

Abstract
The development of methods for the off–on switching of immobilization or presentation of cell-adhesive peptides and proteins during cell culture is important because such surfaces are useful for the analysis of the dynamic processes of cell adhesion and migration. This paper describes a chemically functionalized gold substrate that captures a genetically tagged extracellular matrix protein in response to light. The substrate was composed of mixed self-assembled monolayers (SAMs) of three disulfide compounds containing (i) a photocleavable poly(ethylene glycol) (PEG), (ii) nitrilotriacetic acid (NTA) and (iii) hepta(ethylene glycol) (EG7). Although the NTA group has an intrinsic high affinity for oligohistidine tag (His-tag) sequences in its Ni2+-ion complex, the interaction was suppressed by the steric hindrance of coexisting PEG on the substrate surface. Upon photoirradiation of the substrate to release the PEG chain from the surface, this interaction became possible and hence the protein was captured at the irradiated regions, while keeping the non-specific adsorption of non-His-tagged proteins blocked by the EG7 underbrush. In this way, we selectively immobilized a His-tagged fibronectin fragment (FNIII7–10) to the irradiated regions. In contrast, when bovine serum albumin—a major serum protein—was added as a non-His-tagged protein, the surface did not permit its capture, with or without irradiation. In agreement with these results, cells were selectively attached to the irradiated patterns only when a His-tagged FNIII7-10 was added to the medium. These results indicate that the present method is useful for studying the cellular behavior on the specific extracellular matrix protein in cell-culturing environments.
Keywords: patterning, photocleavage reaction, His-tag, fibronectin, integrin
Introduction
The patterned immobilization of peptides and proteins on material surfaces is important for various bioanalytical and biomedical applications, such as the analysis of ligand–analyte interactions [1] and high-throughput discovery of engineered culture surfaces for stem cell maintenance [2, 3] or selective differentiation [4]. Moreover, functionalized substrates that capture or expose a cell-adhesive peptide on their surfaces in response to an external stimulus, such as heat [5], voltage [6–8] or light [9–11], are useful for the dynamic control of cell adhesion. These substrates, called dynamic substrates, have been applied not only to explore cell migration but also to coculture heterotypic cells [12]. Almost all of the earlier dynamic substrates used linear or cyclic RGD peptides as cell-adhesive ligands. The RGD peptide is a tripeptide of arginine, glycine and aspartate, which is the core recognition sequence in fibronectin, a major extracellular matrix (ECM) protein. This protein interacts with integrin, a membrane protein [13, 14], and hence, the surface permits cell adhesion. Although this small peptide sequence is sufficient for cell adhesion, its biological activity is lower than that of the native protein owing to the absence of complementary domains such as the PHSRN (phenylalanine-histidine-serine-arginine-asparagine) sequence [15, 16]. On the other hand, larger protein fragments containing such complementary sequences exhibit stronger cell adhesion and higher biological activities [17, 18]. Therefore, dynamic substrates will become more physiologically relevant when used in combination with such larger protein fragments instead of the RGD peptides.
To this end, we herein developed a new dynamic substrate that allowed the immobilization of a fragment of a specific ECM protein in response to light (figure 1). The idea is based on our earlier culture substrates, where an antifouling polymer, poly(ethylene glycol) (PEG), was conjugated to the substrate surface via a photocleavable 2-nitrobenzyl ester [19, 20]. Protein adsorption to the surface was suppressed by the PEG brushes, but became possible after removing PEG by photoirradiation; hence, the surface cell adhesiveness changed markedly after the photoirradiation. However, these substrates did not meet the above-described needs in the present form, as they permitted the adsorption of many different proteins after photoirradiation. To endow the substrates with protein selectivity, in this study, we added two new components, i.e. nitrilotriacetic acid (NTA) and EG7, to the surface. The NTA group interacts with an oligohistidine tag (His-tag) peptide sequence in its Ni2+-ion complex, whereas EG7 prevents non-specific adsorption of other proteins. In this surface design, we expected the surface to capture proteins bearing a His-tag sequence in response to light, while suppressing the adsorption of non-His-tagged proteins. In this study, we first optimized the surface compositions of the three components using His-tagged glutathione-S-transferase (GST) as a model protein to realize the above-described working principle. We then evaluated a recombinant fibronectin fragment (FNIII7–10) [18, 21, 22], which encompasses the integrin α5β1 binding domain including RGD and PHSRN sequences, and used its His-tagged protein for cellular patterning. The results strongly support the application of the present strategy for the dynamic control of cell adhesion in cell-culturing environments.
