

Abstract
NiFe2O4@BaTiO3 multiferroic composite particles were produced by a simple hydrothermal method in two steps: preparing NiFe2O4 nanoparticles and then synthesizing core-shell nanocomposites. Multiferroic composite ceramics were sintered from these powders. X-ray diffraction, Raman scattering and energy dispersive x-ray analyses indicated that the core-shell composites with a NiFe2O4 core and BaTiO3 shell were formed in the hydrothermal environment. Different types of sharp interfaces were self-assembled owing to the minimization of direct elastic energy. The saturation magnetization of the composites linearly increased with the NiFe2O4 content while the dielectric constant decreased. A dielectric peak appeared at around 460 °C because of the oxygen vacancies in the BaTiO3 ceramics. It resulted in an enhancement of magnetic permeability in the composites, indicating magnetoelectric coupling that was also observed by direct magnetoelectric measurements.
Keywords: interface, nanocomposite, crystal growth, dielectrics, magnetic properties, hydrothermal method
1. Introduction
Multiferroic materials attract much research interest because of their fundamental physical properties and potential applications [1–6], such as magnetic probes, transducers, novel actuators and sensors. These materials simultaneously possess magnetic, electric and elastic orders and display a magnetoelectric coupling, in which the electric polarization or permittivity can be modified by an applied magnetic field, or a magnetic moment can be induced by an electric field [4–6]. However, these intriguing multiferroic properties in a single phase are observed only in a few systems, and the magnetoelectric coupling usually occurs under extreme conditions, such as very high magnetic fields and/or very low temperatures, because the ferroelectric and magnetic orders have opposite requirements to the 3d-orbital occupancy [1, 2, 7]. Alternatively, magnetoelectric composites possess relatively high magnetoelectric responses originating from the ferroelectric and magnetic phases, which individually do not exhibit multiferroic properties. These magnetoelectric responses are induced by stress mediation between the two phases according to the principles of product properties [4, 5], and therefore depend on the intrinsic properties of each phase and on the mechanical coupling between them. Materials with a high piezoelectric constant such as Pb(Zr,Ti)O3 and BaTiO3 are selected as the ferroelectric phase, whereas Terfenol-D, FeBSiC and ferrites with high magnetostrictive parameters are chosen as the magnetic phase [8–23]. It is easier to tailor the magnetoelectric properties in a composite than in the individual phases by adjusting the volume fractions and interactions among the components. Different connective structures have been tried to enhance the magnetoelectric coupling, including particulate composites [8–14], laminated composites [15–17, 24], multilayers [18, 25], nanofibers [23] and ferrite nanopillars embedded in a ferroelectric matrix [19–22, 26].
Most recently, researchers have turned their interest to magnetoelectric nanocomposites, such as films [17–19, 27] and nanoceramics [9–13, 28], because of the demand for high-density electronic components with high performance, multifunctionality, smaller size and low cost [29]. In nanoscience, surfaces and interfaces play a key role in modulating the effective material properties. Magnetoelectric nanocomposites with a well-matched interface between the magnetic and ferroelectric phases at the atomic scale have been prepared by physical deposition [19–22, 30, 31]. They are an interesting object to investigate the magnetic, ferroelectric, dielectric and magnetoelectric properties. However, it is difficult to prepare magnetoelectric nanocomposites with a well-matched interface between the two phases by wet chemical methods [4].
Some researchers have recently tried new methods of in situ preparation of composite powders to ensure a better microstructural control. Coprecipitation [8, 9], sol–gel [9, 32], gel-combustion [33] and sonochemical methods [12, 34] were employed to improve mixing of the ferroelectric and magnetic phases and obtain homogeneous microstructures. Multiferroic core-shell nanopowders appear to have a better connectivity between the phases, resulting in superior dielectric and magnetoelectric properties [10–12]. The nanocomposite powders exhibit a high chemical activity and can be densified at relatively low temperatures, which avoids the formation of foreign phases at the interface between the two phases during the sintering.
The ratio between piezoelectric and piezomagnetic phases was a main factor influencing the magnetoelectric coefficient [5, 15]. However, the composite ceramics synthesized by the direct mechanical mixing of powders of the two phases exhibited a weak magnetoelectric effect, partly because of the mechanical defects and low percolation threshold (about 18%) in the randomly mixed ferrite grains. The core-shell nanopowder appears to be a better solution to break through the percolation threshold of the magnetostrictive phase [4, 5]. Several core-shell multiferroic composites have been obtained, such as Ni0.5Zn0.5Fe2O4@BaTiO3 [10], Fe2O3@ BaTiO3 [11], BaTiO3@Fe3O4 [12], PZT@NiFe2O4 [13], BaTiO3@Fe2O3 [34] and SrTiO3@BiFeO3 [35]. The main reported magnetoelectric core-shell composites and our results are summarized in table 1. The strain caused by a core and/or shell enhances the magnetoelectric coupling because core materials are wrapped by shell materials through well-defined interfaces [12]. Since the piezoelectric component is far more electrically insulating than the magnetic one, core-shell structures are usually designed to have a ferrite core and ferroelectric shell [9, 10], preventing the ferrite particles from direct contact during sintering. It was proven rather difficult to synthesize microstructured ceramics with a perfect interface between the ferrite core and ferroelectric shell [4]. Nevertheless, a well-matched interface between ferrite and BaTiO3 was produced with a wet chemical method by carefully controlling the experimental conditions [27, 36, 37]. Oxygen vacancies are the most common defects in perovskites and spinels, and are formed during sintering at high temperatures and low partial oxygen pressures [38]. Such defects appearing at the interface will act as charge traps, resulting in a high dielectric loss. Therefore, the sintering temperature is reduced for magnetoelectric core-shell nanopowders compared with the materials prepared by conventional solid-state reaction methods, which suppresses diffusion across the interface.
Table 1.
