

Abstract
Because of the unique combination of their attractive properties, porous ceramics are considered as candidate materials for several engineering applications. The production of porous ceramics from polysiloxane precursors offers advantages in terms of simple processing methodology, low processing cost, and easy control over porosity and other properties of the resultant ceramics. Therefore, considerable research has been conducted to produce various Si(O)C-based ceramics from polysiloxane precursors by employing different processing strategies. The complete potential of these materials can only be achieved when properties are tailored for a specific application, whereas the control over these properties is highly dependent on the processing route. This review deals with processing strategies of polysiloxane-derived porous ceramics. The essential features of processing strategies—replica, sacrificial template, direct foaming and reaction techniques—are explained and the available literature reports are thoroughly reviewed with particular regard to the critical issues that affect pore characteristics. A short note on the cross-linking methods of polysiloxanes is also provided. The potential of each processing strategy on porosity and strength of the resultant SiC or SiOC ceramics is outlined.
Keywords: porous ceramics, polysiloxane, processing
Introduction
Ever since the tailored porosity was identified as an essential property for several engineering components, extensive research had been carried out to explore the potential of various ceramic systems. Owing to their unique combination of properties, such as low density, controlled permeability, high thermal shock resistance, high specific strength and chemical stability at high temperatures, porous ceramics are considered as candidate materials for use in a wide range of applications including filters for diesel particulates, molten metals and hot gases, gas burner media, vacuum chucks, preforms for metal-matrix composites, membrane supports for hydrogen separation, lightweight structural materials and porous bioimplants [1–12].
Preceramic polymers can be converted to ceramic materials upon controlled thermal treatment. In this method, the polymer or alkoxide is first shaped, then cross-linked and/or gelled, and finally converted to the desired ceramic component through pyrolysis at suitably high temperatures. The processing of porous ceramics using preceramic polymers offers many advantages compared to ceramic powders. These include (i) low processing temperatures or low energy consumption for the synthesis compared to high temperatures required for sintering of ceramic powders [13–17], (ii) no additives required for densification [1, 4], (iii) a variety of low-cost plastic-forming techniques can be applied with easy control over rheological properties by modified molecular architecture; important plastic-forming techniques include injection molding, extrusion, resin transfer molding, melt spinning [4, 9, 15], (iv) machining before ceramization can be avoided, thereby reducing tool wear and brittle fracture [1, 5, 10], (v) easy handling before heat treatment, because preceramic polymers can effectively bind the parts at low temperatures [10], (vi) utilization of unique polymeric properties that cannot be found in ceramic powders, such as appreciable plasticity, in situ gas evolution ability, appreciable CO2 solubility, and appreciable solubility of preceramic polymers in organic solvents [9, 10, 18, 19], (vii) nanostructures (wires, belts, tubes, etc) can be created directly during the pyrolysis of catalyst-containing preceramic polymers [10, 11], and (viii) ceramic products containing unique combination of polymer-like nanostructures with ceramic-like properties (hardness, creep resistance and oxidation resistance) can be obtained [6, 9, 10]. Hence, several polymers with different substituents were synthesized, blended and used as precursors for fabricating a variety of porous ceramics such as zirconia, alumina, silica, silicon carbide, silicon oxycarbide, mullite, cordierite, etc. Currently, preceramic polymer-derived porous ceramics are used to produce micro or macro porous materials, ceramic fibers, coatings, joints, nanotubes, micro-gears and micro-tubes for microelectromechanical systems (MEMS), matrices and interfaces in ceramic composites, etc [4, 5, 9, 20, 21].
Since the pioneering work on polycarbosilane by Yajima et al [13, 14], many Si-based polymeric precursors such as polysiloxanes, polysilanes, polysilazanes, polycarbosilanes have been prepared to fabricate a range of ceramic compositions [22, 23]. These polymers, upon controlled pyrolysis, yield ceramic residue through the elimination of organic moieties by breaking of C–H bonds and releasing H2, CH4 or other volatile compounds [22, 24, 25]. Very complex nanostructures with nanocrystalline phases, unique amorphous phases and a free-carbon phase were obtained using preceramic polymers [26–33]. Polysiloxanes are generally denoted as silicones and are superior in heat and chemical resistance when compared to many other polymers. Thus, silicones are widely used as sealants, electric insulator coatings, surface treatments for glass materials, heat resistant oils and chemically stable elastomers [10, 34]. The general synthesis method for the preparation of polysiloxanes comprises the reaction of chloro (organo) silanes with water [10]. Silicones intrinsically maintain their viscoelastic liquid nature over a wide temperature range. For example, glass transition temperatures of polydimethylsiloxane (PDMS) and polymethylphenylsiloxane (PMPhS) are −127 and –86 °C, respectively [34–36]. The cross-linked elastomers can accept relatively high extension without cracks when compared to other polycarbosilane, polysilazane and polysilanes. This particular property is useful to shape the starting materials into desired forms by molding, casting, injection, impregnation or extrusion [34]. The advantages of using polysiloxane precursor for porous ceramics include easy storage and processing, the highest ceramic yield [37], low processing temperature (1000–1200 °C) [38–42], excellent mechanical strength [43–45] despite the amorphous phase, excellent thermal shock resistance [46], good thermochemical stability [47, 48], and excellent creep resistance, better than that of vitreous silica [27]. Accordingly, extensive research has been carried out to fabricate a wide range of porous ceramic structures from polysiloxane precursors by employing various processing methods.
The complete potential of polymer-derived porous ceramics can only be achieved when properties are tailored for a specific application, whereas proper control over the properties is highly dependent on the processing route. The present review aims at providing a comprehensive understanding of several processing strategies and resultant properties of polysiloxane-derived porous ceramics. The group of polysiloxanes addressed in this article includes polysiloxane, polycarbosilane and polysilanes, and the ceramics include SiC, SiOC, mullite or cordierite.
Processing strategies
The simplest approach for the fabrication of porous ceramics is the partial sintering of powder compacts at lower or higher temperatures than optimum, resulting in a porosity of below 60% [49–51]. Many advanced processing strategies have been additionally proposed for the production of polysiloxane-derived porous ceramics. The strategic approaches explained in this article are mainly classified into replica, sacrificial template, direct foaming and reaction technique. The replica, sacrificial template and direct foaming techniques deal mainly with the pore formation, and the reaction technique deals with the matrix. The basic principles of these strategies are schematically presented in figure 1 and a list of porous ceramics of different chemical compositions produced with these strategies is given in table 1. The essential features of each of these strategies are systematically discussed and compared in terms of the microstructural and pore characteristics of the ceramic materials. More importantly, the influence of each processing strategy on microstructure and mechanical properties of the polysiloxane-derived porous ceramics is critically discussed.
Figure 1.
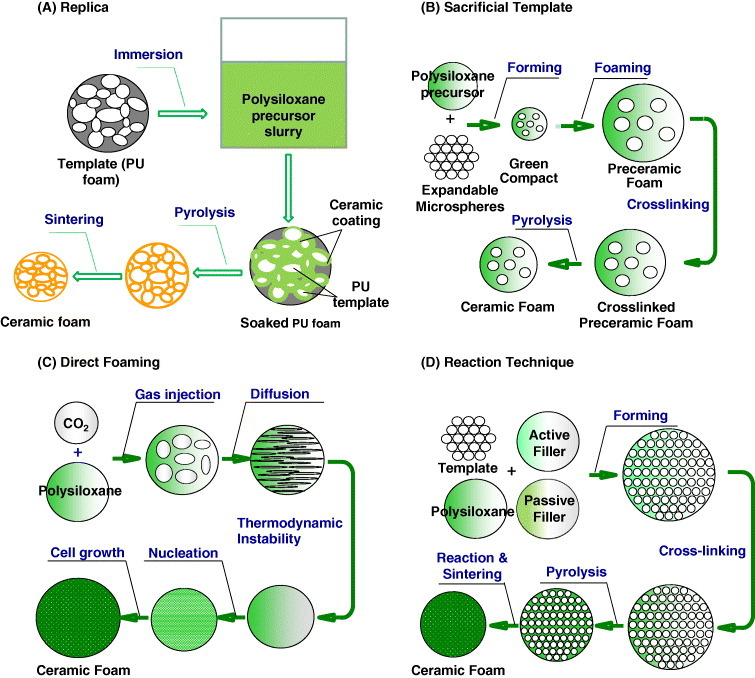
Scheme of processing strategies for the production of polysiloxane-derived porous ceramics: (A) replica, (B) sacrificial template, (C) direct foaming and (D) reaction technique.
Table 1.
The classification of major processing strategies and corresponding compositions of porous ceramics produced from polysiloxane precursors.
| Processing strategy | Composition | Remarks | References |
|---|---|---|---|
| I. Replica | |||
| SiC | Open cells | [57–61, 63] | |
| Cell size: >150 μm | |||
| Porosity range: 85–96% | |||
| SiC–Si3N4 | [57, 59, 60] | ||
| SiC–TiC | [57] | ||
| SiOC/C | [54] | ||
| II. Sacrificial template | |||
| SiC | Open cells | [68, 72] | |
| Cell size: 10–100 μm | |||
| SiOC | Open or closed cells | [69, 70] | |
| Cell density: >104 cells cm-3 | |||
| Porosity range: 21–80% | |||
| Expandable or PMMA templates | SiOC | Open, closed or partially interconnected cells | [67, 73–76] |
| Cell size: 0.5–80 μm | |||
| Cell density: >109 cells cm-3 | |||
| Porosity range: 56–88% | |||
| SiC | [17, 77] | ||
| Expanded (hollow) templates | SiOC | Closed cells | [66] |
| Cell size: <30 μm | |||
| Cell density: >109 cells cm-3 | |||
| Porosity range: 70–87% | |||
| III. Direct foaming | |||
| Foaming by chemical agent | SiOC | Open, closed or interconnected cells | [6, 78, 81–85, 87, 139] |
| Cell size: 80–800 μm | |||
| Porosity range: 75–90% | |||
| SiOC + SiC | Open cells | [82] | |
| Cell size: 100–700 μm | |||
| Foaming using CO2 | |||
| Batch process | SiOC | Closed cells | [79, 90, 92, 96] |
| Cell size: 2–50 μm | |||
| Cell density: 107–1012 cells cm-3 | |||
| Porosity: 45% | |||
| SiC | [90, 91] | ||
| Extrusion process | SiOC | Open or closed cells | [93, 94] |
| Cell density: >107 cells cm-3 | |||
| Porosity range: 27–90% | |||
| IV. Reaction technique | |||
| SiC | Open cells | [18, 97–102, 104–106] | |
| Cell size: 10–45 μm | |||
| Cell density: >109 cells cm-3 | |||
| Porosity range: 32–94% | |||
| Mullite | Partially interconnected open cells | [107–109 ] | |
| Cell size: <20 μm | |||
| Cell density: >109 cells cm-3 | |||
| Porosity range: 32–85% | |||
| Cordierite | Interconnected open cells | [110, 111] | |
| Cell size: 13 um | |||
| Cell density: 109 cells cm-3 | |||
| Porosity range: 11–75% |
Replica
In this method, a porous structure—usually polyurethane (PU) sponge—is coated with or immersed in a polymeric suspension or precursor solution, followed by pyrolysis and high-temperature sintering to produce porous ceramics with the same cell morphology as the original material. The replica technique is widely used for the production of open-cell structures with various pore sizes, porosities and chemical compositions [5]. This strategy results in the production of uniform green coating on the polymer cellular structure as it contains sharp corners at strut edges and hollow ceramic struts, because the polymer is burnt out or decomposed completely. Owing to the limitations in the efficiency of infiltration and excess slurry removal, the cell size generally ranges from 150 μm to a few millimeters [5, 6].
