

Abstract
Hypoxic growth of fungi requires sterol regulatory element‐binding protein (SREBP) transcription factors, and human opportunistic fungal pathogens require SREBP activation for virulence. Proteolytic release of fission yeast SREBPs from the membrane in response to low oxygen requires the Golgi membrane‐anchored Dsc E3 ligase complex. Using genetic interaction arrays, we identified Rbd2 as a rhomboid family protease required for SREBP proteolytic processing. Rbd2 is an active, Golgi‐localized protease that cleaves the transmembrane segment of the TatA rhomboid model substrate. Epistasis analysis revealed that the Dsc E3 ligase acts on SREBP prior to cleavage by Rbd2. Using APEX2 proximity biotinylation, we demonstrated that Rbd2 binds the AAA‐ATPase Cdc48 through a C‐terminal SHP box. Interestingly, SREBP cleavage required Rbd2 binding of Cdc48, consistent with Cdc48 acting to recruit ubiquitinylated substrates. In support of this claim, overexpressing a Cdc48‐binding mutant of Rbd2 bypassed the Cdc48 requirement for SREBP cleavage, demonstrating that Cdc48 likely plays a role in SREBP recognition. In the absence of functional Rbd2, SREBP precursor is degraded by the proteasome, indicating that Rbd2 activity controls the balance between SREBP activation and degradation.
Keywords: Cdc48/p97, intramembrane proteolysis, rhomboid, SREBP, ubiquitin
Subject Categories: Membrane & Intracellular Transport; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Sterol regulatory element‐binding proteins (SREBPs) play a key role in transcriptional regulation of lipid metabolism in response to intracellular levels of cholesterol and fatty acids in mammals (Espenshade & Hughes, 2007). SREBPs are synthesized as inactive precursor proteins containing two hydrophobic transmembrane‐spanning segments for membrane insertion into the endoplasmic reticulum (ER). In response to cholesterol depletion, SREBP cleavage‐activating protein (SCAP) escorts SREBP via COPII vesicles to the Golgi apparatus where it is processed by two proteases: Site‐1 protease (S1P; a membrane‐bound subtilisin‐related serine protease) cleaves in the lumen and Site‐2 protease (S2P; a membrane‐bound zinc metalloprotease) cleaves in the membrane. The proteolytically released N‐terminal segment containing the transactivation domain and the basic helix‐loop‐helix leucine zipper DNA binding (bHLH‐zip) domain travels to the nucleus where it activates transcription of genes encoding enzymes for the biosynthesis and uptake of cholesterol, fatty acids, and triglycerides, as well as activating its own transcription (Shao & Espenshade, 2012).
Sterol regulatory element‐binding proteins are conserved across fungal phyla, including Cryptococcus neoformans, Aspergillus fumigatus, Candida albicans and Schizosaccharomyces pombe (Lane et al, 2001; Chang et al, 2007, 2009; Chun et al, 2007; Willger et al, 2008; Bien & Espenshade, 2010). In S. pombe, the SREBP called Sre1 controls the hypoxic response in addition to lipid homeostasis (Hughes et al, 2005; Todd et al, 2006; Bien & Espenshade, 2010). Under low oxygen conditions, Sre1 is proteolytically cleaved and the N‐terminal transcription factor (Sre1N) is released from the membrane and enters the nucleus to activate hypoxic genes. Importantly, the SREBP oxygen‐responsive pathway is conserved in human opportunistic pathogens, such as C. neoformans and A. fumigatus (Bien & Espenshade, 2010). SREBP is critical for pathogenic fungi to adapt and survive in hypoxic host conditions, which frequently occur at sites of fungal infections (Grahl et al, 2012). Consequently, SREBP is required for C. neoformans and A. fumigatus virulence, and SREBP pathway components are antifungal drug targets.
In addition to Sre1, fission yeast codes for a second SREBP, called Sre2. Sre1 and Sre2 have the same predicted topology, but Sre2 lacks the C‐terminal SCAP binding domain (Hughes et al, 2005). Thus, Sre2 cleavage is not regulated by sterols or oxygen, but is constitutively cleaved (Stewart et al, 2011). Despite these differences in regulation, Sre1 and Sre2 cleavage require the same machinery (Stewart et al, 2011, 2012).
Proteolytic activation of fission yeast SREBP (both Sre1 and Sre2) occurs through a mechanism different than mammalian SREBP. Proteolytic activation requires the Golgi Dsc (defective for SREBP cleavage) E3 ligase, a complex of five subunits, and the AAA‐ATPase Cdc48, the yeast homolog of p97/VCP (Stewart et al, 2011, 2012; Lloyd et al, 2013). Cdc48 binds to the Dsc E3 ligase through the UBX domain of the Dsc5 subunit (Stewart et al, 2012). The Dsc E3 ligase complex possesses E3 ubiquitin ligase activity, and its complex architecture resembles the ER‐associated degradation (ERAD) E3 ligase gp78 (Lloyd et al, 2013; Raychaudhuri & Espenshade, 2015). In ERAD, a membrane‐associated E3 ligase core complex recognizes substrate proteins in the ER and mediates ubiquitinylation (Olzmann et al, 2013). The Cdc48/p97 complex recognizes the poly‐ubiquitin chain and extracts ubiquitinylated substrates to the cytosol for proteasomal degradation. Despite their subunit architectural and functional conservation, the ERAD and Dsc E3 ligases have distinct functional outcomes. While ERAD E3 ligase machinery mediates substrate degradation, current data support a model in which the Dsc E3 ligase ubiquitylates yeast SREBP to direct its subsequent proteolytic activation.
Many unanswered questions stemming from major gaps in our knowledge of how yeast SREBP is activated need to be explored. First, which protease(s) cleaves SREBP? Subunits of the Dsc E3 ligase do not contain predicted protease domains, and S2P is absent from fission yeast (Bien & Espenshade, 2010). Given that activation of SREBP is required for fungal pathogen virulence, identification of an SREBP protease could provide a new drug target. Second, why is Cdc48 required for SREBP cleavage? Point mutants of cdc48 fail to support yeast SREBP cleavage (Stewart et al, 2012). However, the Dsc5 UBX domain that recruits Cdc48 to the Dsc E3 ligase is not required for cleavage, suggesting an independent function for Cdc48 in SREBP cleavage. Finally, is yeast SREBP ubiquitinylation required for cleavage? Our recent studies demonstrate that Dsc E3 ligase activity is required for Golgi localization of the Dsc E3 ligase complex (Raychaudhuri & Espenshade, 2015). Thus, mislocalization of the Dsc E3 ligase confounds the interpretation of dsc mutant data.
In this study, we provide answers to each of these questions. We demonstrate that S. pombe Rbd2 is a Golgi‐resident rhomboid protease that is required for yeast SREBP cleavage activation. Kim and colleagues recently reported an initial genetic characterization of rbd2 showing that Sre1 cleavage activation requires rbd2 in S. pombe, and our results confirm these findings (Kim et al, 2015). Rhomboid proteases are a family of intramembrane serine proteases, whose catalytic site is buried within the membrane bilayer (Urban, 2006; Urban & Dickey, 2011). Conserved in prokaryotes and eukaryotes, rhomboid proteases regulate a wide range of divergent biological processes, such as growth factor signaling, mitochondrial membrane dynamics, protein quality control, and parasite invasion of host cells (Bergbold & Lemberg, 2013; Lemberg, 2013; Rather, 2013). Our epistasis analysis shows that the Dsc E3 ligase complex functions prior to Rbd2, suggesting that Dsc E3‐mediated ubiquitinylation is a prerequisite for Rbd2 function. The C‐terminal, cytosolic tail of Rbd2 binds Cdc48, and disruption of Rbd2–Cdc48 binding blocks SREBP cleavage. Genetic experiments indicate that Cdc48 recruits SREBP to Rbd2 for cleavage, identifying a function for Cdc48 that is independent from its role in binding to the Dsc E3 ligase. The mammalian rhomboid RHDBL4 also binds p97, but in this case, p97 functions after cleavage to promote degradation of cleaved products (Fleig et al, 2012). Interestingly, the absence of functional Rbd2 leads to proteasomal degradation of the uncleaved SREBP precursor form in a Dsc E3‐dependent manner, supporting a model in which the Dsc E3 ligase ubiquitinylates SREBP prior to cleavage by Rbd2. Thus, this study answers key questions regarding the mechanism of yeast SREBP cleavage activation.
Results
Genetic interaction mapping identifies function for rbd2 in SREBP pathway
To identify additional genes involved in SREBP cleavage in S. pombe, we previously carried out an unbiased, high‐throughput genetic interaction screen (Frost et al, 2012). Hierarchical clustering of genetic interaction profile correlations between a set of 1,297 G418‐marked array strains and 579 nourseothricin‐marked query strains revealed functionally related genes, including those involved in dsc‐dependent SREBP pathway (Frost et al, 2012). A gene encoding a putative rhomboid protease, SPCC790.03, was correlated with dsc1‐dsc4 and sre2 (Fig 1A). The absence of sre1 from the cluster was likely due to the fact that the study was performed under normoxic conditions when sre1 is inactive, an observation made in a previous study (Eisenman et al, 2005; Stewart et al, 2011).
Figure 1. Rbd2 is a Golgi‐resident rhomboid protease.
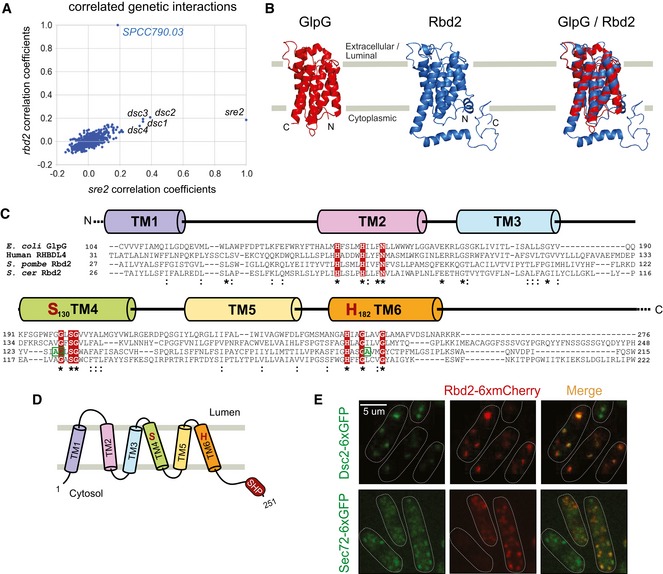
- Plot of correlation coefficients generated from comparison of the genetic profiles from sre2Δ and SPCC790.03Δ (rbd2Δ) to all other profiles from the genetic interaction array. Note the high correlations among dsc1‐4, sre2, and rbd2.
- Protein structural predictions for Schizosaccharomyces pombe Rbd2 (blue) and E. coli GlpG (red). Protein structures were predicted using the Phyre2 server (http://www.sbg.bio.ic.ac.uk/phyre2). Rbd2 model structure was aligned to GlpG using PyMOL.
- Protein sequence alignment of E. coli GlpG, human RHBDL4, S. pombe Rbd2, and S. cerevisiae Rbd2. Alignments were generated using Clustal Omega server (http://www.ebi.ac.uk/Tools/msa/clustalo). Predicted transmembrane domains (TM) and shared sequence motifs were used for alignment. Positions of functionally important, conserved rhomboid amino acid residues (including predicted catalytic dyad serine and histidine) are shaded in red. Residues boxed in green were identified as required for Sre1 activation from a genetic selection and were analyzed in Fig 2E and F. The starting residue numbers of the indicated sequences within their full‐length proteins are given. Asterisks mark residues identical in all sequences, and colons denote conservative substitutions.
- Topology model for S. pombe Rbd2 with the conserved six‐pass TM. Both the N‐ and C‐termini face the cytoplasm. The protease active site serine and histidine form a catalytic dyad between TM4 and TM6. A SHP box, predicted to bind Cdc48, is positioned at the Rbd2 C‐terminus.