Summary of the composition and properties of the multiferroic core-shell nanocomposites.
| Core@Shell | Core-shell preserved | Synthesis method | Interface | Measured properties |
|---|---|---|---|---|
| CoFe2O4@BaTiO3 [8, 9] | Maybe | Coprecipitation/ | Grain boundary | Magnetization linearly increases |
| sol–gel | with CoFe2O4 content. | |||
| α33=1.48 mV (cm Oe)−1 | ||||
| d33=44.6 pC N−1. | ||||
| Ni0.5Zn0.5Fe2O4@BaTiO3 [10] | Maybe | Chemical | Grain boundary | Magnetization, dielectric |
| constant and | ||||
| conductivity versus temperature | ||||
| Fe2O3@BaTiO3 [11] | Yes | Colloidal chemistry/ | Physical | Low magnetization, permittivity |
| solid-state reaction | ||||
| BaTiO3@Fe2O3, | Yes | Sonochemical | Magnetoresistance, magnetization, | |
| BaTiO3@Fe3O4 [12, 34] | magnetodielectric properties; | |||
| magnetization jumps near the BaTiO3 | ||||
| structural phase transition temperatures | ||||
| CoFe2O4@Pb(Zr0.52Ti0.48)O3 [23] | Core-shell | Sol–gel + electrospinning | Physical | Magnetization, piezoresponse and |
| nanofiber | magnetoelectric properties | |||
| Pb(Zr0.52Ti0.48)O3@NiFe2O4 [13] | Yes | Solid-state reaction/ | Magnetization, α33=190 mV (cm Oe)−1 | |
| chemical | ||||
| Ferrite@perovskite [14] | Yes | Hydrothermal + annealing | Physical | Magnetization, resistivity |
| NiFe2O4@BaTiO3∗ | Yes | Two-step hydrothermal | Well matched | Magnetization, magnetodielectric and |
| magnetoelectric properties |
This manuscript.
Nickel ferrites combine a wide range of useful magnetic properties with a relatively low electrical conductivity; they exhibit low eddy current loss in alternating-current applications and have numerous applications in electric devices. BaTiO3 is one of the most useful dielectric materials in microelectronic devices because of its good chemical stability, high dielectric constant and low loss in a wide frequency range. It has a perovskite structure with lattice parameters a = 0.4006 nm and c = 0.4017 nm (powder diffraction file (PDF) No. 89-1428). Meanwhile, NiFe2O4 has a spinel structure with a lattice parameter a = 0.8337 nm (PDF No. 89-4927), approximately twice that of BaTiO3, allowing the formation of a well-matched interface with BaTiO3. In this work, we prepared NiFe2O4@BaTiO3 composite powders with well-matched interfaces by a two-step hydrothermal method, and then sintered the ferroelectric–magnetic composite ceramic. The structural and functional properties were investigated in detail.
2. Experimental procedures
2.1. Synthesis of composites
The chemical reagents used for reactions were Fe(NO3)3·9H2O, Ni(NO3)3·6H2O, Ba(NO3)2, TiCl4 and a NaOH mineralizer. All chemicals were used as received without further purification. High-quality NiFe2O4@BaTiO3 nanoparticles were synthesized by a hydrothermal method in two steps: preparing NiFe2O4 nanoparticles and then covering them with a shell.
Fe(NO3)3·9H2O and Ni(NO3)3·6H2O with a molar ratio of 2:1 were dissolved in distilled water under mechanical stirring and slowly precipitated with NaOH to a final pH of 13. Then, the brown suspension was transferred into an autoclave filled up to 4/5 of its capacity for reaction at 250 °C for 1 h under mechanical stirring. After natural cooling to room temperature, the resultant powders were washed several times with distilled water until the liquid became neutral, and NiFe2O4 nanoparticles were recovered.
TiCl4 was dissolved in ethanol and Ba(NO3)2 in distilled water under mechanical stirring, and the solutions were mixed together with a molar ratio of 1:1.8. NaOH solution was slowly added to coprecipitate metal ions and adjust the pH to 13. Then, the suspension together with the NiFe2O4 nanoparticles was transferred into the autoclave filled up to 4/5 of its capacity. The molar contents of NiFe2O4 in the composites were set to 10, 20, 30, 40 and 50%, and the corresponding samples were named as C10, C20, C30, C40 and C50, respectively. The hydrothermal treatment was performed at pH = 13 and 300 °C for 2 h under mechanical stirring to avoid NiFe2O4 agglomeration. Then, the composite powders were washed several times with distilled water until the liquid became neutral. We separated the NiFe2O4@BaTiO3 nanopowders with a magnet from pure BaTiO3 to ensure the presence of NiFe2O4 in the nanocomposites. For comparison, pure BaTiO3 and NiFe2O4 powders were prepared under the same conditions.
The obtained nanocomposite powders were pressed into different shapes by applying a pressure of about 300 MPa for the electric and magnetic measurements. Tablets 1.5–2 mm in thickness and 6 mm in diameter were used for dielectric measurements. Toroids 1.5 mm in thickness, 8 mm in inner diameter and 18 mm in outer diameter were used for measuring magnetic permeability. The tablets and toroids were sintered in air at 1100 °C for 6 h to obtain xNiFe2O4-(1 − x) BaTiO3 (x = 0, 0.1, 0.2, 0.3, 0.4, 0.5 and 1) ceramics. Then, the tablets were polished to a uniform parallel surface and coated with a silver paste to form electrical contacts. Ten turns of a Cu wire were wound on the toroidal samples for measuring magnetic permeability.
2.2. Characterization
X-ray diffraction (XRD) patterns of the composite powders and ceramics were measured using a Rigaku D/Max 2550 diffractometer with a Cu Kα (λ = 1.5418 Å) source. The lattice parameters, average crystal size and phase ratios of NiFe2O4 and BaTiO3 were calculated with Jade 6.5 software. The morphology of the NiFe2O4-BaTiO3 nanopowders and ceramics was studied by scanning electron microscopy (SEM, FEI-Quanta 200) and transmission electron microscopy (TEM, F30) combined with energy dispersive spectroscopy (EDS). One of the C20 ceramic samples was polished and thinned for investigating the interface between NiFe2O4 and BaTiO3 by high-resolution TEM (HRTEM).
The dielectric properties were measured using an Agilent 4294A complex impedance analyzer in the frequency range of 103–108 Hz. Raman measurements were performed using an ALMEGA-TM dispersive Raman spectrometer and 532 nm laser excitation. Magnetic hysteresis loops were measured with an LDJ9600 vibrating sample magnetometer (VSM). Curie temperature was determined from the change in magnetic permeability upon heating at a rate of 4 °C min−1, and from the change in magnetic moment upon heating at a rate of 10 °C min−1 in a magnetic field of 500 Oe. The magnetoelectric effect was measured using a dynamic method as described in [24]. The composite pellet was poled electrically and then placed in a dc magnetic field that was varied up to 3.6 kOe. An ac magnetic field δH generated by a Helmholtz coil was superimposed on the dc magnetic field, both fields being parallel to the sample plane. The charges generated from the samples were collected by a charge amplifier. Then, the magnetoelectric coefficient was calculated as αE = δE/δH = δV/tδH = δQ/εrε0SδH, where t and S are the thickness and surface area of the sample, E and V are the electric field and voltage induced by the magnetic field, respectively, and εr is the relative permittivity.
