

Abstract
This review on nanoparticles highlights the various biopolymers (proteins and polysaccharides) which have recently revolutionized the world of biocompatible and degradable natural biological materials. The methods of their fabrication, including emulsification, desolvation, coacervation and electrospray drying are described. The characterization of different parameters for a given nanoparticle, such as particle size, surface charge, morphology, stability, structure, cellular uptake, cytotoxicity, drug loading and drug release, is outlined together with the relevant measurement techniques. Applications in the fields of medicine and biotechnology are discussed along with a promising future scope.
Keywords: biopolymer, proteins, polysaccharides, nanoparticles, fabrication, applications
Introduction
Nanotechnology refers to the research and technological developments at atomic, molecular, and macromolecular scales, which lead to the controlled manipulation and study of structures and devices with length scales in the range of 1–100 nm [1]. Nanotechnology, the term probably first coined by Taniguchi in Japan [2], is a branch of manufacturing where dimensions on the order of a nanometer are important. Several researchers emphasized the significance of size and revealed the advantages of nanoparticles over microspheres (>1 μm) [3]. Biological nanoparticles are mainly developed for drug delivery systems as an alternative to liposome technology, in order to overcome the problems related to the stability of these vesicles in biological fluids and during storage [4]. The nanoparticle technology used in the recent years has great significance in improving the efficacy of the drugs. The nanoparticles fit into colloidal drug delivery systems, which offer advantages of drug targeting by modified body distribution [5] as well as the enhancement of the cellular uptake [6], which benefits from reduction of undesired toxic side effects of the free drugs [7]. With their easy accessibility in the body, nanoparticles can be transported via the circulation to different body sites [8], thus aiding in systemic treatments. Nanoparticles can be prepared from a variety of materials such as protein, polysaccharides and synthetic polymers. The choice of materials depends on several factors including (i) size and morphology of the nanoparticles; (ii) surface charge and permeability of the nanoparticle; (iii) degree of biodegradability, biocompatibility and cytotoxicity; (iv) drug loading and release profile desired [9, 10]. This review details the latest developments in protein and polysaccharide nanoparticles; their different methods of fabrication; their characterization in terms of size, surface charge, morphology, stability, structure, cellular uptake, cytotoxicity, drug loading, drug release; and also utilization of these nanoparticles in medicine and biotechnology.
Biopolymer nanoparticles
Biopolymer nanoparticles were first designed using albumin [11] and non-biodegradable synthetic polymers such as polyacrylamide and poly (methylacrylate) [12, 13]. The risks of chronic toxicity due to the intracellular and/or tissue overloading of non-degradable polymers were soon considered as a major limitation for the systemic administration of polyacrylamide and poly (methylacrylate) nanoparticles in humans. As a consequence, the type of nanoparticles that received much attention was designed with synthetic biodegradable polymers including polyalkylcyanoacrylate, poly (lactic-co-glycolic acid) and polyanhydride [14–17].
The therapeutic potential of these biodegradable colloidal systems was investigated for various applications [18–23]. Despite the very interesting results reported in literature, these systems may also be concerned with toxicological problems [24, 25]. There is another limitation for the bionanoparticle-based administration of hydrophilic molecules such as peptides, proteins and nucleic acids (oligonucleotide and genes) which are recognized to have great potential in therapeutics. This limitation is mainly because the polymers forming these nanoparticles are mostly hydrophobic, whereas proteins, peptides and nucleic acids are hydrophilic. This leads to difficulties for the drug to efficiently encapsulated and protected against enzymatic degradation [22, 26]. Therefore, the preparation of nanoparticles using more hydrophilic and naturally occurring materials has been explored [27–29].
The need for developing biodegradable nanoparticles (liposome, virus-like particle (VLP), protein, etc) as effective drug delivery devices was felt years ago [30]. The reason being in addition to the general advantages of nanoparticles, biopolymer nanoparticles in particular offer several advantages, which include the ease of their preparation from well-understood biodegradable polymers and their high stability in biological fluids and during storage [31]. Nanoparticles made of biodegradable polymers like proteins and polysaccharides can act as efficient drug delivery vehicles for sustained, controlled and targeted release, aiming to improve the therapeutic effects and also to reduce the side effects of the formulated drugs [32].
Protein nanoparticles
The first naturally occurring material used for the preparation of nanoparticles consisted of two proteins, albumin and gelatin [11, 16, 33, 34]. Among the colloidal systems, those based on proteins are very promising because they are biodegradable, less immunogenic [35] and non-toxic; they have greater stability in vivo and during storage [36], are relative easy to prepare and to monitor size distribution [37], and their manufacture can be scaled up [38, 39]. In addition, because of the defined primary structure of proteins, the protein-based nanoparticles offer various possibilities for surface modification and covalent drug attachment.
Albumin
Albumin, a protein found in blood plasma, has always been a remarkable molecule owing to its manifold functions and applications. Albumin is a biodegradable, biocompatible and less-immunogenic protein [35, 36, 40]. The paramount function of albumin is in the circulatory system—to aid in transportation, metabolism, and distribution of exogenous and endogenous ligands [40]. It also has an ability to act as an important extracellular antioxidant [42] and to impart protection from free radicals and other harmful chemical agents [43]. These unique attributes of albumin created a premier place for it in the drug therapy from time immemorial. Literature supports the use of modified serum albumin as a selective agent for tumor detection and/or therapy [44] or as a delivery tool of toxic compounds for elimination of Mycobacterium tuberculosis via receptor-mediated drug delivery [45].
Thus, nanotechnology era also employed the well-established properties of albumin, both human serum albumin (HSA) and bovine serum albumin (BSA), for various purposes as the nanoparticle drug (antibodies, interferon gamma, antiviral compounds) targeting carriers [46–48], therapeutic enhancer of anti-cancer drugs [49], modified vehicles for drug delivery across the brain to the central nervous system and also across blood brain barriers [50, 51].
Collagen
Collagen is the structural building material of vertebrates and the most abundant mammalian protein that accounts for 20–30% of total body proteins. Collagen has a unique structure, size and amino acid sequence which results in the formation of triple helix fiber. Collagen is regarded a useful biomaterial because of its excellent biocompatibility, biodegradation and availability [52]. Further, its amenability to modifications paved way for its in numeral applications in nanoparticles fabrication. Those modifications include addition of other proteins, such as elastin, fibronectin and glycosaminoglycans, which results in the improvement of its physicochemical and biological properties [53, 54] as well as control of biodegradability and subsequent release of ligand by use of such crosslinking agents as glutaraldehyde, formaldehyde, ultraviolet and gamma radiation [55]. The biodegradable collagen based nanoparticles are thermally stable, readily sterilizable, can be uptaken by the reticuloendothelial system and enable enhanced uptake of drug molecules into the cells [34, 56].
Gelatin
Gelatin is a natural water-soluble macromolecule resulting from the heat dissolution and partial hydrolysis of collagen. There are two types of gelatin: type-A gelatin is obtained by acid treatment of collagen with the isoelectric point (pI) between 7.0 and 9.0, whereas Type-B gelatin is produced via alkaline hydrolysis of collagen with the pI between 4.8 and 5.0. Gelatin offers a number of advantages over other synthetic polymers including non-irritability, biocompatibility and biodegradability, which makes it one of the desirable materials as carrier molecule [57, 58]. It is a natural macromolecule which is non-toxic and non-carcinogenic, and it shows low immunogenicity and antigenicity [59–62]. Gelatin has large number of functional groups on its surface which aid in chemical crosslinking and derivatization. These advantages led to its application for the synthesis of nanoparticles for drug delivery during the last thirty years [33, 34].
Silk proteins—sericin and fibroin nanoparticles
Silk fibers are primarily made of fibroin and sericin where the structural protein, fibroin is enveloped by the gum-like sticky protein, sericin.
Fibroin
Fibroin—a hydrophobic glycoprotein [63] and one of the ‘core’ proteins—constitutes over 70% of the cocoon. This insoluble protein is almost entirely made of the amino acids glycine, alanine, and serine (-Gly-Ala-Gly-Ala-Gly-Ser-) leading to the formation of antiparallel β-pleated sheet in the fibers [64]. Fibroin is semi-crystalline and consists of two phases: one is the highly crystalline β-pleated sheet phase and the other is non-crystalline phase [65]. Silk fibroin is also histocompatible, less immunogenic and non-toxic [66]. Silk fibroin can be processed into various forms including gels, fibers, membranes, scaffolds, hydrogels and nanoparticles [67–70]. Silk fibroin matrices with high specific surface area, high porosity, good biocompatibility and biodegradability have extensive applications in the field of biomaterials and drug delivery [66, 67]. Silk has been used as suture material for many centuries and it has been described as a biopolymer which evokes minimum foreign body response. Moreover silk-based biomaterials are highly biocompatible with various cell types and promote cell growth and proliferation [71–73].