- Cells carrying a plasmid expressing rbd2‐6xmCherry and either of two Golgi proteins dsc2‐6xGFP (dsc2‐6xGFP rbd2‐6xmCherry) or Sec72‐6xGFP (Sec72‐6xGFP rbd2‐6xmCherry) were imaged by confocal fluorescence microscopy.
SPCC790.03 codes for a putative 251 amino acid (aa) rhomboid protease that is homologous to RBD2 in Saccharomyces cerevisiae. Bioinformatic analysis of S. pombe Rbd2 revealed highly significant shared homology between Rbd2 and GlpG, a well‐studied bacterial member of the rhomboid family of intramembrane proteases (Fig 1B) (Kelley et al, 2015). Furthermore, a multiple sequence alignment revealed that Rbd2 shares significant sequence conservation with rhomboid proteases from other kingdoms (Fig 1C). Based on known rhomboid structures and topologies, Rbd2 has six predicted transmembrane domains (TM) with the N‐ and C‐termini in the cytoplasm (Fig 1D), and its predicted catalytic Ser‐His dyad is positioned in the fourth (S130) and sixth (H182) TM, respectively. In addition, Rbd2 also contains the conserved “GxSG” motif surrounding the catalytic Ser in TM4, the “GxxxG” helix dimerization motif in TM6, and the “HxxxxHxxxN” motif in TM2 characteristic of rhomboid proteases (Fig 1C) (Urban & Baker, 2008; Urban & Shi, 2008). Interestingly, a homology search program also identified a SHP box, a putative Cdc48 interaction domain, in the cytosolic C‐terminus of Rbd2 that has not been described in rhomboid proteases (Fig 1D) (Sato & Hampton, 2006; Kelley et al, 2015). This SHP box is not present in S. cerevisiae Rbd2, suggesting that the two orthologs have distinct functions. The predicted subcellular localization of Rbd2 is the Golgi apparatus (Matsuyama et al, 2006). Consistent with this prediction, Rbd2 fused to six tandem copies of mCherry (Rbd2‐6xmCherry) appeared as puncta in cells that colocalized with two Golgi proteins, Sec72‐6xGFP and Dsc2‐6xGFP (Fig 1E) (Stewart et al, 2011). Our combined structural modeling, bioinformatic analysis, and subcellular localization data suggest that Rbd2 is a S. pombe Golgi‐localized, rhomboid intramembrane protease.
SREBP activation requires Rbd2
To test whether Rbd2 is involved in SREBP proteolytic processing, we generated an rbd2∆ strain. Cells lacking Sre1 activity fail to grow on medium containing cobalt chloride (CoCl2), which mimics hypoxia (Hughes et al, 2005). rbd2∆ cells failed to grow on CoCl2 medium, and growth was rescued by the expression of the cleaved, soluble N‐terminus of Sre1 (Sre1N) (Fig 2A), suggesting that the rbd2∆ growth defect was due to a failure to produce Sre1N. Consistent with this result, rbd2∆ cells failed to accumulate cleaved Sre1N when cells were grown in the absence of oxygen, a condition where Sre1 cleavage was robust in wild‐type cells (Fig 2B, lanes 1–8). As expected, dsc1∆ cells also failed to produce Sre1N (Stewart et al, 2011). Interestingly, unlike dsc1∆ cells, the full‐length Sre1 precursor form decreased in rbd2∆ cells (Fig 2B, lanes 5–13). Taken together, these data suggest that deletion of rbd2 impairs Sre1 activation under hypoxic conditions.
Figure 2. SREBP activation requires rbd2 .
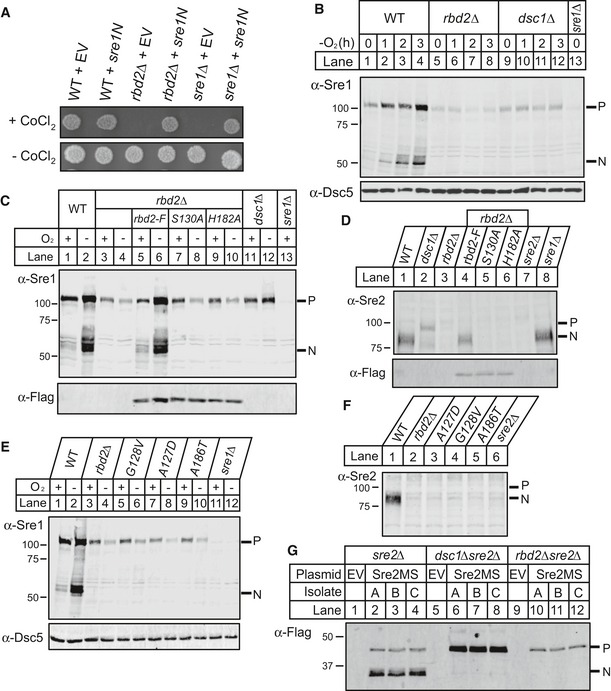
- Wild‐type (WT), rbd2Δ, or sre1Δ yeast (200 cells) containing either empty vector (EV) or a plasmid expressing sre1N (Sre1 amino acids 1–440) were grown on rich medium in the presence or absence of cobalt chloride (CoCl2).
- Western blot was probed with anti‐Sre1 IgG of phosphatase‐treated, whole‐cell lysates from WT and the indicated mutants grown for the indicated time in the absence of oxygen. Dsc5 serves as a loading control and was detected by chemiluminescence. P and N denote Sre1 precursor and cleaved nuclear forms, respectively.
- Indicated yeast strains expressing rbd2 or chromosomal Flag‐tagged rbd2, rbd2‐S130A, or rbd2‐H182A were analyzed for Sre1 cleavage. Western blot was performed using anti‐Sre1 or anti‐Flag IgG of phosphatase‐treated, whole‐cell lysates from cells grown for 3 h in the presence or absence of oxygen.
- Indicated yeast strains expressing rbd2 or chromosomal Flag‐tagged rbd2, rbd2‐S130A, or rbd2‐H182A were analyzed for Sre2 cleavage by Western blot probed with anti‐Sre2 serum or anti‐Flag IgG of phosphatase‐treated, whole‐cell lysates from cells grown in the presence of oxygen.
- Sre1 cleavage assay was performed as in (C). G128V, A127D, and A186T mutant strains were obtained from our previous mutagenesis screening (Stewart et al, 2012). Dsc5 serves as a loading control.
- Sre2 cleavage assay for indicated yeast strains was performed as in (D).
- A model substrate of Sre2 containing amino acids 423–793 (Sre2MS) with an N‐terminal 3xFlag epitope tag was expressed on a plasmid under the control of a CaMV promoter in sre2Δ, dsc1Δsre2Δ, or rbd2Δsre2Δ yeast strains. Whole‐cell lysates from indicated yeast strains were analyzed by Western blot with anti‐Flag antibody. Three independent isolates (A–C) are shown for each strain. Empty vector (EV) transformation serves as a negative control.
To test whether the catalytic dyad residues of Rbd2 (Ser130 and His182) are required for SREBP activation, we assayed Sre1 cleavage in rbd2∆ cells expressing chromosomally Flag‐tagged rbd2, rbd2‐S130A, or rbd2‐H182A. Wild‐type and Rbd2 mutants were expressed equivalently (Fig 2C, bottom panel). While wild‐type Rbd2 supported Sre1 cleavage, catalytically dead Rbd2 mutants did not (Fig 2C, lanes 5–10), indicating Sre1 activation requires Rbd2 protease activity.
Next, we examined effects on activation of the second fission yeast SREBP, Sre2. In wild‐type cells, Sre2 is constitutively cleaved in the presence of oxygen, and the majority of Sre2 exists as Sre2N (Fig 2D, lane 1) (Stewart et al, 2011). Strikingly, neither the precursor nor the cleaved Sre2N form was observed in rbd2∆ cells (Fig 2D, lane 3). Similar to Sre1, Sre2 activation was rescued in rbd2∆ cells by rbd2, but not by the rbd2‐S130A or rbd2‐H182A mutants (Fig 2D, lanes 4–6). Again, the Sre2 precursor accumulated in dsc1∆ cells, but was not detected in the absence of functional Rbd2 (Fig 2D).
In parallel to our genetic interaction studies, we identified point mutations of rbd2 in a genetic selection to identify genes defective for Sre1 cleavage (Stewart et al, 2011). Briefly, each mutant that failed to activate an Sre1 reporter was tested for linkage to rbd2∆, identifying three rbd2‐linked mutants. DNA sequencing of rbd2 in these mutants revealed mutation of rbd2‐A127D, rbd2‐G128A, and rbd2‐A186T. These mutations reside in keystones III and IV, which are key architectural motifs conserved in all rhomboid proteases (Fig 1C, green residues) (Baker & Urban, 2012). Indeed, these rbd2 mutants resembled rbd2∆ cells in Sre1 and Sre2 cleavage assays (Fig 2E and F), confirming that these residues are required for SREBP activation and independently demonstrating that SREBP activation requires Rbd2 rhomboid function.
Finally, Rbd2 was required for processing of a truncated Sre2 model substrate (Sre2MS, aa 423–793), previously shown to be processed like endogenous Sre2 (Cheung & Espenshade, 2013). Cleaved Sre2MS was not detected in rbd2∆ cells (Fig 2G, lanes 10–12). Unlike endogenous Sre2, a small amount of uncleaved Sre2MS was retained in the rbd2∆ cells, possibly due to differences in expression. Uncleaved Sre2MS accumulated in dsc1∆ cells as expected (Fig 2G, lanes 6–8) (Cheung & Espenshade, 2013). Taken together, these results show that Rbd2 activity is required for fission yeast SREBP activation and that precursor forms of SREBP decrease in the absence of functional Rbd2, unlike in dsc1∆ cells.
SREBP precursor is degraded in a proteasome‐ and Dsc E3 ligase‐dependent manner in the absence of rbd2
Uncleaved SREBP precursor decreased in rbd2∆ cells, but not in wild‐type or dsc1∆ cells (Fig 2B–F). We next asked whether disappearance of the precursor was due to proteasomal degradation. To impair proteasome function, we used a temperature‐sensitive allele of mts3 that codes for the 19S proteasome subunit Rpn12 (Gordon et al, 1996). In wild‐type cells, the majority of Sre2 existed as Sre2N, and we observed loss of Sre2 in rbd2∆ cells (Fig 3A, lanes 1–2). Inactivation of mts3 in wild‐type cells resulted in accumulation of both Sre2P and Sre2N (Fig 3A, lane 4). Importantly, inactivation of mts3 in rbd2∆ mts3‐1 double mutant cells showed recovery of the Sre2P but not Sre2N (Fig 3A, lane 3). Consistent with this result, treatment with the 20S proteasome inhibitor bortezomib (Bz) increased both Sre2P and Sre2N in wild‐type cells, but only Sre2P accumulated in rbd2∆ cells (Fig 3B). Finally, the Sre2MS precursor accumulated in rbd2∆ mutant cells upon Bz treatment, but the cleaved form did not (Fig 3C, lanes 7–8). Bz treatment did not change the level of Sre2MS in dsc1∆ cells (Fig 3C, lanes 5–6). Deletion of the ERAD E3 ligases hrd1 or doa10 did not rescue Sre2 levels (Fig EV1A), indicating that these ER‐localized E3 ligases do not target Sre2 for degradation. Taken together, these data indicate that Sre2 precursor is degraded by the proteasome in the absence of Rbd2. Furthermore, proteasome inhibition revealed that Rbd2 initiates SREBP proteolytic processing, because under these conditions, conversion of Sre2 precursor to Sre2N requires Rbd2 (Fig 3A, lanes 3–4 and Fig 3B, lanes 4 and 6).
Figure 3. Uncleaved SREBP is degraded in a proteasome‐dependent and Dsc E3 ligase‐dependent manner in the absence of rbd2 .