3. Results and discussion
3.1. Crystal structure
Figure 1(a) shows the XRD patterns of the as-prepared NiFe2O4, BaTiO3 and NiFe2O4-BaTiO3 powders while figure 1(b) shows the patterns of the corresponding ceramics sintered at 1100 °C. Only NiFe2O4 and BaTiO3 phases are observed, indicating that no intermediate phase formed at the interface between the NiFe2O4 core and BaTiO3 shell either during the hydrothermal synthesis or after sintering. BaTiO3 exhibits a tetragonal structure because the (200), (211) and (202) diffractions are broader than the (111) peak. This means that BaTiO3 has a polar structure in both the powders and sintered composites. However, the absence of splitting in the (002)/(200) doublet indicates that the c/a ratio of the lattice parameters is much lower in our samples than in macroscopic single crystals and coarse-grained ceramics (c/a =1.01). This is a typical feature of nanocrystalline BaTiO3 particles and ceramics [39]. The relative intensity of the NiFe2O4 diffractions varies accordingly with the NiFe2O4 content. Figure 1(c) shows the NiFe2O4 content calculated by fitting the integrated XRD intensities; the values agree well with the nominal composition, implying that the reaction was well completed under the hydrothermal conditions.
Figure 1.

XRD patterns of (a) nanopowders prepared by the hydrothermal method and (b) the corresponding ceramics sintered at 1100 °C for 6 h; dependences on the nominal NiFe2O4 content of (c) the NiFe2O4 content derived from the XRD patterns, (d) lattice parameters and (e) the average crystal size of NiFe2O4 and BaTiO3.
The lattice parameters calculated from the XRD patterns are shown in figure 1(d). The lattice parameters do not vary with composition in the NiFe2O4 and BaTiO3 powders, and change by 1% in ceramics, which may be attributed to the strain between the two phases caused by sintering at high temperature owing to their different shrinking rates. Invariable lattice parameters imply low stress in the nanocomposites. The average crystal size of both phases obtained from the refinement of XRD patterns is plotted in figure 1(e) versus the NiFe2O4 content. The powder crystal size remains constant at about 70 nm for NiFe2O4 and 83 nm for BaTiO3, indicating that NiFe2O4 crystallites remain unchanged during the second step of the synthesis of core-shell composites. It is believed that the crystallite size is a main factor affecting the crystal structure [40]. After sintering at 1100 °C, the BaTiO3 crystal size decreases slightly, while the NiFe2O4 crystal size remains constant at low and increases at high NiFe2O4 concentrations. In the second step of the hydrothermal reaction, several BaTiO3 crystallites may epitaxially grow on one NiFe2O4 nanoparticle. At low NiFe2O4 concentrations, NiFe2O4 particles are isolated homogeneously by BaTiO3 particles that prevent the NiFe2O4 aggregation, whereas at higher concentrations of 40 and 50%, NiFe2O4 particles contact each other, resulting in the NiFe2O4 particle growth during sintering at high temperatures.
Raman spectroscopy is a highly sensitive technique of probing the local crystal structure in materials. Figure 2 shows the Raman spectra of nanocrystallites measured at room temperature for pure BaTiO3 and NiFe2O4 and their composites. There are several Raman-active modes resulting from the tetragonal phase for the as-prepared BaTiO3 nanocrystallites; particularly the bands at 516 and 710 cm−1 correspond to the E(TO) and E(LO) modes in polar BaTiO3, respectively [41]. The polar order of BaTiO3 is strongly depressed as judged from the absence of the 305 cm−1 peak, indicating symmetry with the TiO6 octahedra. The 810 cm−1 peak is specific for cubic BaTiO3 [42, 43]. Its presence in figure 2 implies that the cubic phase appears in the pure BaTiO3 particles because of their composite structure consisting of the inner tetragonal core, the cubic shell, and a gradient lattice-strain layer in between [39]. There are five Raman-active A1g + Eg + 3T2g modes at about 700, 320, 210, 330 and 560 cm−1 in the bulk NiFe2O4; however, these modes are very weak in NiFe2O4 nanoparticles [44, 45] and are not seen in figure 2. The composite exhibits very weak Raman peaks corresponding to the BaTiO3 phase. The weakness of the Raman peaks in the nanocomposites can be explained using the schematic in the top part of figure 2. The BaTiO3 particles have a uniform crystal structure and a significant size of about 300 nm, and therefore exhibit strong Raman signals. The NiFe2O4 core in the nanocomposite effectively splits the original large uniform particle into smaller fragments wherein the signals are reduced owing to the size effects.
Figure 2.
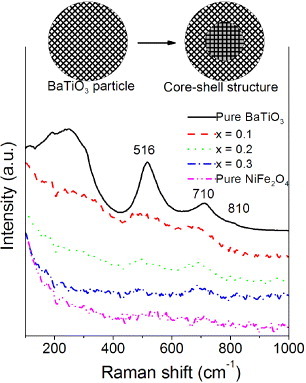
Raman spectra of BaTiO3, NiFe2O4 and their composite powders. The inset shows a schematic of BaTiO3 and core-shell particles.
3.2. Morphology and interface
Figure 3(a) shows a typical SEM image of the hydrothermally synthesized BaTiO3 nanoparticles. The particles are spherical and mostly not aggregated. They have a narrow size distribution of 200–300 nm, similar to previous reports [40, 43, 46]. Figure 3(b) shows a typical TEM image of the precursor NiFe2O4 nanoparticles. Their regular morphology was achieved at pH = 13 owing to the moderate speed of nucleation and crystal growth [47, 48]. Figures 3(c)–(g) show the SEM images of the composites with x = 0.1–0.5, which consist of spherical nanoparticles with a small grain size of 100–250 nm. Even smaller particles can be discerned in sample C50. All the grains contain NiFe2O4 particles because they were selected by a magnet. In fact, all the particles were extracted, indicating that all the BaTiO3 crystallites have grown on the NiFe2O4 particles. BaTiO3 crystallites on NiFe2O4 cannot grow larger than the particles shown in figure 3(a) owing to the lattice mismatch between the two phases. We have investigated the spatial distribution of the two phases on the sample C20 (figure 3(h)). The existence of the core-shell nanostructure in this sample is well supported by the EDS analysis (figure 3(i)): Fe, Ni, Ba and Ti elements are detected in the central part whereas Ba and Ti dominate the outer region. This result confirms the successful preparation of composite powders with a NiFe2O4 core and BaTiO3 shell.
Figure 3.

SEM images of (a) BaTiO3 particles, (b) NiFe2O4 particles and (c–g) C10–C50 nanocomposites; (h) TEM image of sample C20 and (i) EDS spectra acquired at positions 1 and 2 on a typical NiFe2O4@BaTiO3 nanoparticle.