Figure 1.
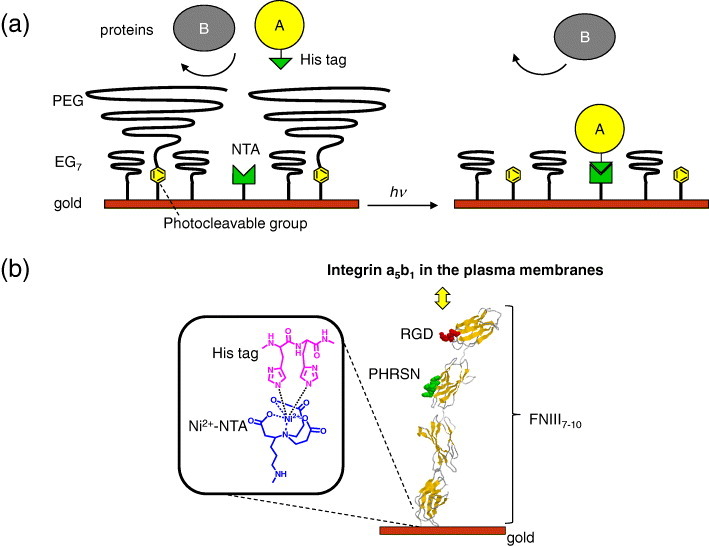
Schematic of a dynamic substrate that captures His-tagged proteins in response to light. (a) The surface of a gold substrate was functionalized with the SAMs of photocleavable PEG, NTA and EG7 ligands. (Left) Before irradiation, the surface prevents adsorption of any proteins because of the presence of PEG brushes. (Right) Photoirradiation cuts off the PEG brushes, and the protein bearing a His-tag (protein A) is immobilized to the NTA group, while keeping the adsorption of non-His-tagged proteins (protein B) blocked by the EG7 underbrushes. (b) A recombinant fibronectin fragment FNIII7–10 bearing a His-tagged sequence is immobilized to the NTA ligand where it mediates cell adhesion by interacting with integrin α5β1 in the plasma membranes.
Experimental details
Reagents and materials
The organic and inorganic compounds used in the study were purchased from Wako (Osaka, Japan), Sigma-Aldrich (St. Louis, MO), or TCI (Tokyo, Japan), unless stated otherwise.
Preparation of gold-sputtered substrate
Gold substrates were deposited on glass using a custom-built sputtering system. Glass cover slips (0.12−0.17 mm thick, Matsunami, Osaka, Japan) were cleaned in piranha solution [1:2 H2O2/H2SO4 (v/v), Caution! Piranha is a vigorous oxidant and should be used with extreme care] for 1 h, rinsed with distilled water and dried with a stream of dry nitrogen. They were first coated with a 10 nm Cr layer to promote adhesion and then with 50 nm of gold.
Organic synthesis
ω-Methoxy-hepta(ethylene glycol) tosylate (EG7-OTs): an aqueous solution of 60% NaH (0.50 g, 13 mmol) was added to 40 ml of dry tetrahydrofuran (THF) solution of ω-methoxy-hepta(ethylene glycol) (EG7-OH, 3.5 g, 10 mmol) on ice and stirred for 60 min at room temperature. p-Toluenesulfonyl chloride (TsCl, 4.6 g, 24 mmol) was added to the solution and stirred for 2 days at room temperature. After condensation of the THF solution, 0.3 M HCl was added, and then the solution mixture was extracted with chloroform. EG7-OTs was purified by silica gel chromatography by first using hexane/ethyl acetate (2:1) as a solvent, and then using chloroform/methanol (98:2). The yield was 3.7 g (73%) and the product contained the following proton nuclear magnetic resonance peaks (1H NMR, CDCl3): 7.80 (d, 2H), 2.35 (d, 2H), 4.16 (t, 2H), 3.70−3.54 (m, 26H), 3.38 (s, 3H) and 2.45 (s, 3H).
ω-Methoxy-hepta(ethylene glycol) azide (EG7-N3): NaN3 (1.7 g, 27 mmol) was added to 13 ml of dry dimethylformamide (DMF) solution of EG7-OTs (0.75 g, 1.5 mmol) in a nitrogen atmosphere and stirred overnight at room temperature. The reaction mixture was distilled under reduced pressure and then dissolved in dichloromethane. The solution was washed with water and 2 M HCl, and EG7-OTs was obtained by the subsequent condensation. The yield was 0.26 g (47%) with the 1H NMR (CDCl3) peaks of 3.69−3.64 (m, 24H), 3.55 (t, 2H) and 3.39−3.38 (m, 5H).