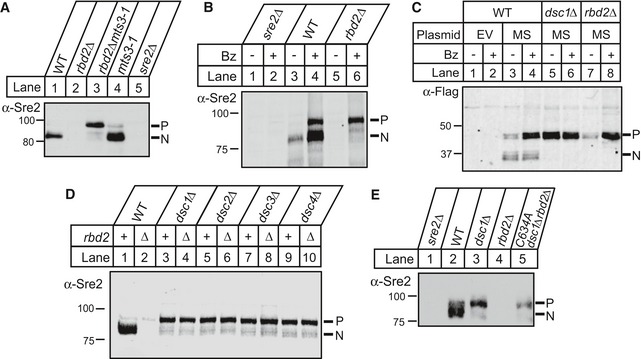
- Wild‐type (WT) and the indicated mutant strains were grown for 3 h at the non‐permissive temperature (36°C) to inactivate Mts3. Phosphatase‐treated, whole‐cell lysates were analyzed by Western blot with anti‐Sre2 serum and imaged using chemiluminescence. P and N denote Sre2 precursor and cleaved nuclear forms, respectively.
- WT, rbd2Δ, and sre2Δ cells were treated with bortezomib (Bz) for 3 h, and phosphatase‐treated, whole‐cell lysates were analyzed by Western blot with anti‐Sre2 serum.
- WT, dsc1Δ, and rbd2Δ cells carrying either empty vector (EV) or a plasmid expressing 3xFlag‐Sre2MS (MS) were treated with bortezomib (Bz) for 3 h. Whole‐cell lysates were analyzed by Western blot with anti‐Flag antibody.
- Whole‐cell lysates from the indicated strains were analyzed by Western blot with anti‐Sre2 serum.
- Catalytically dead Dsc1 E3 ligase (dsc1‐C634A) mutant and other indicated yeast strains were analyzed by Western blot probed with anti‐Sre2 serum and imaged using chemiluminescence.
Figure EV1. Sre1 and Sre2 precursor degradation requires dsc1 .
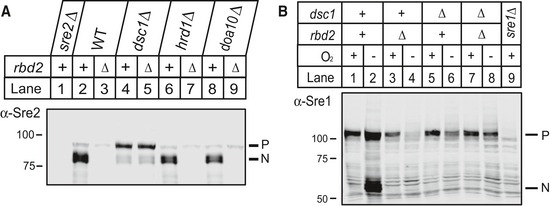
- Western blot of phosphatase‐treated, whole‐cell lysates from indicated strains was probed with anti‐Sre2 IgG. P and N denote Sre2 precursor and cleaved forms, respectively.
- Western blot was probed with anti‐Sre1 IgG of phosphatase‐treated, whole‐cell lysates from wild‐type cells and the indicated mutants grown for 3 h in the presence or absence of oxygen.
Proteasome inhibition did not increase Sre2MS in dsc1∆ cells (Fig 3C, lanes 5–6), suggesting that the Golgi‐localized Dsc E3 ligase may be required for SREBP degradation. Both the Dsc E3 ubiquitin ligase and Rbd2 are required for SREBP activation, and we performed epistasis tests to determine the order of action of these two pathway components. Sre2 decreased in rbd2∆ cells and the precursor accumulated in dsc1∆–dsc4∆ cells (Fig 3D, lanes 1–3, 5, 7, 9). Deletion of dsc1 in rbd2∆ cells restored Sre2 precursor levels to that of the single dsc1∆ mutant (Fig 3D, compare lanes 3 and 4). Identical results were seen for deletion of dsc2–dsc4 (Fig 3D, lanes 5–10). Likewise, Sre1 precursor disappeared under low oxygen in rbd2∆ cells, but was recovered in dsc1∆rbd2∆ cells (Fig EV1B). Thus, the Dsc E3 ligase is required for proteasome‐dependent degradation of SREBP in rbd2∆ cells, demonstrating that the Dsc E3 ligase acts prior to Rbd2 in the biochemical pathway to cleave SREBP. To test specifically whether Sre2 precursor degradation requires Dsc E3 ligase activity, we studied a previously characterized, catalytically dead mutant of Dsc1 with a mutation in the Dsc1 RING domain (dsc1‐C634A) (Raychaudhuri & Espenshade, 2015). As observed for the dsc1 deletion, mutation of the Dsc1 RING domain restored Sre2 precursor in rbd2∆ cells (Fig 3E), showing that the Sre2 precursor degradation requires Dsc RING E3 ligase activity. Collectively, these results suggest that uncleaved SREBP is ubiquitinylated by the Dsc E3 ligase and degraded by the proteasome when Rbd2 function is impaired.
Rbd2 is an active rhomboid intramembrane protease
We next used a heterologous assay with a panel of model rhomboid substrates to examine whether Rbd2 is able to catalyze intramembrane proteolysis directly. This assay relies on co‐transfecting human HEK293T cells with a 3xHA‐tagged rhomboid and a GFP‐tagged substrate and using Western analysis to detect proteolysis. Although this approach has been used to study the protease activity of rhomboid proteases from bacteria, protozoa, plants, and animals, it has never been applied successfully to study yeast rhomboid activity. Transfecting cells with 3xHA‐rbd2 resulted in low levels of protein expression (Fig 4A, lane 2). To circumvent this obstacle, we recoded the entire rbd2 ORF to codon‐optimize it for expression in human cells, and this resulted in > 35‐fold higher expression of 3xHA‐Rbd2 (Fig 4A, lanes 2–3). Despite achieving this robust level of expression (higher than other rhomboid proteases that we used as positive controls), Rbd2 was completely unable to cleave either the Drosophila signaling molecule Spitz, which is a canonical rhomboid substrate, or Plasmodium EBA‐175, which is an “atypical” substrate that is cleaved by many rhomboid proteases that are unable to cleave Spitz (Fig 4B) (Urban & Freeman, 2003; Baker et al, 2006; Baxt et al, 2008).
Figure 4. Rbd2 is an active rhomboid protease.
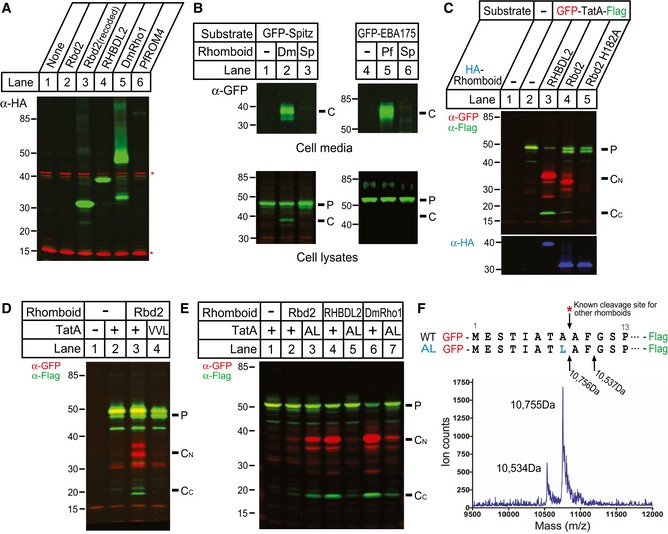
- Human HEK293T cells were transfected with plasmids expressing different 3xHA‐tagged rhomboid proteases: Schizosaccharomyces pombe rbd2, S. pombe rbd2 recoded for human expression, human RHBDL2, D. melanogaster Rho1, and P. falciparum ROM4. Whole‐cell lysates were analyzed by Western blot to detect the HA‐tagged rhomboid. Asterisks indicate loading control bands detected with antibody ab179726 (Abcam).
- Human HEK293T cells were transfected with plasmids expressing the indicated rhomboid substrates and either recoded S. pombe rbd2, D. melanogaster Rho1, or P. falciparum ROM4. Conditioned media and cell lysates were analyzed by Western blot. P and C denote precursor and cleaved products, respectively.
- Human HEK293T cells were transfected with plasmids expressing the GFP‐TatA‐Flag substrate and the indicated rhomboid proteases. Whole‐cell lysates were analyzed by Western blot. P, CN, CC denote precursor, N‐terminal cleaved, and C‐terminal cleaved products, respectively.
- Human HEK293T cells were transfected with plasmids expressing the indicated GFP‐TatA‐Flag substrates and in the absence or presence of recoded S. pombe rbd2. Whole‐cell lysates were analyzed by Western blot.
- Human HEK293T cells were transfected with plasmids expressing the indicated GFP‐TatA‐Flag substrates and different rhomboid proteases. Whole‐cell lysates were analyzed by Western blot.
- Human HEK293T cells were transfected with plasmids expressing the GFP‐TatA (A8L)‐Flag and recoded S. pombe rbd2. C‐terminal cleavage products were purified using anti‐Flag antibodies, and purified protein was analyzed by mass spectrometry.
We therefore next examined cleavage of a third substrate, TatA from the bacterium Providencia stuartii, which is perhaps the most efficient rhomboid substrate known (Moin & Urban, 2012). Co‐transfecting rbd2 resulted in consistent but weak TatA cleavage (Fig 4C, lane 4). To ensure that this was proteolysis, we tagged both termini of TatA and followed processing by Rbd2 or the human rhomboid protease RHBDL2 as a positive control. In each case, we detected both proteolytic fragments, and TatA proteolysis required the predicted catalytic histidine of Rbd2 (Fig 4C, lanes 4–5).
We next examined the substrate requirements for Rbd2 processing of TatA. Rhomboid proteases require helix‐destabilizing residues in the substrate transmembrane segment, and mutating these residues of TatA (GSP, aa 11–13) to VVL abolished processing by Rbd2 (Fig 4D). All rhomboid proteases studied to date cleave after small residues (glycine, alanine, serine) (Urban, 2010), and all rhomboid proteases tested cleave TatA after A8 (Strisovsky et al, 2009). We therefore tested the ability of Rbd2 to cleave TatA harboring the A8L mutant. To our surprise, we found that Rbd2 cleaved the A8L mutant substrate four‐fold more efficiently than wild‐type TatA (Fig 4E, lanes 2–3). However, this mutation nearly abolished processing by other rhomboid proteases (Fig 4E, lanes 4–7). Rhomboid proteases typically have only limited sequence dependence, and thus, mutations can increase proteolysis by shifting the site of cleavage to a new location. To examine whether the A8L mutant was truly being cleaved after a large hydrophobic residue, we immunopurified the C‐terminal cleavage product and determined its size by mass spectrometry (Fig 4F). The predominant cleavage of the TatA A8L mutant was indeed after the leucine at position 8. Interestingly, we discovered a second, minor cleavage site (~20%) after F10, two residues deeper into the TatA transmembrane segment. Notably, this cleavage also occurred between a large and small residue, namely a naturally occurring phenylalanine and glycine. As such, Rbd2 is an active rhomboid intramembrane protease that can cleave after large hydrophobic residues. This unique property has not been observed for any rhomboid protease.
Rbd2 cleavage requires Lys743 in Sre2 luminal loop
Rhomboid proteases cleave type‐I transmembrane substrates (Urban & Freeman, 2003; Ha, 2009) and consistent with this Rbd2 cleaved TatA within its transmembrane segment toward its N‐terminus (Fig 4F). We next asked whether this unique substrate sequence preference was also observed for SREBP. Heterologous Rbd2 cleavage assays using multiple S. pombe SREBP substrate variants failed, possibly due to the lack of Dsc E3 ligase machinery in HEK293 cells. To test directly the requirement for large hydrophobic residues, we assayed cleavage of alanine‐substitution mutants in the N‐terminus of the second predicted transmembrane segment of Sre2 (aa 748–754) (Fig 5A). Processing of each mutant (I748A/Y749A, L750A, F752A, L754A) was normal in wild‐type cells and required both rbd2 and dsc1 (Fig 5B and Appendix Fig S1A), indicating that Sre2MS cleavage does not require these individual large hydrophobic residues. However, we cannot exclude the possibility that one or several of these mutations block cleavage at the wild‐type site but induce a compensating shift in cleavage site, as has been observed in other rhomboid substrates (Strisovsky et al, 2009; Moin & Urban, 2012).
