

Abstract
Conducting polymers hold significant promise as electrode coatings; however, they are characterized by inherently poor mechanical properties. Blending or producing layered conducting polymers with other polymer forms, such as hydrogels, has been proposed as an approach to improving these properties. There are many challenges to producing hybrid polymers incorporating conducting polymers and hydrogels, including the fabrication of structures based on two such dissimilar materials and evaluation of the properties of the resulting structures. Although both fabrication and evaluation of structure–property relationships remain challenges, materials comprised of conducting polymers and hydrogels are promising for the next generation of bioactive electrode coatings.
Keywords: hydrogels, conducting polymers, biomaterials, neural electrodes, hybrid materials
Introduction
Bioelectrodes are an integral component for the delivery of charge and recording of neural activity in a number of neuroprosthetic devices. Electrodes are conventionally fabricated from platinum, platinum alloys and gold. Surface modification of metallic electrodes has been extensively studied with a view to increasing the integration of biological tissue and minimizing foreign body encapsulation at the electrode interface [1–5]. In addition, improved communication between synthetic and biological systems will allow medical devices to perform more efficiently by permitting the use of smaller charges to elicit a neural response [6–9].
Conducting polymers (CPs) have enormous potential for providing a more conducive environment for cell integration [10–13]. CPs exhibit improved impedance characteristics and provide a softer mechanical interface when compared to conventional metal electrodes [1, 13–17]. CPs have an intrinsically unstable conjugated backbone, which when doped with appropriate ions can be used to pass both electronic and ionic charges. Although a number of CP systems have been investigated for use as bioelectrode coatings, studies by Abidian et al [7] and Richardson et al [6, 18] have indicated that the benefits provided by the CP coating following implantation are not maintained in the long-term. In vivo studies have shown that CPs provide low impedance with minimal bilayer capacitance, but as fibrosis proceeds, the growth of non-conductive scar tissue dominates [7, 19]. It is proposed that the provision of appropriate biological molecules will promote the formation of a stable interface prior to the growth of scar tissue.
Several research groups have explored the functionalization of CPs with biological molecules, specifically growth factors [2, 18, 20, 21], cell adhesion proteins [1, 10, 22, 23] and anti-inflammatory drugs [3, 24]. Although bioactivity has been successfully imparted to a variety of CPs, Green et al [1, 2] reported that large peptides and growth factors significantly disrupt the mechanical stability and long-term electrochemical performance of bioactive CP coatings. Additionally, the concentration of the biomolecules that can be incorporated is limited. This in turn reduces the amount of biological signal which can be presented or released from CP matrices. Thus CPs alone may not be sufficient to create a long-term stable interface between synthetic implant devices and neural tissue. More recently, design and development of CP-hydrogel blends have been proposed as an approach to overcoming some of the limitations that CPs used alone may present [3, 25–28]. The underlying premise of this approach is that hydrogels may act to modulate and improve mechanical properties, as well as provide a low-fouling surface and a depot for water soluble, bioactive agents.
Hydrogels are fabricated from hydrophilic polymer chains via chemical or physical crosslinking processes that convert the soluble polymer chains into insoluble polymer networks. Due to their high water content, mechanical properties of hydrogels often closely match those of biological tissues. Hydrogels have been traditionally used for a range of soft tissue replacement applications such as contact lenses, drug delivery vehicles, separation membranes and adhesives [29–32]. Table 1 lists representative natural and synthetic polymers used in medical devices. Where available, the mechanical properties of these are also shown, revealing their similarity to biological tissue.
Table 1.
Representative tensile and shear modulus and % elongation values for selected natural and synthetic hydrogel systems, as well select biological tissues.
| Polymer | E=tensile modulus G'=shear modulus (kPa) | Elongation (%) | References |
|---|---|---|---|
| Hydroxyl ethyl methacrylate (HEMA)–acrylic acid (AA) | E=200–300 | 30–150 | [33] |
| HEMA–epoxy methacrylates | E=300–10 000 | 70–110 | [34] |
| Poly (vinyl alcohol) (PVA) | E=20–1500 | 60–140 | [35, 36] |
| Poly (ethylene glycol) (PEG)–HEMA | E=4000–8000 | 10–30 | [37] |
| (PEG) fumarate | E=15–90 | 30–75 | [38] |
| Acrylamide(AAm)–bis-acrylamide | E=0.05–30, G′= 0.007–19 | – | [39–42] |
| Agarose | E=40 | – | [43] |
| Gellum gum | E=50–200 | 5–15 | [44] |
| Fibrin | G′=0.25–2.15 | – | [42] |
| Collagen | G′=0.002–0.017 | – | [45] |
| Skeletal muscle | E=3–450 | – | [46] |
| Fibroblasts | E=1–3 | – | [47] |
| Retina | E=0.1–0.97 | – | [48] |
| Brain | G′=0.1–5 | – | [42, 49–51] |
Another feature of these hydrophilic polymers is their ability to resist protein deposition and thus act as anti-fouling or anti-fibrotic materials [52, 53]. In neural interfacing applications such as deep brain stimulation, alginate hydrogels have been proposed as materials providing a better mechanical match for neural tissue and potentially capable of modulating fibrotic responses [54]. As a result of their high water content, hydrogels also have excellent diffusive properties. They are commonly used in drug delivery applications, and have been proposed as a means of releasing neurotrophins in neural stimulating electrodes [55]. The release of other growth factors, proteins or drugs could be used to discourage fibrous encapsulation and encourage the growth of neural cells into the implant [56, 57].
It has been proposed that copolymer biomaterials with the benefits of both CPs and hydrogels can meet the essential design requirements of an ideal neural interface [58]. The copolymer must pass charge from a metallic electrode to neural tissue, be soft and flexible, mechanically match electrodes and neural tissue and promote and maintain neural cell integration to the electrode site via biomolecule incorporation. A major challenge is in fabricating a structure that can meet these critical design criteria.
Fabrication routes for CP-hydrogels
Despite both materials being polymers, CPs are usually produced via very different fabrication routes to hydrogels. In order to produce a material with the required physicochemical and biological properties the fabrication routes of both components must be considered.
Conventional CP fabrication
Conducting polymers have an inherently unstable backbone, resulting from the formation of alternate single and double bonds along the monomer units during polymerization. The delocalized pi-bonding electrons, produced across the conjugated backbone, provide an electrical pathway for mobile charge carriers which are introduced through doping. Consequently, the electronic properties, as well as many other physicochemical properties, are determined by the structure of the polymer backbone and the nature and the concentration of the dopant ion. Figure 1 shows the chemical structure of the CP polypyrrole as an example of the typical alternating double bond backbone structure.
Figure 1.
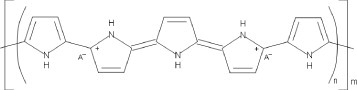
Conducting polymer (polypyrrole) structure defined by conjugated backbone and doped with ionic species A.
Conducting polymers can be synthesized by either electrochemical or chemical processes. A typical electrochemical system is comprised of an aqueous electrolyte solution of monomer units and dopant ions in a three-electrode cell, including a working, counter and reference electrode. Upon application of an electric potential, CP monomers are commonly oxidized to radical cations and form positively charged CP films with concomitant anionic doping on the working electrode (anode). Such electrodeposition is a facile and reproducible method which provides good control over the surface and biological properties of the films. The thickness and morphology of the film are primarily controlled by the amount of electric charge passed between the counter and working electrodes. Additionally, biofunctionality can be easily imparted to CPs during electrodeposition through the incorporation of biological entities such as silk-like protein fragments (SLPF) [59], hyaluronic acid (HA) [60, 61], various laminin peptides [1, 2, 59, 62], enzymes [28, 63–66], polymeric amino acids [67, 68], growth factors [2, 11, 69, 70] and whole cells [71]. Electrodeposition is the most commonly used method for coating conventional metallic bioelectrodes. However, this technique offers limited control over the chain structure of the resulting polymers. Electrodeposited CPs are substantially insoluble owing to the random chain structure, and thus cannot be melt processed and are difficult to post-process.
Conversely, chemical synthesis allows mass production of tailored CPs with improved processability but these CPs lack the simplicity and speed of the electrochemical route. In a conventional chemical oxidative coupling process, a monomer solution in an organic solvent is slowly added to a flask filled with an organic solution of mild Lewis acid catalysts under inert gas. Typical catalysts used are ferric chloride, iron(III) sulfate or cupric chloride. The mixture of precursor and oxidant is then stirred for several hours to promote polymerization. Following polymerization, multi-step purification processes must be performed, such as soxhlet extraction, to remove residual oligomers and oxidants. The doping species of chemically synthesized CPs are generally limited to the small anions, such as chloride and sulphate, associated with the oxidant. Chemically synthesized CPs often have lower electrical conductivity than electrodeposited CPs and thus require dedoping and redoping processes to increase conductivity. Many alternate routes exist for chemical synthesis of CPs and are discussed in detail by Poole-Warren et al [72].
Conjugated aliphatics, including polyacetylene, and benzene derivatives such as polyaniline (PANI), have been largely ruled out for biomedical applications due to their oxidative degradation in air and the cytotoxic nature of their by-products, respectively. Although recent research has shown that the emeraldine salt of PANI (EPANI) can be successfully fabricated in a biocompatible form [73, 74], modern biomedical CPs are typically composed of heterocyclic aromatics, such as derivatives of thiophene and pyrrole [75–78]. Specifically, poly(3,4-ethylenedioxythiophene) (PEDOT) and polypyrrole (PPy) have been widely studied for their superior environmental and electrochemical stability [79–82]. Figure 2 shows the chemical structure of various thiophene derivatives including EDOT, EDOT-MeOH and 3-alkylthiophene.