Although perfect epitaxial orientation for the composite was obtained in thin films [19, 20, 30, 31, 37], it is difficult to observe a homogenous epitaxial relationship between NiFe2O4 and BaTiO3 in composite particles for the following reasons: (i) the irregular NiFe2O4 surfaces hinder the formation of uniform epitaxial BaTiO3; (ii) the BaTiO3 crystallites grow only in one dimension for the films, whereas they grow along any direction out of the NiFe2O4 particle surface in our experiment, resulting in a NiFe2O4–BaTiO3–BaTiO3 triple junction as shown in figure 4(c); (iii) if there exists an epitaxial BaTiO3 on a NiFe2O4 particle in the composite, it is hard to prove that by HRTEM because of the difficulty of thinning the composite ceramic along a low-index direction. Nevertheless, sharp interfaces are observed in figure 4, confirming the absence of impurity phases and defects at the NiFe2O4–BaTiO3 interfaces.
Figure 4.

Typical HRTEM images of interfaces in sample C20: (a, b) two different types of interface and (c) is a triple NiFe2O4–BaTiO3–BaTiO3 interface.
The interface between two phases was characterized by HRTEM in sample C20, which was thinned to several tens of nanometers. Figure 4 shows three typical cross-sectional HRTEM images at the interface. The sharp interfaces between NiFe2O4 and BaTiO3, other than a simple connection through van der Waals interaction [9–13, 28, 35], were obtained via the simple precursor hydrothermal reaction. The low mutual solid solubility between BaTiO3 and NiFe2O4 is an important factor for obtaining clean interfaces. The hydrothermal reaction commonly involves two stages, i.e. nucleation and crystalline growth. At the second step, BaTiO3 tends to grow on NiFe2O4 nanoparticles because this allows the nucleation energy to be saved. The similar lattice parameters of BaTiO3 and NiFe2O4 allow direct deposition of BaTiO3 on NiFe2O4 forming well-matched interfaces. Figures 4(a)–(c) present three typical cross-sectional HRTEM images at different locations of the interface. Figure 4(a) shows the NiFe2O4 (220) and BaTiO3 (110) crystal planes that have similar interplanar distances. Their angles with the interface are, however, different, resulting in a 2% misfit. The lattice mismatch energy at the interface is relaxed through the formation of an asymmetrical interface rather than the dislocations [20, 22, 26, 49]. Figure 4(b) shows the crystal planes of NiFe2O4 (111) and BaTiO3 (110), which have very different interplanar distances. The interface strain energy was relaxed by the planes marked with white lines, which have different densities at two sides of the interface. Figure 4(c) shows a triple interface among the NiFe2O4 (200), BaTiO3 (200) and BaTiO3 (110) crystal planes. The BaTiO3 and NiFe2O4 sides of the interface exhibit different stripe densities that allow us to reduce the lattice strain energy and form clear interfaces. BaTiO3 (200) and BaTiO3 (110) planes connect together through dislocations as indicated by the black arrows in figure 4(c). BaTiO3 (200) lattice planes traverse the dark area out of the NiFe2O4 corner, indicating a perfect self-assembled growth in the hydrothermal environment. Although it is difficult to grow epitaxial BaTiO3 on the NiFe2O4 crystallites as discussed above, sharp interfaces between NiFe2O4 and BaTiO3 with different connections were self-assembled owing to the minimization of the interface elastic energy between the two phases. The conclusion above is general and can be applied to other self-assembled nanocomposite systems.
The ceramics sintered at 1100 °C were observed by SEM. Both the BaTiO3 and NiFe2O4 grains grew after sintering as shown in figures 5(a) and (b). The composites C10, C20 and C30 [figures 5(c)–(e)] maintain a homogeneous microstructure with better mixing of the two phases because the grain growth is suppressed by the very low mutual solid solubility of the phases. The ceramics show much higher density than the materials prepared by solid-state reactions [50, 51]. However, some large crystallites 1–2 μm in size appear in the sample C40 and even larger ones in C50 as shown in figures 5(f) and (e). We analyzed their composition by EDS and found Fe, Ni, Ba, Ti and O in the small grains. However, only Fe, Ni and O elements were detected in the large grains, indicating the NiFe2O4 grain growth. The NiFe2O4 molar ratio is large in these two samples because some NiFe2O4 particles could not be coated by the BaTiO3 grains as shown in figures 3(h) and (g). These NiFe2O4 particles grew larger during the sintering process, as confirmed from the XRD results shown in figure 1(e). In the particulate composite ceramics synthesized by the direct mixing of powders of two phases, the ferrite particles have a low percolation threshold of about 18% [5, 52], over which the relative low resistance NiFe2O4 could degrade the insulating properties of the composite. In this work, the BaTiO3 shell was grown on the NiFe2O4 core to form composite particles. Some NiFe2O4 particles could not be coated at high NiFe2O4 content. Thus, in the composite the coated particles act as insulators, and the insulating properties will be retained if the concentration of remaining uncoated NiFe2O4 crystallites remains below 18%. As a result, the total NiFe2O4 content can be higher than that in the case of direct mixing, and no significant NiFe2O4 agglomeration was observed up to 40% of NiFe2O4 in this study.
Figure 5.

SEM cross-sectional images of ceramics sintered at 1100 °C: (a) BaTiO3, (b) NiFe2O4 and (c–g) C10–C50 nanocomposites; (h) and (i) show EDS spectra acquired at different positions on samples C40 and C50, respectively.
3.3. Electrical properties
We have studied the electrical and magnetic properties of the produced two-phase composites. Figure 6 shows the frequency dependences of the dielectric constant and dielectric loss in NiFe2O4, BaTiO3 and composite ceramics at room temperature. These properties remain almost unchanged below 10 MHz in BaTiO3 and composites. The composites have high dielectric constants of ∼720 for x = 0.10 and ∼530 for x = 0.20. Strong dielectric resonances appear in BaTiO3 and composite ceramics. The dielectric resonance in NiFe2O4 may occur at high frequencies [53], beyond our measurement range. The resonance frequency shifts to higher frequencies with increasing NiFe2O4 content as shown in the top-left inset in figure 6, as in magnetodielectric composites [54–56]. The resonances observed in these ceramics are due to the activation of different piezoelectric vibration modes, which originate from particulate microstructural features and random orientation of the crystallites with respect to the applied field. The presence of the magnetic phase suppresses the piezoelectric resonance [56].
Figure 6.
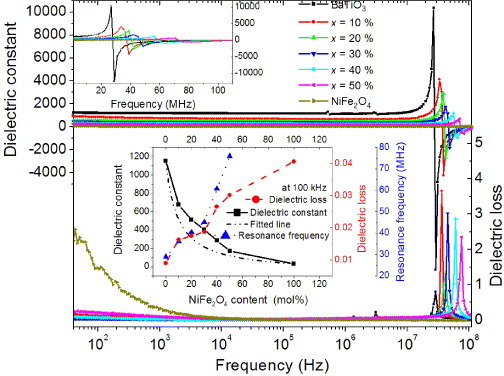
Frequency dependence of dielectric constant and dielectric loss for the samples sintered at 1100 °C. The top-left inset is a magnification of the dielectric peaks. The lower inset shows the dielectric constant, dielectric loss and resonance frequency versus NiFe2O4 content.