Sericin
Sericins—hydrophilic glycoproteins [74] functioning as a ‘glue’—constitute 20–30% of the cocoon [75]. These hot water soluble proteins comprise different polypeptides ranging in weight from 24 to 400 kDa [76–79] and have unusually high serine content (40%) along with significant amounts of glycine (16%) [76, 79]. Sericin is comprised of 35% β-sheet and 63% random coil, and has no α-helical content—hence, its partially unfolded state [80–83]. Sericin nanoparticles, apart from the general advantages of protein nanoparticles, may also offer certain other benefits of the inherent property of sericin. Those include antioxidant [84–87] and antitumor action [88]; enhancement of the bioavailability of such elements as Zn, Mg, Fe, and Ca [89]; as well as suppression of coagulation when sulfated [90]. Sericin is non-toxic to fibroblast cells. Methionine and cysteine content in silk sericin are important factors to promote cell growth and collagen synthesis [91]. Water-soluble silk sericin has no immunogenicity and is also a biocompatible macromolecular protein like silk fibroin [92]. A study of the macrophage response of silk protein concludes that sericin does not usually manifest inflammatory activity when present in soluble form [93]. Recently, Aramwit et al have concluded that sericin promotes wound healing without causing any inflammation [94]. Table 1 shows the milestones in silk protein nanoparticles fabrication and application in chronological order.
Table 1.
Milestones of silk protein nanoparticles, preparation and application in chronological order.
| Protein | Method of fabrication | Particle size (nm) | Remarks | Reference |
|---|---|---|---|---|
| Silk fibroin | Precipitation using water miscible protonic and polar aprotonic organic solvents | 35–125 | Globular insoluble particles well dispersed and stable in aqueous solution | [161] |
| Precipitation using water miscible protonic and polar aprotonic organic solvents | 50–120 | Matrix for immobilization of L-asparaginase | [162] | |
| Microemulsion | 167 | Color dye doped silk fibroin nanoparticles | [163] | |
| Conjugated covalently with insulin using crosslinking reagent glutaraldehyde | 40–120 | Insulin—silk fibroin nanoparticles bioconjugates | [164] | |
| capillary microdot technique | <100 | Sustained and long-term therapeutic delivery of curcumin to breast cancer cells | [165] | |
| Desolvation | 150–170 | Cellular uptake and control release studies | [67] | |
| Silk sericin | Conjugation of sericin with activated PEG | 200–400 | Overcomes its problem of instability in water and insolubility in organic solvents | [166] |
| Sericin–PEG self- assembled through hydrophobic interactions | 204 | Self-assembled nanostructures for immobilization and drug delivery | [167] | |
| Sericin—poly methacrylate core- shell nanoparticles by graft copolymerizing technique | 100–150 | Potential biomedical application as delivery systems | [168] | |
| Self-assembled silk sericin/poloxamer nanoparticles | 100–110 | Nanocarriers of hydrophobic and hydrophilic drugs for targeted delivery | [86] | |
| Self-assembled silk sericin nanostructures | – | Fractal self-assembly of silk protein sericin | a |
aYadavalli V, personal communication.
Keratin
Keratins are a group of cysteine-rich structural proteins that exhibit a high mechanical strength owing to a large number of disulfide bonds. Keratin has been used very recently as nanosuspension that results in ultrathin, transparent keratin coatings to investigate the in vitro cell proliferation behavior as a potential coating material for standard cultivation [95]. The keratin nanosuspension coatings may provide an inexpensive alternative to materials like collagen or fibronectin. Keratin nanosuspension may also find applications in tissue engineering if it is explored further.
Polysaccharide nanoparticles
Polysaccharide-derived nanoparticles and nanostructured surfaces help to improve biocompatibility of cell toxic material, together with new immobilization approaches, which are currently in development for novel bionano-particle-derived pharmaceutical formulations. Nanoparticles from naturally occurring polysaccharides were designed for the administration of peptides, proteins, and nucleic acids [27, 28, 96].
Alginate
Alginate is a naturally occurring, water-soluble, linear unbranched polysaccharide extracted from brown seaweed. Alginate is composed of two types of uronic acids, α-L-guluronic acid and β-D-mannuronic acid. The monomeric units are grouped in three ways: blocks of alternating guluronic and mannuronic residues, blocks of guluronic acids and of mannuronic acids [97, 98]. Alginate has been reported as mucoadhesive, biocompatible, non-immunogenic substance which undergoes dissolution and biodegradation under normal physiological conditions [99]. The solubility of alginate in water depends on the associated cations. Sodium alginate is soluble in water, whereas calcium induces the formation of a gel [97, 100]. Apart from the interaction with calcium, alginate may also form complexes with polycations such as polyenimine (PEI), chitosan, or basic peptides like polylysine and polyarginine [100–102].
Carboxylic groups from the uronic acid confer negative charges to alginate. Chitosan endows nanoparticles with positive surface charge, prolongs the contact time of the active ingredients with the epithelium and enhances absorption via the paracellular transport pathway through the tight junctions [102–104]. Alginate micro and nanoparticles can be easily obtained by inducing gelation with calcium ions [105, 106]. This property can be used to produce a pre-gel consisting of very small aggregates of gel particles, followed by the addition of an aqueous polycationic solution to make a polyelectrolyte complex coating [107]. Poly-L-lysine (PLL), a cationic natural polymer, has been used to combine with alginates to prepare nanoparticles. However, PLL is toxic and immunogenic if injected. Recently, chitosan (CS) was selected as an alternative cationic polymer. Table 2 indicates the milestones of alginate and its composite nanoparticles preparation and applications in chronological order.
Table 2.
Milestones of alginate and its composite nanoparticles preparation and applications in chronological order.
| Polymer | Method of fabrication | Particle size (nm) | Remarks | Reference |
|---|---|---|---|---|
| Alginate | Control of the gel formation of alginate by calcium ions | 250–850 | Evaluation for the drug- loading capacity with doxorubicin as a model drug | [27] |
| Alginate | Gelation in presence of calcium ions and further crosslinking with poly-L-lysine | – | Nanosponges are new antisense oligonucleotide carrier system for specific delivery to lungs, liver and spleen | [169] |
| Sodium alginate and BSA | Emulsion solidification method | 166±34 | Determination of the kinetic parameters of 5-FU sodium alginate-125I BSA nanoparticles metabolism | [170] |
| Calcium alginate | Water-in-oil microemulsion followed by calcium crosslinking of glucoronic acid units | 80 | Examination of the nanoparticles for their potency as carriers for gene delivery | [171] |
| Alginate and chitosan | Ionotropic pre-gelation of alginate with calcium chloride followed by complexation between alginate and chitosan | 764–2209 | Monitor the complexation of contrary charged polyelectrolytes as insulin nanoparticulate carriers | [172] |
| Alginate and chitosan | Induction of a pre-gel with calcium counters ions, followed by polyelectrolyte complex coating with chitosan | 850±88 | Development of an oral insulin delivery system having mild formulation conditions, high insulin entrapment efficiency for gastrointestinal release | [104] |
| Alginate and chitosan | Induction of a pre-gel with calcium counters ions, followed by polyelectrolyte complex coating with chitosan | 750 | In vivo evaluation of the pharmacological activity of the insulin loaded nanoparticles | [173] |
| Alginate | Gelation in presence of calcium ions and further crosslinking with Eudragit E-100 | 200–950 | In vitro release study revealed sustained release of gliclazide from gliclazide loaded alginate nanoparticles | [174] |
| Alginate | Modified coacervation or ionotropic gelation method | 205–572 | Optimization of mucoadhesive nanoparticulate carrier systems for prolonged ocular delivery of the drug | [175] |
Chitosan
Chitosan is the second-abundant naturally occurring polysaccharide. Chitosan is made of randomly distributed β-(1-4)-linked D-glucosamine (deacetylated unit) and N-acetyl-D-glucosamine (acetylated unit). It is produced by deacetylation of chitin extracted from shells of crabs, shrimps and krill [108]. Commercially available chitosan is deacetylated between 66 and 95% and has an average molecular weight between 3.8 and 2000 kDa. Chitosan is linear, hydrophilic, positively charged and has mucoadhesive property [108–110]. It is an excellent biopolymer for preparation of microparticles [8] and nanoparticles [111] owing to its excellent biocompatibility and biodegradability [112, 113]. In vivo, it is degraded by lysozyme [114]. In addition, the amino groups confer to the molecule a high charge density and are readily available for chemical reactions and salt formation with acids. Chitosan is soluble in various acids, can also interact with polyions to form complexes and gels. These properties are exploited in the fabrication of nanoparticles based either on the spontaneous formation of complexes between chitosan and polyions including DNA [96] or on the gelation of a chitosan solution dispersed in a water-in-oil emulsion. Table 3 details the milestones of chitosan and its composite nanoparticle fabrication and applications in chronological order.
Table 3.
Milestones of chitosan and its composite nanoparticle fabrication and applications in chronological order.
| Protein | Method of fabrication | Particle size (nm) | Remarks | Reference |
|---|---|---|---|---|
| Chitosan | Complex coacervation technique | 100–250 | Encapsulate nucleic acids like plasmid DNA for gene delivery for efficient gene transfection | [176] |
| Chitosan | Ionotropic gelation | 213±3 | Encapsulate anticancer agents like cationic anthracycline drug doxorubicin | [177] |
| Chitosan | Ionotropic gelation of Chitosan with TPP anions | 200–1000 | Encapsulate proteins such as bovine serum albumin, tetanus and diphtheria toxoid | [178] |
| Chitosan | AOT/n-hexane reverse micellar system | 30–110 | In bone imaging and targeting purpose | [179] |
| Low molecular weight chitosan | Ionotropic gelation of CS with TPP anions | 350 | Encapsulate vaccines | [180] |
| Chitosan | Complex coacervation technique | 20–500 | Encapsulate nucleic acids (DNA) and to improve the transfection efficiency in vivo and in vitro | [181] |
| Chitosan | Ionotropic gelation | 145–172 | Encapsulate insulin | [182] |
| Chitosan | Ionic gelation of chitosan with tripolyphosphate anions (TPP) | 180–260 | Novel delivery system for ammonium glycyrrhizinate | [160] |
| Chitosan– alginate | Water-in-oil reverse microemulsion | <100 | Encapsulate plasmid DNA for gene delivery for efficient gene transfection | [183] |
| Chitosan | Ionic crosslinking of chitosan solution with TPP | 100–200 | Deliver cholinesterase inhibitor through the nasal mucosa to reach the brain for the treatment of neurodegenerative disease | [184] |
| Lauryl succinyl chitosan | Ionic crosslinking of chitosan solution with TPP | 315–1090 | Release of human insulin as the model protein drug and release kinetics in GI pH | [185] |
Fabrication methodologies
There are three common methods for the preparation of protein and polysaccharide based nanoparticles, namely, emulsification, desolvation and coacervation [115]. Very recently, they were complemented by electrospray drying technique.
