

Abstract
A new series of luminescent 1,4-diazatriphenylene (ATP) derivatives with various peripheral donor units, including phenoxazine, 9,9-dimethylacridane and 3-(diphenylamino)carbazole, is synthesized and characterized as thermally activated delayed fluorescence (TADF) emitters. The influence of the donor substituents on the electronic and photophysical properties of the materials is investigated by theoretical calculations and experimental spectroscopic measurements. These ATP-based molecules with donor–acceptor–donor (D–A–D) structures can reduce the singlet–triplet energy gap (0.04–0.26 eV) upon chemical modification of the ATP core, and thus exhibit obvious TADF characteristics in solution and doped thin films. As a demonstration of the potential of these materials, organic light-emitting diodes containing the D–A–D-structured ATP derivatives as emitters are fabricated and tested. External electroluminescence quantum efficiencies above 12% and 8% for green- and sky-blue-emitting devices, respectively, are achieved.
Keywords: organic light-emitting diodes; 1,4-diazatriphenylene; thermally activated delayed fluorescence; donor–acceptor
Introduction
Organic light-emitting diodes (OLEDs) have received tremendous attention because of their promise for application in full-colour flat-panel displays and next-generation solid-state lighting sources [1, 2]. Continuous development of advanced light-emitting materials is indispensable in progressing these applications. In particular, phosphorescent materials based on organometallic compounds containing iridium or platinum can harvest both singlet and triplet excitons by enhanced intersystem crossing (ISC) via the heavy atom effect and have been successfully used to increase the internal quantum efficiency of OLEDs to nearly 100% [3, 4]. However, the expense and rarity of these precious metal complexes prevent their cost-effective and long-term mass production. Moreover, the stability of current devices with blue-emitting phosphorescent materials is still poor. As an alternative, OLEDs incorporating fluorescent emitters show promising stability and reliability. However, the internal quantum efficiency of fluorescent OLEDs is generally limited to 25% because only singlet excitons are available for light emission.
Recently, an effective triplet-harvesting process that involves up-conversion of triplet (T1) to singlet (S1) excited states, thermally activated delayed fluorescence (TADF) [5–9], has been used to increase the quantum efficiencies of OLEDs without the use of precious metals. OLEDs employing such TADF emitters have an exceptionally high internal quantum efficiency of up to ≍100% [9], which far exceeds the theoretical limit of conventional fluorescent OLEDs and is comparable to that of phosphorescent OLEDs. It is well established that emitters with a small energy gap (ΔEST) between the S1 and T1 states allow efficient reverse ISC, resulting in very high electroluminescence efficiency [9].
In this study, we focus on triphenylene as the central molecular framework to further expand the range of π-conjugated molecular structures that can act as efficient TADF emitters. Triphenylene is a polyaromatic hydrocarbon (PAH) with four fused benzene rings (figure 1), and its derivatives have been utilized as fluorescent emitters [10], hosts [11], hole transport materials [12] and electron transport materials [13] in OLEDs, as well as discotic liquid-crystalline semiconducting materials [14]. To achieve efficient blue TADF emitters, a sufficiently high T1 energy level is required. As presented in figure 1, among the analogous PAHs, triphenylene and its diaza-derivative possess exceptionally high T1 energy levels of up to 2.9 eV [15], so are suitable for use as blue emitter cores. From this point of view, we designed a series of TADF emitters based on 1,4-diazatriphenylene (ATP) as an electron-deficient core (figure 2). Herein, we report the synthesis, photophysical properties, and OLED performance of these ATP-based TADF emitters.
Figure 1.
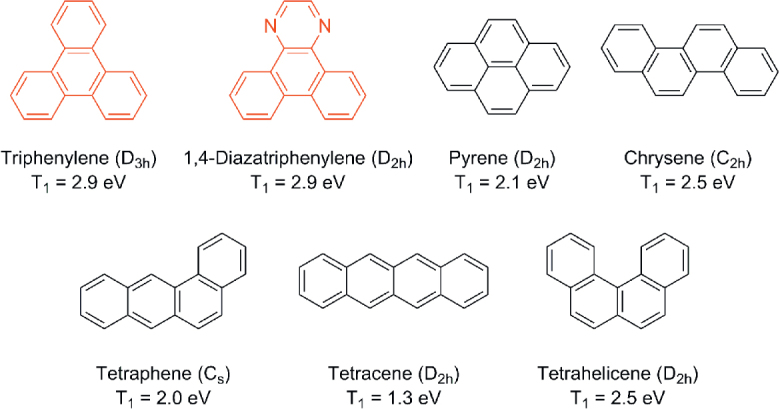
Chemical structures, point groups (in parentheses), and triplet excited energies of PAHs with four fused benzene rings.
Figure 2.

Chemical structures of designed materials based on a 1,4-diazatriphenylene (ATP) core.
Experimental details
Instrumentation and materials
Nuclear magnetic resonance (NMR) spectra were measured on an Avance III 500 spectrometer (Bruker) and JNM-ECP400 spectrometer (JEOL). NMR chemical shifts are reported in parts per million (ppm) using tetramethysilane as an internal standard (TMS = 0.0 ppm). Elemental analyses were carried out with a Yanaco MT-5 CHN corder. UV–vis absorption and photoluminescence (PL) spectra of organic films were measured with a UV-2550 spectrometer (Shimadzu) and FluoroMax-4 spectrofluorometer (Horiba Scientific), respectively. PL quantum yield was measured by an absolute PL quantum yield measurement system (Hamamatsu Photonics C11347-10). Luminescence intensity and lifetime of solutions and organic films were measured with a Quantaurus-Tau system (Hamamatsu Photonics) and streak camera (Hamamatsu Photonics C4334). Organic films were excited by an N2 gas laser (λ = 337 nm, pulse width = 500 ps, repetition rate = 20 Hz) under a vacuum of < 0.4 Pa. Samples were cooled to 4 K with a cryostat (Iwatani Industrial Gases). All quantum chemical calculations were performed using the Gaussian 09 programme package [16]. Geometry optimization was carried out using the M06-2X functional and cc-pVDZ basis set [17, 18]. Low-lying excited singlet and triplet states were computed using the optimized structures with time-dependent density functional theory (TD-DFT) [19, 20] at the M06-2X/cc-pVDZ level.
All reagents and solvents were purchased from Sigma-Aldrich, Tokyo Chemical Industry (TCI) or Wako Pure Chemical Industries, and used as received unless otherwise noted. All reactions were performed under an N2 atmosphere in dry solvents.