The dielectric constant decreases while dielectric loss increases with increasing NiFe2O4 content, as shown in the lower inset in figure 6. This tendency can be modeled using the effective medium approximation and the Maxwell–Garnett equation as follows [57, 58]:
Here ε, εN and εB are the dielectric constants of the composite, NiFe2O4 and BaTiO3, respectively, and is the volume fraction of BaTiO3. Equation (2) fits well the experimental data at 100 kHz over a wide range of compositions as shown in the lower inset in figure 6.
Figure 7 shows the variation of dielectric constant with temperature at 1 kHz, 10 kHz, 100 kHz and 1 MHz for the ceramics with x = 0–1. The dielectric properties differ for our BaTiO3 samples and BaTiO3 ceramics prepared with the solid-state reaction, in which the dielectric constant exhibits a λ-type transition near the Curie temperature of TC ∼120 °C. In contrast, the dielectric constant of BaTiO3, which was hydrothermally synthesized at 300 °C and then sintered at 1100 °C, increases slowly to a maximum at around 100 °C, followed by a decrease, indicating a phase transition. The permittivity peak at around 100 °C confirms the tetragonal structure of the sintered composites. The decrease in the transition temperature compared with that of the bulk material can be explained by the small grain size [39]. With a further increase in temperature, the electrical conductivity increases because of increasing drift mobility of thermally activated electrons. As a result, the dielectric constant and dielectric polarization increase. A new dielectric peak was observed at 460 °C and its detailed mechanism has not been clearly determined. The dielectric relaxation at high temperatures is caused by oxygen vacancies [59–61]. The short-range hopping of oxygen vacancies in the external electric field, similar to reorientation of a dipole, may result in a dielectric relaxation peak [62].
Figure 7.

Variation of dielectric constant with temperature at different frequencies.
The dielectric constant of NiFe2O4 shows a broad peak at 240–500 °C, which shifts towards high temperatures with increasing frequency. Above this peak, the dielectric constant continuously increases with temperature. These observations can be explained as follows. Electron hopping between nickel and iron ions of different chemical valences in the external field produces holes in the bulk of the samples. Then both n- and p-type carriers take part in the conduction. Increasing the temperature excites more charge carriers from their trapping centers. This in turn enhances their contribution to the polarization, resulting in an increase in the dielectric constant ε [63, 64]. As the frequency increases, the electrons cannot keep pace with the rapidly changing field, and thus, the polarization decreases. Consequently, a higher energy is required to restore polarization, which is provided by increasing the temperature. The higher the frequency, the higher the required temperature, and thus, the dielectric maximum shifts towards higher temperatures. The dielectric constant of the composites combines the characteristics of NiFe2O4 and BaTiO3 almost as predicted by the Maxwell–Garnett equation.
The increase in dielectric constant at around 600 °C is related to the magnetic phase transition in NiFe2O4. The electron hopping between Ni2+/Ni3+ and Fe2+/Fe3+ ions in NiFe2O4 causes local displacements in the direction of the applied electric field, which affects the dielectric polarization in ferrites. Such an anomaly is predicted from the thermodynamic theory of simultaneously ferroelectrically and ferromagnetically ordered materials. It can be attributed to phase transitions as an effect of vanishing magnetic order on electric order, and hence, is a signature of magnetoelectric coupling [65]. In ferrites, magnetic ordering is responsible for the change in dielectric constant with temperature [66]. The large increase in the dielectric constant near TC is observed owing to the temperature dependence of domain wall motion [67, 68]. At low temperatures, the mobility of domain walls is hindered, and hence, the dielectric response due to the extrinsic contribution is small. When the temperature is higher than TC, domains disappear after the phase transition, and the dielectric response is also small. Near the magnetic–paramagnetic transition temperature, the thermal energy is comparable to the potential barrier for domain movement; hence, the domain wall motion becomes very active, resulting in a high dielectric constant.
3.4. Magnetic properties
Figure 8(a) shows the magnetic hysteresis loops of powders and nanoceramics. The magnetic characteristics of the powders and ceramics are similar, with thin loops illustrating a soft magnetic behavior. In figure 8(b), the saturation magnetization and coercivity are plotted as functions of NiFe2O4 content. The coercivity of the magnetic nanopowders is related to both the material intrinsic properties and grain size [69]; it remains almost constant at ∼50 Oe, indicating that the NiFe2O4 nanoparticles retain their small size during the BaTiO3 hydrothermal growth. In contrast, the coercivity of the ceramics C10, C20 and C30 significantly increases to 250 Oe. Diffusion of elements may occur and enhance the interfacial interaction during high-temperature sintering. This hinders the rotation of the magnetic domain wall while the magnetic NiFe2O4 particles are magnetized or demagnetized in the external magnetic field, resulting in a high coercivity of the composite ceramics as shown in figure 8.
Figure 8.
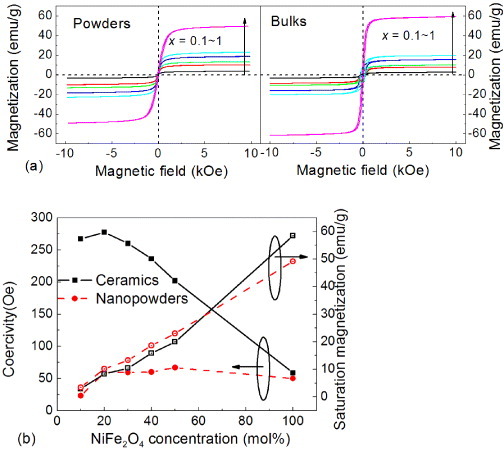
(a) Hysteresis loops of the as-prepared powders and nanocomposite ceramics sintered at 1100 °C; (b) NiFe2O4 concentration dependence of coercivity and saturation magnetization of the powders and ceramics.
The saturation magnetization of the nanopowders increases linearly with the NiFe2O4 content, suggesting that the presence of BaTiO3 phase does not affect the magnetic interaction. The lower magnetization of the composites is due to a better isolation of the magnetic phase by the nonmagnetic part in the core-shell structure, which is consistent with the XRD result shown in figure 1(e). The saturation magnetization of the NiFe2O4 ceramic is higher than that of the nanopowders, which results from the larger grain size or surface effects [69] after sintering at high temperature as shown in figures 3(b) and 5(b). The saturation magnetization is smaller in the composite ceramics than in the nanopowder, which is attributed to the elemental diffusion at the interface during high-temperature sintering. Therefore, the presence of a nonmagnetic perovskite phase and the interface effects do not influence the magnetic properties in the composite powders but slightly reduce the saturation magnetization and enhance the coercivity in the composite ceramics. Although the sharp interfaces between NiFe2O4 and BaTiO3 were obtained, the elemental diffusion between them is difficult to avoid during high-temperature sintering.