Emulsification
The principle of nano-emulsion formation is based on the spontaneous emulsification that occurs on mixing an organic phase and an aqueous phase (scheme 1(a)). The organic phase is a homogeneous solution of oil, lipophilic surfactant and water-miscible solvent, whereas the aqueous phase consists of hydrophilic surfactant and water [116]. This method can be described as the dissolution of hydrophobic substances in an organic solvent which is further emulsified with an aqueous solution at very high shear. This results in the formation of very small droplets (50–100 nm). After emulsification, the organic solvent is removed by evaporation, yielding stable dispersions of solid nanoparticles [117–119]. The major disadvantage of this method is the need to add organic solvent and then to remove it. Besides, residues of organic solvent may cause toxic problems.
Scheme 1.
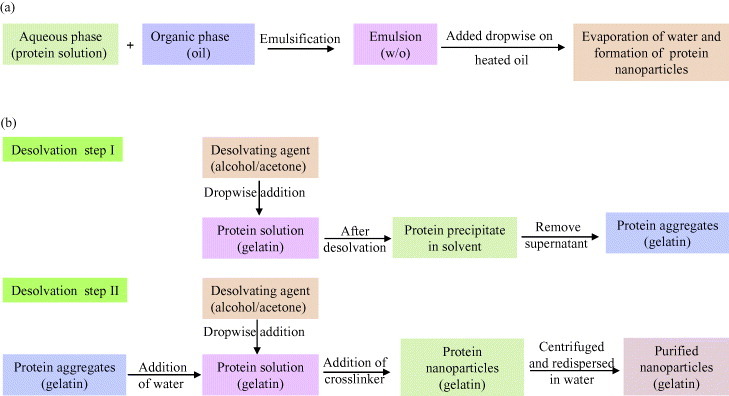
Schematic representation of nanoparticles preparation by (a) emulsification method [38, 119] and (b) two step desolvation method [39, 120].
Desolvation
Marty et al [34] used a different method for fabrication of nanoparticles which involved slow addition of a desolvation factor, such as natural salts or alcohol, to the protein solution. The desolvation factor changes the tertiary structure of protein. On reaching the critical level of desolvation, protein clump will be formed which on crosslinking with a chemical substance (e.g. glutaraldehyde) will result in the formation of nanoparticles.
A variation in the desolvation method was developed by Coester et al [39, 120] as a two-step desolvation process for the synthesis of gelatin nanoparticles (scheme 1(b)). In the first desolvation step, the low molecular gelatin fractions present in the supernatant is removed by decanting, and in the second step, high molecular fractions present in the sediment are redissolved and then desolvated again at pH 2.5. The resulting particles can then be easily purified by centrifugation and redispersion.
Coacervation
The coacervation method is similar to desolvation method; it employs mixing of the aqueous protein solution with organic solvent like acetone or ethanol to yield tiny coacervates. These coacervates are limited by the addition of the crosslinking agent, such as glutaraldehyde, etc [121]. The difference of coacervation and desolvation methods is the various parameters which affect the fabrication process to give desired property to the nanoparticles. These parameters include initial protein concentration, temperature, pH, cross linker concentration, agitation speed, molar ratio of protein/organic solvent and organic solvent adding rate [38].
Electrospray drying
The electrospraying method produces relatively monodisperse and biologically active protein particles. This method involves preparation of protein solution by dissolving the dry powder in an electrosprayable solution. Dispersion of the solution followed by solvent evaporation leaves dry residues collected on suitable deposition substrates. Insulin nanoparticles sized between 88 and 110 nm were prepared by this method [122]. Higher production rate of the nanoparticles also increases their size. The biological activity of the electrosprayed protein-based nanoparticles is not affected by the process conditions [122].
Characterization of nanoparticles
Nanoparticles are generally characterized by their size, morphology and surface charge, using such advanced microscopic techniques as scanning electron microscopy (SEM), transmission electron microscopy (TEM) and atomic force microscopy (AFM). The average particle diameter, their size distribution and charge affect the physical stability and the in vivo distribution of the nanoparticles. Electron microscopy techniques are very useful in ascertaining the overall shape of polymeric nanoparticles, which may determine their toxicity. The surface charge of the nanoparticles affects the physical stability and redispersibility of the polymer dispersion as well as their in vivo performance [31].
Particle size
The major application of nanoparticles is in drug release and drug targeting. It has been found that particle size affects the drug release. Smaller particles offer larger surface area. As a result, most of the drug loaded onto them will be exposed to the particle surface leading to fast drug release. On the contrary, drugs slowly diffuse inside larger particles [123]. As a drawback, smaller particles tend to aggregate during storage and transportation of nanoparticle dispersion. Hence, there is a compromise between a small size and maximum stability of nanoparticles [124].
Another major application of nanoparticles is in the field of biosensors by immobilizing enzymes. Reducing the size of carrier materials generally improves the efficiency of immobilized enzymes, as the smaller particles provide large surface area for surface attachment of enzymes and limit the resistance to the diffusion of substrates. In addition, the physical characteristics of nanoparticles, such as enhanced diffusion and particle motility, can impact the inherent catalytic activity of attached enzymes [125]. Polymer degradation can also be affected by the particle size. For instance, the degradation rate of poly(lactic-co-glycolic acid) was found to increase with increasing particle size in vitro [126].
There are a several tools for determining nanoparticle size as discussed below.
Dynamic light scattering
Currently, the fastest and most popular method of determining particle size is photon-correlation spectroscopy (PCS) or dynamic light scattering (DLS). DLS is widely used to determine the size of Brownian nanoparticles in colloidal suspensions in the nano and submicron ranges [127]. Shining monochromatic light (laser) onto a solution of spherical particles in Brownian motion causes a Doppler shift when the light hits the moving particle, changing the wavelength of the incoming light. This change is related to the size of the particle. It is possible to extract the size distribution and give a description of the particle's motion in the medium, measuring the diffusion coefficient of the particle and using the autocorrelation function. This method has several advantages: the experiment is quick, is almost automatic and does not require extensive experience. Moreover, this method has modest development costs. The advantage of using dynamic scattering is the possibility to analyze samples containing broad distributions of species of widely differing molecular masses (e.g. a native protein and various sizes of aggregates), and to detect very small amounts of the higher mass species (<0.01% in many cases). PCS determines the average particle size and polydispersity index (PI) which is a range of measurement of the particle sizes within measured samples [128]. Observation of larger particles compared to smaller particles reveals that for a given temperature, the larger particles move more slowly than the smaller ones.
Nanoparticle tracking analysis
A slight modification to photon-correlation spectroscopy, nanoparticle tracking analysis (NTA) is a technique developed by NanoSight Ltd to determine the size distribution profile of small particles in a liquid suspension. The technique is used in conjunction with an ultramicroscope which allows small particles in liquid suspension to be visualized moving under Brownian motion. Computer software is then used to track particles’ movements and subsequently estimate their hydrodynamic radius, using the Stokes-Einstein equation. Also, as the samples require minimal preparation, the time required to process one sample is much reduced.
Particle morphology
The size and morphology of nanoparticles exert a profound influence on the physical and chemical properties that determine their interaction with the environment and biological systems. There are certain techniques to analyze the morphology of nanoparticles. Microscopic techniques like SEM, TEM and AFM, along with particle size and distribution analysis, also determine other parameters like morphology or surface roughness of the nanoparticles.
Scanning electron microscope
For SEM characterization, nanoparticles solution should be first converted into a dry powder, which is then mounted on a sample holder followed by coating with a conductive metal, such as gold, using a sputter coater. The sample is then scanned with a focused fine beam of electrons. The surface characteristics of the sample are obtained from the secondary electrons emitted from the sample surface. The nanoparticles must be able to withstand vacuum, and the electron beam can damage the polymer. The mean size obtained by SEM is comparable with results obtained by dynamic light scattering.
Transmission electron microscope
TEM operates on different principle than SEM, yet it often brings same type of data. The sample preparation for TEM is complex and time consuming because of its requirement to be ultra thin for the electron transmittance. The nanoparticles dispersion is deposited onto support grids or films. To make nanoparticles withstand the instrument vacuum and facilitate handling, they are fixed using either a negative staining material, such as phosphotungstic acid or derivatives, uranyl acetate, etc, or by plastic embedding. Alternate method is to expose the sample to liquid nitrogen temperatures after embedding in vitreous ice [129]. The surface characteristics of the sample are obtained when a beam of electrons is transmitted through an ultra thin sample, interacting with the sample as it passes through.