OLED device fabrication and measurements
To measure EL in OLED devices, clean 110 nm-thick indium tin oxide (ITO)-coated glass substrates with a 2 mm stripe pattern were used. The substrates were degreased with distilled water, a neutral detergent, acetone, isopropyl alcohol, and cleaned in a UV–ozone chamber (Nippon Laser and Electrics Laboratory, NLUV253) before being loaded into an evaporation system. Organic layers were thermally evaporated on substrates under a vacuum of < 7 × 10−4 Pa at an evaporation rate of < 0.3 nm s−1. For all of the devices, a cathode aluminium (Al) layer was deposited through a 1 mm diameter opening in a shadow mask. The devices were exposed once to N2 gas after the formation of the organic layers because a metal mask was included to define the cathode area. The current density–voltage–luminance characteristics were evaluated using a Keithley 2400 source metre and an absolute external quantum efficiency measurement system (Hamamatsu, C9920-12).
Synthesis of 9,9-dimethylacridane
The synthetic route to obtain 9,9-dimethylacridane (7) is outlined in scheme 1. A mixture of methyl anthranilate (29.1 g, 0.19 mol), iodobenzene (17 mL, 0.15 mol), K2CO3 (26.7 g, 0.19 mol), and copper powder (1.6 g, 30 mmol) was stirred for 4 h at 180 °C. After cooling to room temperature, the reaction mixture was diluted with EtOAc (100 mL), and filtered through a Celite® pad. The filtrate was concentrated by evaporation. The crude product was purified by column chromatography (silica gel, eluent: hexane/EtOAc = 5:1 v/v) to afford diphenylamine 5 as a colourless oil (yield = 25 g, 72%). 1H NMR (500 MHz, CDCl3): δ 9.47 (brs, 1H), 7.95 (dd, J = 8.0, 1.5 Hz, 1H), 7.44–7.10 (m, 6H) 7.07 (ddd, J = 8.0, 7.5, 1.5 Hz, 1H), 6.71 (td, J = 7.5, 1.5 Hz, 1H), 3.88 (s, 3H).
Scheme 1.

Synthesis of 9,9-dimethylacridane.
MeLi (1.5 M in Et2O, 160 mL) was added dropwise to a solution of 5 (13.6 g, 60 mmol) in Et2O (67 mL) at −80 °C. The mixture was then stirred for 3 h at room temperature. The reaction was quenched by adding water (30 mL). The product was extracted with EtOAc three times, and the combined organic layers were washed with brine and dried over anhydrous MgSO4. After filtration, the filtrate was concentrated by evaporation to afford carbinol 6 as a pale yellow oil (yield = 13.2 g, 97%). The product was used in the next step without further purification. 1H NMR (500 MHz, CDCl3): δ 7.78 (brs, 1H), 7.36 (dd, J = 8.0, 1.5 Hz, 1H), 7.28–7.17 (m, 3H), 7.15 (ddd, J = 8.0, 7.5, 1.5 Hz, 1H), 7.05 (dd, J = 7.5, 1.0 Hz, 2H), 6.92–6.78 (m, 2H), 2.48 (brs, 1H), 2.01 (s, 6H).
MeSO3H (35.1 g, 0.37 mol) was added to a mixture of crude 6 (14.0 g, 62 mmol) and polyphosphoric acid (83.1 g) in toluene (390 mL). The mixture was stirred for 2 h at 60 °C, and then the reaction was quenched by adding water (100 mL). The product was extracted with EtOAc three times, and the combined organic layers were washed with saturated aqueous NaHCO3 and brine, and dried over anhydrous MgSO4. After filtration and evaporation, the product was purified by column chromatography (silica gel, eluent: hexane/EtOAc = 5:1 v/v), washed with hexane, and dried under vacuum to give 9,9-dimethylacridane 7 as colourless crystals (yield = 10.0 g, 78%). 1H NMR (500 MHz, CDCl3): δ 7.38 (dd, J = 8.0, 1.3 Hz, 2H), 7.10 (ddd, J = 8.0, 7.5, 1.3 Hz, 2H), 6.91 (dd, J = 7.5, 7.5 Hz, 2H), 6.69 (d, J = 7.5 Hz, 2H), 6.13 (brs, 1H), 1.58 (s, 6H).
Synthesis of 3-(diphenylamino)carbazole
The synthetic route to obtain 3-(diphenylamino)carbazole (9) is outlined in scheme 2. 4-(N,N-dimethylamino)pyridine (DMAP, 0.80 g, 8.2 mmol) was added to a solution of 3-bromocarbazole (20.3 g, 82 mmol) and Boc2O (24.8 g, 114 mmol) in THF (300 mL). The mixture was stirred for 4 h at room temperature. The reaction was concentrated by evaporation. The residue was purified by column chromatography (silica gel, eluent: toluene/hexane = 1:1 v/v) to afford carbamate 8 as a white solid (yield = 28.7 g, quantitative yield). 1H NMR (500 MHz, CDCl3): δ 8.40 (brd, J = 7.5 Hz, 1H), 8.20 (brd, J = 7.5 Hz, 1H), 7.90 (d, J = 2.0 Hz, 1H), 7.51 (dq, J = 7.5, 0.5 Hz, 1H), 7.40 (dd, J = 8.5, 2.0 Hz, 1H), 7.33 (ddd, J = 7.5, 7.5, 1.0 Hz, 1H), 7.14 (ddd, J = 8.5, 7.5, 1.0 Hz, 1H), 1.39 (s, 9H).
Scheme 2.

Synthesis of 3-(diphenylamino)carbazole.
Pd(OAc)2 (97 mg, 440 μmol) was added to a mixture of 8 (3.0 g, 8.7 mmol), diphenylamine (2.2 g, 13 mmol), t-BuONa (1.0 g, 10 mmol), and P(t-Bu)3 (0.18 g, 0.87 mmol) in toluene (80 mL). The mixture was heated under reflux for 1 h. After cooling to room temperature, the reaction mixture was filtered through a Celite® pad. The filtrate was washed successively with water, saturated aqueous NH4Cl, and brine, and dried over anhydrous MgSO4. After filtration and evaporation, the crude product was used in the next step without further purification. A mixture of the obtained crude product, 1,4-dioxane (50 mL), and hydrochloric acid (36%, 5.0 mL) was stirred for 3 h at 100 °C. After cooling to room temperature, the resulting mixture was neutralized with saturated aqueous NaHCO3. The product was extracted with toluene three times, and the combined organic layers were washed successively with saturated aqueous NaHCO3 and brine, and dried over anhydrous MgSO4. After filtration and evaporation, the crude product was purified by column chromatography (silica gel, eluent: toluene/hexane = 1:1 v/v) to afford 9 as a white solid (yield = 2.1 g, 71%). 1H NMR (500 MHz, toluene-d8): δ 7.94 (d, J = 1.5 Hz, 1H), 7.55 (dd, J = 7.5, 0.5 Hz, 1H), 7.36–7.18 (m, 3H), 7.17–6.98 (m, 9H), 6.94 (dd, J = 8.5, 0.5 Hz, 1H), 6.69 (tt, J = 7.5, 1.0 Hz, 2H), 6.49 (brs, 1H).