Figure 2.
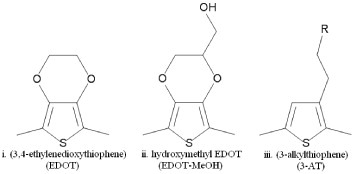
Typical monomer structures used to fabricate (i) Poly(3,4-ethylene dioxythiophene), (ii) Poly(hydroxymethyl-3,4-ethylenedioxythiophene) and (iii) Poly(3-alklythiophene).
Conventional hydrogel fabrication
Hydrogels, by definition, are water insoluble networks. There are many methods to form these insoluble networks, and these methods can be broken down into physical and chemical pathways. Physical crosslinking of hydrogels is generally achieved via hydrogen bonding, ionic or hydrophobic interactions, or inducing crystallinity. These types of crosslinking methods are often achievable in mild conditions and reversible. Alginate is a classic example of a hydrogel that is formed via ionic interactions. Alginate is made up of mannuronic and glucuronic acid residues that readily interact with calcium ions to form a crosslinked hydrogel [83]. Dextran hydrogels can form crosslinked hydrogels in a concentrated solution, and this formation has been reported to be due to crystallization from hydrogen bonding [84]. One example of a crystalline crosslinked polymer is freeze-thawed poly(vinyl alcohol) (PVA). By controlling the number of cycles of heating and freezing, the amount of crystalline regions that are formed within the PVA can be tailored [85]. Another common method of physically forming hydrogels is through hydrophobic interactions. In most of these cases, the polymer molecules are designed to have at least one hydrophilic and one hydrophobic block. The hydrophobic blocks then self assemble in an aqueous solution to form micelles. Many different chemistries have been used to make these types of polymers [86, 87].
For most biomedical applications, chemically crosslinked gels are more relevant. Chemical crosslinking of hydrogels is commonly researched and has been described in detail in several reviews [88–91]. Briefly, chemical crosslinking can be achieved through free radical, condensation, click chemistry or Michael-type addition polymerization. Free radical polymerization involves the generation of radicals through the addition of an initiator and either heat, light or reductive/oxidative chemicals. These radicals then attack an unsaturated bond in the macromer chain. The addition of unsaturations into polymer chains is commonly achieved through the functionalization of the polymers with (meth)acrylates and (meth)acrylamides [92–96]. Free radical polymerization of hydrogels is particularly attractive for many biomedical applications as it can generally be carried out in relatively mild conditions. The polymerization can be done with a wide range of macromers in aqueous solution or in bulk, at room or body temperature, and only very small amounts of the initiator species are required. The major drawback is that the polymerization is random and uncontrolled, as compared to some of the other techniques listed below.
Condensation polymerization and click chemistry involve two complementary chemical species that readily react in the presence of each other, such as amine-carboxylic acid, aldehyde-hydrazide or isocyanate-OH/NH. The reactive chemical species can either be small crosslinking molecules, e.g., glutaraldehyde [58, 92], or attached to a polymer backbone, e.g. polyaldehydes [97]. In these reactions, the amount and the molecular weight of reactive groups have a strong effect on the overall crosslinking density and network properties. When small crosslinking molecules are used, or when these reactions are carried out in organic solvents to limit cross reactions with water, the resulting hydrogels must undergo stringent cleaning processes to ensure any toxic or unreacted molecules are completely removed from the system. This often limits their biomedical applicability to areas where the gels can be pre-formed, cleaned and used at a later time [89].
Controlled polymerizations are very popular methods to make polymer chains, grafts, brushes and star polymers, and have the advantage of generating well defined and mono-disperse polymer chains. One of these methods that is particularly useful in generating crosslinked networks is Michael and Michael-type addition reactions [91, 98]. These reactions generally have good specificity and rate, and can be carried out in mild conditions. The most common Michael-type polymerizations are thiol-ene reactions [98, 99]. While Michael-type addition does result in a controlled network architecture, it suffers from problems with side reactions and incomplete conversion of the functional groups.
All methods mentioned above have their benefits and drawbacks, and the choice of which method to use for network formation for a particular application is highly variable and often depends on the final application. In all the above methods, the polymer chains are chemically bound to one another. In some instances, the hydrogel system is designed to degrade over time, either through the newly formed crosslinks, or the backbone polymer itself. Regardless of whether the system is designed to be non-degradable or degradable, the properties of resulting hydrogel are usually dependant on how the crosslinking was achieved, the extent and efficiency of the crosslinking procedure, and the chemistry of the polymer chains themselves.
In addition, while the chemical crosslinks are the predominant feature of the hydrogels, many hydrogels also have a degree of physical crosslinking. The polymers used are often long chain polymers, and there may be hydrogen bonding, hydrophobic interactions of attached side groups, or entanglement of the chains. All these interactions result in additional crosslinking and affect the overall structure and properties of the gel. However, they are generally present to a much lower extent and are extremely difficult to quantify within the already chemically crosslinked systems. The standard methods for estimating the mesh size of hydrogels rely on swelling and mechanical properties (see below). However, given that these are indirect methods, all crosslinks including both physical and chemical crosslinks are considered using these methods and an average crosslink density of the system is obtained. In general, the degree of crosslinking and the space between crosslinks is one of the biggest determinants of the final network properties.
Routes for producing CP-Hydrogels
A number of routes have been proposed for fabricating materials that incorporate both CPs and hydrogels. Most of these materials have been produced as combinations of two separate phases created by each component. Three categories of CP-hydrogels can be defined by their fabrication route and are depicted in figure 3:
Figure 3.
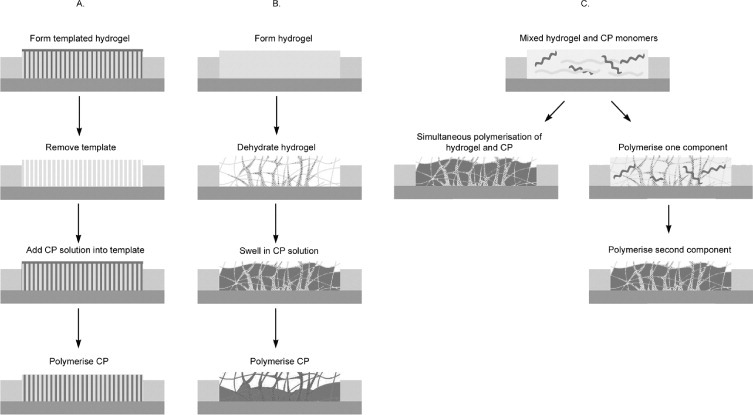
Typical fabrication routes used to produce CP-hydrogels: (A) Templated CP-hydrogels; (B) CPs deposited within a hydrogel matrix and (C) CP-hydrogels formed from mixed precursors—either simultaneously or in a two-step process.
-
A.
Templated CP-hydrogels.
-
B.
CPs deposited within a hydrogel matrix.
-
C.
CP-hydrogels formed from mixed precursors—either simultaneously or in a two-step process.
The properties of these CP-hydrogels are strongly dependent on the concentration and structure of the CP within the hydrogel.
Several combinations of CPs and hydrogels have been formed via these routes and a summary of the copolymers is presented in table 2.
Table 2.
Examples of CP-hydrogel copolymers.
| Route | Hydrogel component | CP component | References |
|---|---|---|---|
| A/B | Wetspun chitosan fibres | PANI | [100] |
| A/B | Methanol treated electrospun PVA fibrils | PANI | [101] |
| A/B | Alginate with electrospun PLGA fibres | PEDOT | [3] |
| B | Poly(2-hydroxyethyl methacrylate) (pHEMA) | PPy | [28, 63, 65, 102] |
| B | P(PY/4PyBA) | [66] | |
| B | PEDOT or PPy | [103] | |
| B | Poly(HEMA-co-PEGMA-co-HMMA-co-SPMA) | P(PY/4PyBA) | [104] |
| B | Alginate | PPy | [27, 105] |
| B | PEDOT | [54] | |
| B | Poly(acrylic acid) (PAA) | PEDOT | [106] |
| B | Poly(2-acrylamido-2-methylpropanesulfonic acid) (PAMPS) | PANI | [107] |
| B | Polyacrylamide (PAAm) | PPy | [108–111] |
| B | PANI | [107, 111–114] | |
| C | PANI | [115] | |
| C | PAAm | BAYTRON-P®(PEDOT:PSS) | [116] |
| C | Chitosan | PANI | [117] |
The electronic properties of CPs generally improve with increasing surface area. While bioelectrode applications demand high surface area of CP films, the poor processability of CPs precludes the use of conventional post-process techniques. Fabrication using a micro or nanostructured template can significantly increase the surface area of the CP, thus overcoming these limitations. CPs polymerized within the confined area of a nanostructured template can provide control over the structure and morphology. Nanostructured hydrogels could potentially be used as templates to synthesize CP-hydrogels with specific structures (category A, figure 3). As an illustration of this approach using a degradable polymer rather than a hydrogel, Abidian et al [3, 4, 7, 25, 118] electrodeposited 100 to 600 nm diameter CP nanofibrils by using electrospun poly(L-lactic acid) (PLLA) and poly(lactic-co-glycolic acid) (PLGA) nanofibres as a template. Either PEDOT or PPy was directly electrodeposited on the gold neural probe coated with the electrospun polymers. The polymer templates were then either removed by soaking the structure in dichloromethane leaving CP nanotubes on the neural probe or degraded for additional controlled drug release functions.