Atomic force microscopy
AFM is yet another tool used to characterize variety of surfaces, including nanoparticles, at the atomic level and it is one of the primary forms of scanning probe microscopes [130]. The prime advantage of AFM is its ability to image non-conducting samples without any specific treatment, thus allowing imaging of delicate biological and polymeric nano and microstructures. AFM requires minimal sample preparation and can be performed in ambient conditions [131]. Scanning with a sharp probe across its surface and then monitoring and compiling the tip-sample interactions provide the images of the sample surface.
Particle stability
The colloidal stability is analyzed through zeta potential of nanoparticles. This potential is an indirect measure of the surface charge. It corresponds to potential difference between the outer Helmholtz plane and the surface of shear. Laser Doppler anemometry is the technique used to measure the zeta potential. It is based on the evaluation of the velocity of particles by the shift caused in the interference fringe, which is produced by the intersection of two laser beams. The electrophoresis mobility is then transformed into zeta potential. Most colloidal particles have negative zeta potential values ranging from about −100 to −5 mV. Surface charges prevent the agglomeration of nanoparticles polymer dispersions because of strong electrostatic repulsion, thereby enhancing the stability of the nanoparticles. The zeta potential can also provide information regarding the nature of material encapsulated within the nanocapsule or coated onto the surface [132].
Particle structure
Analysis of structure changes of the free protein sample and protein nanoparticles is imperative to understand the nature of modifications taking place in the protein in terms of confirmation, folding, chemical bonding, etc, during the synthesis of nanoparticles.
X-ray diffraction
One of the techniques for this purpose is x-ray diffraction (XRD) which is the primary tool for investigating the structure of crystalline materials, from atomic arrangement to crystallite size and imperfections. XRD also analyzes the phase composition, crystallite size and shape, lattice distortions and faulting, composition variations, orientation and in situ structure development of the nanoparticles. Usually, the XRD pattern is obtained by illuminating the sample with an x-rays source (Copper Kα line) with wavelength of 1.54 Å and scanning the diffraction within a certain range of the angle 2θ.
Fourier transform infrared spectroscopy
Another technique to supplement XRD is Fourier transform infrared spectroscopy (FTIR). The advantage of FTIR over crystallographic techniques is its capability to provide information about the structural details of proteins in solution with greater spatial and temporal resolution [133]. Sample used for characterization is usually lyophilized nanoparticles in minute quantities. The basic principle that governs is that the bonds and groups of bonds vibrate at characteristic frequencies. A molecule that is exposed to infrared rays absorbs infrared energy at frequencies which are characteristic to that molecule. FTIR analysis is carried out by illuminating the sample with a modulated IR beam. The sample transmittance and reflectance of the infrared rays at different frequencies is translated into an IR absorption plot, which is then analyzed and matched with known signatures of identified materials in the FTIR library.
Cellular uptake and cytotoxicity
Apart from these characterizations of nanoparticles, it is important to assess their cellular uptake and cytotoxicity, both in vitro and in vivo. Cellular uptake of nanoparticles is determined by tagging the nanoparticles with fluorescent tags like fluorescence isothiocyanate (FITC) followed by incubating these fluorescence-tagged nanoparticles with cells and their visualization under confocal laser scanning microscope [67]. Cytotoxicity analysis is usually performed by incubating nanoparticles with cells and carrying out 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) assay [134], a common method to evaluate toxicity of the biomaterials based on the mitochondrial activity. The underlying principle is the reduction of yellow to purple formazan in living cells [135, 136]. Followed by the addition of a solubilization solution (usually either dimethyl sulfoxide, an acidified ethanol solution, or a solution of the detergent sodium dodecyl sulfate in diluted hydrochloric acid) to dissolve the insoluble purple formazan product into a colored solution. Then, the absorbance of this colored solution is quantified at a certain wavelength (depending on the employed solvent) using a spectrophotometer. These reductions take place only when reductase enzymes are active, and therefore, conversion is often used as a measure of viable cells.
Drug loading and drug release
In addition to the above characterization, bionanoparticles may be evaluated for their property of drug loading and drug release. The drug loading of the nanoparticles is generally defined as the amount of drug bound per mass of polymer (usually moles of drug per mg polymer or mg drug per mg polymer); it could also be given as percentage relative to the polymer. The technique used for this analysis is classical analytical methods like UV spectroscopy or high performance liquid chromatography (HPLC) after ultracentrifugation, ultra filtration, gel filtration, or centrifugal ultrafiltration. The encapsulation efficiency refers to the ratio of the amount of encapsulated/absorbed drug to the total (theoretical) amount of drug used, with regard to the final drug delivery system of the dispersion of nanoparticles. Quantification is performed with the UV spectroscopy or HPLC. Drug release assays are also similar to drug loading assay which is assessed for a period of time to analyze the mechanism of drug release.
Application of nanoparticles in drug delivery
Nanoparticle-based delivery systems have the potential power to improve drug stability, increase the duration of the therapeutic effect and permit enteral or parenteral administration, which may prevent or minimize the drug degradation and metabolism as well as cellular efflux [104, 105, 107, 137]. Protein nanoparticles (figure 1) can transport a number of drugs across the blood brain barrier that normally cannot cross this barrier after intravenous injection. A number of authors have demonstrated a considerable tendency for an accumulation of protein nanoparticles in certain tumors. The binding of a variety of cytotoxic drugs, 5-fluorouracil, paclitaxel and doxorubicin to albumin or gelatin nanoparticles significantly enhanced the efficacy against experimental tumors or human tumors transplanted to nude mice in comparison to free drug.
Figure 1.
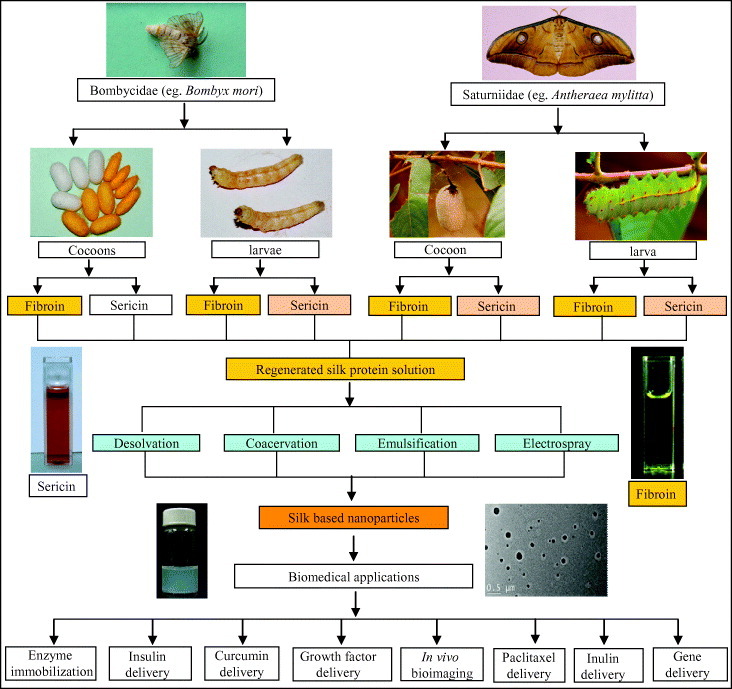
Schematic outline of the preparation of protein nanoparticles and its biomedical applications. In this layout, silk is shown as a representative protein. Silks are mainly obtained from the lepidopteron insects of the family Bombycidae (e.g. Bombyx mori) and Saturniidae (e.g. Antheraea mylitta). Silk proteins can be isolated from the cocoons and mature 5th instar larvae. Silk proteins are of two types: the fibrous protein fibroin and the glue protein sericin. The isolated silk proteins are further processed and engineered to obtain the regenerated silk protein solution. The regenerated silk solutions are then further subjected to processes like desolvation, coacervation, emulsification and electrospraying to obtain the silk-based nanoparticles. These silk-based nanoparticles are further utilized in such biomedical applications as drug delivery, enzyme immobilization, growth factors delivery, in vivo bioimaging, gene delivery, etc [67].
Gelatin nanoparticles loaded with a hydrophilic (pilocarpine HCl) and a hydrophobic (hydrocortisone) drug presented increased bioavailability of topical ophthalmic drug [138] and also for other drugs such as doxorubicin [139]. Later, gelatin nanoparticles were employed for peptide drug delivery system [140]. Nanoparticles provide tremendous potential as gene delivery vehicles. They can easily direct and control gene expression kinetics by altering various processing parameters used to make the nanoparticles, as showed by the works where gelatin was used as efficient carrier system for plasmid vector DNA and proteins as model drugs [141].
Surface modification of gelatin nanoparticles by covalent attachment of biotin-binding proteins, enabling the binding of biotinylated drug targeting ligands by avidin-biotin-complex formation, was also one of the strategies employed [142–144]. Thiolated gelatin nanoparticles were produced to release the incorporated nucleic acid molecule in a highly reducing environment [145]. These nanoparticles can meet specific controlled release needs due to their versatility in chemical modification and crosslinking. This was further demonstrated by Kushibiki and Tabata in 2005 [146] using DNA-containing poly(ethylene glycol)-modified (PEGylated) gelatin nanoparticles. PEGylation of gelatin also proved beneficial to long-circulating delivery system in vivo and also for targeting tumor cells, such as BT/20 human breast cancer cells [147].
Chitosan has been used in the pharmaceutical field in various forms, such as films, beads, intragastric floating tablets, microspheres, and nanoparticles [8, 96, 148–150]. It was employed as a vehicle for drug delivery for sustained release systems [152–155] and mucoadhesive formulations [112, 113, 156]. Chitosan was also used for the improvement of the dissolution rate of poorly soluble drugs [157], drug targeting [158] and enhancement of peptide drug absorption [111, 156].