Synthesis of 6,11-dibromo-1,4-diazatriphenylene (2)
N-Bromosuccinimide (NBS, 27 g, 0.15 mol) was added slowly to a solution of phenanthrenequinone (15 g, 72 mmol) in H2SO4 (95%, 150 mL) at 0 °C. The mixture was stirred for 2 h at room temperature. The reaction mixture was poured into water with crushed ice (ca. 400 mL). The resulting precipitate was collected by filtration. The product was washed with hot EtOAc (300 mL). After filtration, the product was dried under vacuum to provide 1 as an orange solid (yield = 22.4 g, 85%). 1H NMR (500 MHz, CDCl3): δ 8.25 (d, J = 8.5 Hz, 2H), 8.08 (d, J = 2.5 Hz, 2H), 7.96 (dd, J = 8.5, 2.5 Hz, 2H).
Ethylenediamine (20 mL, 0.30 mol) was added dropwise to a suspension of 1 (10 g, 27 mmol) in EtOH (150 mL) at 0 °C. The mixture was heated under reflux for 1 h, and then AcOH (250 mL) was added. The mixture was heated under reflux for further 2 h without N2 flow. After cooling to room temperature, the reaction mixture was poured into water (ca. 100 mL), and the resulting precipitate was collected by filtration. The product was washed successively with hot MeOH (200 mL) and acetone (200 mL). After filtration, the solid product was dried under vacuum to afford 2 as a pale-yellow solid (yield = 5.9 g, 56%). 1H NMR (500 MHz, DMSO-d6): δ 9.23 (d, J = 2.0 Hz, 2H), 9.13 (s, 2H), 8.83 (d, J = 8.5 Hz, 2H), 8.06 (dd, J = 8.5, 2.0 Hz, 2H).
Synthesis of ATP–PXZ, ATP–ACR, and ATP–CZ
ATP–PXZ. Pd(OAc)2 (60 mg, 0.26 mmol) was added to a suspension of 2 (2.0 g, 5.2 mmol), phenoxazine (3.0 g, 16 mmol), P(t-Bu)3 (0.11 g, 0.52 mmol), and K2CO3 (1.5 g, 11 mmol) in toluene (150 mL) at room temperature. The mixture was heated under reflux for 15 h. After cooling, the reaction was quenched by adding MeOH (150 mL). The resulting precipitate was collected by filtration. The product was washed successively with hot MeOH (20 mL) and acetone (20 mL). After filtration, the solid product was dried under vacuum to afford ATP–PXZ as a pale-yellow solid (yield = 2.7 g, 88%). ATP–PXZ was further purified by temperature-gradient sublimation under vacuum. 1H NMR (400 MHz, DMSO-d6): δ 9.17 (d, J = 8.6 Hz, 2H), 9.15 (d, J = 2.2 Hz, 2H), 9.03 (s, 2H), 7.92 (dd, J = 8.6, 2.2 Hz, 2H), 6.80 (dd, J = 7.7, 1.5 Hz, 4H), 6.73 (ddd, J = 7.7, 7.7, 1.5 Hz, 4H), 6.66 (ddd, J = 8.1, 7.7, 1.5 Hz, 4H), 6.10 (dd, J = 8.1, 1.5 Hz, 4H). Elemental analysis (%) calculated for C40H24N4O2: C, 81.07; H, 4.08; N, 9.45; found: C, 81.02; H, 3.98; N, 9.46.
ATP–ACR and ATP–CZ were prepared in a similar manner. Spectroscopic and analytical data for these compounds are described below.
ATP–ACR. Pale-yellow powder (yield = 37%). 1H NMR (400 MHz, DMSO-d6): δ 9.19 (d, J = 8.6 Hz, 2H), 9.12 (d, J = 2.4 Hz, 2H), 9.00 (s, 2H), 7.89 (dd, J = 8.6, 2.4 Hz, 2H), 7.60–7.48 (m, 4H), 7.03–6.89 (m, 8H), 6.42–6.32 (m, 4H), 1.73 (s, 12H). Elemental analysis (%) calculated for C46H36N4: C, 85.68; H, 5.63; N, 8.69; found: C, 85.69; H, 5.62; N, 8.69.
ATP–CZ. Off-white powder (yield = 74%). 1H NMR (400 MHz, DMSO-d6): δ 9.37 (d, J = 2.0 Hz, 2H), 9.20 (d, J = 8.1 Hz, 2H), 9.06 (s, 2H), 8.28 (d, J = 7.8 Hz, 4H), 8.17 (dd, J = 8.1, 2.0 Hz, 2H), 7.57 (d, J = 8.3 Hz, 4H), 7.48 (dd, J = 8.3, 6.8 Hz, 4H), 7.53 (dd, J = 7.8, 6.8 Hz 4H). Elemental analysis (%) calculated for C40H24N4: C, 85.69; H, 4.31; N, 9.99; found: C, 85.64; H, 4.24; N, 9.98.
Synthesis of 7,10-dibromo-1,4-diazatriphenylene (4)
Bromine (13.9 g, 87 mmol) was added to a suspension of phenanthrenequinone (50 g, 0.24 mol) and benzoylperoxide (2.5 g, 6.5 mmol) in nitrobenzene (250 mL) at room temperature. The mixture was stirred and heated to 120 °C. After the formation of gaseous HBr started, further bromine (69.2 g, 0.43 mol) was added dropwise to the mixture. The mixture was stirred for 2 h at 120 °C. After cooling to room temperature, EtOH (250 mL) was added to the reaction mixture. The resulting precipitate was collected by filtration and washed with EtOH. The product was dried under vacuum to afford 3 as an orange solid (yield = 75.5 g, 85%). 1H NMR (500 MHz, CDCl3): δ 8.12 (d, J = 2.0 Hz, 2H), 8.07 (d, J = 8.5 Hz, 2H), 7.67 (dd, J = 8.5, 2.0 Hz, 2H).