The most common method for forming a CP-hydrogel is polymerization of conducting polymers through preformed hydrogel matrices (category B, figure 3). Absorbent hydrogels are formed and dried on a substrate and then reswelled in a CP monomer solution. The CP is polymerized via application of electrical charges or by exposure to a chemical oxidant. CP/pHEMA [28, 63, 65, 66, 102–104], CP/alginate [3, 27, 54, 105], PEDOT/PAA [106], PEDOT/PAMPS [107] and CP/PAAM [107–114] have been synthesized for various biomedical applications. Comparable electrical properties and improved mechanical and biological performances compared with the standalone CP counterparts were reported.
An alternative, less common method reported in literature is the fabrication of CP-hydrogel from mixed precursors (category C, figure 3). In this method, the hydrogel and CP precursors are placed in the same vessel where they are polymerized concurrently [115]. In this study, the CP precursor (aniline), hydrogel precursor (acrylamide) and a hydrogel crosslinker (N,N-methylene bisacrylamide NMBA) were all dissolved in an acidic solution. Then, the oxidant potassium peroxysulfate was added to the mixed solution to polymerize the CP and crosslink the hydrogel.
In some cases, a combination of two fabrication routes has been used to produce interpenetrating networks of nanostructured CP-hydrogels. Ismail et al have fabricated fibrous networks of PANI with PVA and chitosan hydrogels for artificial muscle applications [101, 119]. Aqueous solutions of PVA and chitosan hydrogel were electrospun and wetspun, respectively, to form entangled networks of nanofibres. The resulting fibrillar networks were then reacted with methanol and glutaraldehyde, respectively, to dehydrate the structures. PANI was then chemically synthesized throughout the hydrogel network by swelling the hydrogel in an aniline/HCl solution, followed by exposure to a chemical oxidant solution of ammonium persulfate. The resulting fibrous PANI/PVA gels were mechanically rolled-up to form multilayered cylindrical structures.
Material properties of CP-hydrogels
Selection of an appropriate material for a biomedical application involves consideration of the physical, chemical and biological properties appropriate for the end-use. For such applications as medical electrodes, electrical properties are clearly an important consideration, and the most appropriate solution is often the selection of a combination of two or more different materials. There are many methods of assessing physical properties of a particular material, but not all methods can be employed with equal success across all material types. When CPs and hydrogels are combined, the issue arises as to how to assess and characterize the resulting material which can have very different properties to the individual component polymers. CPs and hydrogels have very different methods for testing their various properties, and converting these techniques for use in the CP-hydrogel is often problematic. In addition, the impact of both the component polymers and the fabrication technique must be assessed across each property. In the end, the critical design criteria for evaluation are physical properties including surface topography, mechanical, electrical and biological properties.
Surface topography
Increased electrode surface area mediates more efficient charge transfer between the biological tissue and synthetic device [120]. Surface roughness is also known to enhance cell adhesion and tissue integration [121, 122]. Electrodeposited CPs typically display very rough surface topography as shown in the PEDOT example shown in figure 4. Surface topography of CPs can be analysed through scanning electron microscopy (SEM); however, the addition of the hydrogel component limits the effectiveness of this analytical method. Since SEM can only be used on samples with a low water content, hydrogels must be dried prior to observation, and the resulting artefacts alter their surface morphology. Despite this, many groups perform SEM on CP-hydrogels. For systems with overlying CP coatings and templates, SEM can provide useful information on the CP feature sizes [3, 118]. However, an analysis which provides for effective quantification of surface features in the hydrated state will provide more useful information on the material characteristics expected for the implanted material.
Figure 4.

Sample SEM images at 1000× magnification: (A) Bare Pt electrode, (B) PEDOT/pTS coated Pt electrode, (C) PEDOT/pTS/NGF coated Pt electrode [11].
Two alternatives for quantifying surface morphology are profilometry and atomic force microscopy (AFM). While profilometry yields quantitative data on surface roughness, conventional stylus profilometry is damaging to both CPs and hydrogels. An ideal surface analytical technique is likely to be found in optical profilometry which can be performed on hydrated substrates with no contact. Additionally, interferometers used for optical profiling allow visualization of CP structures below the hydrogel surface to be visualized.
AFM has been effectively used for quantifying surface roughness and mechanics of CP films on electrodes [123]. Xiao et al obtained AFM images of cells cultured on CP coated electrodes and were able to determine the cell adhesion properties of PEDOT compared to bare gold by dragging the AFM tip across the cell bodies [123]. When applied to CP-hydrogels and performed in the hydrated state, this analysis is likely to yield visual qualification of surface topography, quantification of surface roughness and possible data on cell adherence. Although topography is a key factor in electrical and biological function of electrode coatings, there will be challenges in accurately measuring the surface features of CP-hydrogel combinations given the dissimilar properties of the two materials.
Physical and mechanical characterization
Due to the high water content, hydrogels are generally characterized as relatively low strength and low modulus materials. Some of the more commonly employed methods to assess physical and mechanical properties of hydrogels are examination of swelling behaviour, the equilibrium water content, tensile and compressive strength, diffusivity or mesh size and mass loss. Although the same assessment can be applied to CP-hydrogel materials, correction factors may need to be used to account for the different phases.
In many applications, the water content, or the amount of swelling, of the hydrogel is of great importance as it dictates many mechanical and physical properties of the gels. The degree of swelling can be determined by either a volumetric or mass measurement. The volumetric swelling ratio (Q) is calculated from the mass swelling ratio (q=ms/md) via:
where ms is the mass of the swollen gel, md is the mass of the dried gel, ρpolymer is the polymer density, ρsolvent is the density of the buffer solution. However, combined CP-hydrogel systems may experience difficulties or errors in calculation of this value as the relative amount of each phase is not known and thus the dry mass and overall ‘polymer density’ cannot be split into the CP and hydrogel components. One way to overcome this issue is to use simple evaluation of water uptake or water content in the CP-hydrogel. The swollen and dry weights of the sample were compared by Dai et al [106] and Aouada et al [116]. While this does provide a water content value, it does not allow for more sophisticated calculations, such as mesh size.
The mechanical properties of hydrogels are often hard to accurately measure owing to their low strength, smooth and lubricious surfaces and propensity for defects, such as small air bubbles. Several researchers overcome some of these issues by first drying their hydrogels and testing them in the dry state. However, this is often an unrealistic measure if the hydrogels are intended for use in a wet environment. Compressive modulus of the hydrogels is relatively easy to measure, although since these materials are made up of predominately water, which is an incompressible fluid, the results are only a relative indication of modulus [92]. Much progress has been made in tensile testing of hydrated gels. Sand paper and other rough surfaces have been used to improve the grip strength on the hydrogels [35], while hydrating baths are also used to prevent the drying of the samples during the test. Both of these modifications have improved the accuracy of the data that can be obtained from hydrated gels. However, obtaining these same measurements on the CP-hydrogel polymers is often problematic. Generally, only small amounts or thin films of the co-polymer materials can be produced, which do not have appropriate shapes for mechanical testing. In many cases, the CP is also formed on an underlying conducting substrate or electrode. Thus the CP-hydrogel may not be easily removed from this substrate without major disruptions to the material. In addition, testing cannot be done with CP-hydrogel still attached to the substrate, as substrate is often much stronger than the polymer coating and the results are then skewed towards the substrate properties.
Because hydrogels are completely filled with water, the ‘pores’ or mesh size cannot be simply visualized with SEM or other techniques that are used for solid materials. Instead, other approaches are used that allow calculation of mesh size or of an average molecular weight between crosslinks (Mc). Mesh size may be estimated via evaluating permeability and diffusion using a range of solutes; however, swelling and mechanics are often used as simple tools to estimate the network structure and Mc of a hydrogel. Peppas and Merill derived an equation for a neutral hydrogel in the presence of water that relates Mc to the volumetric swelling ratio (Q) [124, 125],
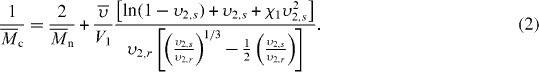 |
Here Mn is the number average molecular weight in the absence of any crosslinking; 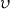 the specific volume of the polymer; V1 the molar volume of the solvent;
the specific volume of the polymer; V1 the molar volume of the solvent; 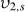 the equilibrium polymer volume fraction;
the equilibrium polymer volume fraction; 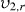 the polymer volume fraction after crosslinking but before swelling; and χ1 is the polymer solvent interaction parameter. Because there are two interaction parameters and specific volumes, and the
the polymer volume fraction after crosslinking but before swelling; and χ1 is the polymer solvent interaction parameter. Because there are two interaction parameters and specific volumes, and the 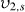 is directly related to the swelling ratio (
is directly related to the swelling ratio (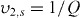 ), the use of this equation will have similar difficulties as those listed above for calculating the swelling ratio. The interaction parameters may also vary significantly in the CP-hydrogel structure as opposed to the homopolymer network, and thus new studies would be needed to obtain correct interaction values. In addition, the calculation of Mn in these systems can be problematic. Some CPs can be solubilized and their Mn obtained. However, any of the CPs that are electrically deposited are insoluble and thus calculating a Mn is extremely difficult. Without these values, the Mc cannot be calculated.