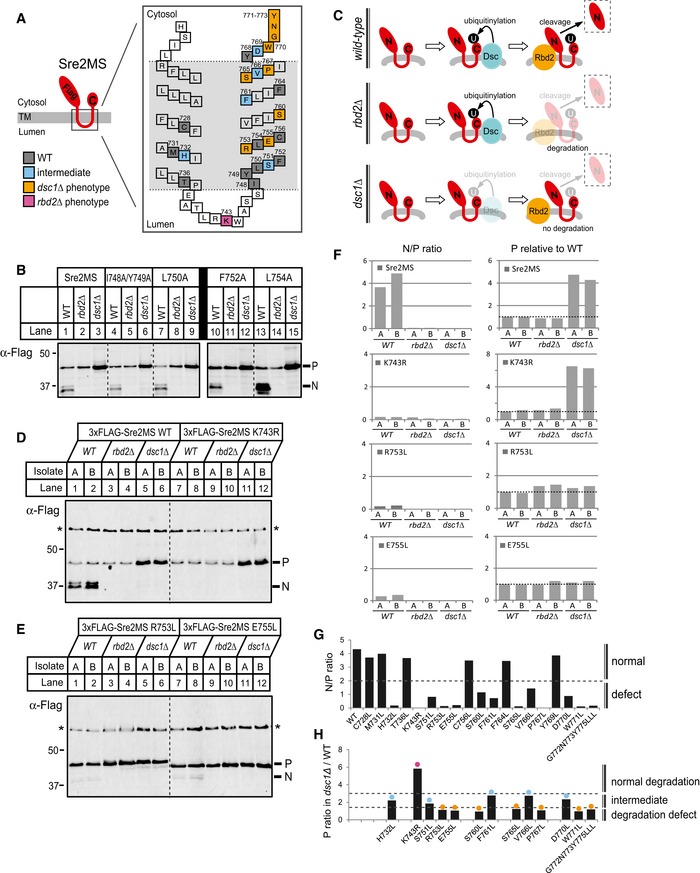
Figure 5. Rbd2 cleavage requires Lys 743.
-
ADiagram of Sre2MS substrate (aa 423–793). The enlarged model shows predicted positions of TM1 and TM2 denoting the 26 amino acids tested for mutagenesis by colored squares. Numbers indicate the number of amino acid residues. Mutants showing a block in cleavage due to defects in Dsc1 function are indicated in orange, Rbd2 function in pink, a combination of defects in Dsc1 and Rbd2 in blue (intermediate), and residues showing normal cleavage are indicated in gray.
-
BWhole‐cell lysates from WT, rbd2Δ, and dsc1Δ cells carrying a plasmid expressing 3xFlag‐Sre2MS WT or indicated mutants were analyzed by Western blot with anti‐Flag antibody. P and N denote Sre2MS precursor and cleaved forms, respectively.
-
CModel outlining the predicted fate of Sre2MS in different yeast strains. U denotes either mono‐ubiquitin or poly‐ubiquitin.
-
D, EWhole‐cell lysates from WT, rbd2Δ, and dsc1Δ cells carrying a plasmid expressing 3xFlag‐Sre2MS WT or indicated mutants were analyzed by Western blot with anti‐Flag antibody. P and N denote Sre2MS precursor and cleaved forms, respectively. Asterisks indicate non‐specific, loading control bands, which were used to normalize the intensities of P and N in (F). Two independent isolates, A and B, are shown for each strain.
-
FIntensities of individual P and N bands from Western blots were quantified using LI‐COR software. Relative intensity of P and N to corresponding non‐specific band (asterisk) in each lane was normalized to that of P level in isolate A of WT strain for each mutagenesis. The ratio of N to P or relative P level was presented in bar graphs.
-
GThe average ratio of N to P level for different Sre2MS mutants in wild‐type cells (Appendix Fig S2) was plotted. Cleavage was scored as normal if the ratio was > 2.
-
HFor Sre2MS mutants showing defects in cleavage, the average fold‐increase in P level in dsc1Δ cells compared to WT cells from two isolates (Appendix Fig S2) was plotted. Mutants were grouped into three phenotypic classes (intermediate, dsc1Δ, and rbd2∆) according to observed precursor accumulation. Precursor accumulation was defined as dsc1Δ if < 1.5 (orange), rbd2Δ if > 3 (pink), and intermediate if in between (blue).
To identify residues important for proteolytic cleavage, we tested 19 additional Sre2MS mutants. We quantified cleavage of each mutant in three different backgrounds: wild type, rbd2Δ, and dsc1Δ (Fig 5C). Sre2MS precursor (P) was processed to a cleaved product (N) in wild‐type cells (Fig 5D, lanes 1–2), and the average N/P ratio was 4–5 (Fig 5F). Unlike for endogenous Sre2 (Fig 2D), we consistently observed unprocessed Sre2MS precursor in wild‐type cells, likely due to higher protein expression. In rbd2Δ cells, Sre2MS precursor was not cleaved to the N form (Fig 5D, lanes 3–4), and the precursor did not accumulate due to Dsc‐dependent proteasomal degradation (Fig 5C). In contrast in dsc1Δ cells, Sre2MS precursor accumulated ~4.5‐fold compared to wild‐type cells (Fig 5D and F), because it is neither cleaved nor degraded due to lack of Dsc E3 ligase function (Fig 5C).
Overall, we observed cleavage defects for 13 of the 19 mutants tested (Fig 5G and Appendix Figs S1 and S2). Since Sre2MS precursor has different fates in rbd2Δ and dsc1Δ cells (Fig 5C), quantification of mutant Sre2MS precursor levels allowed assignment of the step defective in processing to either Dsc‐dependent ubiquitinylation or Rbd2 cleavage. If a Sre2MS mutant is not cleaved due to a failure in Dsc1 ubiquitinylation, we expect that the Sre2MS precursor will not accumulate further in dsc1Δ cells and that the dsc1Δ/wild‐type ratio will equal 1, as seen for the R753L and E755L mutants (Fig 5E and F). In contrast, if mutant Sre2MS precursor is ubiquitinylated by the Dsc E3 ligase but defective for Rbd2 cleavage, we expect the precursor to accumulate in dsc1Δ cells compared to wild‐type cells, as seen for the K743R mutant (Fig 5D, lanes 7–12). To categorize the mutants, we plotted the ratio of Sre2MS precursor in dsc1Δ/wild‐type cells (Fig 5F and H, and Appendix Fig S2). Many amino acid residues appeared to be required for Dsc E3 activity (Fig 5A and H, orange) or for both Dsc and Rbd2 activity (Fig 5A and H, blue). No transmembrane residues were specifically required for Rbd2 activity, but mutation of lysine 743 in the luminal loop to arginine prevented Rbd2‐mediated cleavage (Fig 5A and H, pink). It is not clear whether the lysine is essential for substrate recognition or whether introducing arginine at position 743 interferes with cleavage. This extended mutagenesis study demonstrates that Sre2MS cleavage does not require large hydrophobic residues in the second transmembrane segment, but identifies a luminal lysine residue required for cleavage.
Rbd2 binds Cdc48 through the C‐terminal SHP box
Bioinformatic analysis identified a putative Cdc48‐interaction motif called a SHP box in the cytosolic C‐terminus of Rbd2 (Fig 1D) (Sato & Hampton, 2006). SHP box motifs are present in Cdc48/p97 cofactors such as mammalian UFD1, p47, and yeast Dfm1 (Fig 6A). To test whether Rbd2 binds Cdc48, the Rbd2 C‐terminus (aa 200–251) fused to GST was affinity purified from bacterial lysates and mixed with S. pombe cytosol. After washing, the bound fraction was analyzed for the presence of Cdc48 (Fig 6B). GST‐Rbd2 (aa 200–251) bound Cdc48 to the same extent as the Dsc5 UBX domain, which is a known Cdc48‐binding domain (Fig 6B, compare lanes 8 and 10) (Stewart et al, 2012). Dsc2 UBA domain served as a negative control that binds ubiquitin but not Cdc48 (Fig 6B, compare lanes 9–10) (Lloyd et al, 2013). Truncations of GST‐Rbd2 that lacked the SHP box (Rbd2200–225 or Rbd2200–240) failed to bind Cdc48, while a truncation containing the SHP box (Rbd2225–251) bounds Cdc48 (Fig 6C). Mutational analysis of the Rbd2 SHP box demonstrated that at least two conserved residues, G244 and G246, are critical for Cdc48 binding (Fig 6D). These in vitro experiments established that the Rbd2 C‐terminal SHP box binds Cdc48.
Figure 6. Rbd2 binds Cdc48 through a C‐terminal SHP box.
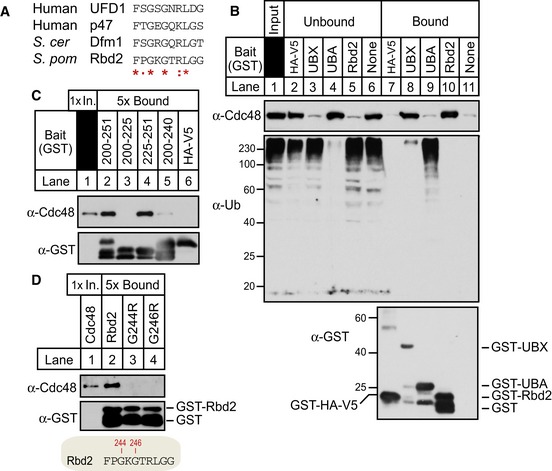
- Alignment of SHP box sequences from human UFD1, human p47, S. cerevisiae Dfm1, and Schizosaccharomyces pombe Rbd2 C‐terminal SHP box (aa 242–251). Asterisks denote residues identical in all sequences, colons mark conservative substitutions, and dots mark semi‐conservative substitutions.
- Recombinant proteins GST‐fused Rbd2 C‐terminus (Rbd2200–251), Dsc5 UBX (Dsc5323–425), Dsc2 UBA (Dsc2298–372), and GST‐HA‐V5 control were bound to GST magnetic beads and incubated with S. pombe cytosol fraction from wild‐type cells. Equivalent amounts of unbound and bound fractions were probed for anti‐Cdc48, anti‐ubiquitin, and anti‐GST IgG.
- GST pull‐down assay was performed as in (B) using truncated forms of Rbd2 C‐terminus; Rbd2200–251, Rbd2200–225, Rbd2225–251, and Rbd2200–240. Unbound (1×) and bound (5×) fractions were analyzed by Western blotting
- Single residue mutants of conserved glycine residues in Rbd2200–251 (G244R and G246R) were tested for Cdc48 binding as in (C).
APEX2 system detects Rbd2–Cdc48 interaction in cells
To test whether Rbd2 and Cdc48 interact in cells, we performed an immunoprecipitation experiment. Flag‐Rbd2 immunoprecipitated endogenous Cdc48 from yeast cells, while mutation of the SHP box (Rbd2‐G246R) decreased Cdc48 binding (Fig 7A, lanes 4–6). As an independent confirmation of this interaction, we employed the APEX2 system (Rhee et al, 2013; Hung et al, 2016). Briefly, APEX2 is an engineered ascorbate peroxidase that generates biotin‐phenoxyl radicals upon addition of biotin‐phenol and hydrogen peroxide (H2O2), resulting in covalent biotinylation of binding partners within a small radius (< 20 nm) (Fig 7B). After cell lysis, proteins labeled with biotin can be isolated with streptavidin beads for identification. We recently optimized the APEX2 system in yeast (Hwang & Espenshade, 2016). To label proximal and interacting proteins of Rbd2 using APEX2 technology, we fused Flag‐APEX2 to the C‐terminus of Rbd2 and overexpressed the fusion protein from a plasmid in rbd2∆ cells. Rbd2‐Flag‐APEX2 supported Sre1 cleavage demonstrating that the fusion protein was functional (Fig EV2A). Wild‐type cells or rbd2∆ cells expressing Rbd2‐Flag‐APEX2 or Rbd2‐G246R‐Flag‐APEX2 were subjected to biotin‐phenol labeling for 1 min, biotinylated proteins were recovered using streptavidin beads, and bound proteins were visualized by Western blotting. Compared to wild‐type cells which contain multiple naturally biotinylated proteins, cells expressing Rbd2‐Flag‐APEX2 and G246R‐Flag‐APEX2 had additional labeled proteins (Fig 7C, lanes 2–3 and 5–6) (Koffas et al, 1998). Western blotting of these samples revealed that Cdc48 was specifically labeled in Rbd2‐Flag‐APEX2 cells (Fig 7C, bottom panels). Mutation of the SHP box prevented labeling of Cdc48, confirming that the SHP box mediates Rbd2 binding to Cdc48. In contrast, the catalytically inactive Rbd2‐S130A mutant still bounds Cdc48 (Fig EV2B). Collectively, these in vitro and in vivo experiments demonstrate that the SHP box mediates Cdc48 binding to Rbd2.
