder M, Marin‐Padilla M, Speck NA (1999) Cbfa2 is required for the formation of intra‐aortic hematopoietic clusters. Development 126: 2563–2575 [DOI] [PubMed] [Google Scholar]
- North TE, Goessling W, Peeters M, Li P, Ceol C, Lord AM, Weber GJ, Harris J, Cutting CC, Huang P, Dzierzak E, Zon LI (2009) Hematopoietic stem cell development is dependent on blood flow. Cell 137: 736–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nüsslein‐Volhard C, Dahm R (2002) Zebrafish: a practical approach. New York: Oxford University Press; [Google Scholar]
- Ogawa S, Kurokawa M, Tanaka T, Mitani K, Inazawa J, Hangaishi A, Tanaka K, Matsuo Y, Minowada J, Tsubota T, Yazaki Y, Hirai H (1996) Structurally altered Evi‐1 protein generated in the 3q21q26 syndrome. Oncogene 13: 183–191 [PubMed] [Google Scholar]
- Palis J, Yoder MC (2001) Yolk‐sac hematopoiesis: the first blood cells of mouse and man. Exp Hematol 29: 927–936 [DOI] [PubMed] [Google Scholar]
- Parsons MJ, Pisharath H, Yusuff S, Moore JC, Siekmann AF, Lawson N, Leach SD (2009) Notch‐responsive cells initiate the secondary transition in larval zebrafish pancreas. Mech Dev 126: 898–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins AS, Fishel R, Jenkins NA, Copeland NG (1991) Evi‐1, a murine zinc finger proto‐oncogene, encodes a sequence‐specific DNA‐binding protein. Mol Cell Biol 11: 2665–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quillien A, Moore JC, Shin M, Siekmann AF, Smith T, Pan L, Moens CB, Parsons MJ, Lawson ND (2014) Distinct Notch signaling outputs pattern the developing arterial system. Development 141: 1544–1552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard C, Drevon C, Canto PY, Villain G, Bollérot K, Lempereur A, Teillet MA, Vincent C, Rosselló Castillo C, Torres M, Piwarzyk E, Speck NA, Souyri M, Jaffredo T (2013) Endothelio‐mesenchymal interaction controls runx1 expression and modulates the notch pathway to initiate aortic hematopoiesis. Dev Cell 24: 600–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert‐Moreno A, Espinosa L, de la Pompa JL, Bigas A (2005) RBPjkappa‐dependent Notch function regulates Gata2 and is essential for the formation of intra‐embryonic hematopoietic cells. Development 132: 1117–1126 [DOI] [PubMed] [Google Scholar]
- Robert‐Moreno A, Guiu J, Ruiz‐Herguido C, Lopez ME, Ingles‐Esteve J, Riera L, Tipping A, Enver T, Dzierzak E, Gridley T, Espinosa L, Bigas A (2008) Impaired embryonic haematopoiesis yet normal arterial development in the absence of the Notch ligand Jagged1. EMBO J 27: 1886–1895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito Y, Kaneda K, Suekane A, Ichihara E, Nakahata S, Yamakawa N, Nagai K, Mizuno N, Kogawa K, Miura I, Itoh H, Morishita K (2013) Maintenance of the hematopoietic stem cell pool in bone marrow niches by EVI1‐regulated GPR56. Leukemia 27: 1637–1649 [DOI] [PubMed] [Google Scholar]
- Saito Y, Morishita K (2015) Maintenance of leukemic and normal hematopoietic stem cells in bone marrow niches by EVI1‐regulated GPR56. Rinsho Ketsueki 56: 375–383 [DOI] [PubMed] [Google Scholar]
- Sato T, Goyama S, Kataoka K, Nasu R, Tsuruta‐Kishino T, Kagoya Y, Nukina A, Kumagai K, Kubota N, Nakagawa M, Arai S, Yoshimi A, Honda H, Kadowaki T, Kurokawa M (2014) Evi1 defines leukemia‐initiating capacity and tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Oncogene 33: 5028–5038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheer N, Campos‐Ortega JA (1999) Use of the Gal4‐UAS technique for targeted gene expression in the zebrafish. Mech Dev 80: 153–158 [DOI] [PubMed] [Google Scholar]
- Scheer N, Groth A, Hans S, Campos‐Ortega JA (2001) An instructive function for Notch in promoting gliogenesis in the zebrafish retina. Development 128: 1099–1107 [DOI] [PubMed] [Google Scholar]
- Solaimani Kartalaei P, Yamada‐Inagawa T, Vink CS, de Pater E, van der Linden R, Marks‐Bluth J, van der Sloot A, van den Hout M, Yokomizo T, van Schaick‐Solerno ML, Delwel R, Pimanda JE, van IJcken WFJ, Dzierzak E (2015) Whole‐transcriptome analysis of endothelial to hematopoietic stem cell transition reveals a requirement for Gpr56 in HSC generation. J Exp Med 212: 93–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzukawa K, Parganas E, Gajjar A, Abe T, Takahashi S, Tani K, Asano S, Asou H, Kamada N, Yokota J (1994) Identification of a breakpoint cluster region 3′ of the ribophorin I gene at 3q21 associated with the transcriptional activation of the EVI1 gene in acute myelogenous leukemias with inv(3)(q21q26). Blood 84: 2681–2688 [PubMed] [Google Scholar]
- Warga RM, Kimmel CB (1990) Cell movements during epiboly and gastrulation in zebrafish. Development 108: 569–580 [DOI] [PubMed] [Google Scholar]
- Wieser R (2007) The oncogene and developmental regulator EVI1: expression, biochemical properties, and biological functions. Gene 396: 346–357 [DOI] [PubMed] [Google Scholar]
- Wiley DM, Kim JD, Hao J, Hong CC, Bautch VL, Jin SW (2011) Distinct signalling pathways regulate sprouting angiogenesis from the dorsal aorta and the axial vein. Nat Cell Biol 13: 686–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimi A, Goyama S, Watanabe‐Okochi N, Yoshiki Y, Nannya Y, Nitta E, Arai S, Sato T, Shimabe M, Nakagawa M, Imai Y, Kitamura T, Kurokawa M (2011) Evi1 represses PTEN expression and activates PI3K/AKT/mTOR via interactions with polycomb proteins. Blood 117: 3617–3628 [DOI] [PubMed] [Google Scholar]
- You LR, Lin FJ, Lee CT, DeMayo FJ, Tsai MJ, Tsai SY (2005) Suppression of Notch signalling by the COUP‐TFII transcription factor regulates vein identity. Nature 435: 98–104 [DOI] [PubMed] [Google Scholar]
- Yuasa H, Oike Y, Iwama A, Nishikata I, Sugiyama D, Perkins A, Mucenski ML, Suda T, Morishita K (2005) Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA‐2 expression. EMBO J 24: 1976–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, He Q, Chen D, Liu W, Wang L, Zhang C, Ma D, Li W, Liu B, Liu F (2015) G protein‐coupled receptor 183 facilitates endothelial‐to‐hematopoietic transition via Notch1 inhibition. Cell Res 25: 1093–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong TP, Childs S, Leu JP, Fishman MC (2001) Gridlock signalling pathway fashions the first embryonic artery. Nature 414: 216–220 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Review Process File