), the use of this equation will have similar difficulties as those listed above for calculating the swelling ratio. The interaction parameters may also vary significantly in the CP-hydrogel structure as opposed to the homopolymer network, and thus new studies would be needed to obtain correct interaction values. In addition, the calculation of Mn in these systems can be problematic. Some CPs can be solubilized and their Mn obtained. However, any of the CPs that are electrically deposited are insoluble and thus calculating a Mn is extremely difficult. Without these values, the Mc cannot be calculated.
The second major approach to estimating network structure of hydrogels is via the rubber elasticity theory which can be used to correlate the compressive modulus to network structure via [126, 127]:
Here K is the compressive modulus, v is Poisson's ratio, ρ is the density of the polymer and T is the temperature of the gel. Many values from the modulus equation are used in the swelling equation (Mn, density, 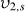 ) and thus either approach for a CP-hydrogel will have the same drawbacks and limitations.
) and thus either approach for a CP-hydrogel will have the same drawbacks and limitations.
In both of the Mc calculations above, the obtained values are estimates based on several assumptions. Mc is a useful measure; it is often used to calculate the mesh size of the network as a direct measurement of the hydrogel mesh size is usually impossible. Also, unless highly controlled polymerization techniques are used, hydrogels generally have a range of mesh sizes due to the variability in the crosslinking efficiency and placement.
It is proposed that the hydrogel component of a CP-hydrogel coating can improve the mechanics of conventional electrodes, minimizing the growth of scar tissue and the impact of strain mismatch which exacerbates foreign body reactions. Mechanical stress and strain of hydrogels are usually assessed by tensile or compression testing, but CPs electrodeposited on substrates are not suitable for these standard techniques. Electrodeposited CPs are generally not soluble and the deposited films must be physically removed from the substrate to obtain free-standing samples. Nevertheless, several studies on various CP samples reported that the tensile moduli of CPs are in the 108–109 Pa range [128–138]. Recently, micro and nanoindentation have been used to provide data on CP mechanics where the coating is assessed on the metallic substrate [14, 139]. If the material can be delaminated from the substrate or produced as a standalone film, then the CP-hydrogels can be assessed by the standard techniques. However, a very limited number of reports exist on the mechanical properties of CP-hydrogels produced for biomedical applications. Aouada et al [115] reported compression moduli of 104 to 105 Pa for PEDOT/PAAM CP-hydrogels with up to 5 vol% of PEDOT. Similarly, Siddhanta and Gangopadhyay [107] reported tensile moduli of 1.5×104 to 1×105 Pa and 30 to 50 Pa for their PANI/PAMPS and PANI/PAAM hydrogels, respectively, with up to 20%, weight in weight, of PANI. The elastic moduli of PEDOT and PANI were previously reported to be between 0.9×108 and 2.8×108 Pa [133–135] and 1.8×108 and 5×109 Pa [136–138], respectively. These results clearly demonstrate that the hydrogel component in this system considerably softens the CP and produces an interface suited to dampening the mechanical difference between synthetic electrodes and the surrounding tissue.
Electrical characterization
Variations in CP components including the choice of monomer, dopant and fabrication parameters can greatly affect the electrical performance of an electrode. The most relevant electrical tests for medical electrode applications are cyclic voltammetry and impedance spectrometry which are used to discern the electrochemical activity and frequency dependant impedance behaviour of materials.
Electroactivity of a material is assessed by application of cyclic voltammetry (CV) across reduction and oxidation (redox) cycles. The size of the redox peaks obtained from the CV spectra are indicative of the charge storage capacity of the electrode [12]. In theory, adding non-conducting polymers should have an insulating effect and result in deterioration of the electrical properties. The charge storage densities of CP-hydrogels can be as high as 560 mC cm−2 [105] for PPy/PSS deposited into an alginate matrix. This compares with the value of 186 mC cm−2 for PPy/PSS alone [81]. More complex structures designed for drug delivery, consisting of layered PEDOT nanotubes, coated with alginate and an additional layer of PEDOT, produced electrodes with a charge carrying capacity of 223 mC cm−2 [3]. Drug loading of a PPy and polyacrylamide gel was shown to decrease charge carrying capacity by reducing ion mobility [111]. However, the hydrogel component was shown to increase the charge transfer properties when compared to the drug loaded CP alone. In general, the addition of a hydrogel component has been shown to increase the charge carrying density of CP coated electrodes [3, 27, 111].
Most CP-hydrogels are reported to have electroactivity upon application of a single cycle of CV where anodic and cathodic peaks are observed. However, several groups stipulate that multiple cycles of CV yields data representative of the long-term electrochemical stability of the proposed electrode materials [1, 2, 81, 82]. As the cycles are repeated, a stable material will have a constant peak size, where a material experiencing degradation of electroactivity will have diminished peak size. Reports by Yamato et al [82] and Cui and Martin [81] have demonstrated that CPs produced from pyrrole suffer from oxidative electrochemical degradation, and by replacing the monomer unit with EDOT, the long-term electroactivity can be increased from 5% of the original activity (for PPy) to 95% (for PEDOT) when cycled over extended periods. However, similar studies have not been carried out on CP-hydrogels. These results clearly highlight the need for assessing long-term electrochemical activity of hydrogels containing CPs.
The impedance of an electrode is vital in determining its performance characteristics in an implantable device for both recording and stimulation applications. Impedance spectroscopy analyses the way in which a material passes charge across a wide range of frequencies. CPs are usually analysed around 1–105 Hz, with particular interest in the biologically relevant frequencies between 102 and 103 Hz [10, 12]. Conventional metal electrodes have higher impedance magnitudes when interfaced with neural tissue than CPs. A typical metal electrode has an impedance approximately two orders of magnitude higher than a PEDOT coated electrode at 1 kHz [105]. This is mainly due to the rough surface topography which increases the available charge transfer area on the electrode. Reports of CPs formed in hydrogel matrices indicate that impedance can be decreased by an additional order of magnitude below that of conventional CP only films [3, 105]. This is thought to be predominantly due to thicker films being grown through the hydrogel, supported by the decrease in impedance correlating with increased CP polymerization time. Other CP-polymer networks designed for increased surface area, such as those comprised of electrospun hydrogels or CP nanotubes, have decreased impedance mediated through the increased charge transfer area [3, 25]. Abidian et al have fabricated PEDOT nanotubes, produced by CP coating PLLA/PLGA nanofibres and have reported a reduction in electrode impedance from 4 MΩ for bare gold, to 1 kΩ for the nanofibres [25]. Removal of the PLGA core further decreased impedance to 800 Ω [25]. Variation in the processing of CP-polymer materials appears to allow designing optimal impedance for implant electrodes.
Biological evaluation
A critical property of any biomaterial is the biological performance. Ideal neural interfacing materials should be designed to promote targeted cell attachment and ingrowth of neural tissue to the electrode interface through release of appropriate biomolecules, such as nerve growth factor [56, 57, 69]. Characterization of the cell response is vital to developing an optimal neural interface material.
In vitro characterization of cell responses has been integral to the development of both hydrogels and CPs. Many CPs have demonstrated negligible toxicity across a wide range of cell types and explants, including clonal neural cells [1, 2, 11, 140], primary neuroblastomas [59], cardiomyocytes [141], glial cells [59] and spiral ganglion explants [20]. It has also been shown that CPs provide a more conducive environment for cell attachment and growth than bare metal electrodes [1, 13], and that CPs loaded with neurotrophins can stimulate neurite growth of target cells [2, 6, 69]. Similarly, many hydrogels have shown in vitro and in vivo compatibility with a wide range of cell types. Although hydrogels characteristically have low protein adsorption and therefore low native cell interactions, many studies have examined addition of biological molecules to promote cell interactions. These include addition of adhesion peptides such as RGD and YIGSR [142–146] which enhance interactions of specific cell types, polysaccharides such as heparin that are able to present growth factors [147, 148], and growth factors themselves [149, 150].
Although several CP-hydrogels have been proposed for biomedical applications, few reports exist on their interactions with cell and tissues. Li et al showed that a PPy-PVA gel attenuated blood protein adsorption and that the surface topography promoted PC12 cell attachment [151]. PANI-gelatine blends have also been assessed for compatibility with rat cardiac myoblasts and were found to promote cell attachment and proliferation to a similar degree as the control tissue culture-treated plastic (TCP) and smooth glass substrates [152]. These assays indicate that CP-hydrogels are likely to be similarly low in toxicity as their component polymers, but further research is required to confirm these results.
In vivo studies are often used to explore the performance of electrode materials in the intended implant scenario and can provide valuable information on the tissue response. An acute in vivo study by Kim et al demonstrated that PEDOT/alginate hydrogels could be used to improve neural recordings from guinea pig cortex [54]. Similarly, Abidian et al implanted PEDOT-PLLA nanotubes in rat cortex and found these results were maintained in chronic applications extending for 7 weeks [118]. However, both studies did not report cell response to materials and hypothesized cell interactions at electrode sites. While it is apparent that CP-hydrogels are promising materials for biomedical applications, significant short-term and long-term in vivo studies including chronic performance of electrodes and the impact on surrounding tissues are required before they can be employed as coatings for medical electrodes.
Future research
Several challenges will be central to eliciting the benefits of hydrogels when used in combination with CPs to improve their mechanical properties and biofunctionality. The main issues include adherence of the CP-hydrogel to the metal electrode substrate, creation of an interpenetrating network without compromising the electrical properties and control of the incorporation and release of biological entities.