Figure 7. APEX2 system detects Rbd2–Cdc48 binding in cells.
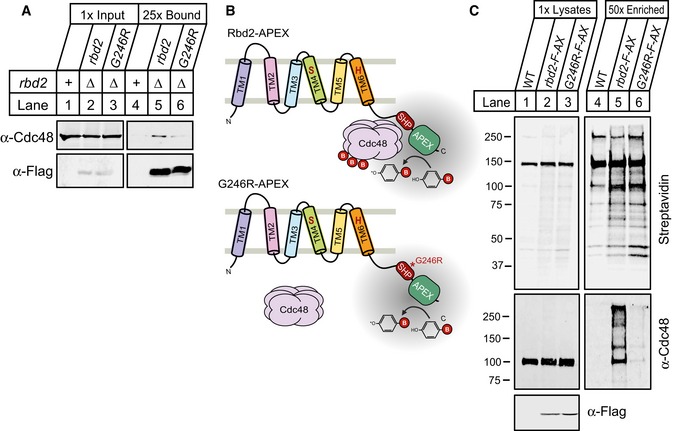
- Detergent‐solubilized yeast extracts from cells containing chromosomal Flag‐tagged rbd2 or rbd2‐G246R in rbd2Δ background were prepared, and proteins associated with Rbd2‐Flag were immunopurified using anti‐Flag magnetic beads. Equal amount of total proteins (lanes 1–3) along with 25× bound fractions (lanes 4–6) were analyzed by Western blot with anti‐Cdc48 serum and anti‐Flag IgG.
- Illustration outlining APEX2‐catalyzed biotin‐phenol labeling. An Rbd2‐Flag‐APEX fusion protein oxidizes biotin‐phenol to biotin‐phenoxyl radical that covalently labels binding partners.
- Biotin‐phenol labeling was performed for 1 min with H2O2 treatment as described in Materials and Methods. WT cells or rbd2Δ cells carrying rbd2‐Flag‐APEX2 (rbd2‐F‐AX) or rbd2‐G246R‐Flag‐APEX2 (G246R‐F‐AX) plasmid were lysed after biotin‐labeling reaction, and proteins were denatured by heating the cells in lysis buffer containing 1% SDS. Biotinylated proteins were then enriched using streptavidin magnetic beads. Lysates and 50×‐enriched eluates were analyzed by Western blot with IRDye 800CW Streptavidin, anti‐Cdc48 serum, or anti‐Flag IgG.
Figure EV2. Rbd2 catalytic dead mutant binds Cdc48.

- WT, rbd2Δ, or two isolates (A and B) of yeast expressing rbd2‐Flag‐APEX2 under control of the constitutive adh1 + promoter in rbd2Δ background were assayed for Sre1 cleavage. Western blot was probed with anti‐Sre1 IgG of whole‐cell lysates from the indicated strains grown for 3 h in the presence or absence of oxygen. P and N denote Sre1 precursor and cleaved forms, respectively. Note that whole‐cell lysates were not treated with phosphatase, and multiple bands of Sre1N represent phosphorylated forms.
- rbd2Δ cells expressing rbd2‐Flag‐APEX2, rbd2‐G246R‐Flag‐APEX2, or rbd2‐S130A‐Flag‐APEX2 from a plasmid were lysed after biotin‐labeling reaction, and proteins were denatured by heating the cells in a lysis buffer containing 1% SDS. Biotinylated proteins were then enriched using streptavidin magnetic beads. Lysates and 20×‐enriched eluates were analyzed by Western blot with IRDye 800CW Streptavidin, anti‐Cdc48 serum, and anti‐Flag IgG.
SREBP activation requires Rbd2 binding to Cdc48
To test whether SREBP activation requires Cdc48 binding to Rbd2, we used four different assays with both Rbd2 SHP box mutants (G244R and G246R): (i) growth on medium containing the hypoxia mimetic CoCl2 (Fig 8A), (ii) Sre1 cleavage assay in the absence of oxygen (Fig 8B), (iii) Sre2 cleavage assay (Fig 8C), and (iv) Sre2MS cleavage assay (Fig 8D). rbd2‐G244R cells exhibited slow growth on CoCl2, reduced Sre1 cleavage, and decreased Sre2N, consistent with partial loss of Rbd2 function. rbd2‐G246R cells showed more severe defects and phenocopied the catalytic mutant Rbd2‐S130A, demonstrating that SREBP activation requires a functional SHP box.
Figure 8. SREBP activation requires Cdc48 binding to Rbd2.
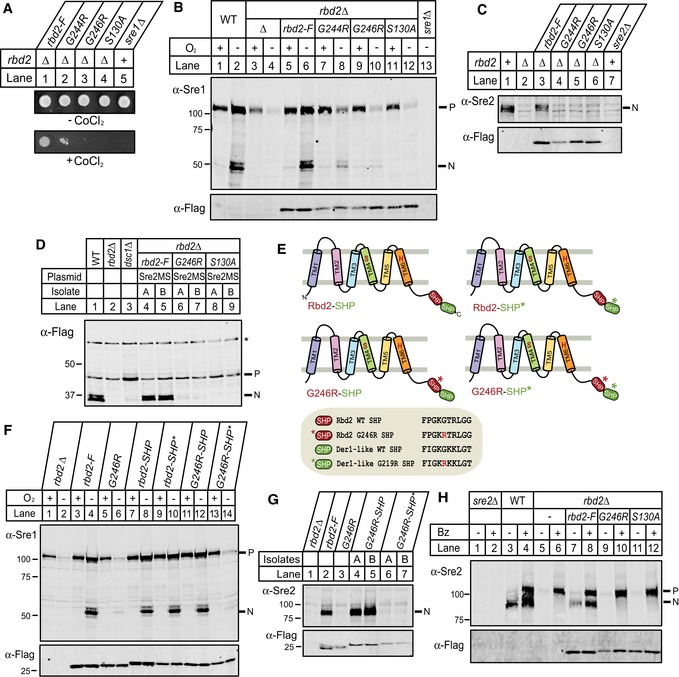
- Yeast strains (200 cells) containing chromosomal Flag‐tagged rbd2, rbd2‐G244R, rbd2‐G246R or rbd2‐S130A in rbd2Δ background, and sre1Δ yeast were grown on rich medium in the absence or presence of cobalt chloride (CoCl2).
- Western blot of phosphatase‐treated, whole‐cell lysates from strains in (A) grown for 3 h in the presence or absence of oxygen was probed with anti‐Sre1 or anti‐Flag IgG. P and N denote Sre1 precursor and cleaved nuclear forms, respectively.
- Sre2 cleavage was assayed by Western blot using anti‐Sre2 serum or anti‐Flag IgG of phosphatase‐treated, whole‐cell lysates from the indicated strains. N denotes Sre2 cleaved nuclear form.
- Whole‐cell lysates from indicated yeast strains carrying a plasmid expressing 3xFlag‐Sre2MS were analyzed by Western blot with anti‐Flag antibody. Two independent isolates, A and B, are shown for each strain. Asterisk denotes non‐specific band.
- Diagram of four Rbd2 chimeras fused to the SHP box from Schizosaccharomyces pombe Der1‐like protein (SPBC365.08c). The SHP boxes of Rbd2 and Der1‐like protein are shaded red and green, respectively. Asterisks denote point mutation in the SHP box.
- Yeast strains containing Flag‐tagged rbd2‐SHP, rbd2‐SHP*, rbd2‐G246R‐SHP, and rbd2‐G246R‐SHP* were generated in rbd2Δ background by chromosomal integration of the rbd2 fusion proteins depicted in (E). Indicated yeast strains were then assayed for Sre1 cleavage as in (B).
- Indicated strains were assayed for Sre2 cleavage as in (C).
- Wild type, sre2Δ, or strains containing chromosomal Flag‐tagged rbd2, rbd2‐G246R, or rbd2‐S130S in rbd2Δ background (lane 7–12) were treated with bortezomib (Bz) for 2 h, and phosphatase‐treated, whole‐cell lysates were analyzed by Western blot with anti‐Sre2 serum or anti‐Flag IgG.
We reasoned that if the Rbd2 SHP box functions to recruit Cdc48, we might restore SREBP cleavage by appending a heterologous SHP box to the Rbd2‐G246R mutant. Fusion of a SHP box from the fission yeast Der1‐like protein (SPBC365.08c) to the C‐terminus of Rbd2‐G246R restored its ability to produce Sre1N to wild‐type levels (Fig 8E, G246R‐SHP; Fig 8F, compare lanes 5–6 to 11–12). In contrast, fusion of a mutant Der1‐like SHP box at the equivalent position (G246R‐SHP*) failed to rescue Sre1 cleavage (Fig 8F, lanes 13–14). Parallel results were obtained in Sre2 cleavage assays (Fig 8G). Furthermore, fusion of the non‐homologous Dsc5 UBX domain that recruits Cdc48 to the Dsc E3 ligase partially rescued Sre1 cleavage (Fig EV3) (Stewart et al, 2012). Thus, SREBP cleavage requires Cdc48 recruitment to Rbd2.
Figure EV3. Fusion of UBX domain to Rbd2‐G246R partially rescues Sre1 cleavage.
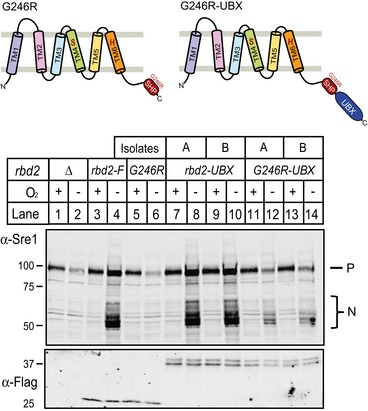
Yeast strains containing Flag‐tagged rbd2‐UBX and rbd2‐G246R‐UBX (diagrammed) were generated in rbd2Δ background by chromosomal integration. Whole‐cell lysates from indicated strains expressing Flag‐tagged Rbd2 variants grown for 3 h in the presence or absence of oxygen were analyzed by Western blotting with anti‐Sre1 or anti‐Flag IgG.
rbd2‐G246R cells failed to generate a detectable SREBP cleavage product despite the presence of an intact catalytic dyad (Fig 8B and D). The inability to detect a cleavage product could be due to a failure to cleave SREBP, or alternatively, a cleaved product may be degraded when Rbd2 fails to recruit Cdc48. To discriminate between these two possibilities, we treated cells with a proteasome inhibitor and examined Sre2 cleavage. As expected, proteasome inhibition resulted in the accumulation of uncleaved Sre2 precursor in both rbd2∆ and rbd2‐S130A cells (Fig 8H, lanes 5–6 and 11–12). rbd2‐G246R cells also accumulated Sre2 precursor, but not a cleaved product, indicating that Cdc48 binding to Rbd2 is required for SREBP cleavage (Fig 8H, lanes 9–10). Thus, the function of Cdc48 is distinct from that shown for p97 with RHBDL4 where p97 acts after cleavage (Fleig et al, 2012). Taken together, these results demonstrate a new function for Cdc48 in mediating Rbd2‐dependent cleavage of yeast SREBPs.