3.5. Magnetoelectric coupling
The temperature dependence of magnetic permeability is a convenient indicator of a phase transition. Figure 9(a) shows the temperature dependence of magnetic permeability measured at 1 MHz. The magnetic permeability falls abruptly when the magnetic state of the ferrite changes from ferromagnetic to paramagnetic around 550 °C. Physically, the thermal agitation is so violent that it reduces the alignment of the magnetic moment along a given axis to zero at TC. The Curie temperature increases with the NiFe2O4 content reaching 565 °C for pure NiFe2O4, which was confirmed by the magnetic moment dependence on temperature as shown in figure 9(b). The decrease in the Curie temperature of the composite is attributed to the diffusion of nonmagnetic elements into the spinel lattice. The nonmagnetic elements weaken the A–B superexchange interaction as a result of the distance increase between the magnetic moments in the A and B sites in the spinel structure. The weaker superexchange interaction is more affected by the thermal motion, resulting in a decrease in the Curie temperature of the composite ceramics. Just below the Curie temperature, magnetic permeability μi increases with temperature to a maximum value because it relates as  to the crystal anisotropy K1, which decreases with temperature (MS is saturation magnetization). Since the temperature dependence is stronger for the crystal anisotropy than magnetization, at the paramagnetic phase transition, the magnetic permeability approaches infinity just below TC and then decreases [70]. This feature is called the Hopkinson peak.
to the crystal anisotropy K1, which decreases with temperature (MS is saturation magnetization). Since the temperature dependence is stronger for the crystal anisotropy than magnetization, at the paramagnetic phase transition, the magnetic permeability approaches infinity just below TC and then decreases [70]. This feature is called the Hopkinson peak.
Figure 9.
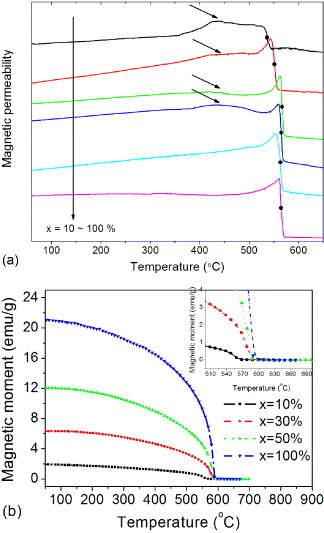
Temperature dependences of (a) magnetic permeability measured at 1 MHz, (b) magnetic moment at 500 Oe for the samples sintered at 1100 °C.
Another broad peak between 420 and 470 °C is observed in the composites C20–C50 having a high BaTiO3 content. This peak does not appear in pure NiFe2O4 and the low-BaTiO3 composite C10. We attribute it to the dielectric change of BaTiO3 shown in figure 7(a). Phase transitions in BaTiO3 could lead to the jump in the magnetization observed in magnetoelectric composites [12, 31, 37]. In our samples, BaTiO3 exhibits a dielectric peak at around 460 °C attributed to oxygen vacancies. Additional free electrons generated in this temperature range should increase the magnetic superexchange interaction, resulting in an enhancement of magnetic permeability.
Figure 10 shows the variation with Hbias of magnetoelectric effects for the composites at 10 kHz. The composites share similar magnetoelectric characteristics, that is, αE increases rapidly to a maximal value and then slowly decreases when the applied magnetic field is increased from zero to 3.6 kOe. The initial rise in magnetoelectric output is attributed to the enhancement of elastic interactions. The magnetoelectric peak indicates that the magnetostriction reaches saturation at a certain value of Hbias, which corresponds to the saturation magnetization in NiFe2O4. We find that the magnetoelectric output decreases with the NiFe2O4 content. A different behavior is observed in laminate composites where the magnetoelectric coefficient increases with increasing piezomagnetic component [15, 25].
Figure 10.
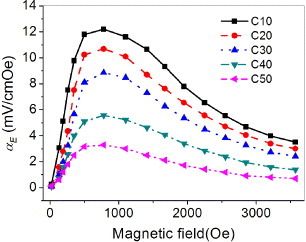
Variation of magnetoelectric coefficient αE with applied magnetic field for the composites at 10 kHz.
4. Conclusions
Core-shell ceramics exhibiting sharp interfaces between the NiFe2O4 core and BaTiO3 shell were prepared by a simple hydrothermal method, as confirmed from Raman spectra and EDS analysis. The good agreement between the measured and nominal molar contents of NiFe2O4 and BaTiO3 indicated an efficient reaction. Different types of interfaces were self-assembled owing to the minimization of direct elastic interactions between the two phases. The saturation magnetization linearly increased with the NiFe2O4 content in the composites. Their dielectric constants combined the properties of NiFe2O4 and BaTiO3 obeying the Maxwell–Garnett relationship. A peak at around 460 °C was observed in the temperature dependence of the dielectric constant and was attributed to the oxygen vacancies in BaTiO3, which also lead to an enhancement of magnetic permeability in the composites. Such an anomaly is a signature of magnetoelectric coupling, which was confirmed by magnetoelectric measurements. Core-shell nanoparticles composed of ferroelectric and ferromagnetic materials are promising candidates for possible applications of nanosized multiferroics and magnetodielectrics.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant Nos. 51172138 and 50972088) and the Fundamental Research Funds for the Central Universities (Grant No. GK201001005).