Ethylenediamine (40 mL, 0.60 mol) was added dropwise to a suspension of 3 (20 g, 55 mmol) in EtOH (300 mL) at 0 °C. The mixture was heated under reflux for 3 h, and then AcOH (250 mL) was added. The mixture was heated under reflux for a further 3 h without N2 flow. After cooling to room temperature, the reaction mixture was added to MeOH (ca. 500 mL), and the resulting precipitate was collected by filtration. The product was washed successively with hot MeOH (200 mL) and acetone (200 mL). After filtration, the solid product was dried under vacuum to afford 4 as a pale-yellow solid (yield = 11.9 g, 56%). 1H NMR (500 MHz, DMSO-d6): δ 9.22 (d, J = 2.0 Hz, 2H), 9.11 (s, 2H), 9.06 (d, J = 8.5 Hz, 2H), 8.03 (dd, J = 8.5, 2.0 Hz, 2H).
Synthesis of m-ATP–PXZ, m-ATP–ACR, m-ATP–CZ, and m-ATP–CDP
m-ATP–PXZ. Pd2(dba)3 · CHCl3 (70 mg, 68 μmol) was added to a suspension of 4 (1.0 g, 2.6 mmol), phenoxazine (1.5 g, 8.1 mmol), cataCXium® PIntB (0.09 g, 0.26 mmol), and t-BuONa (0.57 g, 6.0 mmol) in toluene (60 mL) at room temperature. The mixture was heated under reflux for 12 h. After cooling, the reaction mixture was added to MeOH (150 mL). The resulting precipitate was collected by filtration. The product was washed successively with MeOH (20 mL) and acetone (20 mL). The solid product was dried under vacuum to afford m-ATP–PXZ as a pale-yellow solid (yield = 1.4 g, 91%). This compound was further purified by temperature-gradient sublimation under vacuum. 1H NMR (500 MHz, DMSO-d6): δ 9.42 (d, J = 8.5 Hz, 2H), 9.17 (s, 2H), 9.02 (d, J = 2.0 Hz, 2H), 7.86 (dd, J = 8.5, 2.0 Hz, 2H), 7.12 (dd, J = 7.7, 1.5 Hz, 4H), 6.93 (ddd, J = 8.0, 7.7, 1.5 Hz, 4H), 6.88 (ddd, J = 8.0, 8.0, 1.5 Hz, 4H), 6.33 (dd, J = 8.0, 1.5 Hz, 4H). Elemental analysis (%) calculated for C40H24N4O2: C, 81.07; H, 4.08; N, 9.45; found: C, 81.16; H, 4.00; N, 9.43.
m-ATP–ACR, m-ATP–CZ, and m-ATP–CDP were prepared in a similar manner. Spectroscopic and analytical data for these compounds are described below.
m-ATP–ACR. Pale-yellow powder (yield = 79%). 1H NMR (500 MHz, DMSO-d6): δ 9.51 (d, J = 8.5 Hz, 2H), 9.22 (s, 2H), 9.05 (d, J = 2.0 Hz, 2H), 7.79 (dd, J = 8.5, 2.0 Hz, 2H), 7.50 (dd, J = 8.0, 1.5 Hz, 4H), 6.91 (ddd, J = 8.0, 7.0, 1.5 Hz, 4H), 6.87 (ddd, J = 8.0, 7.0, 1.0 Hz, 4H), 6.15 (dd, J = 8.0, 1.0 Hz, 4H), 1.65 (s, 12H). Elemental analysis (%) calculated for C46H36N4: C, 85.68; H, 5.63; N, 8.69; found: C, 85.60; H, 5.62; N, 8.65.
m-ATP–CZ. Off-white powder (yield = 94%). 1H NMR (500 MHz, DMSO-d6): δ 9.48 (d, J = 8.5 Hz, 2H), 9.33 (d, J = 2.0 Hz, 2H), 9.21 (s, 2H), 8.27 (d, J = 7.5 Hz, 4H), 8.12 (dd, J = 8.5, 2.0 Hz, 2H), 7.59 (d, J = 8.5 Hz, 4H), 7.45 (ddd, J = 8.0, 7.5, 1.0 Hz, 4H), 7.31 (ddd, J = 8.0, 7.5, 1.0 Hz 4H). Elemental analysis (%) calculated for C40H24N4: C, 85.69; H, 4.31; N, 9.99; found: C, 85.77; H, 4.26; N, 9.98.
m-ATP–CDP. Pale-yellow powder (yield = 78%). 1H NMR (500 MHz, DMSO-d6): δ 9.46 (d, J = 8.8 Hz, 2H), 9.35 (d, J = 2.0 Hz, 2H), 9.19 (s, 2H), 8.20 (d, J = 8.0 Hz, 2H), 8.12 (dd, J = 8.8, 2.0 Hz, 2H), 8.05 (d, J = 2.0 Hz, 2H), 7.59 (d, J = 8.5 Hz, 2H), 7.56 (d, J = 8.0 Hz, 2H), 7.41 (ddd, J = 8.5, 8.0, 1.3 Hz, 2H), 7.33–7.15 (m, 12H), 7.08–6.85 (m, 12H). Elemental analysis (%) calculated for C64H42N6: C, 85.88; H, 4.73; N, 9.39; found: C, 85.75; H, 4.66; N, 9.39.
Results and discussion
Molecular design and synthesis
To realize small ΔEST in TADF materials, it is critical to separate the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) to give small exchange energy. Steric hindrance that introduces twisting between donor and acceptor units can successfully achieve this purpose [21–23]. As shown in figure 2, we designed and synthesized a series of donor–acceptor–donor (D–A–D)-type molecules composed of an ATP acceptor core and peripheral aromatic donor moieties such as phenoxazine (PXZ), 9,9-dimethylacridane (ACR), carbazole (CZ) and 3-(diphenylamino)carbazole (CDP). The donor units were attached at 6, 11- and 7,10-positions on the central ATP core to systematically investigate the physicochemical properties of these regioisomers.