Adherence to electrodes
Adherence of surface coatings to electrodes is a major consideration for any electrode modification. Many coating techniques such as platinum black, CPs, multi-walled nanotubes and mixtures of these materials have been limited in their clinical application by poor mechanical adherence. ASTM assays [153] used to determine the adherence of coatings to metallic substrates have shown that conventional CPs can lose up to 51% of their coating when subjected to minor shear forces [154]. With the addition of a biofunctional molecule such as nerve growth factor (NGF), Green et al has reported that delamination of PEDOT from platinum electrodes is significantly increased [1, 2].
Hydrogels are not naturally adherent to smooth surfaces such as metal and glass, and a mechanically stable interface must be obtained to be beneficial in an implantable electrode. Current methods of electrospinning polymers and deposition of CPs through hydrogels are unlikely to create a stable attachment. In non-implant applications, covalent attachment of organic polymers to metallic surfaces has been shown to increase the mechanical stability of the coating [155, 156]. Alternately layered constructs with electrostatic interactions, such as self-assembled polymers, may also provide an increased adherence to the metallic substrate. When neural probes are coated with CP-hydrogel materials, the entire probe is often covered with the hydrogel component before deposition of the CP component over the electroactive segments, leaving large areas between the electrodes coated with hydrogel only [3]. This fabrication method prevents shearing of the individual electrode sites, preserving the mechanical stability of the CP-hydrogel portions.
The underlying electrode surface can also be integral to producing a stable coating. In medical electrodes with Pt-black or iridium oxide coatings, roughening of the underlying electrode surface is used to mediate increased adhesion between the substrate and coating. Although several methods of producing a stable mechanical coating have been proposed, an optimal solution that can provide attachment of CP-hydrogels to many electrode configurations is an important area of on-going research.
Producing an interpenetrating network
In order to gain the full benefits of combining a CP with a hydrogel, the two polymer systems should be completely integrated and an interpenetrating network (IPN) formed. However, obtaining a full IPN is not always an easy task, especially when trying to combine monomers/polymers that have very different properties. The first step in combining the materials is to ensure that they are miscible within each other. Hydrogels, by definition, are highly hydrophilic polymers, whereas the monomers/polymers that make up CPs can range from hydrophobic to hydrophilic. One example of an attempt to overcome this challenge is where Justin and Guiseppi-Elie copolymerized PPy with 4-(3′-pyrrolyl) butyric acid to improve the hydrophilicity of the PPy [104]. They found that the addition of the butyric acid resulted in more favourable polymerization kinetics and allowed the CP to form a more homogeneous blend with the hydrogel [104].
Another challenge to achieving an IPN relates to how the two types of polymer chains are formed and crosslinked. As discussed, CP-hydrogels have been formed via various routes; however, depending on the timing and technique of each polymerization, the end result is often that the CP forms underneath the hydrogel, or that the CP only occupies part of the hydrogel. Both of these scenarios result in a less than ideal situation, and lack of full integration between the two polymer systems. The problems in formation are usually a result of the nature of the networks (i.e. large mesh sizes) and of the vastly different methods of how the polymer networks are formed. Hydrogels generally have an open structure with large mesh sizes, which is highly advantageous for transport and diffusion. While this is property is helpful when trying to load or swell the CP into the hydrogel network, it also means that when forming the CP network, there is plenty of room for the CP molecules to migrate out of the hydrogel and form a separate network with minimal overlap between the two. In addition, when the CP is formed via electrodeposition, there is a strong tendency of the CP to diffuse directly through the hydrogel (which does not assist in its polymerization) and form directly on the electrode surface.
While many researchers have focused on overcoming this issue, it is still an on-going problem that is a challenge with each new CP-hydrogel system that is studied. Therefore, careful consideration of the initial choice of polymer constituents, and meticulous planning and design of the how the CP-hydrogel is going to be formed is a key to obtaining a full IPN.
Control of biomolecule release
Despite being non-conductive, a significant benefit of a hydrogel component in a CP polymer is the ability of hydrogels to be loaded with biomolecules or drugs. Provision of appropriate biomolecules can be used to achieve a specific tissue response at the implant site, such as cell growth and survival, prevention of infection or reduction of inflammation. The water content of hydrogels allow them to carry much higher concentrations of biomolecules than a typical CPs, and hence stimulate a better biological response. An optimal material will allow controlled release of required biomolecules over the required biological time period.
A number of drug release profiles have been obtained, which demonstrate that CP-hydrogel materials can be used to deliver biomolecules and drugs to tissues [25, 108, 111]. Small et al [111] released calcon (solochrome dark blue, MW=416.38 Da) from a Polyacrylamide-PPy gel, and Barthus et al demonstrated release of safranin (MW=350.84 Da) from PPy-polyacrylamide [108]. Both of these studies showed acute release profiles across 250 and 400 min, respectively and it is likely that longer-term profiles would be required for most chronic implants.
Neurotrophins are known to promote growth and survival of neural tissue at the interface of prosthetic devices such as the cochlear implant and bionic eye; however, release of trophic agents is required for as long as possible. Extended periods of delivery are likely to result in greater cell survival and a more stable connection between the synthetic materials and biological tissue. Gillespie et al have shown that release of brain derived neurotrophic factor (BDNF) can rescue diseased spiral ganglion neurons in the deafened cochlea, but cessation of treatment can lead to tissue regression to the diseased state [157]. Shepard et al demonstrated that electrical stimulation can be used to maintain cells following completion of neurotrophin delivery over 28 days [158]. Abidian and Martin hypothesized that dexamethasone incorporated in CP-nanofibres can be released to reduce inflammatory reactions at the site of electrode implantation, but have only published release profiles on the polymer component without the CP [3]. It is important to note that they found only 60% of the dexamethasone was passively released over 5 weeks [3], indicating that polymer nanofibres may release drugs at a rate compatible for mitigating the initial inflammatory response for chronic implants.
It is important to design biofunctional CP-hydrogels with drug release profiles that are relevant to their clinical application. Selection of various components and design of appropriate structure will be integral to establishing drug release characteristics. The suitability of CP-hydrogels to drug release applications and the effect of different fabrication routes on release profiles require substantial understanding and development prior to implementation.
Conclusions
CP-hydrogel co-polymers are promising biomaterials for neural interfaces. The premise underlying the choice of these two material types as an electrode coating is that the CP will act to carry charge whereas the hydrogel will modulate the mechanical properties and enhance the drug carrying capacity and tissue interfacing of the coatings. Most of the studies conducted to date have either used hydrogel coatings layered on top of CPs or have grown CPs within hydrogels following reswelling of dried gels in a CP precursor solution. The former approach may suffer from poor interlayer adhesion and stress concentration at the interface. The latter approach, although closer to a true interpenetrating network, relies on the adhesion of the hydrogel to the electrode surface which may be problematic. A softer neural interface theoretically reduces the effect of strain mismatch at the interface, minimizing the growth of fibrous scar tissue, and thus improves the longevity of the electrode function. Mechanically, all systems incorporating a hydrogel component produce mechanical properties more closely matched to those of tissue. Although improvements in the fabrication of CP-hydrogels are still necessary to obtain an optimal material for electrode coatings, this approach holds significant promise for addressing the limitations that conducting polymer coatings present.