ω-Methoxy-hepta(ethylene glycol) amine (EG7-NH2): Triphenyl phosphine (1.3 g, 5.0 mmol) was added to a tetrahydrofuran (THF) solution of EG7-N3 (0.20 g, 0.55 mmol) and stirred for 3 days at room temperature. The solution was quenched with water and stirred for another day. After washing the aqueous solution with toluene, the aqueous layer was condensed, and then treated by silica gel chromatography (2-propanol/10% NH3=85:15). The yield was 0.10 g (53%) with the 1H NMR (CDCl3) peaks of 3.55−3.42 (m, 28H), 3.29 (s, 3H) and 2.74 (t, 2H).
Disulfide 1 (disulfide bearing a photocleavable PEG): A mixture of bis(12-(4-(1- hydroxyethyl)-2-methoxy-5-nitrophenoxy)dodecyl) disulfide (4) [23], ω-methoxy-poly(ethylene glycol) amine (Mw=2K, 5K, or 12K, MEPA, Nichiyu, Tokyo, Japan) and triethylamine with a 1:2.2:2.2 volume ratio was mixed overnight at room temperature (figure 2(a)). Each reagent was a 5 mM solution in dimethyl sulfoxide (DMSO). The reaction was almost completed as confirmed by a high performance liquid chromatography (data not shown). This mixture was used for the surface modification of the gold substrates without purification. Its 1H NMR (DMSO-d6) peaks were 7.55 (s, 1H), 7.10 (s, 1H), 6.09 (q, 1H), 4.03 (t, 2H), 3.90 (s, 3H), 3.74−3.24 (m, 1081H), 2.97 (q, 2H), 2.68 (t, 2H) and 1.71−1.25 (m, 25H).
Figure 2.
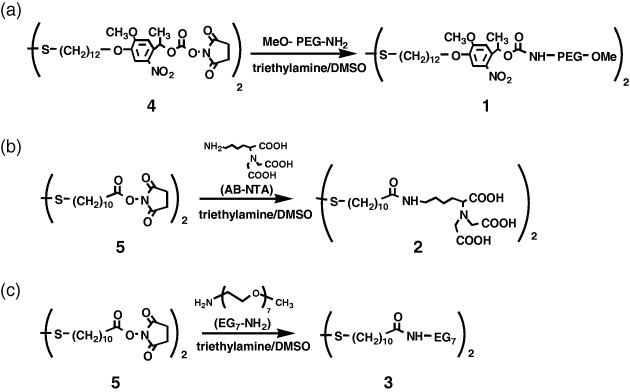
Synthesis of disulfide ligands: (a) PEG-terminated disulfide bearing a photocleavable 2-nitrobenzyl ester, (b) NTA-terminated disulfide and (c) EG7-terminated disulfide.
Disulfide 2 (NTA-terminated disulfide) and disulfide 3 (EG7-terminated disulfide): These conjugates were synthesized in the same way as disulfide 1, except for using disuccinimidyl 11,11′-dithiobisundecanoate (5) (Dojindo, Kumamoto, Japan) instead of 4 (figures 2(b) and (c)). AB-NTA (Dojindo) was used as an NTA ligand. The resulting 1H NMR (DMSO-d6) peaks were: disulfide 2, 3.22 (s, 4H), 3.18 (t, 1H), 2.97 (q, 2H), 2.68 (t, 2H), 2.02 (t, 2H) and 1.62−1.23 (m, 24H); disulfide 3, 3.51−3.16 (m, 31H), 2.68 (t, 2H), 2.04 (s, 2H) and 1.64−1.24 (m, 16H).
Surface modification of gold substrates with disulfide compounds
The above-described disulfide ligands were mixed in a given ratio and diluted to half concentration using DMSO. The gold-sputtered substrate was soaked in this solution and incubated at 30 °C overnight.
Contact angle measurements
Water contact angle measurements were conducted at room temperature using a contact angle meter DropMaster 500 (Kyowa Interface Science, Saitama, Japan). A fixed volume (l μl) of ultrapure water was dropped on the surface and monitored with a charge-coupled device (CCD) camera. The captured images were analyzed using FAMAS software (Kyowa Interface Science) to determine the contact angle. All contact angles are reported as the average of more than three values taken at different points on the surface.