The calculated HOMO and LUMO distributions of the PXZ-functionalized molecules are shown in figure 3. The M06-2X/cc-pVDZ method was used for the calculations because by taking solvent effects into account within the polarizable continuum model, it well reproduced the emission wavelengths of our compounds in organic solvent media. The HOMOs of ATP–PXZ and m-ATP–PXZ are predominantly distributed on the peripheral PXZ units, whereas the LUMOs of both materials are localized over the ATP core. This offers clear spatial separation of the HOMO and LUMO because of the large dihedral angles (ca. 80–90°) between the ATP core and PXZ peripheries. The calculated ΔEST, HOMO and LUMO energy levels, and oscillator strength for the S1 * S0 excitation of these molecules are summarized in table 1. Although the theoretical calculation seems to overestimate ΔEST values, it offers qualitative understanding of the relationship between ΔEST and HOMO–LUMO overlap. ΔEST is calculated to be 0.18 eV for ATP–PXZ and 0.17 eV for m-ATP–PXZ. The ΔEST values of ATP–PXZ and m-ATP–PXZ are somewhat smaller than that of 4CzIPN (ΔEST = 0.3 eV using the same functional), which is a representative TADF material we reported previously [9], so they should be sufficiently small to induce TADF characteristics. Likewise, for ACR-functionalized ATP–ACR and m-ATP–ACR, small ΔEST values of approximately 0.28 and 0.25 eV, respectively, were obtained, suggesting high potential as TADF emitters. In contrast, the CZ substituents do not provide enough twist (i.e., orthogonal configuration) between the donor and acceptor subunits, resulting in a rather large ΔEST of 0.60 eV for both the 6,11-substituted ATP–CZ and 7,10-substituted m-ATP–CZ.
Figure 3.

HOMO and LUMO distributions calculated for ATP–PXZ and m-ATP–PXZ.
Table 1.
Time-dependent density functional theory (TD-DFT) calculation results for ATP-based molecules.
| Compound | ΔEST (eV) | HOMO (eV) | LUMO (eV) | Oscillator strength (f) |
|---|---|---|---|---|
| ATP–PXZ | 0.18 | 6.24 | 1.34 | 0.0006 |
| m-ATP–PXZ | 0.17 | 6.26 | 1.33 | 0.0014 |
| ATP–ACR | 0.28 | 6.29 | 1.24 | 0.0004 |
| m-ATP–ACR | 0.25 | 6.29 | 1.24 | 0.0002 |
| ATP–CZ | 0.60 | 6.64 | 1.26 | 0.2554 |
| m-ATP–CZ | 0.60 | 6.73 | 1.23 | 0.0119 |
| m-ATP–CDP | 0.50 | 6.08 | 1.27 | 0.3633 |
Scheme 3 outlines the synthetic approach used to prepare the designed ATP-based molecules. The dibromo intermediates 1 and 3 were prepared by regioselective bromination of 9,10-phananthrenequinone according to the literature [24–26]. The ionic mechanism led to the 2,7-dibromo derivative 1, whilst the radical mechanism provided the 3,6-dibromo derivative 3. The subsequent cyclization of 1 and 3 with ethylenediamine and aromatization afforded dibromo-1,4-diazatriphenylenes 2 and 4. The target materials were synthesized via palladium-catalyzed aminations of 2 and 4 with two equivalents of the corresponding arylamine (i.e., aromatic donor units) in moderate to high yields (see experimental details). Final materials were purified by repetitive temperature-gradient sublimation before characterization and OLED fabrication.
Scheme 3.

Synthesis of ATP-based D–A–D-type molecules. Reagents and conditions: (a) N-bromosuccinimide, conc. H2SO4, RT, 2 h (85%); (b) ethylenediamine, EtOH, 80 °C, 1 h, then AcOH, Air, 80 °C, 2 h (56% for 2, 56% for 4); (c) Ar2NH, Pd(OAc)2, P(t-Bu)3, K2CO3, toluene, 24 h, 110 °C; (d) Br2, benzoylperoxide, nitrobenzene, 130 °C, 3 h (85%); (e) Ar2NH (donor units), Pd2(dba)3·CHCl3, N-phenyl-2-(di-t-butylphosphino)indole (cataCXium® PIntB), t-BuONa, toluene, 24 h, 110 °C.
PL properties
The photophysical properties of the ATP-based compounds were examined by UV–vis absorption and PL spectroscopies; the results are summarized in table 2. Figure 4 shows UV–vis absorption and PL spectra of the PXZ-functionalized derivatives, ATP–PXZ and m-ATP–PXZ, in toluene. There are two clear absorption bands in the UV–vis spectra: one is an intense absorption at shorter wavelength (< 350 nm) corresponding to the π–π* transition of the ATP core, while the other is a weaker absorption corresponding to an intramolecular charge transfer (CT) transition in the range of ca. 350–450 nm related to the D–A–D structure. Both ATP–PXZ and m-ATP–PXZ exhibit structureless green PL emission with the maximum (λPL) at 546 nm in toluene solution. A slightly smaller Stokes shift is observed for m-ATP–PXZ than ATP–PXZ, which implies that a smaller degree of structural change will take place in the 7,10-substituted derivative upon photoexcitation. Similar intramolecular CT transition characteristics in the S1 states are observed for other ACR-, CZ-, and CDP-functionalized compounds as a result of their D–A–D structures.
Table 2.
Photophysical properties of ATP-based luminescent materials.
| Compound | λabs(nm) sol a | λPL(nm) sol a/film b | ΦPL(%) c sol a/film b | HOMO (eV) d | LUMO (eV) e | S1/T1 (eV) f | ΔEST(eV) g |
|---|---|---|---|---|---|---|---|
| ATP–PXZ | 317, 379 | 546/529 | 24/63 | −5.6 | −3.1 | 2.76/2.67 | 0.09 |
| m-ATP–PXZ | 313, 401 | 546/524 | 30/81 | −5.7 | −3.1 | 2.74/2.70 | 0.04 |
| ATP–ACR | 286, 355 | 503/492 | 26/49 | −5.8 | −3.0 | 2.88/2.76 | 0.16 |
| m-ATP–ACR | 286, 370 | 490/483 | 36/52 | −5.9 | −3.1 | 2.92/2.79 | 0.13 |
| ATP–CZ h | 342, 383 | 451/450 | 48/28 | −5.9 | −3.0 | 3.18/2.82 | 0.36 |
| m-ATP–CZ h | 341, 376 | 449/427 | 40/28 | −5.9 | −3.0 | 3.26/2.82 | 0.44 |
| m-ATP–CDP | 303, 370 | 532/499 | 77/77 | −5.7 | −3.1 | 3.02/2.76 | 0.26 |
Measured in oxygen-free toluene solution.