References
- Green R A, Lovell N H. and Poole-Warren L A. Biomaterials. 2009;30:3637. doi: 10.1016/j.biomaterials.2009.03.043. [DOI] [PubMed] [Google Scholar]
- Green R A, Lovell N H. and Poole-Warren L A. Acta Biomater. 2010;6:63. doi: 10.1016/j.actbio.2009.06.030. [DOI] [PubMed] [Google Scholar]
- Abidian M R. and Martin D C. Adv. Funct. Mater. 2009;19:573. doi: 10.1002/adfm.200801473. [DOI] [Google Scholar]
- Abidian M R. and Martin D C. Biomaterials. 2008;29:1273. doi: 10.1016/j.biomaterials.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilks S J, Richardson-Burn S M, Hendricks J L, Martin D C. and Otto K J. Front. Neuroeng. 2009;2:7. doi: 10.3389/neuro.16.007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson R T.et al2009Biomaterials 302614. 10.1016/j.biomaterials.2009.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abidian M R, Salas L G, Yazdan-Shahmorad A, Marzullo T C, Martin D C. and Kipke D R. Proc. 3rd Int. IEEE EMBS Conf. on Neural Engineering (Kohala Coast, Hawaii, USA).2007. [Google Scholar]
- Seymour J. and Kipke D R. Mater. Res. Soc. Symp. Proc. 2006;926:1. [Google Scholar]
- Polikov V S, Tresco P A. and Reichert W M. J. Neurosci. Methods. 2005;148:1. doi: 10.1016/j.jneumeth.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Kim D H, Richardson-Burns S M, Povlich L, Abidan M R, Spanninga S, Hendricks J L. and Martin D C. In: Indwelling Neural Implants: Strategies for Contending with the in vivo Environment. Reichert W M, editor. Boca Raton, FL: CRC Press; 2007. p. p 177. [PubMed] [Google Scholar]
- Green R A, Suaning G J, Poole-Warren L A. and Lovell N H. Proc. 4th Int. IEEE EMBS Conf. on Neural Engineering (Antalya, Turkey); 2009. p. p 60. [Google Scholar]
- Green R A, Lovell N H, Wallace G G. and Poole-Warren L A. Biomaterials. 2008;29:3393. doi: 10.1016/j.biomaterials.2008.04.047. [DOI] [PubMed] [Google Scholar]
- Cui X and Martin D C. 2003. Sensors Actuators A 103 384 10.1016/S0924-4247(02)00427-2 [DOI] [Google Scholar]
- Yang J. and Martin D C. J. Mater. Res. 2006;21:1124. doi: 10.1557/jmr.2006.0145. [DOI] [Google Scholar]
- Xiao Y, Cui X. and Martin D C. J. Electroanal. Chem. 2004;573:43. doi: 10.1016/j.jelechem.2004.06.024. [DOI] [Google Scholar]
- Baek S, Green R A, Lovell N H and Poole-Warren L A. 2009. Proc. 3rd Indo-Australian Conf. on Biomaterials, Implants, Tissue Engineering and Regenerative Medicine/19th Annual Australasian Society for Biomaterials and Tissue Engineering Conf. (Sydney, Australia) [Google Scholar]
- Ding J, Price W E, Ralph S F. and Wallace G G. Synth. Met. 2000;110:123. doi: 10.1016/S0379-6779(99)00277-5. [DOI] [Google Scholar]
- Richardson R T.et al2007Biomaterials 28513. 10.1016/j.biomaterials.2006.09.008 [DOI] [PubMed] [Google Scholar]
- Ludwig K A, Uram J D, Yang J. and Martin D C. J. Neural Eng. 2006;3:59. doi: 10.1088/1741-2560/3/1/007. [DOI] [PubMed] [Google Scholar]
- Thompson B C, Moulton S E, Ding J, Richardson R, Cameron A, O'Leary S, Wallace G G. and Clark G M. J. Control. Release. 2006;116:285. doi: 10.1016/j.jconrel.2006.09.004. [DOI] [PubMed] [Google Scholar]
- Evans A J, Thompson B C, Wallace G G, Millard R, O'Leary S J, Clark G M, Shepherd R K and Richardson R T. 2009. J. Biomed. Mater. Res. A 91 241. [DOI] [PubMed] [Google Scholar]
- Gus'kova O A, Khalatur P G, Bauerle P. and Khokhlov A R. Chem. Phys. Lett. 2008;461:64. doi: 10.1016/j.cplett.2008.06.058. [DOI] [Google Scholar]
- Cui X, Wiler J, Dzaman M, Altschuler R A. and Martin D C. Biomaterials. 2003;24:777. doi: 10.1016/S0142-9612(02)00415-5. [DOI] [PubMed] [Google Scholar]
- Wadhwa R, Lagenaur C F. and Cui X T. J. Control. Release. 2006;110:531. doi: 10.1016/j.jconrel.2005.10.027. [DOI] [PubMed] [Google Scholar]
- Abidian M R, Kim D H. and Martin D C. Adv. Mater. 2006;18:405. doi: 10.1002/adma.200501726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekramul Mahmud H N M, Kassim A, Zainal Z. and Wan Mahmood Mat Y. J. Appl. Polym. Sci. 2006;100:4107. doi: 10.1002/app.23327. [DOI] [Google Scholar]
- Kim D H, Abidan M. and Martin D C. J. Biomed. Mater. Res. 2004;71:577. doi: 10.1002/jbm.a.30124. [DOI] [PubMed] [Google Scholar]
- Brahim S. and Guiseppi-Elie A. Electroanalysis. 2005;17:556. doi: 10.1002/elan.200403109. [DOI] [Google Scholar]
- Christensen L, Breiting V, Janssen M, Vuust J. and Hogdall E. Aesthetic Plast. Surg. 2005;29:34. doi: 10.1007/s00266-004-0113-6. [DOI] [PubMed] [Google Scholar]
- Drury J L. and Mooney D J. Biomaterials. 2003;24:4337. doi: 10.1016/S0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- Elisseeff J, Puleo C, Yang F. and Sharma B. Orthod. Craniofac. Res. 2005;8:150. doi: 10.1111/j.1601-6343.2005.00335.x. [DOI] [PubMed] [Google Scholar]
- Peppas N A, Hilt J Z, Khademhosseini A. and Langer R. Adv. Mater. 2006;18:1345. doi: 10.1002/adma.200501612. [DOI] [Google Scholar]
- Johnson B D, Beebe D J and Crone W. 2004. Mater. Sci. Eng. C 24 575 10.1016/j.msec.2003.11.002 [DOI] [Google Scholar]
- Wang J Q. and Wu W H. Eur. Polym. J. 2005;41:1143. doi: 10.1016/j.eurpolymj.2004.11.034. [DOI] [Google Scholar]
- Martens P, Blundo J, Nilasaroya A, Odell R A, Cooper-White J. and Poole-Warren L A. Chem. Mater. 2007;19:2641. doi: 10.1021/cm0626381. [DOI] [Google Scholar]
- Schmedlen K H, Masters K S. and West J L. Biomaterials. 2002;23:4325. doi: 10.1016/S0142-9612(02)00177-1. [DOI] [PubMed] [Google Scholar]
- Lee W F. and Lin W J. J. Polym. Res.— Taiwan. 2002;9:23. doi: 10.1023/A:1020671105481. [DOI] [Google Scholar]
- Temenoff J S, Athanasiou K A, LeBaron R G. and Mikos A G. J. Biomed. Mater. Res. 2002;59:429. doi: 10.1002/jbm.1259. [DOI] [PubMed] [Google Scholar]
- Flanagan L A C A, Ju Y-E, Marg B, Osterfield M. and Janmey P A. Neuroreport. 2002;13:2411. doi: 10.1097/00001756-200212200-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach J B, Brown X Q, Jacot J G, DiMilla P A. and Wong J Y. J. Neural Eng. 2007;4:26. doi: 10.1088/1741-2560/4/2/003. [DOI] [PubMed] [Google Scholar]
- Lo C-M, Wang H-B, Dembo M. and Wang Y-L. 2000;79:144. doi: 10.1016/S0006-3495(00)76279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges P C, Miller W J, Meaney D F, Sawyer E S. and Janmey P A. Biophys. J. 2006;90:3012. doi: 10.1529/biophysj.105.073114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley C T, Thorpe S D, O'Brien F J, Robinson A J. and Kelly D J. J. Mech. Behav. Biomed. Mater. 2009;2:512. doi: 10.1016/j.jmbbm.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Teratsubo M, Tanaka Y. and Saeki S. Carbohydr. Polym. 2002;47:1. doi: 10.1016/S0144-8617(00)00338-6. [DOI] [Google Scholar]
- Willits R K. and Skornia S L. J. Biomater. Sci., Polym. Ed. 2004;15:1521. doi: 10.1163/1568562042459698. [DOI] [PubMed] [Google Scholar]
- Morrow D A, Haut Donahue T L, Odegard G M. and Kaufman K R. J. Mech. Behav. Biomed. Mater. 2010;3:124. doi: 10.1016/j.jmbbm.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahaffy R E, Shih C K, MacKintosh F C. and Käs J. Phys. Rev. Lett. 2000;85:880. doi: 10.1103/PhysRevLett.85.880. [DOI] [PubMed] [Google Scholar]
- Lu Y-B.et al2006Proc. Natl Acad. Sci. USA 10317759. 10.1073/pnas.0606150103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gefen A, Gefen N, Zhu Q, Raghupathi R. and Margulies S S. J. Neurotrauma. 2003;20:1163. doi: 10.1089/089771503770802853. [DOI] [PubMed] [Google Scholar]
- Gefen A. and Margulies S S. J. Biomech. 2004;37:1339. doi: 10.1016/j.jbiomech.2003.12.032. [DOI] [PubMed] [Google Scholar]