Cdc48 acts as a substrate adaptor for Rbd2
Rhomboid proteases exhibit weak binding affinity for substrates (Dickey et al, 2013). Given that Cdc48 cofactors bind ubiquitin (Stolz et al, 2011), we hypothesized that Cdc48 plays a role in SREBP recognition by recruiting SREBP substrate to Rbd2 through cofactor binding. If this was true, we reasoned that increasing the concentration of Cdc48‐binding defective Rbd2 enzyme would bypass the requirement for Cdc48. To test our hypothesis, we performed Sre1 cleavage analysis in yeast cells expressing rbd2‐Flag‐APEX2, rbd2‐G246R‐Flag‐APEX2, or rbd2‐S130A‐Flag‐APEX2 under control of the constitutive adh1 + promoter in rbd2Δ cells. Indeed, overexpression of rbd2‐G246R fully restored Sre1 cleavage, while overexpression of catalytically dead rbd2‐S130A did not (Fig 9A). In addition, Sre1 precursor levels increased with time in rbd2‐G246R overexpressing cells, indicating that cleaved Sre1N was transcriptionally active and promoted sre1 transcription through a well‐characterized positive feedback loop (Todd et al, 2006). Consistent with the Western blot results, rbd2‐G246R overexpression rescued growth of rbd2Δ cells on CoCl2 while rbd2‐S130A did not (Fig 9B). Compared to rbd2‐Flag expressed from its endogenous promoter, we observed ~14‐fold higher expression of rbd2‐Flag‐APEX2, rbd2‐G246R‐Flag‐APEX2, and rbd2‐S130A‐Flag‐APEX2 using the adh1 + promoter (Fig 9C).
Figure 9. Overexpression of Rbd2 bypasses requirement for Cdc48 binding to Rbd2.
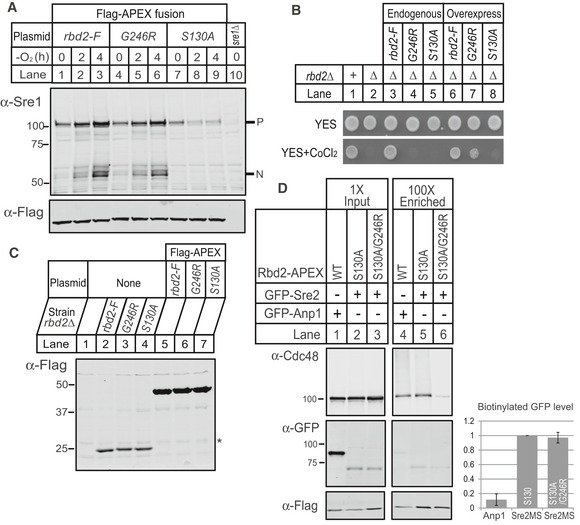
- Western blot of phosphatase‐treated, whole‐cell lysates from yeast strains carrying rbd2‐Flag‐APEX2 (rbd2‐F), rbd2‐G246R‐Flag‐APEX2 (G246R), or rbd2‐S130A‐Flag‐APEX2 (S130A) plasmid in rbd2Δ background and sre1Δ yeast was probed with anti‐Sre1 or anti‐Flag IgG. P and N denote Sre1 precursor and cleaved nuclear forms, respectively.
- Indicated yeast strains (200 cells) containing chromosomal Flag‐tagged rbd2, rbd2‐G246R, or rbd2‐S130A in rbd2Δ background (lane 3–5) or yeast strains (200 cells) carrying rbd2‐Flag‐APEX2, rbd2‐G246R‐Flag‐APEX2, or rbd2‐S130A‐Flag‐APEX2 plasmid in rbd2Δ background (lane 6–8) were grown on rich medium in the absence or presence of cobalt chloride (CoCl2).
- Comparison of Rbd2 protein levels from overexpression system to endogenous expression. Western blot of whole‐cell lysates from yeast strains containing chromosomal Flag‐tagged rbd2, rbd2‐G246R, or rbd2‐S130A in rbd2Δ background and yeast strains carrying rbd2‐Flag‐APEX2, rbd2‐G246R‐Flag‐APEX2, or rbd2‐S130A‐Flag‐APEX2 plasmid in rbd2Δ background was probed with anti‐Flag IgG. For quantification, the intensity of Flag band in each lane was normalized to the non‐specific band (asterisk).
- rbd2Δ cells expressing indicated plasmids of GFP‐Anp1 or GFP‐Sre2MS together with rbd2‐Flag‐APEX2, rbd2‐S130A‐Flag‐APEX2, or rbd2‐S130A/G246R‐Flag‐APEX2 were treated with Bz at a concentration of 1 mM for 2 h prior to biotin‐labeling reaction. After labeling termination, cells were lysed and denatured as described in Materials and Methods. Biotinylated proteins were then enriched using streptavidin magnetic beads. Lysates (1× input) and 100×‐enriched eluates were analyzed by Western blot with IRDye 800CW Streptavidin, anti‐Flag IgG, anti‐Cdc48 serum, and anti‐GFP IgG. Intensity of GFP band in enriched sample was normalized to that in the corresponding input sample, then normalized to the Flag intensity in enriched sample. Relative intensities of biotinylated GFP level are shown in bar graph. Error bars denote standard deviation from three biological replicates.
Lastly using the proximity biotinylation technique, we tested whether Rbd2 and SREBP interact using catalytically dead Rbd2 to capture substrate‐enzyme binding. Proximity biotinylation experiments using endogenous expression of Rbd2‐APEX2 failed to detect an interaction with the known binding partner Cdc48, so we overexpressed Rbd2 fusion proteins using the adh1 + promoter. Rbd2‐S130A‐APEX2 labeled both Cdc48 and GFP‐Sre2MS (Fig 9D, lane 5). Sre2 labeling by Rbd2‐S130A‐APEX2 was specific insomuch as we observed 10‐fold lower labeling of the Golgi mannosyltransferase Anp1 (Fig 9D, lane 4). Consistent with the ability of overexpressed Rbd2‐G246R to support SREBP cleavage, we detected equal labeling of GFP‐Sre2MS in cells overexpressing Rbd2‐S130A/G246R‐APEX2 (Fig 9D, lanes 5–6). As expected, Rbd2‐S130A/G246R‐APEX2 showed reduced Cdc48 labeling (Fig 9D, lane 6). Collectively, these results demonstrate that increasing the concentration of Rbd2 enzyme bypasses the requirement of Cdc48 binding for Sre1 cleavage, defining a role for Cdc48 in substrate recruitment.
Discussion
Rbd2 is the first SREBP protease identified in fission yeast (Kim et al, 2015). Using genetic interaction mapping, we independently identified Rbd2 as a Golgi‐localized rhomboid protease required for proteolytic activation of yeast SREBPs. Deleting rbd2 or mutating its catalytic residues prevented cleavage of both Sre1 and Sre2 (Figs 2 and 3). Our mammalian cell‐based, rhomboid cleavage assays using heterologous substrates showed that Rbd2 is a bona fide rhomboid protease with a conserved, Ser‐His catalytic dyad (Fig 4). Notably, Rbd2 showed novel substrate specificity: Rbd2 cleaved neither the canonical D. melanogaster substrate Spitz nor an alterative P. falciparum substrate EBA175, but cleaved P. stuartii TatA, the most efficient rhomboid substrate (Strisovsky et al, 2009; Moin & Urban, 2012). To date, characterized rhomboid proteases cleave substrates in the membrane after residues with small side chains (Strisovsky et al, 2009; Urban, 2010). Although Rbd2 cleavage of TatA required substrate helix‐destabilizing residues, Rbd2 preferentially cleaved TatA substrates after large hydrophobic side chains (Leu and Phe) (Fig 4F), revealing a unique property not observed for any other rhomboid protease.
Mammalian SREBPs are activated by regulated intramembrane proteolysis (Brown et al, 2000). The serine protease S1P cleaves SREBP in the luminal loop to produce a type‐II (Nin‐Cout), single transmembrane intermediate that is subsequently cleaved in the membrane by the zinc metalloprotease S2P. Rhomboid proteases preferentially cleave transmembrane substrates with a type‐I (Nout‐Cin) orientation (Urban & Freeman, 2003; Ha, 2009). Despite the sequence requirements observed for Rbd2 in TatA cleavage, mutation of any single large hydrophobic residue in the N‐terminal end of TM2 did not block Sre2MS cleavage (Fig 5B). However, the inability to map the Sre2MS cleavage site(s) prevented us from determining whether the mutants support cleavage by the shifting cleavage site to other large, hydrophobic residues. Such cleavage site shifts in substrate mutants are common (Strisovsky et al, 2009; Moin & Urban, 2012). Our previous mutagenesis studies showed that Sre2MS cleavage required multiple residues in its type‐I oriented TM2, while we found no essential residues in the type‐II oriented TM1 (Cheung & Espenshade, 2013). Here, we assigned functions for these residues to either Dsc E3 ligase activity or Rbd2 cleavage. Lysine 743 in the luminal loop was the only residue required for Rbd2‐mediated Sre2MS cleavage, while others were involved in Dsc E3 ligase function (Fig 5 and Appendix Figs S1 and S2). Precisely how this luminal lysine residue in Sre2 promotes cleavage remains to be determined.
Should Rbd2 cleave SREBP between the transmembrane segments, it would functionally replace S1P by initiating SREBP activation and yield a type‐II intermediate requiring additional processing. Importantly, the active Sre1N and Sre2N terminate at a cytosolic position, ~10 amino acids prior to TM1 (Stewart et al, 2011; Cheung & Espenshade, 2013). These observations suggest that SREBP activation requires a second protease. We previously proposed that SREBP activation required the proteasome (Stewart et al, 2011), but results presented in this study suggest otherwise. In the earlier study, we used a temperature‐sensitive mutation in mts3 to impair 19S proteasome function, and we observed decreased levels of Sre1N under low oxygen (Stewart et al, 2011). However, in that experiment, mts3‐1 cells required a 3‐h preincubation at non‐permissive temperature, and Sre1 precursor levels also decreased during the course of this experiment, suggesting that proteasome inactivation had indirect effects on SREBP cleavage. In the current study, we observed that cleavage of Sre2 was not inhibited in mts3‐1 cells grown for three hours at the non‐permissive temperature (Fig 3A). Furthermore, acute proteasome inhibition with bortezomib did not block Sre2 processing (Fig 3B). Collectively, these data suggest that an undiscovered protease, rather than the proteasome, functions in SREBP proteolytic processing downstream of Rbd2, and that fission yeast may employ a new variation of RIP to activate SREBPs.
Parallel studies in A. fumigatus recently identified an Rbd2 homolog, RbdA/B, as required for SREBP activation, demonstrating that this function is conserved in ascomycetous fungi (Dhingra et al, 2016; Vaknin et al, 2016). In addition, studies in Aspergillus nidulans found that SREBP activation required both the Dsc E3 ligase and the signal peptide peptidase SppA, an aspartyl protease (Bat‐Ochir et al, 2016). Taken together, these recent Aspergillus studies support a model for SREBP cleavage in which Rbd2 initiates activation followed by SppA cleavage. However, unlike A. nidulans SREBP cleavage, S. pombe Sre1 cleavage did not require Ypf1, the S. pombe signal peptide peptidase homolog, or two other aspartyl proteases Yps1 or Sxa1 (Fig EV4), leaving unknown the identity of the second SREBP protease in fission yeast.
Figure EV4. Sre1 cleavage does not require signal peptide peptidase ypf1 .

Western blot of phosphatase‐treated, whole‐cell lysates from wild‐type cells and the indicated mutants grown for 3 h in the presence or absence of oxygen was probed with anti‐Sre1 IgG. P and N denote Sre1 precursor and cleaved forms, respectively.