Plasmid construction and extraction of His-tagged GST and FNIII7–10
Two types of His-tagged protein were used in this study (figure 3(a)). For the expression of His-tagged GST, pET41a vector (Merck, Whitehouse Station, NJ) was used without modification. By transforming BL21 (Merck) with this plasmid DNA, a recombinant GST bearing His-tag at the C-terminal side (GST-His) was produced. The cell lysate was applied to a glutathione sepharose column (GE Healthcare, Uppsala, Sweden), and the protein was eluted with glutathione (10 mM).
Figure 3.
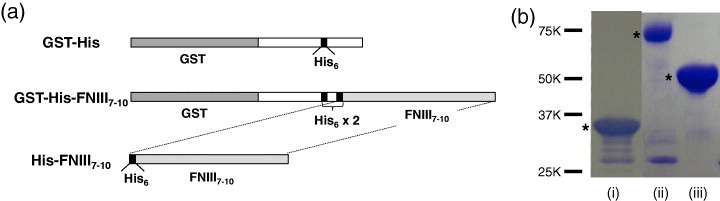
His-tagged proteins used in this study. (a) Schematic of the constructs. (b) Sodium dodecyl sulfate polyacrylamide gel electrophoresis of purified His-tagged proteins: (i) GST-His, (ii) GST-His-FNIII7–10 and (iii) His-FNIII7–10. The gels were stained with Coomassie Brilliant Blue. Molecular markers are also shown as reference. The His-tagged proteins are marked by asterisks.
The cDNA for His-tagged FNIII7–10 (His-FNIII7–10) was produced by a standard polymerase chain reaction (PCR) using appropriate primers, one of which included a cDNA sequence of hexahistidine (CACCATCACCATCACCAT) and cDNA of FNIII7–10 in the XA3 plasmid as a template [18]. The PCR product was inserted between EcoRV and HindIII sites of pET41a. After purification of the GST fusion protein (GST-His6-FNIII7–10) in the same way as GST-His, the GST tag was removed using thrombin (Merck).
The protein concentrations of both final products, i.e. GST-His and His-FNIII7–10, were determined by the Bradford method using a Bio-Rad protein assay reagent (Bio-Rad, Hercules, CA) with bovine serum albumin (BSA) as a standard. The purity of the protein was evaluated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (figure 3(b)).
Patterning His-tagged proteins and their immunofluorescence detection
The substrate was placed upside down in a glass-bottom dish filled with water and irradiated using an inverted fluorescence microscope (IX81-PAFM, Olympus, Tokyo, Japan) through a stripe-patterned photomask inserted at the field diaphragm, as described previously [24, 25]. After irradiation for 10 J cm−2 (e.g. 500 mW cm−2 for 20 s) at 365 nm, the substrate was reversed and incubated with either His-tagged proteins or BSA (50 μg ml−1 each) for 2 h, and then washed with phosphate buffer saline (PBS, pH 7.4). The substrates were incubated with the corresponding primary antibodies, biotinylated anti-rabbit IgG antibodies, and then stained with FluoroSpheres NeutrAvidin labeled microspheres (0.04 μm, yellow-green, Invitrogen, Carlsbad, CA). Fluorescence images were captured with a cooled CCD camera (Retiga-Exi, Q-Imaging, Burnaby, BC, Canada) under the fluorescence microscope, using the following set of barrier filters (Omega Optical, Brattleboro, VT): 485DF15, 505DRLPXR, 510ALP. The obtained fluorescence images were analyzed using the MetaMorph image processing system (Molecular Devices, Downingtown, PA).
Cell culture and patterning
NIH3T3 cells (RIKEN) were cultured in Dulbecco's modified Eagle's medium (DMEM, SIGMA) containing 10% newborn calf serum (Invitrogen), 100 units ml−1 penicillin and 100 μg ml−1 streptomycin (Invitrogen) in 5% CO2 at 37 °C. The substrate was soaked in a normal culture medium, with or without His-FNIII7–10 (50 μg ml−1), and then irradiated in a circular or stripped pattern. NIH3T3 cells were allowed to attach in the same medium and the phase-contrast images were captured after a given time. As a control experiment, plasma fibronectin (pFN, BD Biosciences, Franklin Lakes, NJ) was added instead of His-FNIII7–10. A soluble GRGDS peptide (100 μM, Peptide Institute Inc., Osaka, Japan) was added in the medium to see the involvement of the RGD sequence in cell adhesion. Immunofluorescence experiments for the identification of integrin subtypes were performed using the methods described previously [18]. In most of the cell experiments, the content of disulfide 2 was increased from 0.5 to 5% to increase the immobilized amount of His-FNIII7–10, hence, making the surface more cell-adhesive.