- Shafieian M, Darvish K K. and Stone J R. J. Biomech. 2009;42:2136. doi: 10.1016/j.jbiomech.2009.05.041. [DOI] [PubMed] [Google Scholar]
- Na L, Zhongzhou L. and Shuguang X. J. Membr. Sci. 2000;169:17. doi: 10.1016/S0376-7388(99)00327-0. [DOI] [Google Scholar]
- Li H, Liu Y C, Shu X Z, Gray S D. and Prestwich G D. Biomacromolecules. 2004;5:895. doi: 10.1021/bm034463j. [DOI] [PubMed] [Google Scholar]
- Kim D-H, Wiler J A, Anderson D J, Kipke D R. and Martin D C. Acta Biomater. 2010;6:57. doi: 10.1016/j.actbio.2009.07.034. [DOI] [PubMed] [Google Scholar]
- Winter J O, Cogan S F. and Rizzo J F. J. Biomater. Sci. Polym. Ed. 2007;18:1031. doi: 10.1163/156856207781494403. [DOI] [PubMed] [Google Scholar]
- Lu Y, Wang D, Li T, Zhao X, Cao Y, Yang H. and Duan Y Y. Biomaterials. 2009;30:4143. doi: 10.1016/j.biomaterials.2009.04.030. [DOI] [PubMed] [Google Scholar]
- Jhaveri S J, Hynd M R, Dowell-Mesfin N, Turner J N, Shain W. and Ober C K. Biomacromolecules. 2009;10:174. doi: 10.1021/bm801101e. [DOI] [PubMed] [Google Scholar]
- Dai W S. and Barbari T A. J. Membr. Sci. 1999;156:67. doi: 10.1016/S0376-7388(98)00330-5. [DOI] [Google Scholar]
- Cui X Y, Lee V A, Raphael Y, Wiler J A, Hetke J F, Anderson D J. and Martin D C. J. Biomed. Mater. Res. 2001;56:261. doi: 10.1002/1097-4636(200108)56:2<261::AID-JBM1094>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Asplund M, von Holst H. and Inganas O. Biointerphases. 2008;3:83. doi: 10.1116/1.2998407. [DOI] [PubMed] [Google Scholar]
- Collier J H, Camp J P, Hudson T W. and Schmidt C E. J. Biomed. Mater. Res. 2000;50:574. doi: 10.1002/(SICI)1097-4636(20000615)50:4<574::AID-JBM13>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Stauffer W R. and Cui X T. Biomaterials. 2006;27:2405. doi: 10.1016/j.biomaterials.2005.10.024. [DOI] [PubMed] [Google Scholar]
- Brahim S, Narinesingh D. and Guiseppi-Elie A. Biosens. Bioelectron. 2002;17:53. doi: 10.1016/S0956-5663(01)00262-7. [DOI] [PubMed] [Google Scholar]
- Asberg P. and Inganas O. Biosens. Bioelectron. 2003;19:199. doi: 10.1016/S0956-5663(03)00220-3. [DOI] [PubMed] [Google Scholar]
- Brahim S, Narinesingh D. and Guiseppi-Elie A. Biosens. Bioelectron. 2002;17:973. doi: 10.1016/S0956-5663(02)00089-1. [DOI] [PubMed] [Google Scholar]
- Brahim S, Wilson A M, Narinesingh D, Iwuoha E. and Guiseppi-Elie A. Microchim. Acta. 2003;143:123. doi: 10.1007/s00604-003-0065-6. [DOI] [Google Scholar]
- Li H C. and Khor E. Polym. Int. 1994;35:53. doi: 10.1002/pi.1994.210350105. [DOI] [Google Scholar]
- Song H K, Toste B, Ahmann K, Hoffman-Kim D. and Palmore G T R. Biomaterials. 2006;27:473. doi: 10.1016/j.biomaterials.2005.06.030. [DOI] [PubMed] [Google Scholar]
- Kim D H, Richardson-Burns S M, Hendricks J L, Sequera C. and Martin D C. Adv. Funct. Mater. 2006;17:1. [Google Scholar]
- Hodgson A J, John M J, Campbell T, Georgevich A, Woodhouse S, Aoki T, Ogata N. and Wallace G G. In: Smart Structures and Materials 1996: Smart Materials Technologies and Biomimetics. Crowson A, editor. Bellingham, WA: SPIE Optical Engineering Press; 1996. [Google Scholar]
- Richardson-Burns S M, Hendricks J L, Foster B, Povlich L K, Kim D H. and Martin D C. Biomaterials. 2007;28:1539. doi: 10.1016/j.biomaterials.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole-Warren L A, Lovell N H, Baek S. and Green R A. Expert Rev. Med. Devices. 2010;7:35. doi: 10.1586/erd.09.58. [DOI] [PubMed] [Google Scholar]
- Kamalesh S, Tan P, Wang J, Lee T, Kang E-T. and Wang C-H. J. Biomed. Mater. Res. 2000;52:467. doi: 10.1002/1097-4636(20001205)52:3<467::AID-JBM4>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Wang H J, Ji L W, Li D F and Wang J Y. 2008. J. Phys. Chem. B 112 2671 10.1021/jp0750957 [DOI] [PubMed] [Google Scholar]
- Shreyas S R. and Jessica W. Front. Neuroeng. 2009;2:6. [Google Scholar]
- Cogan S F. Annu. Rev. Biomed. Eng. 2008;10:275. doi: 10.1146/annurev.bioeng.10.061807.160518. [DOI] [PubMed] [Google Scholar]
- Guimard N K, Gomez N. and Schmidt C E. Prog. Polym. Sci. 2007;32:876. doi: 10.1016/j.progpolymsci.2007.05.012. [DOI] [Google Scholar]
- Ateh D D, Navsaria H A. and Vadgama P. J. R. Soc. Interface. 2006;3:741. doi: 10.1098/rsif.2006.0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G C. and Pickup P G. PCCP Phys. Chem. Chem. Phys. 2000;2:1255. [Google Scholar]
- Tourillon G. and Garnier F. J. Electrochem. Soc. 1983;130:2042. doi: 10.1149/1.2119517. [DOI] [Google Scholar]
- Cui X and Martin D C. 2003. Sensors Actuators B 89 92 10.1016/S0925-4005(02)00448-3 [DOI] [Google Scholar]
- Yamato H, Ohwa M. and Wernet W. J. Electroanal. Chem. 1995;397:163. doi: 10.1016/0022-0728(95)04156-8. [DOI] [Google Scholar]
- Lim F. and Sun A. Science. 1980;210:908. doi: 10.1126/science.6776628. [DOI] [PubMed] [Google Scholar]
- Stenekes R J H, Talsma H. and Hennink W E. Biomaterials. 2001;22:1891. doi: 10.1016/S0142-9612(00)00375-6. [DOI] [PubMed] [Google Scholar]
- Stauffer S R. and Peppast N A. Polymer. 1992;33:3932. doi: 10.1016/0032-3861(92)90385-A. [DOI] [Google Scholar]
- Mecke A, Dittrich C. and Meier W. Soft Matter. 2006;2:751. doi: 10.1039/b605165k. [DOI] [PubMed] [Google Scholar]
- Smart T, Lomas H, Massignani M, Flores-Merino M V, Perez L R. and Battaglia G. Nano Today. 2008;3:38. doi: 10.1016/S1748-0132(08)70043-4. [DOI] [Google Scholar]
- Van Tomme S R, Storm G. and Hennink W E. Int. J. Pharm. 2008;355:1. doi: 10.1016/j.ijpharm.2008.01.057. [DOI] [PubMed] [Google Scholar]
- Hennink W E. and van Nostrum C F. Adv. Drug Deliv. Rev. 2002;54:13. doi: 10.1016/S0169-409X(01)00240-X. [DOI] [PubMed] [Google Scholar]
- Nguyen K T. and West J L. Biomaterials. 2002;23:4307. doi: 10.1016/S0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- Mespouille L, Hedrick J L. and Dubois P. Soft Matter. 2009;5:4878. doi: 10.1039/b910041p. [DOI] [Google Scholar]
- Martens P. and Anseth K S. Polymer. 2000;41:7715. doi: 10.1016/S0032-3861(00)00123-3. [DOI] [Google Scholar]
- Martens P, Holland T. and Anseth K S. Polymer. 2002;43:6093. doi: 10.1016/S0032-3861(02)00561-X. [DOI] [Google Scholar]
- van Dijk-Wolthuis W N E, Hoogeboom J A M, van Steenbergen M J, Tsang S K Y. and Hennink W E. Macromolecules. 1997;30:4639. doi: 10.1021/ma9704018. [DOI] [Google Scholar]
- Franssen O, Vos O P. and Hennink W E. J. Control. Release. 1997;44:237. doi: 10.1016/S0168-3659(96)01527-1. [DOI] [Google Scholar]
- Hennink W E, Franssen O, van Dijk-Wolthuis W N E. and Talsma H. J. Control. Release. 1997;48:107. doi: 10.1016/S0168-3659(97)00047-3. [DOI] [Google Scholar]
- Draye J P, Delaey B, Van de Voorde A, Van Den Bulcke A, Bogdanov B. and Schacht E. Biomaterials. 1998;19:99. doi: 10.1016/S0142-9612(97)00164-6. [DOI] [PubMed] [Google Scholar]
- Vernon B, Tirelli N, Bachi T, Haldimann D and Hubbell J A. 2003. J. Biomed. Mater. Res. A 64 447 10.1002/jbm.a.10369 [DOI] [PubMed] [Google Scholar]
- Rydholm A E, Held N L, Benoit D S W, Bowman C N and Anseth K S. 2008. J. Biomed. Materials Res. A 86 23 10.1002/jbm.a.31526 [DOI] [PubMed] [Google Scholar]
- Ismail Y A, Chang J, Shin S R, Mane R S, Han S H. and Kim S J. J. Electrochem. Soc. 2009;156:A313. doi: 10.1149/1.3077597. [DOI] [Google Scholar]
- Ismail Y A, Shin M Y and Kim S J. 2009. Sensors Actuators B 136 438 10.1016/j.snb.2008.10.052 [DOI] [Google Scholar]
- Brahim S, Slaughter G. and Guiseppi-Elie A. Proc. SPIE. 2003;5053:1. doi: 10.1117/12.484748. [DOI] [Google Scholar]
- Alba N A, Justin G, Wadhwa R, Sun M, Sclabassi R J. and Cui X T. Mater. Res. Soc. Symp. Proc. 2008;1065:27. [Google Scholar]
- Justin G. and Guiseppi-Elie A. Biomacromolecules. 2009;10:2539. doi: 10.1021/bm900486d. [DOI] [PubMed] [Google Scholar]