Inspired by the early work of Schwartzwalder and Somers [2], many investigations explored the usage of several templates such as wood [52, 53], coral [54, 55], polymeric foams [56–59], etc, to prepare porous ceramics for use in a variety of applications [5]. The porous ceramics prepared from polymeric foams and ceramic slurries possess thin struts containing central holes [58]. Edirsinghe and co-workers developed a simple method where polysilane solution was substituted for the ceramic slurry to produce porous SiC ceramics with improved strength [56–59]. The polysilanes were dissolved in dichloromethane to form polymeric precursor solutions in which a PU sponge was immersed to form pre-foams. These pre-foams were subsequently heated at temperatures ranging from 900 to 1300 °C for 2 h in flowing nitrogen to produce open-cell SiC ceramics with the same morphology and sizes of original PU sponge cells. The essential steps in the foam-ceramic slurry method, namely removal of excess ceramic slurry from polymeric sponge and evaporation of volatile slurry after drying, are not required in the foam-polymeric precursor method [56–59]. In contrast to ceramic suspension-derived reticulated structures, porous ceramics obtained from preceramic polymers contain homogeneous microstructures with crack-free struts due to the improved wetting of the sponge and/or partial melting of the cross-linked polymers during pyrolysis [56–59]. A representative microstructure of the reticulated ceramics consisted of well-defined open-cell structures and defect-free struts as shown in figure 2(A) [58]. Furthermore, shrinkage can be controlled by varying the concentration of polysilanes in the precursor solutions [57–60] and/or second phase [56]. It is possible that the expansion and gas evolution of the polymer during heating lead to the significant stresses that can cause macroscopic cracks on the resultant ceramic struts. Thus, components produced by this route are usually reported to possess low mechanical strength. The optimum concentration [56] and repeated coating [61, 62] of polymeric precursor slurry significantly affect the strength of the resultant porous SiC ceramics. Vogt et al prepared macroporous nitrogen-based silicon carbide (NBSiC) ceramics by dipping the PU foams several times into the polysiloxane (MK polymer) precursor slurry [62]. After first coating cycle, the PU foam was thermally decomposed at 1000 °C in nitrogen, leaving hollow struts inside the ceramic structure, and simultaneously MK polymer was pyrolyzed. The polysiloxane acts as an additional binding phase at elevated temperatures according to the following equation and allows an appropriate handling and re-infiltration of the foam structures before nitridation at 1400 °C or re-crystallization at 1850 °C [56].
The repeated coating approach is believed to eliminate the cracks in struts resulting in stronger NBSiC ceramics.
Figure 2.

(A) Macroporous cellar structure of SiC ceramics after pyrolysis of polysilane-infiltrated polyurethane (PU) foams (reproduced with permission from [59] © 2000 Springer). (B) The crack-free SiOC–C ceramic structure developed using extracted, maleic acid anhydrate (MA)-modified and polymethylhydrosiloxane (PMHS)-infiltrated pine wood compounds after pyrolysis at 800 °C in a nitrogen atmosphere (reproduced with permission from [54] © 2004 Elsevier).
Greil and co-workers investigated the possibility of producing porous SiOC–C ceramic composites using a native wood template and preceramic polymer [53, 63, 64]. First, the wood-polysiloxane composite was prepared by infiltration and reaction of native wood preforms with a polymethylhydrosiloxane (PMHS) precursor. The pyrolysis of the cured material in an inert atmosphere at 800 °C yielded biomorphous SiOC–C composites [53, 63]. It was reported that the esterification of wood with maleic acid anhydrate (MA) facilitates the penetration of PMHS into the wood cell wall, resulting in high ceramic yield after pyrolysis [53]. The extracted MA-modified PMHS-infiltrated wood compounds yielded an additional porosity and void formation that prevent cracking during pyrolysis (see figure 2(B)).
Sacrificial template
In this method, a biphasic composite comprising a precursor matrix and homogeneously dispersed template is first prepared by impregnating previously consolidated preforms of the sacrificial material with preceramic polymer. It is followed by extraction of the template material. Finally, pyrolysis and/or sintering of the composite generate a porous structure. The cross-linking of macromolecules at a slightly lower temperature (than that used in pyrolysis) is recommended to pre-consolidate the continuous matrix phase so that the porous structure is not collapsed during the extraction step [5, 65]. A brief note on the significance and influence of cross-linking step on processing of polymer-derived ceramics is presented in a later section 3.
In principle, the sacrificial template method generates a negative copy of the original template, as opposed to the positive copy obtained from the previous replica method. Thus, removal of sacrificial phase does not cause flaws in struts, and the mechanical strength of the porous structures is higher of those produced via replica method. The usually reported compressive strengths of the sacrificial-templated porous structures vary in the range of 10–300 MPa [5]. Based on the type, volume fraction and size of the template, the porosity can be varied from 20 to 90% and the pore size from 1 to 100 μm. This method is capable of producing tailored porosity, pore size distribution and pore morphology of the resultant ceramic component by using an appropriate pore-forming template material. Polymer-derived porous ceramics were fabricated using a wide variety of templates: salts, liquids, metals/ceramics, synthetic/natural organics, etc [5]. Highly porous and homogeneous ceramic structures can be produced from preceramic precursors by sacrificial template strategy, whereas it is difficult to produce porous ceramic products with homogenous microstructure using powder processing. For example, a high template content of 90% led to the larger number of cells, resulting in porosity as high as 88% of SiOC ceramics from polycarbosilane [66] or polysiloxane [65] precursors.
Fitzgerald et al produced homogeneously distributed open-cell SiC foams with controlled cell sizes varying roughly between 10 and 100 μm, where polycarbosilane was pressure infiltrated into a porous sodium chloride compact [67]. The resulting polycarbosilane foam was cured and finally pyrolyzed to convert into ceramic. Kim et al dispersed a chemical blowing agent in polysiloxane precursor and foamed it by compression molding [68]. The porous preceramic polymer was pyrolyzed at 1100 °C in nitrogen to yield porous silicon oxycarbide ceramics with a porosity of 60% and a cell density greater than 104 cells cm-3. Low-density polyethylene (LDPE) can also be used as a sacrificial agent to produce controlled and uniformly distributed open-celled or major close-celled porous silicon oxycarbide ceramic structures from a polysiloxane precursor [69]. If the polyolefin content is high enough to make the polyolefin/preceramic polymer co-continuous then the porous ceramic after pyrolysis would be fully open-celled. However, if the polyolefin content is low then the separately isolated polyolefin phase could be dispersed in the matrix of the preceramic polymer, and a close-celled porous ceramic structure could be produced [69]. The polycarbosilane/camphene solution prepared at 60 °C upon thermally induced phase separation during freezing can produce highly aligned pore morphology [70]. Wang et al prepared porous SiC ceramics using polymethylsilane (PMS) precursor and glass filters, carbon nanotube, carbon fiber or silica templates [71]. The synthesis procedures included the infiltration of the templates with appropriate concentration of the preceramic polymer, their curing, and pyrolysis and template removal. Sacrificial materials such as salts, ceramics and other metallic particles are usually extracted by chemical means. For example, salt templates can be extracted by repeated washing the foam with water, and ceramic particles, metallic particles or fibers can be removed by acid etching [5, 67–71].
Colombo et al proposed a strategy where silicone resin powder mixed with filled poly (methyl methacrylate) (PMMA) microbeads as sacrificial templates [72] was used to produce open-cell microcellular SiOC ceramics. The compression strength decreased with increasing cell size. When 50 wt% PMMA was added, the resultant SiOC ceramics after pyrolysis exhibited a maximum bending strength of 24 MPa and a maximum elastic modulus of 16 GPa [73]. Closed-cell SiOC ceramic foams with porosity ranging from 56 to 85% were also developed which involved (i) forming a compact using a mixture of preceramic polymer (polyalkylsiloxane) and expandable microspheres, (ii) forming the compact by heating, (iii) cross-linking the foamed body and (iv) transforming the foamed body into a ceramic foam by pyrolysis [74]. A representative microstructure of porous SiOC ceramics is presented in figure 3(A). It was further demonstrated that the addition of inert fillers leads to higher porosity and larger cell size due to the beneficial effect of fillers in expansion. All specimens containing fillers had cell densities greater than 109 cells cm-3 and cell size smaller than 35 μm [75].
Figure 3.

(A) SiOC-based open cell structure obtained using 20% polysiloxane and 80% expandable microsphere (461DU40). The specimen was pyrolyzed at 1200 °C for 1 h in nitrogen. (B) Typical microstructure of porous SiOC ceramics obtained using 20% polysiloxane and 80% expanded microspheres (461DE20d70). The green compacts were cross-linked at 180 °C in air followed by pyrolysis at 1200 °C for 1 h in nitrogen.
The pyrolysis temperature has significant effect on the shape and size of pores of the resultant ceramics [76–79]. It was demonstrated that the features of the produced ceramic foams were dependent on the composition and the structure of the pyrolyzed precursors. The polymers with higher functionalities gave higher pyrolytic yields due to cross-linking or branched structures formed during polymerization and pyrolysis [17, 76, 79]. The higher microbead content in the polycarbosilane precursor led to a larger number of cells, lower bulk density, and higher porosity, while enhanced densification of strut at high-temperature pyrolysis resulted in flexural strength as high as ∼30 MPa at 70% porosity [66, 78].
Kim et al developed a simplified process where expanded (hollow) PMMA microspheres were used to produce closed-cell microcellular SiOC ceramics with cell densities greater than 109 cells cm-3 and cell size smaller than 30 μm from a polyalkylsiloxane precursor [65]. The strategy involved: (i) forming preceramic foams using a mixture of preceramic polymer and expanded microspheres by pressing, (ii) cross-linking the formed body and (iii) transforming the body into microcellular ceramics by pyrolysis. There was no need of heating the compact before cross-linking as was the case when expandable microspheres were used. By controlling the expanded microsphere content, it was possible to produce closed-cell, microcellular SiOC ceramics with porosities ranging from 70 to 87% [65]. Representative microstructure of porous SiOC ceramics obtained using 20% polysiloxane and 80% expanded microspheres reveals homogenously distributed closed-cell morphology (figure 3(B)). The lack of macroscopic defects and small cell size (30 μm) led to the superior strength of the ceramics. Typically, the compressive strength of the microcellular ceramics with 0.3 relative density (70% porosity) was 100 MPa [65].