Results and discussion
Design rationale of mixed SAMs
The substrate was composed of mixed SAMs of three disulfide compounds: (i) PEG-terminated disulfide with photocleavable 2-nitrobenzyl ester (1), (ii) NTA-terminated disulfide (2) and (iii) hepta(ethylene glycol) (EG7)-terminated disulfide (3). Each compound was synthesized in one step by the amide coupling of corresponding amine compounds and disulfides bearing succinimidyl ester (figure 2). The three disulfide ligands were mixed in a given ratio in DMSO and applied to the gold substrate. Throughout this study, we use this nominal mixing ratio and note that the actual ratio on the substrate surface could be different.
Disulfide 1 is an analogue of photocleavable PEG silanes used in our previous studies [19, 20]. Its disulfide group allows the functionalization of the gold surface and the 2-nitrobenzyl group endows the molecule with a photocleavable feature. The ligand exhibited a dose-dependent release from the gold surface under near-UV irradiation, as confirmed by contact angle measurement (figure 4). The reaction almost saturated within 45−120 s that corresponds to the irradiation dose of 4.5−12 J cm−2. Therefore, we irradiated the substrates with the dose of 10 J cm−2 in all of the succeeding experiments.
Figure 4.
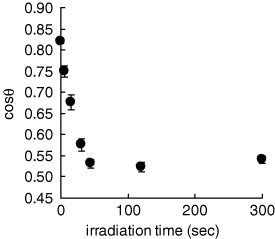
Contact angle on the gold substrate modified with disulfide ligand 1 versus irradiation time (in PBS, at λ=365 nm and 100 mW cm−2). Error bars show standard deviations among three different substrates.
The disulfides 2 and 3 are new components distinct from our previous photoactivatable substrates: 2 interacts with His-tagged proteins upon its coordination with Ni2+ ion, whereas 3 suppresses the non-specific adsorption of other non-His-tagged proteins (figure 1, right). The two disulfide underbrushes are covered by PEG of disulfide 1 that prevents His-tagged proteins from accessing the substrate surface before photoirradiation (figure 1, left). This PEG coverage is removed by the photoirradiation, making the Ni2+-NTA complex accessible to the His-tagged proteins in the bulk solution (figure 1, right). To realize this working principle, the molecular weight and/or density of PEG of disulfide 1 should be sufficiently high to block the interaction of the His-tagged proteins before irradiation (requirement 1), whereas a sufficient density of EG7 in disulfide 3 is required for the reduction of the non-specific adsorption of non-His-tagged proteins to the irradiated surface (requirement 2). However, when we increase the content of disulfide 1 , the content of 3 decreases, which demands the optimization of the surface composition of the mixed SAMs.
Optimization of mixed SAM composition
We first prepared substrates with various surface compositions and examined the effect of the PEG molecular weight (2K, 5K or 12K) and the content of disulfide 1 on the blocking of the immobilization of His-tagged proteins in the non-irradiated regions (requirement 1) in PBS. The content of disulfide 2 was kept constant at 0.5%. GST-His was used as a model protein and its immobilized amount in PBS was detected by the immunofluorescence method. As expected, the adsorption of GST-His to the non-irradiated regions was blocked at high PEG contents (figure 5(a)), and the amount of PEG required for the complete blocking of GST-His immobilization decreased with increasing molecular weight of PEG. It is noteworthy that PEG12K prevented the immobilization of GST-His to the non-irradiated surface even when its content was reduced to 10%. Since there are trade-off requirements for the higher contents for both photocleavable PEG and EG7 (vide supra), we concluded that PEG12K was the most suitable agent for our purpose.
Figure 5.