6 wt%-doped film in a host matrix (host = CBP for ATP–PXZ; mCP for m-ATP–PXZ, ATP–ACR, m-ATP–CZ, and m-ATP–CDP; mCBP for m-ATP–ACR and ATP–CZ).
Absolute PL quantum yield evaluated using an integrating sphere.
Determined by photoelectron yield spectroscopy of neat films.
Deduced from HOMO and optical energy gap (Eg).
Singlet (S1) and triplet (T1) energy estimated from onset wavelength of the emission spectra of doped films at 300 and 5 K, respectively.
ΔEST = S1 − T1.
Non-TADF material.
Figure 4.

UV–vis absorption and PL spectra of ATP–PXZ and m-ATP–PXZ in toluene.
Next, we studied the delayed fluorescence characteristics of the ATP-based materials by means of transient PL decay measurements. As reported in previous studies [5–9, 21–23], TADF materials in N2-saturated solution typically show two distinct components of prompt decay originating from fluorescence, and delayed decay based on TADF. In this series of materials, ATP–PXZ, m-ATP–PXZ, ATP–ACR, m-ATP–ACR and m-ATP–CDP 6 are found to show noticeable TADF emission components in N2-saturated solution. Figure 5 presents the transient PL decay curves for ATP–PXZ in toluene before and after N2 bubbling. The PL profile exclusively exhibits rapid decay before N2 bubbling with an emission lifetime of τ = 31 ± 2 ns. In contrast, after N2 bubbling, ATP–PXZ exhibits both a nano-second-scale prompt component (τp = 73 ± 3 ns) and micro-second-scale delayed component (τd = 1.9 ± 0.1 μs) at room temperature (300 K). The latter long-tail emission should originate from TADF. In addition, a PL quantum yield (ΦPL) of 24 ± 1% was obtained after N2 bubbling, which is six times higher than that measured before bubbling (ΦPL = 4 ± 1%), because of suppression of excited state energy transfer to triplet oxygen molecules. Meanwhile, CZ-functionalized derivatives ATP–CZ and m-ATP–CZ did not show obvious TADF characteristics in solution regardless of N2 bubbling, which is ascribed to their relatively large ΔEST (tables 1 and 2).
Figure 5.
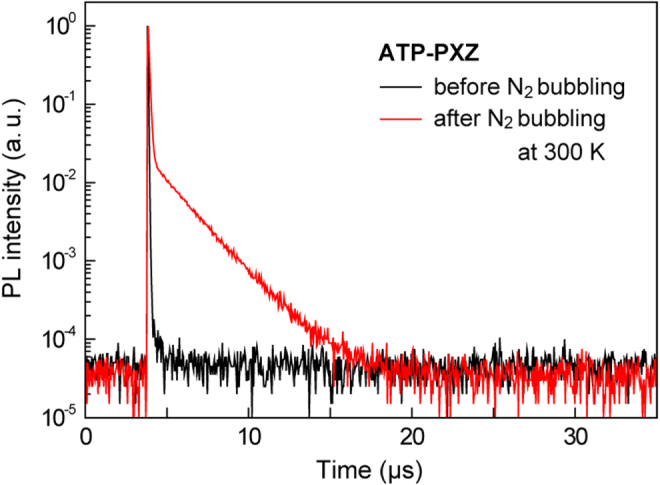
Transient PL decay curves for ATP–PXZ in toluene before and after N2 bubbling.
The PL properties of the ATP-based TADF emitters were also studied in doped thin films with an amorphous host matrix. We used 4,4′-bis(9-carbazolyl)-1,1′-biphenyl (CBP; T1 = 2.7 eV), 1,5-bis(9-carbazolyl)benzene (mCP; T1 = 2.9 eV) and 3,3′-bis(9-carbazolyl)-1,1′-biphenyl (mCBP; T1 = 2.8 eV) as host materials on account of their wide energy gaps and high T1 energy levels [27, 28]. Figure 6 depicts the PL spectra of 6 wt%-emitter:host codeposited thin films. In the doped films, sky-blue to green emissions were observed with λPL ranging from 483 to 529 nm. The λPL positions of the codeposited thin films are hypsochromically shifted by about 10–30 nm compared with those measured in toluene solution. Compared with their solution states, ΦPL of all of these compounds increased considerably in the solid state (ΦPL = 49–81%), most likely because the fixation of fragmental rotation and conformational changes can suppress the non-radiative decay to some degree. We also investigated the transient PL characteristics of the emitter:host codeposited thin films. As displayed in figure 7, a 6 wt%-ATP–PXZ:CBP film clearly exhibits both prompt and delayed PL components. Because the prompt fluorescence and delayed emission spectra are coincident, the long-tail emission should originate from TADF. The overall ΦPL of the 6 wt%-ATP–PXZ:CBP film was 63 ± 1% at room temperature (table 2), in which the prompt (ΦF) and TADF (ΦTADF) efficiencies were estimated to be 14% and 49%, respectively.
Figure 6.
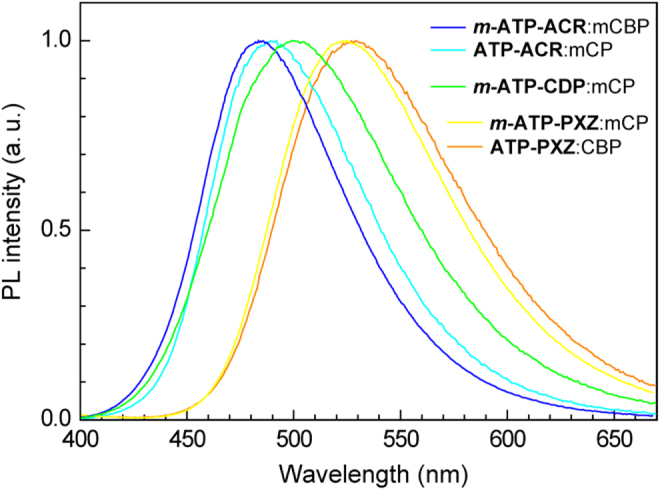
PL spectra of 6 wt%-emitter:host codeposited thin films measured at room temperature.
Figure 7.