Direct foaming
In direct foaming, bubbles are generated in the polymeric precursor solution to create a stable foam structure which upon heating yields porous ceramics [1, 4, 77, 78, 80, 81]. This is an easy, cheap and fast way to prepare both open and closed-cell structures with a wide range of cell dimensions and wide range of porosity or relative density. Often, these structures contain cell walls with interconnected porosity, offering added advantage with unique permeability that can allow finer adjustment of fluid transport within the structure [79, 82]. The other important advantage is that the ceramic struts are rather dense and defect-free, providing stronger foams with the compressive strength as high as ∼30 MPa for a relative density of 0.3 [6]. However, this method has difficulty in producing material with narrow cell size distribution, and materials often have anisotropic structure [6]. The foam structure can be obtained by blowing an agent that can be volatile liquid, solid or a gas. The volatile liquid can be Freon or pentane, while solid agents like CaCO3 powder decompose at high temperature to generate bubbles. The gas can be added by mechanical stirring or injection or it can be developed in situ by chemical reactions such as oxidation of SiC, SiC filler or water vapor from the cross-linking of silicone resins or other chemicals [6].
In direct foaming, three important processes are involved that essentially control the development and stability of foams: (i) drainage of liquid through cell edges until an equilibrium is reached, (ii) Ostwald ripening and coalescence of bubbles leading to the coarsening of some bubbles and shrinkage or disappearance of other bubbles and (iii) film rupture when the cell wall becomes thin and weak due to the drainage and coarsening, ultimately leading to the collapse of the liquid foams [1, 5, 6]. The total porosity of directly foamed ceramics is proportional to the amount of gas incorporated into the suspension or slurry, and the pore size is determined by the stability of the wet foam prior to setting [5]. Thus, foam stabilization is the crucial step in the direct foaming method. Accordingly, strategies like using several surfactants and solid particles have been tried to stabilize the liquid foam towards retaining the cellular morphology in ceramic precursors [5]. However, the usage of polymeric precursor allows generation of foam structure without requiring additional setting agents [4, 6]. Porous SiC, SiOC and SiNC compounds were accordingly produced using solely the thermosetting properties of silicon-based polymers in combination with in situ blowing agents, either in the presence of surfactants [4] or by applying the pressure-drop technique [78].
The research group of Colombo developed a method where preceramic polymers were incorporated into organic precursor solutions and foamed. The foamed mixtures were subsequently pyrolyzed to produce porous ceramics [6, 77, 80, 81]. Thus, SiC, SiOC and SiNC based porous ceramics were developed by direct co-foaming of preceramic polymers (usually silicone resin) mixed with PU precursors (polyols and isocyanates), the latter serve as blowing aids and structural templates [72, 80–84]. This method offers substantial flexibility in fabricating foams with the desired cell size, degree of cell opening, cell morphology, bulk density, thermal, elastic and mechanical properties using different types of PU precursors and ceramic/metallic fillers [6, 82]. For example, completely open-celled, interconnected (cell walls containing holes), and closed-cell foams can be obtained using flexible, semi-rigid and rigid PU precursors, respectively [74, 75]. On the other hand, the residual carbon content (from PU) can be removed by direct foaming of the preceramic polymer with liquid blowing agent like pentane or Freon, thereby improving high-temperature properties of the foams [6, 85]. Microcellular ceramics with a cell size of about 8 μm were fabricated using PMMA microbeads as sacrificial templates [82]. Direct foaming method produces foams of high green strength and enables easy machining after drying. The electrical conductivity of polymer-derived porous ceramics depends on the type and amount of fillers and the type of precursor polymers used [85]. A combination of proper selection of the foaming and pyrolysis processes with appropriate fillers is believed to produce porous ceramic structures with a large degree of variation in functionality and properties [6, 85, 86]. It was also reported that a phenyl methyl poly (silsesquioxane) melt containing small amounts of ethoxy and hydroxyl groups can be foamed by an in situ blowing technique above 200 °C without using any additional blowing agent [87, 88].
In an innovative approach, Kim et al demonstrated that an extremely fine and uniformly distributed cellular or microcellular structure can be developed by implementing the thermodynamic instability principle in a foaming system to produce the desired cell morphology such as controllable cell size, cell density and cell distribution [78, 84]. The proposed strategy involved: (i) saturating preceramic polymers using gaseous, liquid, or supercritical CO2, (ii) nucleating and growing a large number of bubbles using thermodynamic instability via a rapid pressure drop or heating and (iii) transforming the microcellular preceramics into ceramics by pyrolysis and optional sintering [89, 95]. A low-temperature curing step of polycarbosilane powder involving cross-linking is suggested to avoid any chance of collapsing the porous structure during pyrolysis or sintering [90]. Microcellular SiC or SiOC ceramics having cell densities above 109 cells cm-3 and cell sizes below 10 μm were fabricated using polycarbosilane and/or polysiloxane precursors [89, 90]. However, this method produces a major closed-cell structure [78, 89, 90].
This strategy is further extended to manipulate the open-cell content by controlling the LDPE content and compounding conditions in a compounder element such as an inner mixer. The compounded polysiloxane-LDPE blends were foamed using gaseous CO2 followed by burning out the sacrificial polyolefin phase during pyrolysis to produce cellular or microcellular SiOC ceramics [91]. SiOC ceramics with a wide range of porosities from 27 to 90%, and cell densities from 107 to 109 cells cm-3 were produced from extruded blends of polysiloxane and polymer microbeads [92]. While direct pyrolysis of blends resulted in open-cell ceramics, the combined process of foaming with gaseous CO2 and subsequent pyrolysis yielded closed-cell ceramics [92]. A representative microstructure of the SiOC-based closed-cell structure obtained using 50% polysiloxane and 50% polymer microbeads is shown in figure 4(A). When compounded blends of polysiloxane, LDPE and polymer microbeads were foamed and pyrolyzed, SiOC ceramics with homogeneous open-cell structures were obtained [93] (figure 4(B)). The proper selection of cross-linking conditions is essential to promote a stable foam structure [94]. The compounding temperature of the polysiloxane could be decreased by ∼20 °C when metallocene polyethylene (mPE) was used instead of LDPE and the undesirable thermal cross-linking of polysiloxane was minimized [95].
Figure 4.

Microstructures of porous ceramics produced via the direct foaming technique: (A) SiOC-based closed-cell structure obtained using 50% polysiloxane and 50% poly(methyl methacrylate-co-ethylene glycol dimethacrylate) microbeads (∼20 μm) and (B) SiOC-based open-cell structure obtained using 54% polysiloxane, 36% poly(methyl methacrylate-co-ethylene glycol dimethacrylate) microbeads (∼20 μm), and 10% low-density polyethylene. The raw materials were compounded using a counter-rotating twin-screw extruder. The extruded samples were saturated with gaseous CO2 at room temperature for 24 h under a pressure of 5.5 MPa. Thermodynamic instability was then introduced by lowering the pressure at a rate of 3.9 MPa s-1. The foamed samples were cross-linked by doping a catalyst and subsequently heating the specimen up to 200 °C. The specimen was then pyrolyzed at 1200 °C for 1 h in argon.
Reaction technique for matrix
Though the reaction synthesis is an old technique to fabricate structural ceramics, it is only recently identified that a combination of previously discussed strategies and reaction synthesis can develop cellular or microcellular ceramics with tailored porosity and strength. Uniformly distributed open-cell microcellular SiC ceramics with cell densities greater than 109 cells cm-3 and cell size smaller than 20 μm were developed by carbothermal reduction of polysiloxane-polymer microbeads-derived SiOC ceramic foams [96]. The particular strategy involved (i) fabricating a preceramic foam from a mixture of polysiloxane, phenol resin (as carbon source), polymer microbeads (as sacrificial template), SiC (an optional filler), and Al2O3–Y2O3 (an optional sintering additive); (ii) cross-linking (at 180–200 °C in air) the polysiloxane in the formed body; (iii) transforming the polysiloxane and phenol resin by pyrolysis at 1100–1200 °C in nitrogen into SiOC and C, respectively; and (iv) synthesizing SiC by carbothermal reduction at 1450–1650 °C in argon or nitrogen [96, 97]. Figure 5(A) shows an open-cell microcellular SiC ceramics obtained via carbothermal reduction of SiOC foams, using 67% polysiloxane, 13% phenol resin, 3% Y2O3, 2% Al2O3, and 15% expandable microspheres. It was demonstrated that an optimum combination of expandable microsphere and inert filler (mostly SiC) contents can result in ceramics with porosity varying between 60 and 95% [96, 97]. The expandable microsphere content above 30 mass% and inert filler content beyond 30% led to the collapse of the structure due to the coalescence of cells during processing [98]. The subsequent sintering at 1800–1950 °C resulted in porous SiC ceramics with high strength [98]. The decreased porosity by densification and strut strengthening by necking resulted in a ceramic with a typical compressive strength of ∼290 MPa and flexural strength of ∼100 MPa at ∼40% porosity [98]. The impingement of growing grains with the SiC fillers during sintering led to a finer microstructure and improved strength of the porous SiC ceramics [99]. Hollow microspheres with proper sintering additive composition can result in producing partly interconnected, open-cell SiC ceramics with high strength [100, 101]. It was also demonstrated that the microstructure and strength could be further improved by a judicious selection of processing conditions and starting ratio of SiC powder to polysiloxane-derived SiC (SiC: PDSiC) [18]. The porosity increased with increasing PDSiC content that can be attributed to the weight loss according to the following reactions [18, 49, 96, 102]:
The PDSiC ceramic specimens showed a more homogeneous microstructure than the powder-processed ceramics. The high-temperature strength reduction caused by the formation of free Si in the high-temperature reaction between SiC and sintering additives (Al2O3) could be effectively suppressed by the addition of excess carbon [103]. In another work, the expandable microspheres were in situ foamed during extrusion at 130 °C, thus eliminating the additional foaming treatment [104]. Furthermore, porous SiC ceramics with cell size of 10–45 μm and adjusted grain size and grain morphology can be developed upon annealing of extruded-pyrolyzed-carbothermally reduced foams at high temperatures (1750 or 1950 °C) for 1–6 h in argon [105].
Figure 5.

Microstructure of porous ceramics produced via the reaction method: (A) porous SiC ceramics produced using 67% polysiloxane, 13% phenol resin, 3% Y2O3, 2% Al2O3, and 15% expandable microspheres (461DU40). The green compact was expanded at 138 °C and subsequently cross-linked at 180 °C, pyrolyzed at 1200 °C for 1 h and further heat-treated at 1650 °C for 2 h in argon; (B) porous cordierite obtained using 23.5% polysiloxane, 32.7% talc, 26.3% Al2O3, 2.5% kaolin, and 15% expandable microspheres. The green compact was expanded at 138 °C and subsequently cross-linked at 180 °C in air. The cross-linked compacts were heat-treated at 1350 °C for 4 h in air.