Prior to these studies, it was known that SREBP cleavage required cdc48, but Cdc48's role was unclear because its binding to the Dsc E3 ligase was not required for SREBP cleavage (Stewart et al, 2012). Here, we demonstrate that SREBP cleavage requires Cdc48 binding to the C‐terminal SHP box in Rbd2 (Fig 8). Cells expressing an Rbd2 mutant defective for Cdc48 binding accumulated Sre2 precursor with proteasome inhibition rather than cleaved products (Fig 8H). Thus, Cdc48 functions in Rbd2‐dependent cleavage. The human rhomboid protease RHBDL4 binds ubiquitinylated substrates through a ubiquitin‐interacting motif and cleaves them in the ER to promote ERAD (Fleig et al, 2012). Interestingly, RHBDL4 also binds to p97/Cdc48 but through a different motif, called VBM. p97/Cdc48 binding to RHBDL4 facilitates extraction of cleaved products for proteasomal degradation. Cdc48 and its cofactors bind to ubiquitin (Stolz et al, 2011). In a function similar to that for the RHBDL4 ubiquitin‐interacting motif, we propose that Cdc48 with its cofactors is a receptor for ubiquitin, thereby recruiting ubiquitinylated SREBPs to Rbd2. While p97 functions after RHDBL4 cleavage, here Cdc48 functions in recognizing substrate prior to the cleavage event. Supporting this model, overexpression of Rbd2 that cannot bind Cdc48 rescued Sre1 cleavage, bypassing the requirement for Cdc48 (Fig 9).
As noted, activation of A. fumigatus SREBP requires a rhomboid protease (Dhingra et al, 2016; Vaknin et al, 2016). Our bioinformatic analysis identified a conserved SHP box in the cytosolic C‐terminus of A. fumigatus RbdB (Fig EV5A) (Sato & Hampton, 2006). GST‐RbdB (aa 207–272) bound to Cdc48 in vitro, but the binding was ~7‐fold lower than GST‐Rbd2 (Fig EV5B). Reduced binding to RbdB may be due to an asparagine substitution at the G246 position in the Rbd2 SHP box (Fig EV5A). S. cerevisiae Rbd2 which does not contain a conserved SHP box was used as a negative control. Testing whether Cdc48 binding to RbdB is also required for A. fumigatus SREBP, cleavage activation is an important next step to further understand how Cdc48 functions with rhomboid proteases.
Figure EV5. Aspergillus fumigatus RbdB contains a conserved SHP box.

- Alignment of SHP box sequences from Schizosaccharomyces pombe Rbd2 C‐terminal SHP box (aa 242–251) and A. fumigatus RbdB C‐terminal SHP box (aa 263–272). Asterisks denote identical residues, colon marks conservative substitution, and dots mark semi‐conservative substitutions
- Recombinant proteins GST‐fused S. pombe Rbd2 C‐terminus (aa 200–251), A. fumigatus RbdB C‐terminus (aa 211–272), S. cerevisiae Rbd2 C‐terminus (aa 192–262), and GST were bound to GST magnetic beads and incubated with S. pombe cytosol fraction from wild‐type cells. 5× bound fractions were probed for anti‐Cdc48 and anti‐GST IgG. The S. pombe cytosol fraction was used for 1× input (In.) loading.
The Dsc1 subunit of the Dsc E3 ligase contains a RING domain and functions as an ubiquitin ligase with the E2 ubiquitin‐conjugating enzyme Ubc4 (Raychaudhuri & Espenshade, 2015). Mutation of the Dsc1 RING domain, deletion of dsc1‐dsc5, or mutation of ubc4 blocks SREBP cleavage (Stewart et al, 2011, 2012). In addition, Sre2 binds to the Dsc E3 ligase (Stewart et al, 2011; Lloyd et al, 2013). Together, these data suggest that SREBPs are substrates for the Dsc E3 ubiquitin ligase. However, recently we found that inhibition of Dsc E3 ligase activity prevents ER‐to‐Golgi transport of the Dsc complex, confounding the interpretation of these dsc mutant phenotypes (Raychaudhuri & Espenshade, 2015). Here, we observed that in rbd2∆ cells, SREBPs are degraded by the proteasome in a Dsc‐dependent manner (Figs 3 and EV1). Although SREBP ubiquitinylation remains to be shown, these data provide additional support for SREBPs as Dsc E3 ligase substrates. Interestingly, preliminary studies indicate that cytosolic lysines in Sre2MS are not required for cleavage, suggesting that SREBP ubiquitinylation may involve non‐classical sites. Ubc4 conjugates mono‐ubiquitin to proteins, while proteasomal degradation typically requires poly‐ubiquitin chains (Seino et al, 2003; Rodrigo‐Brenni & Morgan, 2007). Whether other ubiquitinylation enzymes are required for SREBP degradation in rbd2∆ cells and whether these enzymes are also required for SREBP cleavage remains to be tested. However, our studies using hrd1∆ and doa10∆ cells indicate that these two ERAD E3 ligases are not required for cleavage or degradation (Fig EV1A).
Combined with our past work, these new findings lead us to speculate on a new model for the activation of Sre1 (Fig 10). Following oxygen‐regulated transport by the SCAP homolog Scp1 to the Golgi, Sre1 is ubiquitinylated by the Dsc E3 ligase complex. Rbd2 cleaves ubiquitinylated Sre1 in the second transmembrane segment or luminal juxtamembrane region in a reaction that requires substrate recruitment by Cdc48. This Sre1 intermediate is then cleaved by a second, unidentified protease to produce the soluble, active transcription factor. Sre2 activation follows a similar pathway, but ER‐to‐Golgi transport is constitutive. In the absence of Rbd2 cleavage (Fig 10 dashed box), ubiquitinylated SREBPs are degraded by the proteasome. Thus, changes in Rbd2 activity will control the fate of SREBP substrates between protein processing and degradation. This mechanism provides a new control point for the regulation of SREBP activity in fungi. In mammals, a related regulatory mechanism exists: when S1P is inhibited, SCAP is targeted to the lysosome for degradation, leading to reduced pathway activity (Shao & Espenshade, 2014).
Figure 10. Model for yeast SREBP cleavage.
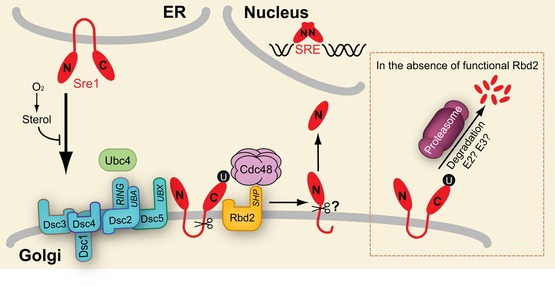
Under low sterol and oxygen conditions, Sre1 exits the ER and travels to the Golgi. Golgi‐localized Sre1 is ubiquitinylated by E2 Ubc4 and Dsc E3 ligase. Ubiquitinylated Sre1 is recruited by Cdc48 and cleaved by Rbd2. The intermediate cleaved product is further processed by a second, unknown protease to generate Sre1N that traffics to the nucleus to activate transcription. When Rbd2 is inactive (dashed box), ubiquitinylated Sre1 is subjected to proteasomal degradation. SRE, sterol regulatory element; U, either mono‐ubiquitin or poly‐ubiquitin.
In summary, convergent evidence from studies in S. pombe and A. fumigatus demonstrates that SREBP activation requires a rhomboid protease, Rbd2 and RbdA/B, respectively (Bat‐Ochir et al, 2016; Dhingra et al, 2016; Vaknin et al, 2016). Fungal SREBPs are required for growth under hypoxic conditions and for virulence of human opportunistic pathogens (Bien & Espenshade, 2010). In addition, SREBP is required for virulence of Penicillium digitatum, a major citrus fruit fungal pathogen (Liu et al, 2015) and P. digitatum contains an rbd2 homolog (ORF name, PDIP_45580). Thus, identification of a fungal SREBP protease opens the door to development of rhomboid inhibitors that could act as antifungals to improve both clinical and agricultural outcomes.
Materials and Methods
Materials and standard methods (cell culture, plasmid construction, linkage analysis, Western blotting, immunoprecipitation, in vitro binding assay, microscopy, and mass spectrometry) are described in the Appendix Supplementary Methods. Yeast strains and oligonucleotide primers used in this study may be found in Appendix Tables S1 and S2, respectively.
Yeast strain construction
The rbd2‐integrating plasmids contained rbd2 +, rbd2‐S130A‐3xFlag, rbd2‐H182A‐3xFlag, rbd2‐G244R‐3xFlag, rbd2‐G246R‐3xFlag, rbd2‐SHP‐3xFlag, or rbd2‐SHP*‐3xFlag coding sequence flanked by 600 bp of upstream genomic sequence and 600 bp of downstream genomic sequence with a his3 + marker for chromosomal reconstitution of histidine auxotrophy. Plasmids containing rbd2 wild type and mutants were linearized with AscI and transformed into rbd2Δ strain (PEY1681) and selected for minimal medium lacking histidine to generate strains carrying integrated wild‐type and mutant alleles of rbd2 at the his3 + locus. Isolates expressing Rbd2‐3xFlag were confirmed by Western blotting. The rbd2‐Flag‐APEX2 plasmid and its derivatives were transformed into rbd2Δ strain and selected for minimal medium lacking leucine to generate strains carrying rbd2‐Flag‐APEX2, rbd2‐S130A‐Flag‐APEX2, rbd2‐G246R‐Flag‐APEX2, or rbd2‐G246R/S130A‐Flag‐APEX2.
Genetic interaction study
The genetic interaction study was performed as described (Frost et al, 2012). Included in the query set for the study were the dsc genes and multiple alleles of cdc48. Hierarchical clustering profiles were analyzed for genetic interaction profiles similar to the dsc genes.
Biotin‐phenol labeling
The original biotin‐phenol labeling protocol for mammalian cells was modified for yeast (Hung et al, 2016; Hwang & Espenshade, 2016). Exponentially growing cells (5 × 108) were harvested and washed with water. Cell pellets were resuspended in 1 ml of 1.2 M sorbitol dissolved in H2O. Biotin‐phenol (2.5 mM) was added, and the cells were incubated for another 1 h at room temperature. H2O2 (1 mM) was added for 1 min to initiate biotin‐phenol labeling, and cells were quickly spun down. To stop the reaction, the solution was aspirated off and cell pellets were then washed four times with a quenching solution consisting of 5 mM Trolox, 10 mM sodium azide, and 10 mM sodium ascorbate in 1.2 M sorbitol dissolved in H2O. Cell pellets were washed with 1.2 M sorbitol once more and lysed by addition of 27 mM NaOH, 1% (v/v) 2‐mercaptoethanol for 10 min on ice. Total protein was precipitated with trichloroacetic acid (20% w/v) followed by acetone wash. Proteins were solubilized in 200 μl lysis buffer (25 mM Tris pH 7.4, 150 mM NaCl, 1% SDS) containing Complete Protease Inhibitor EDTA free by sonication. After denaturation by heating at 75°C for 15 min, protein samples were either directly analyzed by Western blotting or subjected to enrichment of biotinylated proteins. For enrichment, proteins (1–2 mg) were first dialyzed in dialysis buffer (25 mM Tris pH 7.4, 150 mM NaCl, 0.2% SDS) using 3.5K MWCO dialysis tubing (Thermoscientific) for 2–3 h at room temperature, and then incubated with 50–100 μl streptavidin‐coated magnetic beads for 2–3 h at room temperature. Streptavidin beads were then washed with 0.3% SDS‐containing wash buffer (25 mM Tris pH 7.4, 150 mM NaCl, 0.3% SDS) six times followed by 2 × wash with 2 M urea/50 mM Tris pH 7.4, 1 × wash with 1 M KCl, and 1 × with wash buffer. Biotinylated proteins were eluted by incubating the beads with 30 μl 2 × SDS sample loading buffer and heating to 75°C for 15 min.
Author contributions
JH, DR, and PJE designed and performed experiments except as noted. SR and HG contributed to Figs 3 and 6, respectively. SU and LC performed experiments in Fig 4. AF performed genetic interaction study. JH, DR, SU, and PJE wrote the manuscript with contributions from the other authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
This work was supported by grants from the National Institutes of Health (R01HL077588, PJE; R01AI066025, SU; T32GM007445, DR HG LC), the Howard Hughes Medical Institute (SU), and the David and Lucile Packard Foundation (SU). We are grateful to all members of the Espenshade and Urban laboratories for insightful discussions and general assistance.