The advantage of chitosan nanoparticles is its hydrophilic nature which results in its longer circulation in blood as observed by Allemann et al [17] for the hydrophilic nanoparticles. Therefore, hydrophilic systems not only control the rate of drug administration that prolongs the duration of the therapeutic effect, but also deliver the drug to specific sites [160]. Surface-modified chitosan nanoparticles are also suitable for the entrapment and controlled release of proteins and vaccines, such as ethylene oxide propylene oxide block copolymer [96].
Conclusions
Nanoparticles can be developed from a variety of materials such as synthetic polymers, proteins and polysaccharides. Biopolymeric nanomaterials are advantageous compared to the synthetic polymers, as they are biodegradable, biocompatible and non-toxic in nature.
Biopolymeric nanoparticles (proteins and polysaccharides) are fabricated by four different types of methods, namely emulsification, desolvation, coacervation and electrospray drying.
Biopolymeric nanoparticles are generally characterized in terms of size and morphology (by DLS, SEM, TEM and AFM techniques); particle stability (DLS and Zeta analyzer); particle structure (XRD and FTIR), cytotoxicity and biocompatibility.
Biopolymeric nanoparticles hold enormous applications as vehicle for administration of drugs and vaccines. Bioactive molecules within the nanoparticles can target specific sites (tumors) and deliver at those desired locations.
Future scope
Several challenges remain for further development of biopolymer-based nanoparticles:
The preparations strategies of protein and polysaccharide based nanostructures need to be enhanced for obtaining evenly dispersed and stable nanoparticles.
The properties and applications of protein as well as polysaccharide based nanoparticles are still in the developing stage for drug delivery and therapeutic purposes.
The cytotoxicity, biodegradability, biocompatibility and immune response of these biopolymeric nanoparticles need to be evaluated for their potential applications in clinical drug delivery.
The development and the production of the biopolymeric nanoparticles are still at the laboratory scale, biologists and chemical engineers need to collaborate for the large-scale production of these nanoparticles.
The combination of bioengineering, chemical modification and nanomaterials sciences in designing nano-particulate structures will improve the pharmacokinetics and biodistribution of the drug delivery system.
Acknowledgements
Our laboratory is financially supported by Department of Biotechnology (Indo-Australian Biotechnology Fund), Department of Science and Technology (Indo-Russian Joint programme), Government of India and Indo-US Science and Technology Forum, New Delhi.
References
- Gao J H. and Xu B. Nano Today. 2009;4:37. doi: 10.1016/j.nantod.2008.10.009. [DOI] [Google Scholar]
- Taniguchi N. Ann. CIRF. 1983;2:573. doi: 10.1016/S0007-8506(07)60185-1. [DOI] [Google Scholar]
- Meclean S, Processer E, O'Malley D, Clark N, Ramtoola Z. and Brayden D. Eur. J. Pharm. Sci. 1998;6:153. doi: 10.1016/S0928-0987(97)10007-0. [DOI] [PubMed] [Google Scholar]
- Oppenheim R C. Int. J. Pharm. 1981;8:217. doi: 10.1016/0378-5173(81)90100-9. [DOI] [Google Scholar]
- Kreuter J. Pharm. Acta Helv. 1983;58:217. [PubMed] [Google Scholar]
- Schafer V, von Briesen H, Andreesen R, Steffan A M, Royer C, Troster S, Kreuter J. and Rubsamen-Waigmann H. Pharm. Res. 1992;9:541. doi: 10.1023/A:1015852732512. [DOI] [PubMed] [Google Scholar]
- Narayani R. and Rao K P. Int. J. Pharm. 1993;95:85. doi: 10.1016/0378-5173(93)90393-T. [DOI] [Google Scholar]
- Berthold A, Cremer K. and Kreuter J. J. Control. Release. 1996;39:17. doi: 10.1016/0168-3659(95)00129-8. [DOI] [Google Scholar]
- Kreuter J. Nanoparticles in Colloidal Drug Delivery Systems. New York: Marcel Dekker; 1994. p. p 219. [Google Scholar]
- Kreuter J. J. Drug Target. 1995;3:171. doi: 10.3109/10611869509015940. [DOI] [PubMed] [Google Scholar]
- Scheffel U, Rhodes B A, Natarajan T K. and Wagner H N. J. Nucl. Med. 1972;13:498. [PubMed] [Google Scholar]
- Birrenbach G. and Speiser P. J. Pharm. Sci. 1976;65:1763. doi: 10.1002/jps.2600651217. [DOI] [PubMed] [Google Scholar]
- Kreuter J. and Speiser P P. Infect. Immun. 1976;13:204. doi: 10.1128/iai.13.1.204-210.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvreur P, Kante B, Roland M, Guiot P, Bauduin P. and Speiser P. J. Pharm. Pharmacol. 1979;31:311. doi: 10.1111/j.2042-7158.1979.tb13510.x. [DOI] [PubMed] [Google Scholar]
- Gurny R. Drug Dev. Ind. Pharm. 1981;7:1. doi: 10.3109/03639048109055684. [DOI] [PubMed] [Google Scholar]
- Vauthier-Holtzscherer C, Benabbou S, Spenlehauer G, Veillard M. and Couvreur P. STP Pharm. Sci. 1991;1:109. [Google Scholar]
- Allemann E, Leroux J C, Gurny R. and Doelker E. Pharm. Res. 1993;10:1732. doi: 10.1023/A:1018970030327. [DOI] [PubMed] [Google Scholar]
- Mathiowitz E.et al1997Nature 386410. 10.1038/386410a0 [DOI] [PubMed] [Google Scholar]
- Couvreur P. and Vauthier C. J. Control. Release. 1991;17:187. doi: 10.1016/0168-3659(91)90058-L. [DOI] [PubMed] [Google Scholar]
- Couvreur P. and Vauthier C. In: Drug Absorption Enhancement Concepts, Limitations and Trends. de Boer A G, editor. Leiden Amsterdam: Harwood Academic; 1994. p. p 457. [Google Scholar]
- Couvreur P, Dubernet C. and Puisieux F. Eur. J. Pharm. Biopharm. 1995;41:2. [Google Scholar]
- Fattal E, Vauthier C, Aynie I, Nakada Y, Lambert G, Malvy C. and Couvreur P. J. Control. Release. 1998;53:137. doi: 10.1016/S0168-3659(97)00246-0. [DOI] [PubMed] [Google Scholar]
- Labhasetwar V, Song C. and Levy R J. Adv. Drug Deliv. Rev. 1997;24:63. doi: 10.1016/S0169-409X(96)00483-8. [DOI] [Google Scholar]
- Maassen S, Fattal E, Müller R H. and Couvreur P. STP Pharma. 1993;3:11. [Google Scholar]
- Fernandez-Urrusuno R, Fattal E, Porquet D, Feger J. and Couvreur P. Toxicol. Appl. Pharmacol. 1995;130:272. doi: 10.1006/taap.1995.1032. [DOI] [PubMed] [Google Scholar]
- Emile C, Bazile D, Herman F, Helene C. and Veillard M. Drug Deliv. 1996;3:187. doi: 10.3109/10717549609029449. [DOI] [PubMed] [Google Scholar]
- Rajaonarivony M, Vauthier C, Couarraze G, Puisieux F. and Couvreur P. J. Pharm. Sci. 1993;82:912. doi: 10.1002/jps.2600820909. [DOI] [PubMed] [Google Scholar]
- Wang N. and Wu X S. Pharm. Dev. Technol. 1997;2:135. doi: 10.3109/10837459709022618. [DOI] [PubMed] [Google Scholar]
- Calvo P, Remunan-Lopez C, Vila-Jato J L. and Alonso M J. Pharm. Res. 1997;14:1431. doi: 10.1023/A:1012128907225. [DOI] [PubMed] [Google Scholar]
- Kumaresh S S, Tejraj M A, Anandrao R K. and Walter E R. J. Control. Release. 2001;70:1. doi: 10.1016/S0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- Mua L and Seowc P H. 2006. Colloids Surf. B 47 90 10.1016/j.colsurfb.2005.08.016 [DOI] [Google Scholar]
- Couvreur P, Gref R, Andrieux K. and Malvy C. Prog. Solid State Chem. 2006;34:231. doi: 10.1016/j.progsolidstchem.2005.11.009. [DOI] [Google Scholar]