The thermo-oxidational degradation of polysiloxane at 700 °C in air leads to the development of highly reactive amorphous SiO2, which reacts with Al2O3 fillers at ∼1370 °C to facilitate mullite formation [106, 107]. Kim et al adapted this approach to prepare partially interconnected open-cell microcellular mullite ceramics with cell densities greater than 109 cells cm-3 and cell size smaller than 20 μm from an Al2O3-filled polysiloxane [108]. The adopted strategy involved (i) fabricating a body formed by combining polysiloxane, Al2O3 filler, polymer microbead templates, and Y2O3 sintering additive; (ii) cross-linking the polysiloxane in the formed body; (iii) pyrolyzing it to transform polysiloxane into SiO2; and (iv) synthesizing mullite by reacting SiO2 and Al2O3 at 1500–1600 °C. The following reactions are believed to occur during pyrolysis and synthesis of mullite:
Higher microbead content, lower sintering temperature and a smaller amount of Y2O3 led to a high porosity. Typically, a compressive strength of ∼90 MPa was obtained at ∼40% porosity [108].
In a similar method, cordierite (2MgO·2Al2O3·5SiO2) ceramics consisting of interconnected open-cell structures with cell densities of ∼109 cells cm-3 and cell sizes of ∼13 μm were obtained from talc and Al2O3-filled polysiloxane [109]. The proposed strategy for making the microcellular cordierite ceramics involved three steps: (i) fabricating ceramic-filled preceramic foams by heating a mixture of polysiloxane, expandable microspheres, talc (3MgO·4SiO2·H2O), and alumina in a mold; (ii) cross-linking the foamed body; and (iii) transforming the body into microcellular cordierite ceramics by sintering at 1300–1400 °C for 10 h [109, 110]. A typical microstructure of porous cordierite ceramics obtained using 23.5% polysiloxane, 32.7% talc, 26.3% Al2O3, 2.5% kaolin and 15% expandable microsphere is shown in figure 5(B). The porosity could be controlled in the range of 60–75% by manipulating the packing density. A wider range of porosity of 11–72% could be obtained by adjusting the template content, sintering temperature and the sintering additive composition [110]. A compressive strength of ∼150 MPa was recorded at 35% porosity when 6 wt% kaolin was added as the sintering additive.
The pore characteristics of polysiloxane-derived porous ceramics can be effectively controlled by employing different processing strategies. The replica strategy generally produces open-cell macroporous structures, whereas ceramics with tailored porosity, pore size and its distribution can be obtained by the sacrificial template or reaction technique. Direct foaming or reaction technique is preferred to produce micro or macrocellular structures with interconnected porosity [6, 10]. Macrocellular foams with a cell size ranging between 100 and 600 μm were fabricated from methyl polysiloxane using a direct foaming approach, whereas microcellular foams, with a cell size of about 8 μm, were fabricated using PMMA microbeads as sacrificial templates [82]. SiOC ceramics with hierarchical porosity can also be produced either by controlled pyrolysis, deposition of various meso-porous layers, etching or the addition of suitable fillers [19, 111–113]. The pore size, pore morphology and the specific surface areas of polymer-derived ceramic bodies strongly depend on the composition of the preceramic material and on the maximum pyrolysis temperature. Fast pyrolysis can lead to crack formation and a loss of specific surface area at temperatures above 600 °C, whereas slow pyrolysis can preserve mesopores up to 1200 °C combined with high surface areas [83]. The pore size and morphology of the cellular structures change with the addition of template materials. Owing to the easy solubility in ethanol, polysiloxane can be uniformly coated on the templates leading to a highly homogeneous closed-cell microcellular porous SiOC structure [65]. The pore morphology of the SiC ceramic structure fabricated by carbothermal reduction and subsequent sintering of polysiloxane-derived SiC changed from spherical to irregular shape as the hollow microsphere content was increased. This change was due to the greater opportunity for contact between the microspheres in the compacts [100]. The volume fractions of both submicron- and micron-size pores decreased significantly with increasing sintering temperature [100].
Cross-linking of polysiloxanes
It is known that most of the conventional polymers are difficult to use as ceramic precursors because of the decomposition of their main chain during pyrolysis. However, cross-linked polymer structures can impart high strength for handling or molding of component before pyrolysis. The degree of cross-linking of preceramic polymers can significantly affect their plastic forming capability, leading to hindered flow at processing temperature and formation of residual porosity [114, 115]. Thus, cross-linking is an important step in achieving high ceramic yields from preceramic polymers to produce a variety of ceramic products.
The synthesis of heat-resistant ceramic foams by using viscoelastic nature of silicone resins was widely studied. Because silicone resins can accept various kinds of fillers, plasticizers, blowing agents and cross-linking agents, a proper control over cell size, cell density and cell connectivity is fundamentally possible by employing simple plastic-foaming methods [34]. Excellent dimensional stability after the pyrolysis of the polysiloxane-derived porous ceramics is associated with the thermosetting nature and high ceramic yield of silicones. A dense cross-linked structure is necessary to prevent the bond rearrangement during heating and to increase the ceramic yield. As compared with the  (351.5 kJ mol−1),
(351.5 kJ mol−1),  bond (347.7 kJ mol−1) and
bond (347.7 kJ mol−1) and 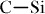 bonds (290.0 kJ mol−1), the high energy of the flexible
bonds (290.0 kJ mol−1), the high energy of the flexible  bond (369.0 kJ mol−1) is promising for the high ceramic yield of silicones [34].
bond (369.0 kJ mol−1) is promising for the high ceramic yield of silicones [34].
This section briefly reviews the studies where the vital role of cross-linking of polysiloxanes was highlighted. The available cross-linking methods can be categorized into (i) heat treatment, (ii) catalysis and (iii) non-conventional treatment by laser or ionizing radiation. Polysiloxane-derived SiC-based preceramic or ceramic composites produced by different cross-linking methods are listed in table 2.
Table 2.
List of polysiloxane-derived ceramic compositions produced by different cross-linking methods.
| Cross-linking method | Ceramic composition | References |
|---|---|---|
| Heat treatment | ||
| SiOC | [95] | |
| SiC | [117–119] | |
| SiC fibers | [14] | |
| SiC nanoparticles | [120, 121] | |
| Si–O–C | [122] | |
| Catalysis | ||
| Boron-containing | SiC | [123–125] |
| catalyst | ||
| Metallocene | SiC | [126, 127] |
| Chlorine-containing | SiC | [128] |
| catalyst | ||
| Amine catalyst | SiOC | [70, 75, 81, 82, 96] |
| condensation | ||
| Laser treatment | ||
| SiC nanopowders | [129, 130] | |
| Radiation | ||
| SiC or SiOC | [131–134] |
The cross-linking by heat treatment is widely used probably because of the simplicity. The simple thermal curing of reflux-treated PMS yields high SiC ceramic yields without using catalysts or high pressures [116–118]. The evolution of Si–Si3 cross-linked PMS structure via methylsilane formation at ∼250 °C leads to high ceramics yields up to 89% [117]. Yajima et al suggested that cross-linking of polycarbosilane occurs after the Si-H bonds are cleaved at ∼450 °C [13]. SiC nanoparticles and 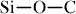 fiber were produced from polymethylsilsesquioxane (PMSQ) precursors [119–121] by advanced cross-linking methods. For example, melt spinning on PMSQ at 130–180 °C followed by metal chloride (SiCl4, Si(CH3)Cl3, TiCl4 or BCl3) vapor curing yields
fiber were produced from polymethylsilsesquioxane (PMSQ) precursors [119–121] by advanced cross-linking methods. For example, melt spinning on PMSQ at 130–180 °C followed by metal chloride (SiCl4, Si(CH3)Cl3, TiCl4 or BCl3) vapor curing yields 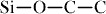 fibers [121]. This new technique can be applicable to the processing of porous polysiloxane-derived ceramics.
fibers [121]. This new technique can be applicable to the processing of porous polysiloxane-derived ceramics.
Cross-linking can largely alternate the average molecular weight of precursor polymers and, in turn, influence the rheological properties and foaming behavior of the blends. If the degree of cross-linking is too low then the melt strength and viscosity of polymer will be insufficient to promote a well-distributed porous structure. In contrast, too high a degree of cross-linking will also negatively affect the cell formation. Wang et al reported that the viscosity increased at higher rate when polysiloxane was cross-linked at higher temperature. In their study, cross-linking of polysiloxane at 160 °C for 30 min and subsequent CO2 foaming yielded a preceramic structure with fine uniform cells [94].
The processability of the precursor can be improved by doping the preceramic polymer with a proper catalyst. Boron-containing catalysts have been actively studied as cross-linking agents for improving the ceramics yields [122–124]. Metallocene catalysts promote the dehydration of the  species at high pressure and thereby improve residual SiC yields [125, 126]. The highly reactive nature of
species at high pressure and thereby improve residual SiC yields [125, 126]. The highly reactive nature of 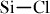 bond in chlorine-containing polysilanes enables the cross-linking of polycarbosilanes to take place partly in the same temperature range where transformation of polysilanes to polycarbosilanes occurs [127]. Amine-catalyzed condensation of
bond in chlorine-containing polysilanes enables the cross-linking of polycarbosilanes to take place partly in the same temperature range where transformation of polysilanes to polycarbosilanes occurs [127]. Amine-catalyzed condensation of 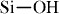 groups was used in cross-linking of polysiloxanes [80, 81, 83, 84]. Kim et al cross-linked the expanded compacts of polysiloxane/PMMA [74], extruded blends of polysiloxane/LDPE [69], and CO2-foamed blends of polysiloxane/mPE [95, 100] by introducing aminoalkylalkoxysilanes as the condensation catalyst and subsequently annealing in air, prior to pyrolysis. In this way, they produced closed or open-cell SiOC porous structures.
groups was used in cross-linking of polysiloxanes [80, 81, 83, 84]. Kim et al cross-linked the expanded compacts of polysiloxane/PMMA [74], extruded blends of polysiloxane/LDPE [69], and CO2-foamed blends of polysiloxane/mPE [95, 100] by introducing aminoalkylalkoxysilanes as the condensation catalyst and subsequently annealing in air, prior to pyrolysis. In this way, they produced closed or open-cell SiOC porous structures.