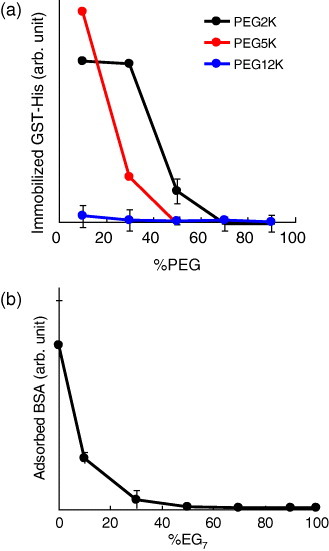
Effect of the surface composition on reducing undesired protein immobilization/adsorption in PBS. (a) Effect of the PEG chain lengths and content on the immobilization of GST-His to the non-irradiated regions. (b) Effect of the EG7 content on the adsorption of BSA to the irradiated regions. The immobilized/adsorbed proteins were detected by the immunofluorescence methods, and the amount of each protein was evaluated from its fluorescence intensity. The %NTA was 0.5%.
Next, we evaluated the effect of EG7 on the blocking of the undesired adsorption of non-His-tagged proteins to the irradiated regions (requirement 2) in PBS. BSA was used as a representative non-His-tagged protein because it is a major protein in serum and firstly adsorbs to the materials surface in cell culture environments. The adsorption of BSA to the irradiated regions was very high on the substrate without EG7 (figure 5(b), %EG7=0), indicating no protein selectivity with our previous photoactivatable substrates [19, 20]. However, increasing the EG7 content of the substrate resulted in a monotonic decrease in the non-specific adsorption of BSA to the irradiated regions (figure 5(b)). BSA adsorption became almost negligible when the EG7 content reached 50%, indicating that the substrate has a potential for the selective immobilization of His-tagged ECM proteins in response to light.
On the basis of these results, we used three substrates with the EG7 contents of 50, 70 and 90% for photopatterning His-FNIII7–10. The content of disulfide 2 was kept constant at 0.5%, and the amounts of photocleavable PEG 1 were 49.5, 29.5 and 9.5%, respectively. With all the substrates, we were able to align the His-FNIII7–10 along the irradiated striped patterns (figures 6(a)–(c)). However, their immobilized amounts decreased significantly with increasing EG7 content. A similar trend was also observed with GST-His (data not shown). The lower immobilization of His-tagged proteins on the substrates was attributed to the unfavorable complex formation of Ni2+-NTA and the His-tag sequence within dense EG7 underbrushes, as the molecular length of disulfide 2 is shorter than that of 3. On the basis of these results, we used two substrates with EG7 contents of 50 and 70% for subsequent cell studies.
Figure 6.
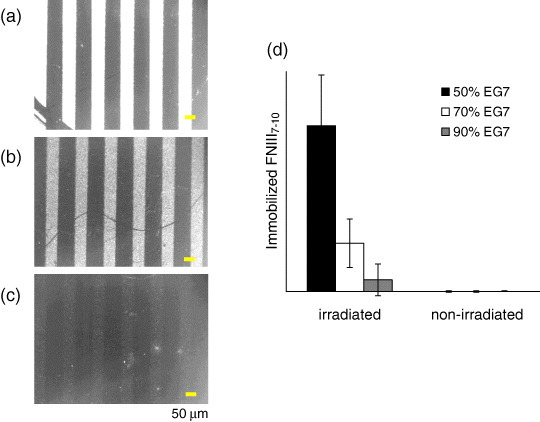
Photopatterning of His-FNIII7–10 on the substrates with different EG7 contents in PBS. (a–c) Immunofluorescence detection of FNIII7–10 immobilized to the (a) 50%, (b) 70% and (c) 90% EG7 substrate. Scale bars represent 50 μm. (d) Amount of immobilized FNIII7–10 to the irradiated and non-irradiated regions of (a–c). The %NTA was 0.5%.
Cell patterning
We used the substrates for the in situ patterning of NIH3T3 cells in a normal serum-containing medium. When the culture medium contained 50 μg ml−1 His-FNIII7–10, the cells were selectively attached to the irradiated circular region on the 70% EG7 substrates (figure 7(a)). The substrates allowed cell adhesion neither in the absence of His-FNIII7–10 (figure 7(b)) nor when plasma fibronectin was added at the same weight concentration (figure 7(c)). These results demonstrate that the surface captured His-FNIII7–10 only when the substrate was irradiated, and this immobilization was specific to the protein bearing a His-tag sequence. On the other hand, the cells adhered to the 50% EG7 substrates regardless of whether the medium contained His-FNIII7–10 or not (figures 7(d) and (e)). The His-FNIII7–10-independent cellular patterning on this substrate was not attributed to the different geometries (circle versus stripe) or sizes (300 μm versus 50 μm) of the patterns used in these experiments, because we also observed FNIII7–10-dependent cellular alignment in the same striped pattern on the 70% EG7 substrate (data not shown). Rather, the results indicate that the EG7 content of 50% was sufficient to block BSA adsorption in PBS, but insufficient to block the adsorption of some ECM proteins under the cell culture condition.