- Kreuter J. Pharm. Acta Helv. 1978;53:33. [PubMed] [Google Scholar]
- Marty J J, Oppenheimer R C. and Speiser P. Pharm. Acta Helv. 1978;53:17. [PubMed] [Google Scholar]
- Rubino O P, Kowalsky R. and Swarbrick J. Pharm. Res. 1993;10:1059. doi: 10.1023/A:1018979126326. [DOI] [PubMed] [Google Scholar]
- Lin W, Coombes G A, Garnett C, Davies C, Schacht E, Davis S. and Illun L. Pharm. Res. 1994;11:1588. doi: 10.1023/A:1018957704209. [DOI] [PubMed] [Google Scholar]
- MacAdam A B, Shafi Z B, James S L, Marriott C. and Martin G P. Int. J. Pharm. 1997;151:47. doi: 10.1016/S0378-5173(97)04886-2. [DOI] [PubMed] [Google Scholar]
- Jahanshahi M, Najafpour G. and Rahimnejad M. Afr. J. Biotechnol. 2008;7:362. [Google Scholar]
- Coester C, Nayyar P. and Samuel J. Eur. J. Pharm. Biopharm. 2006;62:306. doi: 10.1016/j.ejpb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Lin W, Garnett M C, Davies M C, Bignotti F, Ferruti P, Davis S S. and Illum L. Biomaterials. 1997;18:559. doi: 10.1016/S0142-9612(96)00176-7. [DOI] [PubMed] [Google Scholar]
- Carter D C. and Ho J X. Adv. Protein Chem. 1994;45:153. doi: 10.1016/S0065-3233(08)60640-3. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Biochem. Pharmacol. 1988;37:569. doi: 10.1016/0006-2952(88)90126-8. [DOI] [PubMed] [Google Scholar]
- Emerson T E. Crit. Care Med. 1989;17:690. doi: 10.1097/00003246-198907000-00020. [DOI] [PubMed] [Google Scholar]
- Sinn H, Schrenk H H, Friedrich E A, Schilling U. and Maier-Borst W. Nucl. Med. Biol. 1990;17:819. doi: 10.1016/0883-2897(90)90031-u. [DOI] [PubMed] [Google Scholar]
- Majumdar S. and Basu S K. Antimicrob. Agents Chemother. 1991;35:135. doi: 10.1128/aac.35.1.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa Y, Takayama K, Ueda H, Machida Y. and Nagai T. Drug Des. Deliv. 1987;2:99. [PubMed] [Google Scholar]
- Segura S, Espuelas S, Renedo M J. and Irache J M. Drug Dev. Ind. Pharm. 2005;31:271. doi: 10.1081/ddc-52063. [DOI] [PubMed] [Google Scholar]
- Irache J M, Merodio M, Arnedo A, Camapanero M A, Mirshahi M. and Espuelas S. Mini-Rev. Med. Chem. 2005;5:293. doi: 10.2174/1389557053175335. [DOI] [PubMed] [Google Scholar]
- Santhi K, Dhanaraj S A, Joseph V, Ponnusankar S. and Suresh B. Drug Dev. Ind. Pharm. 2002;28:1171. doi: 10.1081/DDC-120014584. [DOI] [PubMed] [Google Scholar]
- Kreuter J, Hekmatara T, Dreis S, Vogel T, Gelperina S. and Langer K. J. Control. Release. 2007;118:54. doi: 10.1016/j.jconrel.2006.12.012. [DOI] [PubMed] [Google Scholar]
- Merodio M, Irache J M, Eclancher F, Mirshahi M. and Villarroya H. J. Drug Target. 2000;8:289. doi: 10.3109/10611860008997907. [DOI] [PubMed] [Google Scholar]
- Lynn A K, Yannas I V and Bonfield W. 2004. J. Biomed. Mater. Res. B: Appl. Biomater. B 71 343. [DOI] [PubMed] [Google Scholar]
- Barbani N, Giusti P, Lazzeri L, Polacco G. and Pizzirani G. J. Biomater. Sci. Polym. Ed. 1995;7:461. doi: 10.1163/156856295X00535. [DOI] [PubMed] [Google Scholar]
- Lefebvre F, Gorecki S, Bareille R, Amedee J, Bordenave L. and Rabaud M. Biomaterials. 1992;13:28. doi: 10.1016/0142-9612(92)90091-2. [DOI] [PubMed] [Google Scholar]
- Ruderman R J, Wade C W R, Shepard W D. and Leonard F. J. Biomed. Mater. Res. 1973;7:263. doi: 10.1002/jbm.820070213. [DOI] [PubMed] [Google Scholar]
- Bender A, von Briesen H, Kreuter J, Duncan I B. and Rubsamen-Waigmann H. Antimicrob. Agents Chemother. 1996;40:1467. doi: 10.1128/aac.40.6.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwick H G. and Heide K. Bibl. Haematol. 1969;3:111. doi: 10.1159/000384833. [DOI] [PubMed] [Google Scholar]
- Ward A G. and Courts A. The Science and Technology of Gelatin. New York: Academic; 1977. [Google Scholar]
- Bajpai A K. and Choubey J. J. Mater. Sci: Mater. Med. 2006;17:345. doi: 10.1007/s10856-006-8235-9. [DOI] [PubMed] [Google Scholar]
- Stevens K R, Einerson N J, Burmania J A. and Kao W J. J. Biomater. Sci. Polym. Ed. 2002;13:1353. doi: 10.1163/15685620260449741. [DOI] [PubMed] [Google Scholar]
- Marios Y, Chakfe N, Deng X, Marios M, How T, King W. and Guidoin R. Biomaterials. 1995;16:1131. doi: 10.1016/0142-9612(95)93576-Y. [DOI] [PubMed] [Google Scholar]
- DiSilvio L, Courtney-Harris R G. and Downes S. J. Mater. Sci., Mater. Med. 1994;5:819. doi: 10.1007/BF00213141. [DOI] [Google Scholar]
- Sinohara H, Asano Y. and Fukui A. Biochem. Biophys. Acta. 1971;237:273. doi: 10.1016/0304-4165(71)90317-5. [DOI] [PubMed] [Google Scholar]
- Asakura T. and Kaplan D L. In: Encyclopedia of Agricultural Science vol 4, Arntzen C J, Ritter E M, editors. New York: Academic; 1994. p. p 1. [Google Scholar]
- Lotz B. and Colonna-Cesari F. Biochemie. 1979;61:205. doi: 10.1016/S0300-9084(79)80067-X. [DOI] [PubMed] [Google Scholar]
- Altman G H, Diaz F, Jakuba C, Calabro T, Horan R L, Chen J, Lu H, Richmond J. and Kaplan D L. Biomaterials. 2003;24:401. doi: 10.1016/S0142-9612(02)00353-8. [DOI] [PubMed] [Google Scholar]
- Kundu J, Chung Y I, Kim Y H, Tae G and Kundu S C. 2010. Int. J. Pharm. doi: 10.1016/j.ijpharm.2009.12.052 [DOI] [PubMed] [Google Scholar]
- Vepari C. and Kaplan D L. Prog. Polym. Sci. 2007;32:991. doi: 10.1016/j.progpolymsci.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu J, Patra C and Kundu S C. 2008. Mater. Sci. Eng. C 28 1376 10.1016/j.msec.2008.03.004 [DOI] [Google Scholar]
- Kundu J, Dewan M, Ghoshal S. and Kundu S C. J. Mater. Sci., Mater. Med. 2008;19:2679. doi: 10.1007/s10856-008-3398-1. [DOI] [PubMed] [Google Scholar]
- Kaplan D L, Mello C M, Arcidiacono S, Fossey S, Senecal K. and Muller W. In: Protein-Based Materials. McGrath K, Kaplan D L, editors. Boston: Birkhauser; 1997. p. p 103. [Google Scholar]
- Sofia S, McCarthy M B, Gronowicz G. and Kaplan D L. J. Biomed. Mater. Res. 2001;54:139. doi: 10.1002/1097-4636(200101)54:1<139::AID-JBM17>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Shao Z. and Vollrath F. Nature. 2002;418:741. doi: 10.1038/418741a. [DOI] [PubMed] [Google Scholar]
- Sinohara H. Comput. Biochem. Physiol. 1979;6311:87. doi: 10.1016/0305-0491(79)90239-6. [DOI] [Google Scholar]
- Kundu S C, Dash B C, Dash R. and Kaplan D L. Prog. Polym. Sci. 2008;33:998. doi: 10.1016/j.progpolymsci.2008.08.002. [DOI] [Google Scholar]
- Gamo T, Inokuchi T. and Laufer H. Insect Biochem. Mol. Biol. 1977;7:285. [Google Scholar]
- Tokutake S. Biochem. J. 1980;187:413. doi: 10.1042/bj1870413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasu Y, Yamada H. and Tsubouchi K. Biosci. Biotechnol. Biochem. 2002;66:2715. doi: 10.1271/bbb.66.2715. [DOI] [PubMed] [Google Scholar]
- Michaille J J, Couble P, Prudhomme J C. and Garel A. Biochemie. 1986;68:1165. doi: 10.1016/S0300-9084(86)80060-8. [DOI] [PubMed] [Google Scholar]
- Tsukada M. and Bertholon G. Bull. Sci. Inst. Text. Fr. 1981;10:141. [Google Scholar]
- Mandal B B, Priya A S. and Kundu S C. Acta Biomater. 2009;5:3007. doi: 10.1016/j.actbio.2009.03.026. [DOI] [PubMed] [Google Scholar]
- Dash B C, Mandal B B. and Kundu S C. J. Biotechnol. 2009;144:321. doi: 10.1016/j.jbiotec.2009.09.019. [DOI] [PubMed] [Google Scholar]