References
- Eerenstein W, Mathur N D. and Scott J F. Nature. 2006;442:759. doi: 10.1038/nature05023. [DOI] [PubMed] [Google Scholar]
- Wang K F, Liu J-M. and Ren Z F. Adv. Phys. 2009;58:321. doi: 10.1080/00018730902920554. [DOI] [Google Scholar]
- Vaz C A, Hoffman J, Ahn C H. and Ramesh R. Adv. Mater. 2010;22:2900. doi: 10.1002/adma.200904326. [DOI] [PubMed] [Google Scholar]
- Ma J, Hu J, Li Z. and Nan C-W. Adv. Mater. 2011;23:1062. doi: 10.1002/adma.201003636. [DOI] [PubMed] [Google Scholar]
- Nan C-W, Bichurin M I, Dong S, Viehland D. and Srinivasan G. J. Appl. Phys. 2008;103:045001. doi: 10.1063/1.2836410. [DOI] [Google Scholar]
- Fiebig M. J. Phys. D: Appl Phys. 2005;38:R123. doi: 10.1088/0022-3727/38/8/R01. [DOI] [Google Scholar]
- Hill N A. J. Phys. Chem. B. 2000;104:6694. doi: 10.1021/jp000114x. [DOI] [Google Scholar]
- Corral-Flores V, Bueno-Baques D, Carrillo-Flores D. and Matutes-Aquino J A. J. Appl. Phys. 2006;99:045001. doi: 10.1063/1.2165147. [DOI] [Google Scholar]
- Corral-Flores V, Bueno-Baqués D. and Ziolo R F. Acta Mater. 2010;58:764. doi: 10.1016/j.actamat.2009.09.054. [DOI] [Google Scholar]
- Curecheriu L P, Buscaglia M T, Buscaglia V, Mitoseriu L, Postolache P, Ianculescu A. and Nanni P. J. Appl. Phys. 2010;107:045001. doi: 10.1063/1.3340844. [DOI] [Google Scholar]
- Buscaglia M T, Buscaglia V, Curecheriu L, Postolache P, Mitoseriu L, Ianculescu A C, Vasile B S, Zhe Z. and Nanni P. Chem. Mater. 2010;22:4740. doi: 10.1021/cm1011982. [DOI] [Google Scholar]
- Koo Y S, Song K M, Hur N, Jung J H, Jang T H, Lee H J, Koo T Y, Jeong Y H, Cho J H. and Jo Y H. Appl. Phys. Lett. 2009;94:045001. doi: 10.1063/1.3073751. [DOI] [Google Scholar]
- Islam R A, Bedekar V, Poudyal N, Liu J P. and Priya S. J. Appl. Phys. 2008;104:045001. doi: 10.1063/1.3013437. [DOI] [Google Scholar]
- Liu R, Zhao Y, Huang R, Zhao Y. and Zhou H. J. Mater. Chem. 2010;20:10665. doi: 10.1039/c0jm02602f. [DOI] [Google Scholar]
- Zhou J-P, Zhao W, Guo Y-Y, Liu P. and Zhang H-W. J. Appl. Phys. 2009;105:045001. doi: 10.1063/1.3097769. [DOI] [Google Scholar]
- Wu T, Emmons M, Chung T-K, Sorge J. and Carman G P. J. Appl. Phys. 2010;107:045001. doi: 10.1063/1.3362919. [DOI] [Google Scholar]
- Zhou J-P, He H, Shi Z. and Nan C-W. Appl. Phys. Lett. 2006;88:045001. doi: 10.1063/1.2162262. [DOI] [Google Scholar]
- He H C, Wang J, Zhou J-P. and Nan C-W. Adv. Funct. Mater. 2007;17:1333. doi: 10.1002/(ISSN)1616-3028. [DOI] [Google Scholar]
- Zheng H. Science. 2004;303:661. doi: 10.1126/science.1094207. [DOI] [PubMed] [Google Scholar]
- Tan Z, Slutsker J. and Roytburd A L. J. Appl. Phys. 2009;105:045001. doi: 10.1063/1.3056160. [DOI] [Google Scholar]
- Zheng H, Straub F, Zhan Q, Yang P L, Hsieh W K, Zavaliche F, Chu Y H, Dahmen U. and Ramesh R. Adv. Mater. 2006;18:2747. doi: 10.1002/(ISSN)1521-4095. [DOI] [Google Scholar]
- Levin I, Li J, Slutsker J. and Roytburd A L. Adv. Mater. 2006;18:2044. doi: 10.1002/(ISSN)1521-4095. [DOI] [Google Scholar]
- Xie S, Ma F, Liu Y. and Li J. Nanoscale. 2011;3:3152. doi: 10.1039/c1nr10288e. [DOI] [PubMed] [Google Scholar]
- Zhou J-P, Meng L, Xia Z-h, Liu P. and Liu G. Appl. Phys. Lett. 2008;92:045001. doi: 10.1063/1.2841660. [DOI] [Google Scholar]
- Islam R A, Ni Y, Khachaturyan A G. and Priya S. J. Appl. Phys. 2008;104:045001. doi: 10.1063/1.2966597. [DOI] [Google Scholar]
- Zhan Q, Yu R, Crane S P, Zheng H, Kisielowski C. and Ramesh R. Appl. Phys. Lett. 2006;89:045001. doi: 10.1063/1.2364692. [DOI] [Google Scholar]
- Luo H. J. Am. Chem. Soc. 2007;129:14132. doi: 10.1021/ja075764u. [DOI] [PubMed] [Google Scholar]
- Mornet S, Elissalde C, Bidault O, Weill F, Sellier E, Nguyen O. and Maglione M. Chem. Mater. 2007;19:987. doi: 10.1021/cm0616735. [DOI] [Google Scholar]
- Yang H, Wang H, He L, Shui L. and Yao X. J. Appl. Phys. 2010;108:045001. doi: 10.1063/1.3490782. [DOI] [Google Scholar]
- Zhou J-P, Wang P-F, Qiu Z-C, Zhu G-Q. and Liu P. J. Cryst. Growth. 2008;310:508. doi: 10.1016/j.jcrysgro.2007.10.066. [DOI] [Google Scholar]
- Tian H F, Qu T L, Luo L B, Yang J J, Guo S M, Zhang H Y, Zhao Y G. and Li J Q. Appl. Phys. Lett. 2008;92:045001. doi: 10.1063/1.2844858. [DOI] [Google Scholar]
- Dandan W, Weihua G, Haijin D. and Ming L. J. Phys. D: Appl Phys. 2007;40:5002. doi: 10.1088/0022-3727/40/16/037. [DOI] [Google Scholar]
- Iordan A R, Airimioaiei M, Palamaru M N, Galassi C, Sandu A V, Ciomaga C E, Prihor F, Mitoseriu L. and Ianculescu A. J. Eur. Ceram. Soc. 2009;29:2807. doi: 10.1016/j.jeurceramsoc.2009.03.031. [DOI] [Google Scholar]
- Koo Y S, Bonaedy T, Sung K D, Jung J H, Yoon J B, Jo Y H, Jung M H, Lee H J, Koo T Y. and Jeong Y H. Appl. Phys. Lett. 2007;91:045001. doi: 10.1063/1.2817940. [DOI] [Google Scholar]