Figure 7.

Patterning of NIH3T3 cells on the substrates in a serum-containing medium. (a–c) Cell adhesion to the 70% EG7 substrate (a) with or (b) without His-FNIII7–10 (50 μg ml-1) or (c) with pFN (50 μg ml-1). The substrates were irradiated in a circular pattern. (d, e) Cell adhesion on the 50% EG7 substrate (d) with or (e) without His-FNIII7–10 (50 μg ml-1). The substrates were irradiated in a striped pattern. Cells were allowed to attach for 4 h and phase-contrast images were obtained after removing the unattached cells. The %NTA was 5% for (a–c) and 0.5% for (d) and (e). Scale bars represent 50 μm.
We also evaluated whether the observed photoinduced cell adhesion was mediated by the interaction of the immobilized FNIII7–10 and integrin α5β1, which is a typical receptor for the binding of fibronectin. When an excess soluble GRGDS peptide was added to the medium containing serum and His-FNIII7–10, the cell adhesion was completely suppressed (figure 8(a)), indicating that cell adhesion was mediated by the interaction of the RGD sequence of FNIII7–10 and integrin at the plasma membranes. To identify integrin subtypes involved in this interaction, the cells attached to the irradiated region via FNIII7–10 were cross-linked with DTSSP (Pierce), and the FNIII7–10−integrin complex was analyzed using antibodies specific to α5 and αv subtypes of integrin [18]. We observed the strong accumulation of integrin α5 to the cell-substrate interface but almost none of the αv subtype (figures 8(b)−(e)). The results demonstrate that His-FNIII7–10 was immobilized at the irradiated regions and the mediated cell adhesion occurred via the FNIII7–10−integrin α5β1 interactions.
Figure 8.

Identification of proteins involved in the cell adhesion to the photoirradiated regions on the 70% EG7 substrate. (a) Effect of a soluble GRGDS peptide on the cell pattern formation. Cells were seeded in a serum-containing medium supplemented with 50 μg ml-1 His-FNIII7–10 and an excess GRGDS (100 μM) and allowed to attach for 6 h. (b, d) Immunofluorescence detection of the integrin (b) α5 and (d) αv subtypes. The integrin-FNIII7–10 complex was cross-linked and then detected using the corresponding antibodies by the method described previously [18]. (c, e) Phase-contrast images of the cells shown in (b) and (d). The %NTA was 5%. Scale bars represent 50 μm.
Conclusions
In this study, we developed a dynamic substrate that selectively captured genetically tagged proteins in response to light. The substrate surface was composed of mixed monolayers of disulfide ligands bearing photocleavable PEG, NTA and EG7 ligands. Before photoirradiation, PEG blocked the interaction between His-tagged proteins in the bulk solution and the NTA group on the substrate surface, but this interaction was activated upon photocleaving PEG from the surface. Meanwhile, the non-specific adsorption of other non-His-tagged proteins was suppressed by the EG7 underbrushes. By optimizing the surface composition and using a His-tagged fibronectin fragment (His-FNIII7–10), we produced a substrate that changed from non-cell-adhesive to cell-adhesive in response to light. Cell adhesion to the substrate was mainly mediated by His-FNIII7–10 supplemented to the culture medium, and hence, integrin α5β1 was accumulated at the cell-substrate interfaces. The present method will be useful for the dynamic analysis of cell behavior, including cell spreading and cell migration, on the specific ECM protein in cell-culturing environments.