The heat treatment approach to convert polymers to ceramics has inherent limitations such as the shape changes, polymer degradation, etc, before the occurrence of preferred chemical reactions. It has been recently identified that unconventional methods like laser treatment [128, 129] and ionizing radiation by means of electron beam or gamma rays [130–133] can also be used to treat the preceramic polymers. These processes offer advantage of processing at ambient temperature, which is crucial for polymers with low softening, melting, or decomposition temperatures. The high degree of reactivity in the  and
and  groups of silazane precursors can be utilized in rapid cross-linking by laser treatment to produce SiC or Si3N4 nanoparticles [129]. Radiation processing of silicon-containing polymers generates free radicals in the bulk of the material, which in addition to cross-links also form new bonds. If the irradiation is performed in the presence of oxygen, then peroxides are formed that on further decomposition produce cross-linking bonds [131]. However, a prior cross-linking step is often necessary to minimize the possibility of crack formation due to weight loss and shrinkage during pyrolysis [132, 133].
groups of silazane precursors can be utilized in rapid cross-linking by laser treatment to produce SiC or Si3N4 nanoparticles [129]. Radiation processing of silicon-containing polymers generates free radicals in the bulk of the material, which in addition to cross-links also form new bonds. If the irradiation is performed in the presence of oxygen, then peroxides are formed that on further decomposition produce cross-linking bonds [131]. However, a prior cross-linking step is often necessary to minimize the possibility of crack formation due to weight loss and shrinkage during pyrolysis [132, 133].
Mechanical properties
Whereas the strength of ceramics produced from ceramic precursors using polymer replica is considerably evaluated [6], the data for polysiloxane-derived porous ceramics are scarce. Nagrejo and Edirishingwe reported that immersion of suitable concentration of polysilanes in PU foams yielded highly porous (85–96%) SiC, SiC-Si3N4 and SiC-TiC foams with a compressive strength ranging from 1.1 to 1.6 MPa [56]. Appropriate combination of repeated re-infiltration and pre-sintering steps has been suggested to increase the strut thickness and to fill up the hollow struts thereby strengthening the cell structure [61, 62]. The porous SiC structure derived from coating of polysiloxane on PU foams under nitridation at 1400 °C showed an increase in the compressive strength from 0.3 MPa after two coating cycles to 1 MPa after four coating cycles [62]. However, increasing the sintering temperature to 1800 °C caused re-crystallization of amorphous SiC and reduction of strength [62]. Flexural strength of SiC powder-derived porous SiC ceramics synthesized with PU replica was reported to be lower than 2 MPa [134], but no information is available on flexural strength of polysiloxane-derived porous ceramics produced by the replica method.
Sacrificial template strategy produces porous ceramics with high strength. The reported flexural strength of porous SiC ceramics made by the replica technique, reaction method, and gel casting were 1.5 MPa at 70% porosity [134, 135], ∼4 MPa at 73% porosity [136], and ∼5 MPa at 80% porosity [134], respectively. The respective cell sizes were 0.3–3 mm [135, 137], 10–20 μm [134] and 100–400 μm [135]. Jin and Kim fabricated highly porous microcellular (5–8 μm cell size) Si(O)C ceramics with flexural strength of ∼30 MPa at 70% porosity and ∼6 MPa at 80% porosity by pyrolyzing at 1400 °C a cross-linked body consisting of a polycarbosilane precursor, polymer microbeads sacrificial template and polysiloxane binder [66]. Colombo et al [72] reported that the compressive strength of microcellular SiOC ceramics decreased with increasing cell size. The highly homogeneous microstructure with small cell size can be associated with the high strength levels of polymer-derived ceramics produced by the sacrificial template strategy. A high compressive strength of 100 MPa in SiOC ceramics was recorded at 70% porosity (0.3 relative density) when cross-linked compacts of polysiloxane and expanded (hollow) microspheres were pyrolyzed [65].
The compressive and flexural strengths of SiOC ceramics produced by direct foaming of polysiloxane and blown PU templates vary in the range of 1–30 and 0.6–15 MPa, respectively [81, 86, 138, 139]. The superior strength was attributed to the lack of macroscopic defects in the struts and cell walls, such as the hollow features that are typical of conventionally manufactured reticulated foams [6, 86]. A flexural strength of 13 MPa can be maintained during a long-term annealing at 1100 °C in air, and the modification in strength at 1200 °C was attributed to the evolution of intrastrut porosity. The latter was caused by the oxidation of residual free carbon, which was accompanied by devitrification of the SiOC and/or the passivating silica scale [86]. With low PU content (5.25:1 weight ratio of silicone resin to PU), the small-size carbon-rich areas are embedded within the dense SiOC matrix and the material strength can be retained after oxidation at 1200 °C [138]. Addition of submicron SiC powder to the SiOC amorphous matrix had only minor effect on crushing strength and elastic modulus of the foams [81]. The microcellular foams possessed a 2–5 times higher crushing strength than macrocellular foams of similar density because of the reduced probability to encounter a flaw having the critical dimension [65, 82].
Ceramics of SiC, mullite or cordierite having a wide range of strength from 5 to 300 MPa and porosities from 10 to 75% can be produced by the reaction technique. The carbothermal reduction and subsequent sintering of SiOC foams, templated from hollow microspheres or polymer microbeads, resulted in open-cell SiC ceramics possessing a compressive strength of 240–290 MPa and a flexural strength of 60–100 MPa at 40% porosity [103, 105]. The proper composition of starting mixture of SiC powder and polysiloxane-derived SiC resulted in a flexural strength of 57 MPa at 50% porosity [18]. As the chemistry of sintering additives affects densification of struts and pore structure, specimens prepared using AlN+Y2O3 additives showed the highest strength of 34 MPa, at a porosity of 56% [101]. The addition of SiC fillers resulted in finer microstructure with less weight loss and less shrinkage that increased the flexural strength of porous SiC ceramics from 34 MPa to 42 MPa after sintering at 1900 °C [99]. When hollow microspheres were used as templates, carbothermal reduction and subsequent sintering of carbon-filled polysiloxane rendered porous SiC ceramics with a flexural strength of 60 MPa and a compressive strength of 240 MPa at ∼40% porosity [100]. For comparison, flexural strengths of ∼10 MPa at ∼50% porosity [140] and ∼28 MPa at ∼44% porosity [141] were reported in reaction-bonded porous SiC ceramics and 17 MPa at ∼61% porosity in sintered porous SiC ceramics [142]. A compressive strength of ∼160 MPa was also reported at 40% porosity in porous SiC ceramics prepared using polymer microbeads as the sacrificial templates [143]. The superior strength was attributed to both the lack of macroscopic defects and the presence of a rigid strut with a well-developed grain structure.
The collection of compressive and flexural strength data for various polysiloxane-derived porous ceramics produced via different processing strategies is shown as a function of porosity in figures 6 and 7. The porosity varied from 10 to 96%. Among the four processing strategies, the replica technique yields polysiloxane-derived porous ceramics with poor strength, whereas the reaction technique provides ceramics with high strength. Based on various processing parameters, the available data indicates a minimum compressive strength of 1 MPa [56] with 96% porosity for the porous ceramics fabricated by the replica technique, and a minimum flexural strength of 0.6 MPa with 90% porosity for the porous ceramics fabricated by direct foaming [86]. A maximum compressive strength of 300 MPa [100] with 34% porosity and a maximum flexural strength of 144 MPa with 32% porosity [18] were recorded for the porous ceramics developed by the reaction technique. Figures 6 and 7 also indicate that the reaction technique offers porous ceramics with wide ranges of the compressive strength (from 5 to 300 MPa), and flexural strength (from 5 to 32 MPa). The sacrificial template strategy provided porous ceramics with the compressive strength varying from 10 to 100 MPa and the flexural strength between 1 and 40 MPa, whereas direct foaming rendered porous ceramics with compressive strength ranging from 0.7 to 30 MPa and flexural strength from 0.6 to 15 MPa. This data suggests that the variation of the processing strategy significantly influences the strength of the porous ceramics with a given porosity. Based on the specific application, an appropriate selection of the processing strategy is necessary to control the strength levels of the ceramics with the desired porosity.
Figure 6.
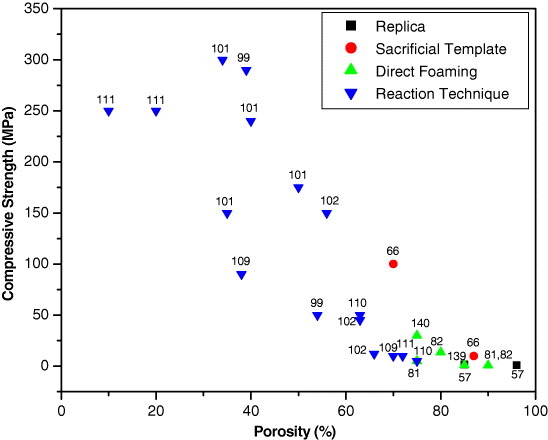
Compressive strength as a function of porosity of polysiloxane-derived porous ceramics produced via different strategies. Data points are labeled with the corresponding reference numbers.
Figure 7.
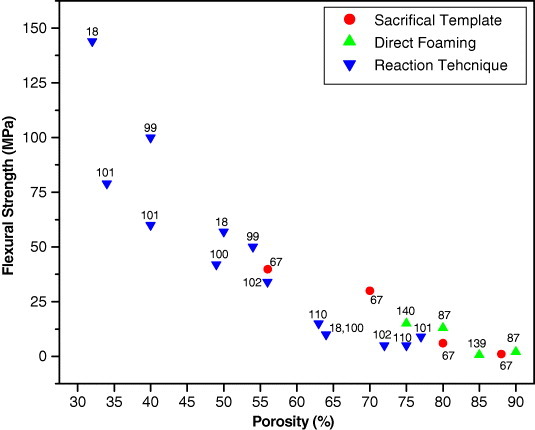
Flexural strength as a function of porosity of polysiloxane-derived porous ceramics produced via different strategies. Data points are labeled with the corresponding reference numbers.
It is evident from figures 6 and 7 that the flexural or compressive strength generally decreases with increasing porosity of polysiloxane-derived porous ceramics. The processing strategy has a minimal effect on the porosity dependence of strength. This tendency has also been observed in many other porous ceramics [98, 134, 144] and can be attributed to the higher probability of the pore coalescence under load at higher porosities. The pore coalescence increases the defect size and reduces the strength. According to a model proposed by Gibson and Ashby [3], the relative strength of a cellular material showing a brittle crushing behavior is related to its relative density through the following expression
Here σo and ρo are, respectively, strength and true density of the cell-wall material, σ and ρ are, respectively, strength and density of the cellular material, and C is a dimensionless constant. The value of the exponent m is 1.5 for open-cell foams, and it ranges from 1 to 1.5 for closed-cell foams, depending on the volume of the solid contained in the cell edges. The strength data of porous ceramics derived from polysiloxanes is in agreement with the above relation, but different m values are reported. For example, the m value was 2.22 in the compressive strength and density relation for closed-cell SiOC ceramics produced using hollow microsphere templates [59], whereas m values of 1, 2 and 3.6 were reported when different flexible PU foams were blown in the polysiloxane solution [77, 81, 86]. The m value for the flexural strength was 2.3 in direct foaming [86]. Similarly, m of 3.1 was reported for the mullite ceramics produced by the reaction synthesis of alumina with polysiloxane-derived silica [108]. The observed deviations in the m value suggest a more pronounced influence of density on the strength than that predicted by the model. This can probably be attributed to the following factors. The model assumed a constant cell size, whereas macrostructural inhomogeneities (distribution of cell sizes) are often found in ceramic foams. The presence of mixed types of cells—closed and open cells—cannot be avoided and the stress distribution associated with the hybrid cell walls was not considered. The strength can also be influenced by other important parameters, such as a possible variation in the strut strength as a result of dissimilar cell density [65]. The unexpectedly low flexural strength of specimens with high density is often attributed to the presence of an abnormally large amount of voids or microdefects in the struts [86].