- Bunning T J, Jiang H, Adams W W, Crane R L, Farmer B. and Kaplan D L. In: Silk Polymers-Material Science and Biotechnology: ACS Symposium Series 544. Kaplan D L, Adams W W, Farmer B, Viney C, editors. Washington, DC: American Chemical Society; 1993. p. p 353. [Google Scholar]
- Mandal B B. and Kundu S C. Nanotechnology. 2009;20:355101. doi: 10.1088/0957-4484/20/35/355101. [DOI] [PubMed] [Google Scholar]
- Suzuki N.et al2004Biofactors 21329. 10.1002/biof.552210164 [DOI] [PubMed] [Google Scholar]
- Dash R, Acharya C, Bindu P C. and Kundu S C. Biochem. Mol. Biol. Rep. 2008;41:236. doi: 10.5483/bmbrep.2008.41.3.236. [DOI] [PubMed] [Google Scholar]
- Dash R, Mandal M, Ghosh S K. and Kundu S C. Mol. Cell. Biochem. 2008;311:111. doi: 10.1007/s11010-008-9702-z. [DOI] [PubMed] [Google Scholar]
- Zhaorigetu S, Masahiro S, Watanabe H. and Kato N. Biosci. Biotechnol. Biochem. 2001;65:2181. doi: 10.1271/bbb.65.2181. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Yamada H. and Kato N. Nutr. Res. 2000;20:1505. doi: 10.1016/S0271-5317(00)80031-7. [DOI] [Google Scholar]
- Tamada Y, Sano M, Niwa K, Imai T. and Yoshino G. J. Biomater. Sci. Polym. Ed. 2004;15:971. doi: 10.1163/1568562041526469. [DOI] [PubMed] [Google Scholar]
- Aramwit P, Kanokpanont S, De-Eknamkul W, Kamei K. and Srichana T. J. Biomater. Sci. Polym. Ed. 2009;20:1295. doi: 10.1163/156856209X453006. [DOI] [PubMed] [Google Scholar]
- Zhang Y Q. Biotechnol. Adv. 2002;20:91. doi: 10.1016/S0734-9750(02)00003-4. [DOI] [PubMed] [Google Scholar]
- Zhang Y Q, Ma Y, Xia Y Y, Shen W D, Mao J P. and Xue R Y. J. Control. Release. 2006;115:307. doi: 10.1016/j.jconrel.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Aramwit P, Kanokpanont S, De-Eknamkul W. and Srichana T. J. Biosci. Bioengg. 2009;107:556. doi: 10.1016/j.jbiosc.2008.12.012. [DOI] [PubMed] [Google Scholar]
- Reichl S. Biomaterials. 2009;30:6854. doi: 10.1016/j.biomaterials.2009.08.051. [DOI] [PubMed] [Google Scholar]
- Calvo P, Remunan-Lopez C, Vila-Jato J L. and Aloso M J. J. Appl. Polym. Sci. 1997;63:125. doi: 10.1002/(SICI)1097-4628(19970103)63:1<125::AID-APP13>3.0.CO;2-4. [DOI] [Google Scholar]
- Gacesa P. Carbohydr. Polym. 1988;8:161. doi: 10.1016/0144-8617(88)90001-X. [DOI] [Google Scholar]
- Guisely K B. Enzyme Microb. Technol. 1989;11:706. doi: 10.1016/0141-0229(89)90119-1. [DOI] [Google Scholar]
- Gombotz W R. and Wee S F. Adv. Drug Deliv. Rev. 1998;31:267. doi: 10.1016/S0169-409X(97)00124-5. [DOI] [PubMed] [Google Scholar]
- Rees D A. and Welsh E J. Angew. Chem. Int. Ed. Engl. 1977;16:214. doi: 10.1002/anie.197702141. [DOI] [Google Scholar]
- Takahashi T, Takayama K, Machida Y. and Nagai T. Int. J. Pharm. 1990;61:35. doi: 10.1016/0378-5173(90)90041-2. [DOI] [Google Scholar]
- Bystricky S, Malovikova A. and Sticzay T. Carbohydr. Polym. 1991;15:299. doi: 10.1016/0144-8617(91)90044-D. [DOI] [Google Scholar]
- Kotze A F, Thanou M M, Luebetaen H L, de Boer A G, Verhoef J C. and Junginger H E. J. Pharm. Sci. 1999;88:253. doi: 10.1021/js980233c. [DOI] [PubMed] [Google Scholar]
- Sarmento B, Ribeiro A, Veiga F, Sampaio P, Neufeld R. and Ferreira D. Pharm. Res. 2007;24:2198. doi: 10.1007/s11095-007-9367-4. [DOI] [PubMed] [Google Scholar]
- Pan Y, Li Y J, Zhao H Y, Zheng J M, Xu H, Wei G, Hao J S. and Cui F D. Int. J. Pharm. 2002;249:139. doi: 10.1016/S0378-5173(02)00486-6. [DOI] [PubMed] [Google Scholar]
- Mladenovska K, Cruaud O, Richomme P, Belamie E, Raicki R S, Venier-Julienne M C, Popovski E, Benoit J P. and Goracinova K. Int. J. Pharm. 2007;345:59. doi: 10.1016/j.ijpharm.2007.05.059. [DOI] [PubMed] [Google Scholar]
- De S. and Robinson D. J. Control. Release. 2003;89:101. doi: 10.1016/S0168-3659(03)00098-1. [DOI] [PubMed] [Google Scholar]
- Kas H S. J. Microencapsul. 1997;14:689. doi: 10.3109/02652049709006820. [DOI] [PubMed] [Google Scholar]
- Berscht P C, Nies B, Liebendorfer A. and Kreuter J. Biomaterials. 1994;15:593. doi: 10.1016/0142-9612(94)90209-7. [DOI] [PubMed] [Google Scholar]
- Hirano S, Seino H, Akiyama Y. and Nonaka I. Polym. Eng. Sci. 1988;59:897. [Google Scholar]
- Jin C. Chin. J. Biomed. Engg. 1996;15:102. [Google Scholar]
- Lehr C M, Bouwstra J A, Schacht E H. and Junginger H E. Int. J. Pharm. 1992;78:43. doi: 10.1016/0378-5173(92)90353-4. [DOI] [Google Scholar]
- Luelen H L, Lehr C M, Rentel C O, Noach A B J, Boer A G, Verhoef J C. and Junginger H E. J. Control. Release. 1994;29:329. doi: 10.1016/0168-3659(94)90078-7. [DOI] [Google Scholar]
- Nordtveit R J, Vårum K M. and Smidsr⊘d O. Carbohydr. Polym. 1996;29:163. doi: 10.1016/0144-8617(96)00003-3. [DOI] [Google Scholar]
- Langer K, Balthasar S, Vogel V, Dinauer N, Von Briesen H. and Schubert D. Int. J. Pharm. 2003;257:169. doi: 10.1016/S0378-5173(03)00134-0. [DOI] [PubMed] [Google Scholar]
- Bouchemal K, Briançon S, Perrier E. and Fessi H. Int. J. Pharm. 2004;280:241. doi: 10.1016/j.ijpharm.2004.05.016. [DOI] [PubMed] [Google Scholar]
- Sjöström B, Kaplun A, Talmon Y. and Cabane B. Pharm. Res. 1995;12:39. doi: 10.1023/A:1016278302046. [DOI] [PubMed] [Google Scholar]
- Gao Z, Shukla A J, Johnson J R. and Crowley W R. Pharm. Res. 1995;12:857. doi: 10.1023/A:1016209020160. [DOI] [PubMed] [Google Scholar]
- Jahanshahi M. and Babaei Z. Afr. J. Biotechnol. 2008;7:4926. [Google Scholar]
- Coester C J, Langer K, Von Briesen H. and Kreuter J. J. Microencapsul. 2000;17:187. doi: 10.1080/026520400288427. [DOI] [PubMed] [Google Scholar]
- Lin W, Coombes A, Davies M, Davis S. and Illum L. J. Drug Target. 1993;1:237. doi: 10.3109/10611869308996081. [DOI] [PubMed] [Google Scholar]
- Gomez A, Bingham D, De Juan L. and Tang K. Mater. Res. Soc. Symp. Proc. 1999;550:101. [Google Scholar]
- Redhead H M, Davis S S. and Illum L. J. Control. Release. 2001;70:353. doi: 10.1016/S0168-3659(00)00367-9. [DOI] [PubMed] [Google Scholar]
- Jahanshahi M, Sanati M H, Minuchehr Z, Hajizadeh S. and Babaei Z. Am. Inst. Phys. 2007;228:929. [Google Scholar]
- Betancor L. and Luckarift H R. Trends Biotechnol. 2008;26:566. doi: 10.1016/j.tibtech.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Dunne M, Corrigan O I. and Ramtoola Z. Biomaterials. 2000;21:1659. doi: 10.1016/S0142-9612(00)00040-5. [DOI] [PubMed] [Google Scholar]
- Berne B J. and Pecora R. Dynamic Light Scattering. New York: Wiley; 1975. [Google Scholar]
- Takahashi K, Kato H, Saito T, Matsuyama S. and Kinugasa S. Part. Part. Syst. Charact. 2008;25:31. doi: 10.1002/ppsc.200700015. [DOI] [Google Scholar]
- Amzallag A, Vaillant C, Jacob M, Unser M, Bednar J, Kahn J, Dubochet J, Stasiak A. and Maddocks J H. Nucleic Acids Res. 2006;34:125. doi: 10.1093/nar/gkl675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard C R. Chem. Educ. 1996;5:1. doi: 10.1007/s00897960059a. [DOI] [Google Scholar]
- Magonov S N. Appl. Spectrosc. Rev. 1993;28:1. doi: 10.1080/05704929308021499. [DOI] [Google Scholar]
- Mohanraj V J. and Chen Y. Trop. J. Pharm. Res. 2006;5:561. [Google Scholar]
- Berthomieu C. and Hienerwadel R. Photosynth. Res. 2009;101:157. doi: 10.1007/s11120-009-9439-x. [DOI] [PubMed] [Google Scholar]