- Luo J. and Maggard P A. Adv. Mater. 2006;18:514. doi: 10.1002/(ISSN)1521-4095. [DOI] [Google Scholar]
- Ren S, Laver M. and Wuttig M. Appl. Phys. Lett. 2009;95:045001. doi: 10.1063/1.3241999. [DOI] [Google Scholar]
- Liu B, Sun T, He J. and Dravid V P. ACS Nano. 2010;4:6836. doi: 10.1021/nn101952q. [DOI] [PubMed] [Google Scholar]
- Xu J. and Itoh M. Chem. Mater. 2005;17:1711. doi: 10.1021/cm050145a. [DOI] [Google Scholar]
- Hoshina T, Wada S, Kuroiwa Y. and Tsurumi T. Appl. Phys. Lett. 2008;93:045001. doi: 10.1063/1.3027067. [DOI] [Google Scholar]
- Lee B W. and Cho S-B. J. Electroceram. 2004;13:379. doi: 10.1007/s10832-004-5129-4. [DOI] [Google Scholar]
- Shiratori Y, Pithan C, Dornseiffer J. and Waser R. J. Raman Spectrosc. 2007;38:1288. doi: 10.1002/(ISSN)1097-4555. [DOI] [Google Scholar]
- Clark I J, Takeuchi T, Ohtori N. and Sinclair D C. J. Mater. Chem. 1999;9:83. doi: 10.1039/a805756g. [DOI] [Google Scholar]
- Boulos M, Guillemet-Fritsch S, Mathieu F, Durand B, Lebey T. and Bley V. Solid State Ionics. 2005;176:1301. doi: 10.1016/j.ssi.2005.02.024. [DOI] [Google Scholar]
- Ahlawat A, Sathe V G, Reddy V R. and Gupta A. J. Magn. Magn. Mater. 2011;323:2049. doi: 10.1016/j.jmmm.2011.03.017. [DOI] [Google Scholar]
- Ahlawat A. and Sathe V G. J. Raman Spectrosc. 2011;42:1087. doi: 10.1002/jrs.2791. [DOI] [Google Scholar]
- Wei Lu S, Lee B I, Lin Wang Z. and Samuels W D. J. Cryst. Growth. 2000;219:269. doi: 10.1016/S0022-0248(00)00619-9. [DOI] [Google Scholar]
- Lv L, Bian X-B, Zhou J-P, Zhu G-Q, Liu P, Chen X-Z. and Liu Q. J. Synth. Cryst. 2010;39:977. [Google Scholar]
- Bucko M M. and Haberko K. J. Eur. Ceram. Soc. 2007;27:723. doi: 10.1016/j.jeurceramsoc.2006.04.052. [DOI] [Google Scholar]
- Slutsker J, Levin I, Li J, Artemev A. and Roytburd A L. Phys. Rev. B. 2006;73:045001. doi: 10.1103/PhysRevB.73.184127. [DOI] [Google Scholar]
- Mitoseriu L, Buscaglia V, Viviani M, Buscaglia M T, Pallecchi I, Harnagea C, Testino A, Trefiletti V, Nanni P. and Siri A S. J. Eur. Ceram. Soc. 2007;27:4379. doi: 10.1016/j.jeurceramsoc.2007.02.167. [DOI] [Google Scholar]
- Singh D, Gupta M. and Gupta R. Phys. Rev. B. 2002;65:045001. doi: 10.1103/PhysRevB.65.064432. [DOI] [Google Scholar]
- Pecharromán C. and Moya J S. Adv. Mater. 2000;12:294. doi: 10.1002/(ISSN)1521-4095. [DOI] [Google Scholar]
- Praveena K, Sadhana K, Bharadwaj S. and Murthy S R. J. Magn. Magn. Mater. 2009;321:2433. doi: 10.1016/j.jmmm.2009.02.138. [DOI] [Google Scholar]
- Shen J, Zhou J, Li B. and Li L. Mater. Lett. 2006;60:1071. doi: 10.1016/j.matlet.2005.10.076. [DOI] [Google Scholar]
- Bai Y, Zhou J, Sun Y, Li B, Yue Z, Gui Z. and Li L. Appl. Phys. Lett. 2006;89:045001. doi: 10.1063/1.2349283. [DOI] [Google Scholar]
- Ciomaga C E, Dumitru I, Mitoseriu L, Galassi C, Iordan A R, Airimioaei M. and Palamaru M N. Scr. Mater. 2010;62:610. doi: 10.1016/j.scriptamat.2010.01.005. [DOI] [Google Scholar]
- Gittleman J I. and Abeles B. Phys. Rev. B. 1977;15:3273. doi: 10.1103/PhysRevB.15.3273. [DOI] [Google Scholar]
- Ruppin R. Optics Commun. 2000;182:273. doi: 10.1016/S0030-4018(00)00825-7. [DOI] [Google Scholar]
- Patankar K K, Joshi S S. and Chougule B K. Phys. Lett. A. 2005;346:337. doi: 10.1016/j.physleta.2005.06.099. [DOI] [Google Scholar]
- Kang B S, Choi S K. and Park C H. J. Appl. Phys. 2003;94:1904. doi: 10.1063/1.1589595. [DOI] [Google Scholar]
- Yu Z, Ang C, Vilarinho P M, Mantas P Q. and Baptista J L. J. Appl. Phys. 1998;83:4874. doi: 10.1063/1.367286. [DOI] [Google Scholar]
- Ang C, Yu Z. and Cross L E. Phys. Rev. B. 2000;62:228. doi: 10.1103/PhysRevB.62.228. [DOI] [Google Scholar]
- Ahmed M A, Okasha N. and Gabal M. Mater. Chem. Phys. 2004;83:107. doi: 10.1016/j.matchemphys.2003.09.008. [DOI] [Google Scholar]
- Verma A, Thakur O P, Prakash C, Goel T C. and Mendiratta R G. Mater. Sci. Eng. B. 2005;116:1. doi: 10.1016/j.mseb.2004.08.011. [DOI] [Google Scholar]
- Kumar M. J. Appl. Phys. 2007;101:045001. doi: 10.1063/1.2404795. [DOI] [Google Scholar]
- Deverin J A. Ferroelectrics. 1978;19:9. doi: 10.1080/00150197808237831. [DOI] [Google Scholar]
- Devan R S, Deshpande S B. and Chougule B K. J. Phys. D: Appl. Phys. 2007;40:1864. doi: 10.1088/0022-3727/40/7/004. [DOI] [Google Scholar]
- Uniyal P. and Yadav K L. J. Alloys Compd. 2010;492:406. doi: 10.1016/j.jallcom.2009.10.275. [DOI] [Google Scholar]
- Lv L, Zhou J-P, Liu Q, Zhu G, Chen X-Z, Bian X-B. and Liu P. Physica E. 2011;43:1798. doi: 10.1016/j.physe.2011.06.014. [DOI] [Google Scholar]
- Hossain A K M A, Mahmud S T, Seki M, Kawai T. and Tabata H. J. Magn. Magn. Mater. 2007;312:210. doi: 10.1016/j.jmmm.2006.09.030. [DOI] [Google Scholar]