Summary
Polysiloxane precursors offer unique advantages in processing of porous ceramics. The state-of-the-art technology for producing porous Si(O)C-based ceramics from polysiloxane precursors is discussed in this review. The replica strategy is widely used to produce open-cell macroporous SiC or SiOC structure with cell sizes usually larger than 150 μm and porosities ranging from 85 to 96%. This strategy results in weak ceramics with compressive strength below 2 MPa. The precursor concentration, coating efficiency, template type and pyrolysis temperature significantly affect the microstructure and strength of the resultant ceramics.
The sacrificial template strategy can control the porosity, pore size distribution, and pore morphology of the resultant SiOC or SiC ceramics. The type and amount of pore-forming template and its removal after pyrolysis can vary the porosity from 20 to 90%, cell size from 1 to 100 μm, and cell density from 104 to 1010 cells cm-3. The proper selection of expanded or expandable microsphere templates is essential to produce the desired openness of the cell structure. The strength of these ceramics is higher compared to those obtained by the replica method.
Direct foaming with chemical blowing agents or self-blowing of sacrificial templates offers SiC, SiOC or SiC + SiOC cellular structures with cell sizes ranging from 80 to 800 μm, and porosities between 75 and 90%. The compressive strength as high as 30 MPa at a porosity of 75% has been achieved. The generation and stabilization of the foam are the keys to obtaining the desired porosity and pore size. Foaming using supercritical CO2 is an effective method to produce cellular or microcellular structures with controlled and uniformly distributed porosity.
The carbothermal reduction and subsequent sintering of SiOC ceramics obtained with other strategies produce uniformly distributed open-cell SiC structure. The porosity of SiC ceramics ranges from 30 to 90% and the cell size from 10 to 45 μm with cell densities exceeding 109 cells cm-3. The starting composition, fillers and sintering additives are important for producing stronger SiC ceramics with desired porosity levels. A compressive strength of ∼290 MPa and a flexural strength of 100 MPa have been obtained for SiC ceramics with 40% porosity. The reaction technique has also been extended to produce interconnected or partially interconnected open-cell structures of mullite and cordierite ceramics. The cross-linking of preceramic polymers prior to pyrolysis is necessary in preventing collapse of cellular structures and in achieving high yields of the ceramics.
The processing of polysiloxane-derived porous ceramics initially started from the replica technique and has been continuously improving with the development of other strategies towards meeting the technological requirements in several engineering applications. Each strategy results in different range of microstructural or pore characteristics that are useful for specific applications. For example, predominantly closed-cell structures are required for materials used for thermal insulation, whereas open interconnected cell structures are preferred for uses involving fluid transport such as catalysts and filters. However, such application-specific porous ceramic compositions are possible to fabricate by a particular processing strategy and there is an immediate need for versatile strategies that allow tuning the microstructure, pore characteristics, and properties and that can be additionally applied to ceramic materials of different compositions. A combination of the existing processing strategies is also one of the prospective directions for future work in producing the polysiloxane-derived porous ceramics with sufficient control over the final microstructural and pore characteristics and other properties. Although porosity and other properties of the resultant ceramics can be effectively controlled with the simple processing methodology, mass-production is required to fabricate large-sized or complex-shaped porous objects. The future work in the processing of polysiloxane-derived porous ceramics should be directed towards establishing strategies that are inexpensive, versatile and can produce complex-shaped objects. In one of such recent study, porous SiOC ceramics were fabricated from polysiloxane-hollow microsphere blend using inexpensive method of steam chest molding and pyrolysis [145].
Acknowledgment
This study was supported by the National Research Foundation of Korea (NRF) grant (No. 2009-0083986) funded by the Korean government (MEST).
References
- Sepulveda P. and Binner J G P. J. Eur. Ceram. Soc. 1999;19:2059. doi: 10.1016/S0955-2219(99)00024-2. [DOI] [Google Scholar]
- Schwartzwalder K, Somers H and Somers A V. 1963US Patent No. 3090094 [Google Scholar]
- Gibson L J. and Ashby M F. Cellular Solids, Structure and Properties. 2nd edn. Cambridge: Cambridge University Press; 1999. [Google Scholar]
- Colombo P. and Hellmann J R. Mater. Res. Innovat. 2002;6:260. doi: 10.1007/s10019-002-0209-z. [DOI] [Google Scholar]
- Studart A R, Gonzenbach U T, Tervoort E. and Gauckler L J. J. Am. Ceram. Soc. 2006;89:1771. doi: 10.1111/j.1551-2916.2006.01044.x. [DOI] [Google Scholar]
- Colombo P. 2006. Phil. Trans. R. Soc. A 364 109 10.1098/rsta.2005.1683 [DOI] [PubMed] [Google Scholar]
- Scheffler M. and Colombo P. Cellular Ceramics: Structure, Manufacturing, Properties and Applications. Weinheim: Wiley-VCH; 2005. [Google Scholar]
- Rice R W. Porosity of Ceramics. New York: Marcel Dekker; 1998. [Google Scholar]
- Kim Y W, Jin Y J, Eom J H, Song I H. an an and Kim H D. J. Mater Sci. 2010;45:2808. doi: 10.1007/s10853-010-4270-5. [DOI] [Google Scholar]
- Colombo P, Mera G, Riedel R. and Soraru G D. J. Am. Ceram. Soc. 2010;93:1805. [Google Scholar]
- Vakifahmetoglu C, Pippel E, Woltersdorf J. and Colombo P. J. Am. Ceram. Soc. 2010;93:959. doi: 10.1111/j.1551-2916.2009.03448.x. [DOI] [Google Scholar]
- Yang Y, Guo Q, He X, Shi J, Liu L. and Zhai G. J. Eur. Ceram. Soc. 2010;30:113. doi: 10.1016/j.jeurceramsoc.2009.08.006. [DOI] [Google Scholar]
- Yajima S, Hayashi and Omori M. Chem. Lett. 1975;931 [Google Scholar]
- Yajima S, Okamura K, Hayashi J. and Omori M. J. Am. Ceram. Soc. 1976;59:324. doi: 10.1111/j.1151-2916.1976.tb10975.x. [DOI] [Google Scholar]
- Cranstone W R I, Bushnell-Watson S M. and Sharp J H. J. Mater. Res. 1995;10:2659. doi: 10.1557/JMR.1995.2659. [DOI] [Google Scholar]
- Hemida A T, Birot M, Pillot J P, Dunogues J. and Pailler R. J. Mater. Sci. 1997;32:3475. doi: 10.1023/A:1018637204036. [DOI] [Google Scholar]
- Bao X, Nangrejo M R. and Edirisinghe M J. J. Mater. Sci. 1999;34:2495. doi: 10.1023/A:1004666326039. [DOI] [Google Scholar]
- Chae S H, Kim Y W, Song I H. and Kim H D. J. Eur. Ceram. Soc. 2009;29:2867. doi: 10.1016/j.jeurceramsoc.2009.03.027. [DOI] [Google Scholar]
- Colombo P. J. Eur. Ceram. Soc. 2008;28:1389. doi: 10.1016/j.jeurceramsoc.2007.12.002. [DOI] [Google Scholar]
- Kroke E, Li Y L, Konetschny C, Lecomte E, Fasel C an and Riedel R. 2000. Mater. Sci. Eng. R 26 97 10.1016/S0927-796X(00)00008-5 [DOI] [Google Scholar]
- Raj R. Am. Ceram. Soc. Bull. 2001;80:25. [Google Scholar]
- Baney R. and Chandra G. Encyclopedia of Polymer Science and Engineering. New york: Wiley; 1988. [Google Scholar]
- Burns G T, Taylor R B, Xu Y, Zangil A. an and Zank G A. Chem. Mater. 1992;4:1313. doi: 10.1021/cm00024a035. [DOI] [Google Scholar]
- Riedel R. In: Processing of Ceramics. Brook R J, editor. Wurzburg, Germany: VCH; 1996. [Google Scholar]
- Bouillon E.et al1991J. Mater. Sci. 261333. 10.1007/BF00544474 [DOI] [Google Scholar]
- Soraru G D, Modena S, Guadagnino E. an an and Colombo P. J. Am. Ceram. Soc. 2002;85:1529. doi: 10.1111/j.1151-2916.2002.tb00308.x. [DOI] [Google Scholar]
- Rouxel T, Soraru G D. and Vicens J. J. Am. Ceram. Soc. 2001;84:1052. doi: 10.1111/j.1151-2916.2001.tb00789.x. [DOI] [Google Scholar]