The EMBO Journal (2016) 35: 2332–2349
References
- Baker RP, Wijetilaka R, Urban S (2006) Two Plasmodium rhomboid proteases preferentially cleave different adhesins implicated in all invasive stages of malaria. PLoS Pathog 2: e113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker RP, Urban S (2012) Architectural and thermodynamic principles underlying intramembrane protease function. Nat Chem Biol 8: 759–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bat‐Ochir C, Kwak JY, Koh SK, Jeon MH, Chung D, Lee YW, Chae SK (2016) The signal peptide peptidase SppA is involved in sterol regulatory element‐binding protein cleavage and hypoxia adaptation in Aspergillus nidulans . Mol Microbiol 100: 635–655 [DOI] [PubMed] [Google Scholar]
- Baxt LA, Baker RP, Singh U, Urban S (2008) An Entamoeba histolytica rhomboid protease with atypical specificity cleaves a surface lectin involved in phagocytosis and immune evasion. Genes Dev 22: 1636–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergbold N, Lemberg MK (2013) Emerging role of rhomboid family proteins in mammalian biology and disease. Biochim Biophys Acta 1828: 2840–2848 [DOI] [PubMed] [Google Scholar]
- Bien CM, Espenshade PJ (2010) Sterol regulatory element binding proteins in fungi: hypoxic transcription factors linked to pathogenesis. Eukaryot Cell 9: 352–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Ye J, Rawson RB, Goldstein JL (2000) Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100: 391–398 [DOI] [PubMed] [Google Scholar]
- Chang YC, Bien CM, Lee H, Espenshade PJ, Kwon‐Chung KJ (2007) Sre1p, a regulator of oxygen sensing and sterol homeostasis, is required for virulence in Cryptococcus neoformans . Mol Microbiol 64: 614–629 [DOI] [PubMed] [Google Scholar]
- Chang YC, Ingavale SS, Bien C, Espenshade P, Kwon‐Chung KJ (2009) Conservation of the sterol regulatory element‐binding protein pathway and its pathobiological importance in Cryptococcus neoformans . Eukaryot Cell 8: 1770–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung R, Espenshade PJ (2013) Structural requirements for sterol regulatory element‐binding protein (SREBP) cleavage in fission yeast. J Biol Chem 288: 20351–20360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun CD, Liu OW, Madhani HD (2007) A link between virulence and homeostatic responses to hypoxia during infection by the human fungal pathogen Cryptococcus neoformans . PLoS Pathog 3: e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra S, Kowlaski CH, Thammahong A, Beattie SR, Bultman KM, Cramer RA (2016) RbdB, a rhomboid protease critical for SREBP activation and virulence in Aspergillus fumigatus . mSphere 1: e00035‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey SW, Baker RP, Cho S, Urban S (2013) Proteolysis inside the membrane is a rate‐governed reaction not driven by substrate affinity. Cell 155: 1270–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman HC, Nosanchuk JD, Webber JB, Emerson RJ, Camesano TA, Casadevall A (2005) Microstructure of cell wall‐associated melanin in the human pathogenic fungus Cryptococcus neoformans . Biochemistry 44: 3683–3693 [DOI] [PubMed] [Google Scholar]
- Espenshade PJ, Hughes AL (2007) Regulation of sterol synthesis in eukaryotes. Annu Rev Genet 41: 401–427 [DOI] [PubMed] [Google Scholar]
- Fleig L, Bergbold N, Sahasrabudhe P, Geiger B, Kaltak L, Lemberg MK (2012) Ubiquitin‐dependent intramembrane rhomboid protease promotes ERAD of membrane proteins. Mol Cell 47: 558–569 [DOI] [PubMed] [Google Scholar]
- Frost A, Elgort MG, Brandman O, Ives C, Collins SR, Miller‐Vedam L, Weibezahn J, Hein MY, Poser I, Mann M, Hyman AA, Weissman JS (2012) Functional repurposing revealed by comparing S. pombe and S. cerevisiae genetic interactions. Cell 149: 1339–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon C, McGurk G, Wallace M, Hastie ND (1996) A conditional lethal mutant in the fission yeast 26 S protease subunit mts3+ is defective in metaphase to anaphase transition. J Biol Chem 271: 5704–5711 [DOI] [PubMed] [Google Scholar]
- Grahl N, Shepardson KM, Chung D, Cramer RA (2012) Hypoxia and fungal pathogenesis: to air or not to air? Eukaryot Cell 11: 560–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha Y (2009) Structure and mechanism of intramembrane protease. Semin Cell Dev Biol 20: 240–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL, Todd BL, Espenshade PJ (2005) SREBP pathway responds to sterols and functions as an oxygen sensor in fission yeast. Cell 120: 831–842 [DOI] [PubMed] [Google Scholar]
- Hung V, Udeshi ND, Lam SS, Loh KH, Cox KJ, Pedram K, Carr SA, Ting AY (2016) Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat Protoc 11: 456–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Espenshade PJ (2016) Proximity‐dependent biotin labeling in yeast using the engineered ascorbate peroxidase APEX2. Biochem J 473: 2463–2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10: 845–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ha HJ, Kim S, Choi AR, Lee SJ, Hoe KL, Kim DU (2015) Identification of Rbd2 as a candidate protease for sterol regulatory element binding protein (SREBP) cleavage in fission yeast. Biochem Biophys Res Commun 468: 606–610 [DOI] [PubMed] [Google Scholar]
- Koffas MA, Ramamoorthi R, Pine WA, Sinskey AJ, Stephanopoulos G (1998) Sequence of the Corynebacterium glutamicum pyruvate carboxylase gene. Appl Microbiol Biotechnol 50: 346–352 [DOI] [PubMed] [Google Scholar]
- Lane S, Zhou S, Pan T, Dai Q, Liu HP (2001) The basic helix‐loop‐helix transcription factor Cph2 regulates hyphal development in Candida albicans partly via Tec1. Mol Cell Biol 21: 6418–6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberg MK (2013) Sampling the membrane: function of rhomboid‐family proteins. Trends Cell Biol 23: 210–217 [DOI] [PubMed] [Google Scholar]
- Liu J, Yuan Y, Wu Z, Li N, Chen Y, Qin T, Geng H, Xiong L, Liu D (2015) A novel sterol regulatory element‐binding protein gene (sreA) identified in penicillium digitatum is required for prochloraz resistance, full virulence and erg11 (cyp51) regulation. PLoS One 10: e0117115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd SJ, Raychaudhuri S, Espenshade PJ (2013) Subunit architecture of the Golgi Dsc E3 ligase required for sterol regulatory element‐binding protein (SREBP) cleavage in fission yeast. J Biol Chem 288: 21043–21054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuyama A, Arai R, Yashiroda Y, Shirai A, Kamata A, Sekido S, Kobayashi Y, Hashimoto A, Hamamoto M, Hiraoka Y, Horinouchi S, Yoshida M (2006) ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe . Nat Biotechnol 24: 841–847 [DOI] [PubMed] [Google Scholar]
- Moin SM, Urban S (2012) Membrane immersion allows rhomboid proteases to achieve specificity by reading transmembrane segment dynamics. eLife 1: e00173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olzmann JA, Kopito RR, Christianson JC (2013) The mammalian endoplasmic reticulum‐associated degradation system. Cold Spring Harb Perspect Biol 5: a013185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rather P (2013) Role of rhomboid proteases in bacteria. Biochim Biophys Acta 1828: 2849–2854 [DOI] [PubMed] [Google Scholar]
- Raychaudhuri S, Espenshade PJ (2015) Endoplasmic reticulum exit of golgi‐resident defective for SREBP cleavage (Dsc) E3 ligase complex requires its activity. J Biol Chem 290: 14430–14440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY (2013) Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339: 1328–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigo‐Brenni MC, Morgan DO (2007) Sequential E2s drive polyubiquitin chain assembly on APC targets. Cell 130: 127–139 [DOI] [PubMed] [Google Scholar]
- Sato BK, Hampton RY (2006) Yeast Derlin Dfm1 interacts with Cdc48 and functions in ER homeostasis. Yeast 23: 1053–1064 [DOI] [PubMed] [Google Scholar]
- Seino H, Kishi T, Nishitani H, Yamao F (2003) Two ubiquitin‐conjugating enzymes, UbcP1/Ubc4 and UbcP4/Ubc11, have distinct functions for ubiquitination of mitotic cyclin. Mol Cell Biol 23: 3497–3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, Espenshade PJ (2012) Expanding roles for SREBP in metabolism. Cell Metab 16: 414–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, Espenshade PJ (2014) Sterol regulatory element‐binding protein (SREBP) cleavage regulates Golgi‐to‐endoplasmic reticulum recycling of SREBP cleavage‐activating protein (SCAP). J Biol Chem 289: 7547–7557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart EV, Nwosu CC, Tong Z, Roguev A, Cummins TD, Kim DU, Hayles J, Park HO, Hoe KL, Powell DW, Krogan NJ, Espenshade PJ (2011) Yeast SREBP cleavage activation requires the Golgi Dsc E3 ligase complex. Mol Cell 42: 160–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart EV, Lloyd SJ, Burg JS, Nwosu CC, Lintner RE, Daza R, Russ C, Ponchner K, Nusbaum C, Espenshade PJ (2012) Yeast sterol regulatory element‐binding protein (SREBP) cleavage requires Cdc48 and Dsc5, a ubiquitin regulatory X domain‐containing subunit of the Golgi Dsc E3 ligase. J Biol Chem 287: 672–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolz A, Hilt W, Buchberger A, Wolf DH (2011) Cdc48: a power machine in protein degradation. Trends Biochem Sci 36: 515–523 [DOI] [PubMed] [Google Scholar]
- Strisovsky K, Sharpe HJ, Freeman M (2009) Sequence‐specific intramembrane proteolysis: identification of a recognition motif in rhomboid substrates. Mol Cell 36: 1048–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd BL, Stewart EV, Burg JS, Hughes AL, Espenshade PJ (2006) Sterol regulatory element binding protein is a principal regulator of anaerobic gene expression in fission yeast. Mol Cell Biol 26: 2817–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S, Freeman M (2003) Substrate specificity of rhomboid intramembrane proteases is governed by helix‐breaking residues in the substrate transmembrane domain. Mol Cell 11: 1425–1434 [DOI] [PubMed] [Google Scholar]
- Urban S (2006) Rhomboid proteins: conserved membrane proteases with divergent biological functions. Genes Dev 20: 3054–3068 [DOI] [PubMed] [Google Scholar]
- Urban S, Baker RP (2008) In vivo analysis reveals substrate‐gating mutants of a rhomboid intramembrane protease display increased activity in living cells. Biol Chem 389: 1107–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S, Shi Y (2008) Core principles of intramembrane proteolysis: comparison of rhomboid and site‐2 family proteases. Curr Opin Struct Biol 18: 432–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S (2010) Taking the plunge: integrating structural, enzymatic and computational insights into a unified model for membrane‐immersed rhomboid proteolysis. Biochem J 425: 501–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban S, Dickey SW (2011) The rhomboid protease family: a decade of progress on function and mechanism. Genome Biol 12: 231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaknin Y, Hillmann F, Iannitti R, Ben Baruch N, Sandovsky‐Losica H, Shadkchan Y, Romani L, Brakhage A, Kniemeyer O, Osherov N (2016) Identification and characterization of a novel Aspergillus fumigatus rhomboid family putative protease, RbdA, involved in hypoxia sensing and virulence. Infect Immun 84: 1866–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willger SD, Puttikamonkul S, Kim KH, Burritt JB, Grahl N, Metzler LJ, Barbuch R, Bard M, Lawrence CB, Cramer RA Jr (2008) A sterol‐regulatory element binding protein is required for cell polarity, hypoxia adaptation, azole drug resistance, and virulence in Aspergillus fumigatus . PLoS Pathog 4: e1000200 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File