- Kim D H, Abidan M. and Martin D C. Mater. Res. Soc. Symp. Proc. 2004;1:F5.5.1. [Google Scholar]
- Dai T, Qing X, Lu Y. and Xia Y. Polymer. 2009;50:5236. doi: 10.1016/j.polymer.2009.09.025. [DOI] [Google Scholar]
- Siddhanta S K. and Gangopadhyay R. Polymer. 2005;46:2993. doi: 10.1016/j.polymer.2005.01.084. [DOI] [Google Scholar]
- Barthus R C, Lira L M. and Torresi S I C D. J. Braz. Chem. Soc. 2008;19:630. doi: 10.1590/S0103-50532008000400004. [DOI] [Google Scholar]
- Gilmore K, Hodgson A J, Luan B, Small C J. and Wallace G G. Polym. Gels Netw. 1994;2:135. doi: 10.1016/0966-7822(94)90032-9. [DOI] [Google Scholar]
- Kim B C, Spinks G M, Wallace G G. and John R. Polymer. 2000;41:1783. doi: 10.1016/S0032-3861(99)00308-0. [DOI] [Google Scholar]
- Small C J, Too C O. and Wallace G G. Polym. Gels Netw. 1997;5:251. doi: 10.1016/S0966-7822(96)00044-5. [DOI] [Google Scholar]
- Lira L M, Barthus R C. and Cordoba De Torresi S I. Pharmacoelectrochemistry—210th ECS Meet. vol 3. Princeton, NJ: Electrochemical Society; 2007. p. p 105. [Google Scholar]
- Lira L M and Cordoba de Torresi S I. 2008. Sensors Actuators B 130 638 10.1016/j.snb.2007.10.020 [DOI] [Google Scholar]
- Lira L M. and de Torresi S I C. Electrochem. Commun. 2005;7:717. doi: 10.1016/j.elecom.2005.04.027. [DOI] [Google Scholar]
- Tang Q W, Wu J H, Sun H, Lin J M, Fan S J. and Hu D. Carbohydr. Polym. 2008;74:215. doi: 10.1016/j.carbpol.2008.02.008. [DOI] [Google Scholar]
- Aouada F A, Guilherme M R, Campese G M, Girotto E M, Rubira A F. and Muniz E C. Polym. Test. 2006;25:158. doi: 10.1016/j.polymertesting.2005.11.005. [DOI] [Google Scholar]
- Tiwari A. and Gong S Q. Electroanalysis. 2008;20:1775. doi: 10.1002/elan.200804237. [DOI] [Google Scholar]
- Abidian M R, Ludwig K A, Marzullo T C, Martin D C. and Kipke D R. Adv. Mater. 2009;21:3764. doi: 10.1002/adma.200900887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail Y A, Shin S R, Shin K M, Yoon S G, Shon K, Kim S I and Kim S J. 2008. Sensors Actuators B 129 834 10.1016/j.snb.2007.09.083 [DOI] [Google Scholar]
- Bauerdick S, Burkhardt C, Kern D P. and Nisch W. Biomed. Microdevices. 2003;5:93. doi: 10.1023/A:1024526626016. [DOI] [Google Scholar]
- Fan Y W, Cui F Z, Hou S P, Xu Q Y, Chen L N. and Lee I S. J. Neurosci. Methods. 2002;120:17. doi: 10.1016/S0165-0270(02)00181-4. [DOI] [PubMed] [Google Scholar]
- Hallab N J, Bundy K J, O'Connor K, Moses R L. and Jacobs J J. Tissue Eng. 2001;7:55. doi: 10.1089/107632700300003297. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Martin D C, Cui X. and Shenai M. Appl. Biochem. Biotechnol. 2006;128:117. doi: 10.1385/ABAB:128:2:117. [DOI] [PubMed] [Google Scholar]
- Hassan C M, Doyle F J, III, Peppas N A. Macromolecules. 1997;30:6166. doi: 10.1021/ma970117g. [DOI] [Google Scholar]
- Peppas N A. Hydrogels in Medicine and Pharmacy. Vol I–III. Boca Raton, FL: CRC Press; 1986. [Google Scholar]
- Flory P J. Principles of Polymer Chemistry. Ithaca, NY: Cornell University Press; 1953. [Google Scholar]
- Anseth K S, Bowman C N. and Brannon-Peppas L. Biomaterials. 1996;17:1647. doi: 10.1016/0142-9612(96)87644-7. [DOI] [PubMed] [Google Scholar]
- Otero T F, López Cascales J J and Vázquez Arenas G. 2007. Mater. Sci. Eng. C 27 18 10.1016/j.msec.2005.11.002 [DOI] [Google Scholar]
- Pytel R Z, Thomas E L. and Hunter I W. Polymer. 2008;49:2008. doi: 10.1016/j.polymer.2008.01.053. [DOI] [Google Scholar]
- Buckley L J, Roylance D K and Wnek G E. 1987. J. Polym. Sci. B 25 2179 10.1002/polb.1987.090251011 [DOI] [Google Scholar]
- Murray P, Spinks G M, Wallace G G. and Burford R P. Synth. Met. 1998;97:117. doi: 10.1016/S0379-6779(98)00119-2. [DOI] [Google Scholar]
- Murray P, Spinks G M, Wallace G G. and Burford R P. Synth. Met. 1997;84:847. doi: 10.1016/S0379-6779(96)04177-X. [DOI] [Google Scholar]
- Lang U. and Dual J. Mech. Behav. Mater. X. 2007;345–346:1189. [Google Scholar]
- Lang U, Naujoks N. and Dual J. Synth. Met. 2009;159:473. doi: 10.1016/j.synthmet.2008.11.005. [DOI] [Google Scholar]
- Lang U. and Dual J. POLYTRONIC 2004. 4th IEEE Int. Conf. on Polymers and Adhesives in Microelectronics and Photonics; 2004. p. p 230. [Google Scholar]
- Pomfret S J, Adams P N, Comfort N P. and Monkman A P. Adv. Mater. 1998;10:1351. doi: 10.1002/(SICI)1521-4095(199811)10:16<1351::AID-ADMA1351>3.0.CO;2-8. [DOI] [Google Scholar]
- Smela E, Lu W. and Mattes B R. Synth. Met. 2005;151:25. doi: 10.1016/j.synthmet.2005.03.009. [DOI] [Google Scholar]
- MacDiarmid A G, Min Y, Wiesinger J M, Oh E J, Scherr E M. and Epstein A J. Synth. Met. 1993;55:753. doi: 10.1016/0379-6779(93)90147-O. [DOI] [Google Scholar]
- Passeri D, Bettucci A, Biagioni A, Rossi M, Alippi A, Tamburri E, Lucci M, Davoli I. and Berezina S. Ultramicroscopy. 2009;109:1417. doi: 10.1016/j.ultramic.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Schmidt C E, Shastri V R, Vacanti J P. and Langer R. Proc. Natl Acad. Sci. USA. 1997;94:8948. doi: 10.1073/pnas.94.17.8948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C W, Lee M F, Yang Y F. and Wu C C. IEEE Trans. Eng. Med. Biol. 2005;27:2619. doi: 10.1109/IEMBS.2005.1617006. [DOI] [PubMed] [Google Scholar]
- LaNasa S M. and Bryant S J. Acta Biomater. 2009;5:2929. doi: 10.1016/j.actbio.2009.05.011. [DOI] [PubMed] [Google Scholar]
- Pierschbacher M D. and Ruoslahti E. Nature. 1984;309:30. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- DeLong S A, Moon J J. and West J L. Biomaterials. 2005;26:3227. doi: 10.1016/j.biomaterials.2004.09.021. [DOI] [PubMed] [Google Scholar]
- Merrett K, Griffith C M, Deslandes Y, Pleizier G. and Sheardown H. J. Biomater. Sci. Polym. Ed. 2001;12:647. doi: 10.1163/156856201316883467. [DOI] [PubMed] [Google Scholar]
- Patel P N, Gobin A S, West J L. and Patrick C W. Tissue Eng. 2005;11:1498. doi: 10.1089/ten.2005.11.1498. [DOI] [PubMed] [Google Scholar]
- Nilasaroya A, Poole-Warren L A, Whitelock J M. and Martens P J. Biomaterials. 2008;29:4658. doi: 10.1016/j.biomaterials.2008.08.011. [DOI] [PubMed] [Google Scholar]
- Yamaguchi N. and Kiick K L. Biomacromolecules. 2005;6:1921. doi: 10.1021/bm050003+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie-Barbick J E, Moon J J. and West J L. J. Biomater. Sci. Polym. Ed. 2009;20:1763. doi: 10.1163/156856208X386381. [DOI] [PubMed] [Google Scholar]
- Seliktar D, Zisch A H, Lutolf M P, Wrana J L and Hubbell J A. 2004. J. Biomed. Mater. Res. A 68 704 10.1002/jbm.a.20091 [DOI] [PubMed] [Google Scholar]
- Li Y, Neoh K G, Cen L. and Kang E T. Langmuir. 2005;21:10702. doi: 10.1021/la0514314. [DOI] [PubMed] [Google Scholar]
- Li M, Guo Y, Wei Y, MacDiarmid A G. and Lelkes P I. Biomaterials. 2006;27:2705. doi: 10.1016/j.biomaterials.2005.11.037. [DOI] [PubMed] [Google Scholar]
- ASTM2007D3359-02: Standard Test Methods for Measuring Adhesion by Tape Test [Google Scholar]
- Green R A. Thesis University of New South Wales.2009. [Google Scholar]
- Krysinski P, Jackowska K, Mazur M. and Tagowska M. Electrochim. Acta. 2000;46:231. doi: 10.1016/S0013-4686(00)00577-6. [DOI] [Google Scholar]
- Adenier A, Cabet-Deliry E, Lalot T, Pinson J. and Podvorica F. Chem. Mater. 2002;14:4576. doi: 10.1021/cm0211397. [DOI] [Google Scholar]
- Gillespie L N, Clark G M, Bartlett P F. and Marzella P L. J. Neurosci. Res. 2003;71:785. doi: 10.1002/jnr.10542. [DOI] [PubMed] [Google Scholar]
- Shepherd R K, Coco A. and Epp S B. Hear. Res. 2008;242:100. doi: 10.1016/j.heares.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]