- Rouxel T, Massouras G. and Soraru G D. J. Sol-Gel Sci. Technol. 1999;14:87. doi: 10.1023/A:1008779915809. [DOI] [Google Scholar]
- Riedel R, Kienzle A, Dressler W, Ruwisch L, Bill J. and Aldinger F. Nature. 1996;382:796. doi: 10.1038/382796a0. [DOI] [Google Scholar]
- Riedel R, Ruswisch L M, An L N. and Raj R. J. Am. Ceram. Soc. 1998;81:3341. doi: 10.1111/j.1151-2916.1998.tb02780.x. [DOI] [Google Scholar]
- Soraru G D. and Suttor D. J. Sol-Gel. Sci. Technol. 1999;14:69. doi: 10.1023/A:1008775830830. [DOI] [Google Scholar]
- Turquat C, Kleebe H J, Gregori G, Walter S. and Soraru G D. J. Am. Ceram. Soc. 2001;84:2189. doi: 10.1111/j.1151-2916.2001.tb00986.x. [DOI] [Google Scholar]
- Monthioux M. and Delverdier O. J. Eur. Ceram. Soc. 1996;16:721. doi: 10.1016/0955-2219(95)00186-7. [DOI] [Google Scholar]
- Narsawa M. Materials. 2010;3:3518. doi: 10.3390/ma3063518. [DOI] [Google Scholar]
- Yim A and Pierre L E St. 1969. J. Polym. Sci. B 8 237 10.1002/pol.1969.110070313 [DOI] [Google Scholar]
- Polmanteer K E, Hunter M J, Holms D R, Bumm C W. an and Smith D I. J. Appl. Polym. Sci. 1959;1:3. doi: 10.1002/app.1959.070010102. [DOI] [Google Scholar]
- Soraru G D, Campostrini R, Maurina S. and Babonneau F. J. Am. Ceram. Soc. 1997;80:999. doi: 10.1111/j.1151-2916.1997.tb02933.x. [DOI] [Google Scholar]
- Klonczynski A, Schneider G, Riedel R. and Theissmann R. Adv. Eng. Mater. 2004;6:64. doi: 10.1002/adem.200300525. [DOI] [Google Scholar]
- Thunemann M, Herzog A, Vogt U. and Beffort O. Adv. Eng. Mater. 2004;6:167. doi: 10.1002/adem.200300513. [DOI] [Google Scholar]
- Zhu S, Ding S, Xi H. and Wang R. Mater. Lett. 2005;59:595. doi: 10.1016/j.matlet.2004.11.003. [DOI] [Google Scholar]
- Ma Y, Ma Q S, Suo J. and Chen Z H. Ceram. Int. 2008;34:253. doi: 10.1016/j.ceramint.2006.08.018. [DOI] [Google Scholar]
- Cordelair J. and Greil P. J. Eur. Ceram. Soc. 2000;20:1947. doi: 10.1016/S0955-2219(00)00068-6. [DOI] [Google Scholar]
- Soraru G D, Dallapiccola E. and D'Andrea G. J. Am. Ceram. Soc. 1996;79:2074. doi: 10.1111/j.1151-2916.1996.tb08939.x. [DOI] [Google Scholar]
- Shah S P. and Raj R. Acta Mater. 2002;50:4093. doi: 10.1016/S1359-6454(02)00206-9. [DOI] [Google Scholar]
- Esfehanian M, Oberacker R, Fett T. and Hoffmann M J. J. Am. Ceram. Soc. 2008;91:3803. doi: 10.1111/j.1551-2916.2008.02730.x. [DOI] [Google Scholar]
- Colombo P, Hellmann J R. and Shelleman D L. J. Am. Ceram. Soc. 2002;85:2306. doi: 10.1111/j.1151-2916.2002.tb00452.x. [DOI] [Google Scholar]
- Soraru G D, Modena S, Guadagnino E, Colombo P, Egan J. and Pantano C. J. Am. Ceram. Soc. 2002;85:1529. doi: 10.1111/j.1151-2916.2002.tb00308.x. [DOI] [Google Scholar]
- Latournerie J, Dempsey P, Hourlier-Bahloul D. and Bonnet J P. J. Am. Ceram. Soc. 2006;89:1485. doi: 10.1111/j.1551-2916.2005.00869.x. [DOI] [Google Scholar]
- Fukushima M, Zhou Y, Miyazaki H, Yoshizawa Y, Hirao K, Iwamoto Y, Yamazaki S. and Nagano T. J. Am. Ceram. Soc. 2006;89:1523. doi: 10.1111/j.1551-2916.2006.00931.x. [DOI] [Google Scholar]
- Shan S Y, Yang J F, Gao J Q, Zhang W H, Jin Z H, Janssen R. and Ohji T. J. Am. Ceram. Soc. 2005;88:2594. doi: 10.1111/j.1551-2916.2005.00444.x. [DOI] [Google Scholar]
- Arita H, Castano V M. and Wilkinson D S. J. Mater. Sci. Mater. Med. 1995;6:19. doi: 10.1007/BF00121241. [DOI] [Google Scholar]
- Herzog A, Klingner R, Vogt U. and Graule T. J. Am. Ceram. Soc. 2004;87:784. doi: 10.1111/j.1551-2916.2004.00784.x. [DOI] [Google Scholar]
- Zollfrank C, Kladny R, Sieber H. and Greil P. J. Eur. Ceram. Soc. 2004;24:479. doi: 10.1016/S0955-2219(03)00202-4. [DOI] [Google Scholar]
- White R A, White E W. and Weber J N. Science. 1972;176:922. doi: 10.1126/science.176.4037.922. [DOI] [PubMed] [Google Scholar]
- Skinner D P, Newnham R E. and Cross L E. Mater. Res. Bull. 1978;13:599. doi: 10.1016/0025-5408(78)90185-X. [DOI] [Google Scholar]
- Nangrejo M R. and Edirisinghe M J. J. Porous Mater. 2002;9:131. doi: 10.1023/A:1020834509443. [DOI] [Google Scholar]
- Bao X, Nangrejo M R. and Edirisinghe M J. J. Mater. Sci. 2000;35:4365. doi: 10.1023/A:1004805023228. [DOI] [Google Scholar]
- Nangrejo M R, Bao X. and Edirisinghe M J. J. Mater. Sci. Lett. 2000;19:787. doi: 10.1023/A:1006725023801. [DOI] [Google Scholar]
- Nangrejo M R, Bao X. and Edirisinghe M J. J. Eur. Ceram. Soc. 2000;20:1777. doi: 10.1016/S0955-2219(00)00046-7. [DOI] [Google Scholar]
- Nangrejo M R, Bao X. and Edirisinghe M J. J. Inorg. Mater. 2001;3:37. doi: 10.1016/S1466-6049(00)00096-9. [DOI] [Google Scholar]
- Zhu X, Jiang D, Tan S. and Zhang Z. J. Am. Ceram. Soc. 2001;84:1654. doi: 10.1111/j.1151-2916.2001.tb00895.x. [DOI] [Google Scholar]
- Vogt U F, Gyorfy L, Herzog A, Graule T. and Plesch G. J. Phys. Chem. Solids. 2007;68:1234. doi: 10.1016/j.jpcs.2006.12.008. [DOI] [Google Scholar]
- Greil P. J. Eur. Ceram. Soc. 2001;21:105. doi: 10.1016/S0955-2219(00)00179-5. [DOI] [Google Scholar]
- Zollfrank C, Kladny R, Motz G, Sieber H. an and Greil P. Ceram. Trans. 2001;114:43. [Google Scholar]
- Kim Y W, Jin Y J, Chun Y S, Song I H. and Kim H D. Scr. Mater. 2005;53:921. doi: 10.1016/j.scriptamat.2005.06.032. [DOI] [Google Scholar]
- Jin Y J. and Kim Y W. J. Mater. Sci. 2010;45:282. doi: 10.1007/s10853-009-3993-7. [DOI] [Google Scholar]
- Fitzgerald T J, Michaud V J. and Mortensen A. J. Mater. Sci. 1995;30:1037. doi: 10.1007/BF01178442. [DOI] [Google Scholar]
- Kim Y, Lee K H, Lee S H. and Park C B. J. Ceram. Soc. Japan. 2003;111:863. doi: 10.2109/jcersj.111.863. [DOI] [Google Scholar]
- Wang C, Wang J, Park C B. and Kim Y W. J. Ceram. Proc. Res. 2009;10:238. [Google Scholar]
- Yoon B H, Lee E J, Kim H E. and Koh Y H. J. Am. Ceram. Soc. 2007;90:1753. doi: 10.1111/j.1551-2916.2007.01703.x. [DOI] [Google Scholar]
- Wang H, Sung I, Li X. and Kim D. J. Porous Mater. 2004;11:265. doi: 10.1023/B:JOPO.0000046353.24308.86. [DOI] [Google Scholar]
- Colombo P, Bernardo E. and Biasetto B. J. Am. Ceram. Soc. 2004;87:152. doi: 10.1111/j.1551-2916.2004.00152.x. [DOI] [Google Scholar]
- Shibuya M, Takahashi T. and Koyama K. Compos. Sci. Technol. 2007;67:119. doi: 10.1016/j.compscitech.2006.03.022. [DOI] [Google Scholar]
- Kim Y W, Kim S H, Kim H D. and Park C B. J. Mater. Sci. 2004;39:5647. doi: 10.1023/B:JMSC.0000040071.55240.85. [DOI] [Google Scholar]
- Kim S H, Kim Y W. and Park C B. J. Mater. Sci. 2004;39:3513. doi: 10.1023/B:JMSC.0000026964.88284.ab. [DOI] [Google Scholar]
- Fukushima M, Zhou Y, Yoshizawa Y I, Miyazaki H. and Hirao K. J. Ceram. Soc. Japan. 2006;114:571. doi: 10.2109/jcersj.114.571. [DOI] [Google Scholar]
- Colombo P. and Modesti M. J. Am. Ceram. Soc. 1999;82:573. doi: 10.1111/j.1151-2916.1999.tb01803.x. [DOI] [Google Scholar]
- Kim Y W, Kim S H, Xu X, Choi C H, Park C B. and Kim H D. J. Mater. Sci. Lett. 2002;21:1667. doi: 10.1023/A:1020820608722. [DOI] [Google Scholar]
- Innocentini M D M, Sepulveda P, Salvini V R. and Pandolfelli V C. J. Am. Ceram. Soc. 1998;81:3349. doi: 10.1111/j.1151-2916.1998.tb02782.x. [DOI] [Google Scholar]
- Colombo P, Griffoni M. and Modesti M. J. Sol-Gel Sci. Technol. 1998;13:195. doi: 10.1023/A:1008604800737. [DOI] [Google Scholar]
- Colombo P, Griffoni M. and Modesti M. J. Sol-Gel Sci. Technol. 1999;14:103. doi: 10.1023/A:1008736100788. [DOI] [Google Scholar]
- Colombo P. and Bernardo E. Compos. Sci. Technol. 2003;63:2353. doi: 10.1016/S0266-3538(03)00268-9. [DOI] [Google Scholar]
- Schmidt H, Koch D, Grathwohl G. and Colombo P. J. Am. Ceram. Soc. 2001;84:2252. doi: 10.1111/j.1151-2916.2001.tb00997.x. [DOI] [Google Scholar]
- Biasetto L, Francis A, Palade P, Principi G. and Colombo P. J. Mater. Sci. 2008;43:4119. doi: 10.1007/s10853-007-2224-3. [DOI] [Google Scholar]
- Colombo P, Gambaryan-Roisman T, Scheffler M, Buhler P. and Greil P. J. Am. Ceram. Soc. 2001;84:2265. doi: 10.1111/j.1151-2916.2001.tb01000.x. [DOI] [Google Scholar]