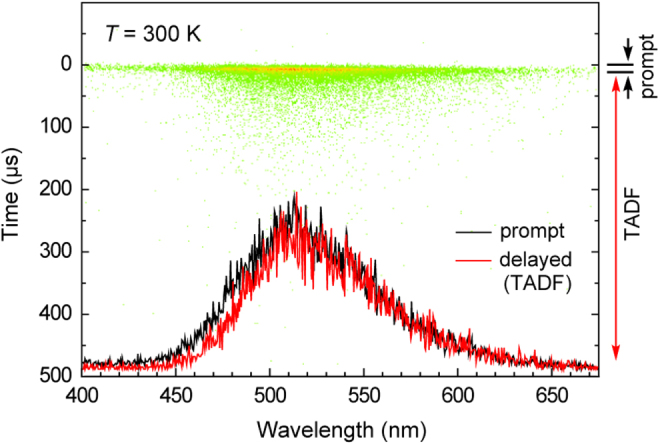
Streak image and PL spectra of a 6 wt%-ATP–PXZ:CBP film taken at 300 K showing the prompt (fluorescence, black) and delayed (TADF, red) components. Each dot in the streak image corresponds to the photon count of PL.
TADF-OLED characteristics
The performances of ATP-based TADF emitters were investigated by fabricating multilayer OLEDs with the device configurations of ITO (100 nm)/α-NPD (35 nm)/6 wt%-emitter:host (15 nm)/TPBi (65 nm)/LiF (0.8 nm)/Al (70 nm) for green emitters (ATP–PXZ and m-ATP–PXZ), and ITO (100 nm)/α-NPD (35 nm)/mCP (10 nm)/6 wt%-emitter:host (15 nm)/PPT (10 nm)/TPBi (40 nm)/LiF (0.8 nm)/Al (70 nm) for sky-blue emitters (ATP–ACR, m-ATP–ACR and m-ATP-CDT). We used 4,4′-bis[N-(1-naphthyl)-N-phenylamino]-1,1′-biphenyl (α-NPD) as a hole-transporting layer and 1,3,5-tris(N-phenylbenzimidazol-2-yl)benzene (TPBi) as an electron-transporting layer. For sky-blue TADF-OLEDs, thin layers (10 nm) of mCP and 2, 8-bis(diphenylphosphoryl) dibenzo-[b,d]thiophene (PPT) [29] with high T1 energies were inserted to suppress triplet exciton quenching of the sky-blue emitters with T1 of ca. 2.8 eV at the interfaces in the devices.
Figure 8 depicts the external quantum efficiency (ηext) versus current density (J) curves and current density–voltage–luminance (J–V–L) characteristics of the fabricated TADF-based OLEDs. Device performances are also summarized in table 3. The green-emitting OLEDs containing ATP–PXZ and m-ATP–PXZ display higher ηext (11.7% and 12.6%, respectively) than those of the sky-blue-emitting OLEDs with ATP–ACR and m-ATP–ACR (7.5% and 8.7%, respectively), because of the higher ΦPL of the PXZ-functionalized ATPs in doped thin films than those with ACR. The ATP–PXZ-based OLED shows a maximum current efficiency (ηc) of 37.9 cd A−1 and power efficiency (ηp) of 24.8 lm W−1. Moreover, as listed in table 3, their EL maxima (λEL) are similar to the corresponding λPL, confirming that EL emission originates from the same intramolecular CT excited (S1) states of the D–A–D-structured ATP derivatives.
Figure 8.

(a) External EL quantum efficiency versus current density curves and (b) current density–voltage–luminance (J–V–L) characteristics of TADF–OLEDs with device configurations of ITO/α-NPD/6 wt%-emitter:host/TPBi/LiF/Al for ATP–PXZ and m-ATP–PXZ, and ITO/α-NPD/mCP/6 wt%-emitter:host/PPT/TPBi/LiF/Al for ATP–ACR and m-ATP–ACR.
Table 3.
Performance of TADF-based OLEDs a.
| Emitter | Host | λEL (nm) | Von (V) | Lmax (cd m−2) | ηc (cd A−1) | ηp (lm W−1) | ηext (%) |
|---|---|---|---|---|---|---|---|
| ATP–PXZ | CBP | 529 | 3.2 | 23 600 | 37.9 | 24.8 | 11.7 |
| m-ATP–PXZ | mCP | 516 | 3.4 | 21 000 | 34.0 | 24.3 | 12.6 |
| ATP–ACR | mCP | 496 | 4.8 | 2300 | 11.5 | 5.7 | 7.5 |
| m-ATP–ACR | mCBP | 486 | 4.8 | 3240 | 13.1 | 6.2 | 8.7 |
| m-ATP–CDP | mCP | 499 | 4.8 | 3290 | 13.4 | 6.4 | 7.5 |
Device configurations: ITO/α-NPD/6 wt%-emitter:host/TPBi/LiF/Al for ATP–PXZ and m-ATP–PXZ, and ITO/α-NPD/mCP/6 wt%-emitter:host/PPT/TPBi/LiF/Al for ATP–ACR, m-ATP–ACR, and m-ATP–CDP. λEL = EL emission maximum; Von = turn-on voltage at 1 cd m−2; Lmax = maximum luminance; ηc = current efficiency at 100 cd m−2; ηP = power efficiency at 100 cd m−2; ηext = maximum external EL quantum efficiency.
In the TADF-based OLEDs under electrical excitation, the triplet excitons are directly generated by carrier recombination and successively converted to the S1 state via efficient reverse ISC. Accordingly, the theoretical maximum of internal EL quantum efficiency (ηint) can be given by the following equation [6]:
where ηr,S denotes the singlet-exciton production rate (25%), ηr,T represents the triplet-exciton production rate (75%), and ΦISC (≍86%) is the efficiency of ISC. We estimate that ηint for the ATP–PXZ-based device is 58%, so the theoretical maximum ηext is approximately 12% assuming a light out-coupling efficiency of 20%. The experimental ηext of 11.7% obtained for the ATP–PXZ-based device is consistent with the theoretical value, suggesting that an appropriate charge carrier balance and effective triplet exciton confinement are attained in the emitting layer.
Conclusions
We have reported the synthesis, characterization and OLED application of a series of luminescent ATP-based D–A–D-type materials incorporating various donor substituents. The twisting between the electron-accepting ATP core and electron-donating peripheral substituents gives rise to very small ΔEST values (0.04–0.26 eV), especially for PXZ-, ACR- and CDP-functionalized derivatives. All of these materials exhibit high PL quantum yields (49–81%) as well as obvious TADF characteristics in codeposited thin films with an appropriate host material. TADF-based OLEDs employing ATP–PXZ and m-ATP–PXZ as a green emitter exhibit a high external EL quantum efficiency of approximately 12%. These results demonstrate the potential of donor-functionalized triphenylenes as emitters in high-performance TADF-based OLEDs.