References
- Meredith J C, Karim A. and Amis E J. MRS Bull. 2002;27:330. doi: 10.1557/mrs2002.101. [DOI] [Google Scholar]
- Kato K, Sato H. and Iwata H. Langmuir. 2005;21:7071. doi: 10.1021/la050893e. [DOI] [PubMed] [Google Scholar]
- Derda R, Musah S, Orner B P, Klim J R, Li L. and Kiessling L L. J. Am. Chem. Soc. 2010;132:1289. doi: 10.1021/ja906089g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Chan E W L. and Yousaf M N. J. Am. Chem. Soc. 2010;132:2614. doi: 10.1021/ja907187f. [DOI] [PubMed] [Google Scholar]
- Ebara M, Yamato M, Aoyagi T, Kikuchi A, Sakai K. and Okano T. Biomacromolecules. 2004;5:505. doi: 10.1021/bm0343601. [DOI] [PubMed] [Google Scholar]
- Yousaf M N, Houseman B T. and Mrksich M. Proc. Natl. Acad. Sci. USA. 2001;98:5992. doi: 10.1073/pnas.101112898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo W S, Yousaf M N. and Mrksich M. J. Am. Chem. Soc. 2003;125:14994. doi: 10.1021/ja038265b. [DOI] [PubMed] [Google Scholar]
- Yeo W S. and Mrksich M. Langmuir. 2006;22:10816. doi: 10.1021/la061212y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmuro-Matsuyama Y. and Tatsu Y. Angew. Chem., Int. Ed. Engl. 2008;47:7527. doi: 10.1002/anie.v47:39. [DOI] [PubMed] [Google Scholar]
- Petersen S, Alonso J M, Specht A, Duodu P, Goeldner M. and del Campo A. Angew. Chem., Int. Ed. Engl. 2008;47:3192. doi: 10.1002/(ISSN)1521-3773. [DOI] [PubMed] [Google Scholar]
- Liu D, Xie Y, Shao H. and Jiang X. Angew. Chem., Int. Ed. Engl. 2009;48:4406. doi: 10.1002/(ISSN)1521-3773. [DOI] [PubMed] [Google Scholar]
- Nakanishi J, Takarada T, Yamaguchi K. and Maeda M. Anal. Sci. 2008;24:67. doi: 10.2116/analsci.24.67. [DOI] [PubMed] [Google Scholar]
- Akiyama S K. and Yamada K M. J. Biol. Chem. 1985;260:402. [PubMed] [Google Scholar]
- Ruoslahti E. Annu. Rev. Cell Dev. Biol. 1996;12:697. doi: 10.1146/annurev.cellbio.12.1.697. [DOI] [PubMed] [Google Scholar]
- Aota S, Nomizu M. and Yamada K M. J. Biol. Chem. 1994;269:24756. [PubMed] [Google Scholar]
- Garcia A J, Schwarzbauer J E. and Boettiger D. Biochemistry. 2002;41:9063. doi: 10.1021/bi025752f. [DOI] [PubMed] [Google Scholar]
- Wang H, He Y, Ratner B D and Jiang S Y. 2006. J. Biomed. Mater. Res. A 77A 672 10.1002/(ISSN)1552-4965 [DOI] [PubMed] [Google Scholar]
- Petrie T A, Capadona J R, Reyes C D. and Garcia A J. Biomaterials. 2006;27:5459. doi: 10.1016/j.biomaterials.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Kikuchi Y, Nakanishi J, Nakayama H, Shimizu T, Yoshino Y, Yamaguchi K, Yoshida Y. and Horiike Y. Chem. Lett. 2008;37:1062. doi: 10.1246/cl.2008.1062. [DOI] [Google Scholar]
- Kaneko S, Nakayama H, Yoshino Y, Fushimi D, Yamaguchi K, Horiike Y. and Nakanishi J. Phys. Chem. Chem. Phys. 2011;13:4051. doi: 10.1039/c0cp02013c. [DOI] [PubMed] [Google Scholar]
- Cutler S M. and Garcia A J. Biomaterials. 2003;24:1759. doi: 10.1016/S0142-9612(02)00570-7. [DOI] [PubMed] [Google Scholar]
- Petrie T A, Raynor J E, Reyes C D, Burns K L, Collard D M. and Garcia A J. Biomaterials. 2008;29:2849. doi: 10.1016/j.biomaterials.2008.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi J, Nakayama H, Shimizu T, Ishida H, Kikuchi Y, Yamaguchi K. and Horiike Y. J. Am. Chem. Soc. 2009;131:3822. doi: 10.1021/ja809236a. [DOI] [PubMed] [Google Scholar]
- Nakanishi J, Kikuchi Y, Takarada T, Nakayama H, Yamaguchi K. and Maeda M. J. Am. Chem. Soc. 2004;126:16314. doi: 10.1021/ja044684c. [DOI] [PubMed] [Google Scholar]
- Nakanishi J, Kikuchi Y, Takarada T, Nakayama H, Yamaguchi K. and Maeda M. Anal. Chim. Acta. 2006;578:100. doi: 10.1016/j.aca.2006.04.059. [DOI] [PubMed] [Google Scholar]