- Colombo P, Hellmann J R. and Shelleman D L. J. Am. Ceram. Soc. 2001;84:2245. doi: 10.1111/j.1151-2916.2001.tb00996.x. [DOI] [Google Scholar]
- Gambaryan-Roisman T, Scheffler M, Buhler P. and Greil P. Ceram. Trans. 2000;108:121. [Google Scholar]
- Pantano C G, Singh A K. and Zhang H J. J. Sol-Gel. Sci. Technol. 1999;14:7. doi: 10.1023/A:1008765829012. [DOI] [Google Scholar]
- Kim Y W, Kim S H, Wang C. and Park C B. J. Am. Ceram. Soc. 2003;86:2231. doi: 10.1111/j.1151-2916.2003.tb03641.x. [DOI] [Google Scholar]
- Kim Y W. and Park C B. Compos. Sci. Technol. 2003;63:2371. doi: 10.1016/S0266-3538(03)00270-7. [DOI] [Google Scholar]
- Wang C, Wang J, Park C B. and Kim Y W. J. Mater. Sci. 2007;42:2854. doi: 10.1007/s10853-006-0229-y. [DOI] [Google Scholar]
- Kim Y W, Wang C. and Park C B. J. Ceram. Soc. Japan. 2007;115:419. doi: 10.2109/jcersj.115.419. [DOI] [Google Scholar]
- Eom J H. and Kim Y W. Met. Mater. Int. 2007;13:521. doi: 10.1007/BF03027913. [DOI] [Google Scholar]
- Wang C, Wang J, Park C B. and Kim Y W. J. Mater. Sci. 2004;39:4913. doi: 10.1023/B:JMSC.0000035335.92101.7c. [DOI] [Google Scholar]
- Wang C, Wang J, Park C B. and Kim Y W. J. Ceram. Proc. Res. 2009;10:66. [Google Scholar]
- Kim Y W, Kim S H, Song I H, Kim H D. and Park C B. J. Am. Ceram. Soc. 2005;88:2949. doi: 10.1111/j.1551-2916.2005.00509.x. [DOI] [Google Scholar]
- Jang D H, Kim Y W, Song I H, Kim H D. and Park C B. J. Ceram. Soc. Japan. 2006;114:549. doi: 10.2109/jcersj.114.549. [DOI] [Google Scholar]
- Eom J H, Kim Y W, Song I H and Kim H D. 2007. Mater. Sci. Eng. A 464 129 10.1016/j.msea.2007.03.076 [DOI] [Google Scholar]
- Chae S H. and Kim Y W. J. Mater. Sci. 2009;44:1404. doi: 10.1007/s10853-009-3264-7. [DOI] [Google Scholar]
- Eom J H, Kim Y W, Song I H. and Kim H D. J. Eur. Ceram. Soc. 2008;28:1029. doi: 10.1016/j.jeurceramsoc.2007.09.009. [DOI] [Google Scholar]
- Eom J H. and Kim Y W. J. Mater. Sci. 2009;44:4482. doi: 10.1007/s10853-009-3638-x. [DOI] [Google Scholar]
- Grande T, Sommerset H, Hagen E, Wiik K. and Einarsrud M A. J. Am. Ceram. Soc. 1997;80:1047. doi: 10.1111/j.1151-2916.1997.tb02945.x. [DOI] [Google Scholar]
- Eom J H, Kim Y W. and Kim K J. J. Ceram. Soc. Japan. 2010;118:102. doi: 10.2109/jcersj2.118.102. [DOI] [Google Scholar]
- Kim Y W, Eom J H, Wang C. and Park C B. J. Am. Ceram. Soc. 2008;91:1361. doi: 10.1111/j.1551-2916.2008.02280.x. [DOI] [Google Scholar]
- Eom J H, Kim Y W. and Narisawa M. J. Ceram. Process. Res. 2008;9:176. [Google Scholar]
- Suttor D, Kleebe H J. and Ziegler G. J. Am. Ceram. Soc. 1997;80:2541. doi: 10.1111/j.1151-2916.1997.tb03156.x. [DOI] [Google Scholar]
- Kolitsch U, Seifert H J, Ludwig T. and Aldinger F. J. Mater. Res. 1999;14:447. doi: 10.1557/JMR.1999.0064. [DOI] [Google Scholar]
- Kim Y W, Kim H D. and Park C B. J. Am. Ceram. Soc. 2005;88:3311. doi: 10.1111/j.1551-2916.2005.00597.x. [DOI] [Google Scholar]
- Song I H, Kim M J, Kim H D. and Kim Y W. Scr. Mater. 2006;54:1521. doi: 10.1016/j.scriptamat.2005.12.039. [DOI] [Google Scholar]
- Jang D H, Kim Y W. and Kim H D. J. Ceram. Soc. Japan. 2007;115:52. doi: 10.2109/jcersj.115.52. [DOI] [Google Scholar]
- Vakifahmetoglu C, Colombo P, Pauletti A, Martin C F. and Babonneau F. Int. J. Appl. Ceram. Technol. 2010;7:528. [Google Scholar]
- Yeon S H, Reddington P, Gogotsi Y, Fischer, Vakifahmetoglu C. and Colombo P. Carbon. 2010;48:201. doi: 10.1016/j.carbon.2009.09.004. [DOI] [Google Scholar]
- Vakifahmetoglu C, Menapace I, Hirsch A, Biasetto L, Hauser R, Riedel R. and Colombo P. Ceram. Int. 2010;35:3281. doi: 10.1016/j.ceramint.2009.05.022. [DOI] [Google Scholar]
- Haug R, Weinmann M, Bill J. and Aldinger F. J. Eur. Ceram. Soc. 1999;19:1. doi: 10.1016/S0955-2219(98)00167-8. [DOI] [Google Scholar]
- Konetschny C, Galusek D, Reschke S, Fasel C. and Riedel R. J. Eur. Ceram. Soc. 1999;19:2789. doi: 10.1016/S0955-2219(99)00070-9. [DOI] [Google Scholar]
- Narisawa M, Iseki T, Katase Y. and Okamura K. J. Am. Ceram. Soc. 2003;86:227. doi: 10.1111/j.1151-2916.2003.tb00004.x. [DOI] [Google Scholar]
- Iseki T, Narisawa M, Katase Y, Oka K, Dohmaru T. and Okamura K. Chem. Mater. 2001;13:4163. doi: 10.1021/cm000744b. [DOI] [Google Scholar]
- Iseki T, Narisawa M, Okamura K, Oka K. and Dohmaru T. J. Mater. Sci. Lett. 1999;18:185. doi: 10.1023/A:1006603610905. [DOI] [Google Scholar]
- Narisawa M, Yasuda H, Mor R, Mabuchu H, Oka K. and Kim Y W. J. Ceram. Soc. Japan. 2008;116:121. doi: 10.2109/jcersj2.116.121. [DOI] [Google Scholar]
- Kim K J, Lee S, Lee J H, Roh M H, Lim K Y. and Kim Y W. J. Am. Ceram. Soc. 2009;92:424. doi: 10.1111/j.1551-2916.2008.02913.x. [DOI] [Google Scholar]
- Narisawa M, Sumimoto R I, Kita K I, Kado H, Mabuchi H. and Kim Y W. J. Appl. Poly. Sci. 2009;114:2600. doi: 10.1002/app.30802. [DOI] [Google Scholar]
- Kho J G, Min D S. and Kim D P. J. Mater. Sci. Lett. 2000;19:303. doi: 10.1023/A:1006754428380. [DOI] [Google Scholar]
- Boury B, Bryson N. and Soula G. Chem. Mater. 1998;10:297. doi: 10.1021/cm970443a. [DOI] [Google Scholar]
- Boury B, Bryson N. and Soula G. Appl. Organomet. Chem. 1999;13:419. doi: 10.1002/(SICI)1099-0739(199906)13:6<419::AID-AOC855>3.0.CO;2-8. [DOI] [Google Scholar]
- Seyferth D, Wood T G, Tracy H J. and Robisoe J L. J. Am. Ceram. Soc. 1991;75:1300. doi: 10.1111/j.1151-2916.1992.tb05578.x. [DOI] [Google Scholar]
- Zhang Z, Babonneau F, Laine R M, Mu Y, Harrod J F. and Rahn J A. J. Am. Ceram. Soc. 1991;74:670. doi: 10.1111/j.1151-2916.1991.tb04080.x. [DOI] [Google Scholar]
- Martin H P, Muller E, Richter R, Roewer G. and Brendler E. J. Mater. Sci. 1997;32:1381. doi: 10.1023/A:1018577111536. [DOI] [Google Scholar]
- Jakubenas K. and Marcus H L. J. Am. Ceram. Soc. 1995;78:2263. doi: 10.1111/j.1151-2916.1995.tb08653.x. [DOI] [Google Scholar]
- Gonsalves K E, Strutt P R, Xiao T D. and Klemens P G. J. Mater. Sci. 1992;27:3231. doi: 10.1007/BF01116018. [DOI] [Google Scholar]
- Okamura K. and Seguchi T. J. Inorg. Organomet. Polym. 1992;2:171. doi: 10.1007/BF00696544. [DOI] [Google Scholar]
- Sugimoto M, Shimoo T, Okamura K. and Seguchi T. J. Am. Ceram. Soc. 1995;78:1849. doi: 10.1111/j.1151-2916.1995.tb08898.x. [DOI] [Google Scholar]
- Pivin C, Colombo P. and Tonidandel M. J. Am. Ceram. Soc. 1996;79:1967. doi: 10.1111/j.1151-2916.1996.tb08021.x. [DOI] [Google Scholar]
- Wach R A, Sugimoto M. and and Y. J. Am. Ceram. Soc. 2007;90:275. doi: 10.1111/j.1551-2916.2006.01376.x. [DOI] [Google Scholar]
- Zhu X, Jiang D. and Tan S. Mater. Sci. Eng. A. 2002;323:232. doi: 10.1016/S0921-5093(01)01352-1. [DOI] [Google Scholar]
- Mouazer R, Mullens S, Thijs I, Luyten J. and Buekenhoudt A. Adv. Eng. Mater. 2005;7:1124. doi: 10.1002/adem.200500163. [DOI] [Google Scholar]
- Zhang Z, Wang F, Yu X, Wang Y, Yan Y, Li K. and Luan Z. J. Am. Ceram. Soc. 2009;92:260. doi: 10.1111/j.1551-2916.2008.02842.x. [DOI] [Google Scholar]
- Yao X, Tan S, Zhang X, Huang Z. and Jiang D. J. Mater. Sci. 2007;42:4960. doi: 10.1007/s10853-006-0473-1. [DOI] [Google Scholar]
- Takahashi T, Munstedt H, Modesti M. and Colombo P. J. Eur. Ceram. Soc. 2001;21:2821. doi: 10.1016/S0955-2219(01)00220-5. [DOI] [Google Scholar]
- Colombo P. Adv. Eng. Mater. 1999;1:203. doi: 10.1002/(SICI)1527-2648(199912)1:3/4<203::AID-ADEM203>3.0.CO;2-M. [DOI] [Google Scholar]
- Ding S, Zeng Y P an and Jiang D. 2006. Mater. Sci. Eng. A 425 326 10.1016/j.msea.2006.03.075 [DOI] [Google Scholar]
- Ding S, Zhu S, Zeng Y. and Jiang D. Ceram. Int. 2006;32:461. doi: 10.1016/j.ceramint.2005.03.024. [DOI] [Google Scholar]
- Chi W, Jiang D, Huang Z. and Tan S. Ceram. Int. 2004;30:869. doi: 10.1016/j.ceramint.2003.10.006. [DOI] [Google Scholar]
- Lee S H. and Kim Y W. J. Kor. Ceram. Soc. 2006;43:458. doi: 10.4191/KCERS.2006.43.8.458. [DOI] [Google Scholar]
- Chun Y S. and Kim Y W. Met. Mater. Int. 2005;11:351. doi: 10.1007/BF03027504. [DOI] [Google Scholar]
- Kim Y W, Eom J H, Park C B, Zhai W, Guo Y and Balasubramanian M. 2010. J. Am. Ceram. Soc. at press [Google Scholar]