Acknowledgments
This work was supported in part by the Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST) and International Institute for Carbon Neutral Energy Research (WPI-I2CNER) sponsored by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
Footnotes
m-ATP–CDP with ΔEST of 0.26 eV exhibits both a nano-second-scale prompt component (τp = 50 ± 2 ns) and micro-second-scale delayed component (τd = 18.1 ± 0.3 μs) in oxygen-free toluene solution at 300 K, demonstrating its TADF properties.
References
- Bredas J L, Marder S R. and Riechmanis E. Chem. Mater. 2011;23:309. doi: 10.1021/cm1034694. [DOI] [Google Scholar]
- Grimsdale A C, Chan K L, Martin R E, Jokisz P G. and Holmes A B. Chem. Rev. 2009;109:897. doi: 10.1021/cr000013v. [DOI] [PubMed] [Google Scholar]
- Adachi C, Baldo M A, Thompson M E. and Forrest S R. J. Appl. Phys. 2001;90:5048. doi: 10.1063/1.1409582. [DOI] [Google Scholar]
- O’Brien D F, Baldo M A, Thompson M E. and Forrest S R. Appl. Phys. Lett. 1999;74:442. doi: 10.1063/1.123055. [DOI] [Google Scholar]
- Endo A, Sato K, Yoshimura K, Kai T, Kawada A, Miyazaki H. and Adachi C. Appl. Phys. Lett. 2011;98:083302. doi: 10.1063/1.3558906. [DOI] [Google Scholar]
- Lee S Y, Yasuda T, Nomura H. and Adachi C. Appl. Phys. Lett. 2012;101:093306. doi: 10.1063/1.4749285. [DOI] [Google Scholar]
- Zhang Q, Li L, Shizu K, Huang S, Hirata S, Miyazaki H. and Adachi C. J. Am. Chem. Soc. 2012;134:14706. doi: 10.1021/ja306538w. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Shizu K, Miyazaki H. and Adachi C. Chem. Commun. 2012;48:11392. doi: 10.1039/c2cc36237f. [DOI] [PubMed] [Google Scholar]
- Uoyama H, Goushi K, Shizu K, Nomura H. and Adachi C. Nature. 2012;492:234. doi: 10.1038/nature11687. [DOI] [PubMed] [Google Scholar]
- Wettach H, Jester S S, Colsmann A, Lemmer U, Rehmann N, Meerholz K. and Hðger S. Synth. Met. 2010;160:691. doi: 10.1016/j.synthmet.2010.01.003. [DOI] [Google Scholar]
- Togashi K, Yasuda T. and Adachi C. Chem. Lett. 2013;42:383. doi: 10.1246/cl.121247. [DOI] [Google Scholar]
- Park J-Y, Kim J M, Lee H, Ko K-Y, Yook K S, Lee J Y. and Baek Y G. Thin Solid Films. 2011;519:5917. doi: 10.1016/j.tsf.2011.03.022. [DOI] [Google Scholar]
- Togashi K, Nomura S, Yokoyama N, Yasuda T. and Adachi C. J. Mater. Chem. 2012;22:20689. doi: 10.1039/c2jm33669c. [DOI] [Google Scholar]
- Adam D, Schumacher P, Simmerer J, Häussling L, Siemensmeyer K, Etzbach K H, Ringsdorf H. and Haarer D. Nature. 1994;371:141. doi: 10.1038/371141a0. [DOI] [Google Scholar]
- Zander M. Phosphorimetry. New York: Elsevier; 1968. [Google Scholar]
- Frisch M J. Wallingford, CT: Gaussian; 2009. Gaussian 09, Revision C.01. [Google Scholar]
- Bauernschmitt R. and Ahlrichs R. Chem. Phys. Lett. 1996;256:454. doi: 10.1016/0009-2614(96)00440-X. [DOI] [Google Scholar]
- Casida M E, Jamorski C, Casida K C. and Salahub D R. J. Chem. Phys. 1998;108:4439. doi: 10.1063/1.475855. [DOI] [Google Scholar]
- Zhao Y. and Truhlar D. Theor. Chem. Acc. 2008;120:215. doi: 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Dunning J T H. J. Chem. Phys. 1989;90:1007. doi: 10.1063/1.456153. [DOI] [Google Scholar]
- Nakagawa T, Ku S-Y, Wong K-T. and Adachi C. Chem. Commun. 2012;48:9580. doi: 10.1039/c2cc31468a. [DOI] [PubMed] [Google Scholar]
- Méhes G, Nomura H, Zhang Q, Nakagawa T. and Adachi C. Angew. Chem. Int. Ed. 2012;51:11311. doi: 10.1002/anie.201206289. [DOI] [PubMed] [Google Scholar]
- Nasu K, Nakagawa T, Nomura H, Lin C-J, Cheng C-H, Tseng M-R, Yasuda T. and Adachi C. Chem. Commun. 2013;49:10385. doi: 10.1039/c3cc44179b. [DOI] [PubMed] [Google Scholar]
- Unver E K, Tarkuc S, Udum Y A, Tanyel C. and Toppare L. J. Polym. Sci. A. 2010;48:1714. doi: 10.1002/pola.v48:8. [DOI] [Google Scholar]
- Brunner K, van Dijken A, Bðrner H, Bastiaansen J J A M, Kiggen N M M. and Langeveld B M W. J. Am. Chem. Soc. 2004;126:6035. doi: 10.1021/ja049883a. [DOI] [PubMed] [Google Scholar]
- Kobin B, Grubert L, Blumstengel S, Henneberger F. and Hecht S. J. Mater. Chem. 2012;22:4383. doi: 10.1039/c2jm15868j. [DOI] [Google Scholar]
- Gong S, He X, Chen Y, Jiang Z, Zhong C, Ma D, Qin J. and Yang C. J. Mater. Chem. 2012;22:2894. doi: 10.1039/c1jm14903b. [DOI] [Google Scholar]
- Holmes R, Forrest S, Tung Y, Kwong R, Brown J, Garon S. and Thompson M. Appl. Phys. Lett. 2003;82:2422. doi: 10.1063/1.1568146. [DOI] [Google Scholar]
- Cai X, Padmaperma A B, Sapochak L S, Vecchi P A. and Burrows P E. Appl. Phys. Lett. 2008;92:083308. doi: 10.1063/1.2885117. [DOI] [Google Scholar]