- Wilson A P. In: Cytotoxicity and Viability Assays in Animal Cell Culture: A Practical Approach vol 1, 3rd edn. Masters J R W, editor. London: Oxford University Press; 2000. [Google Scholar]
- Dobrucki B T. J. Cytometry. 2002;47:236. doi: 10.1002/cyto.10080. [DOI] [PubMed] [Google Scholar]
- Mosmann T. J. Immunol. Methods. 1983;65:55. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Florence A. Pharm. Res. 1997;14:259. doi: 10.1023/A:1012029517394. [DOI] [PubMed] [Google Scholar]
- Vandervoort V. and Ludwig A. Eur. J. Pharm. Biopharm. 2004;57:251. doi: 10.1016/S0939-6411(03)00187-5. [DOI] [PubMed] [Google Scholar]
- Leo E, Arletti R, Forni F. and Cameroni R. Farmaco. 1997;52:385. [PubMed] [Google Scholar]
- Li J K, Wang N. and Wu X S. J. Microencapsul. 1998;15:163. doi: 10.3109/02652049809006846. [DOI] [PubMed] [Google Scholar]
- Mladenovska K, Kumbaradzi E F, Dodov G M, Makraduli L. and Goracinova K. Int. J. Pharm. 2002;242:247. doi: 10.1016/S0378-5173(02)00167-9. [DOI] [PubMed] [Google Scholar]
- Wartlick H, Michaelis K, Balthasar S, Strebhardt K, Kreuter J. and Langer K. J. Drug Target. 2004;12:461. doi: 10.1080/10611860400010697. [DOI] [PubMed] [Google Scholar]
- Langer K, Coester C, Weber C, von Briesen H. and Kreuter J. Eur. J. Pharm. Biopharm. 2000;49:303. doi: 10.1016/S0939-6411(00)00068-0. [DOI] [PubMed] [Google Scholar]
- Coester C, Kreuter J, von Briesen H. and Langer K. Int. J. Pharm. 2000;196:147. doi: 10.1016/S0378-5173(99)00409-3. [DOI] [PubMed] [Google Scholar]
- Kommareddy S. and Amiji M. Bioconjug. Chem. 2005;16:1423. doi: 10.1021/bc050146t. [DOI] [PubMed] [Google Scholar]
- Kushibiki T. and Tabata Y. J. Biomater. Sci. Polym. 2005;16:1447. doi: 10.1163/156856205774472326. [DOI] [PubMed] [Google Scholar]
- Kaul G. and Amiji M. Pharm. Res. 2002;197:1061. doi: 10.1023/A:1016486910719. [DOI] [PubMed] [Google Scholar]
- Felt O, Buri P. and Gurny R. Drug Dev. Ind. Pharm. 1998;24:979. doi: 10.3109/03639049809089942. [DOI] [PubMed] [Google Scholar]
- Giunchedi P, Genta I, Conti B, Muzzarelli R A A. and Conte U. Biomaterials. 1998;19:157. doi: 10.1016/S0142-9612(97)00181-6. [DOI] [PubMed] [Google Scholar]
- Illum L. Pharm. Res. 1998;15:1326. doi: 10.1023/A:1011929016601. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wu Q, Wang Y N. and Ma J B. Acta Chem. Sin. 2003;61:614. [Google Scholar]
- Hou W M, Miyazaki S, Takada M. and Komai T. Chem. Pharm. Bull. 1985;33:3986. doi: 10.1248/cpb.33.3986. [DOI] [PubMed] [Google Scholar]
- Kawashima Y, Handa T, Kasai A, Kasai A, Takenaka H, Lin S Y. and Ando Y. J. Pharm. Sci. 1985;74:264. doi: 10.1002/jps.2600740308. [DOI] [PubMed] [Google Scholar]
- Miyazaki S, Yamaguchi H, Yokouchi C, Takada M. and Hou W M. Chem. Pharm. Bull. 1988;36:4033. doi: 10.1248/cpb.36.4033. [DOI] [PubMed] [Google Scholar]
- Shiraishi S, Imai T. and Otagiri M. J. Control. Release. 1993;25:217. doi: 10.1016/0168-3659(93)90080-O. [DOI] [Google Scholar]
- Illum L, Farraj N F. and Davis S S. Pharm. Res. 1994;11:1186. doi: 10.1023/A:1018901302450. [DOI] [PubMed] [Google Scholar]
- Miyazaki S, Ishi K. and Nadai T. Chem. Pharm. Bull. 1981;29:3067. doi: 10.1248/cpb.29.3067. [DOI] [PubMed] [Google Scholar]
- Gallo J M. and Hassan E E. Pharm. Res. 1988;5:300. doi: 10.1023/A:1015978704810. [DOI] [PubMed] [Google Scholar]
- Hassan E E, Parish R C. and Gallo J M. Pharm. Res. 1992;9:390. doi: 10.1023/A:1015803321609. [DOI] [PubMed] [Google Scholar]
- Wu Y, Yang W, Wang C, Hu J. and Fu S. Int. J. Pharm. 2005;295:235. doi: 10.1016/j.ijpharm.2005.01.042. [DOI] [PubMed] [Google Scholar]
- Zhang Y Q, Shen W D, Xiang R L, Zhuge L J, Gao W J. and Wang W B. J. Nanopart. Res. 2007;9:885. doi: 10.1007/s11051-006-9162-x. [DOI] [Google Scholar]
- Zhang Y Q, Xiang R L, Yan H B. and Chen X X. Chem. J. Chin. Univ. 2008;29:628. [Google Scholar]
- Myung S J, Kim H S, Kim Y, Chen P. and Jin H J. Macromol. Res. 2008;16:604. [Google Scholar]
- Yan H B, Zhang Y Q, Ma Y L. and Zhou L X. J. Nanopart. Res. 2009;11:1937. doi: 10.1007/s11051-008-9549-y. [DOI] [Google Scholar]
- Gupta V, Aseh A, Ríos C N, Aggarwal B B. and Mathur A B. Int. J. Nanomedicine. 2009;4:115. doi: 10.2147/ijn.s5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M. and Yamada H. Sen-i Gakkaishi. 1992;48:305. [Google Scholar]
- Cho K Y.et al2003Int. J. Biol. Macromol. 3236. 10.1016/S0141-8130(03)00023-0 [DOI] [PubMed] [Google Scholar]
- Song Y, Jin Y, Sun J. and Wei D. Polym. Int. 2006;55:1350. doi: 10.1002/pi.2093. [DOI] [Google Scholar]
- Aynie I, Vauthier C, Chacun H, Fattal E. and Couvreur P. Antisense Nucleic Acid Drug Dev. 1999;9:301. doi: 10.1089/oli.1.1999.9.301. [DOI] [PubMed] [Google Scholar]
- Yi Y M, Yang T Y. and Pan W M. World J. Gastroenterol. 1999;5:57. doi: 10.3748/wjg.v5.i1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J O. and Peng C A. Macromol. Symp. 2004;219:147. doi: 10.1002/masy.200550113. [DOI] [Google Scholar]
- Sarmento B, Ferreira D, Veiga F. and Ribeiro A. Carbohydr. Polym. 2006;66:1. doi: 10.1016/j.carbpol.2006.02.008. [DOI] [Google Scholar]
- Sarmento B, Ribeiro A J, Veiga F, Ferreira D C. and Neufeld R J. J. Nanosci. Nanotechnol. 2007;7:2833. doi: 10.1166/jnn.2007.609. [DOI] [PubMed] [Google Scholar]
- Sonavane G S. and Devarajan P V. J. Biomed. Nanotech. 2007;3:160. doi: 10.1166/jbn.2007.005. [DOI] [Google Scholar]
- Motwani S K, Chopra S, Talegaonkar S, Kohli K, Ahmad F J. and Khar R K. Eur. J. Pharm. Biopharm. 2008;68:513. doi: 10.1016/j.ejpb.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Mao H Q.et al2001J. Control. Release 70399. 10.1016/S0168-3659(00)00361-8 [DOI] [PubMed] [Google Scholar]
- Janes K A, Fresneau M P, Marazuela A, Fabra A. and Alonso M J. J. Control. Release. 2001;73:255. doi: 10.1016/S0168-3659(01)00294-2. [DOI] [PubMed] [Google Scholar]
- Soppimath K S, Aminabhavi T M, Kulkarni A R. and Rudzinski W E. J. Control. Release. 2001;70:1. doi: 10.1016/S0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- Banerjee T, Mitra S, Kumar Singh A, Kumar Sharma R. and Maitra A. Int. J. Pharm. 2002;243:93. doi: 10.1016/S0378-5173(02)00267-3. [DOI] [PubMed] [Google Scholar]
- Vila A, Sanchez A, Janes K A, Behrens I, Kissel T. and Vila-Jato J L. Eur. J. Pharm. Biopharm. 2004;57:123. doi: 10.1016/j.ejpb.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Mansouri S, Lavigne P, Corsi K, Benderdour M, Beaumont E. and Fernandes J C. Eur. J. Pharm. Biopharm. 2004;57:1. doi: 10.1016/S0939-6411(03)00155-3. [DOI] [PubMed] [Google Scholar]
- Ma Z, Lim T M. and Lim L Y. Int. J. Pharm. 2005;293:271. doi: 10.1016/j.ijpharm.2004.12.025. [DOI] [PubMed] [Google Scholar]
- You J O, Liu Y C. and Peng C A. Int. J. Nanomedicine. 2006;1:173. doi: 10.2147/nano.2006.1.2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma B V, Ali M, Baboota S. and Ali J. Indian J. Pharm. Sci. 2007;69:712. [Google Scholar]
- Rekha M R. and Sharma C P. J. Control. Release. 2009;135:144. doi: 10.1016/j.jconrel.2009.01.011. [DOI] [PubMed] [Google Scholar]