

Abstract
Magnetostructural correlations in antiperovskite manganese nitrides were investigated systematically for stoichiometric and solid solution Mn3Cu1−xAxN (A = Co, Ni, Zn, Ga, Ge, Rh, Pd, Ag, In, Sn or Sb). This class of nitrides is attracting great attention because of their giant negative thermal expansion, which is achieved by doping Ge or Sn into the A site as a relaxant of the sharp volume contraction on heating (spontaneous volume magnetostriction ωs) because of the magnetovolume effects. The physical background of large ωs and mechanism of how the volume contraction becomes gradual with temperature are central concerns for the physics and applications of these nitrides. An entire dataset of thermal expansion, crystal structure and magnetization demonstrates that the cubic triangular antiferromagnetic state is crucial for large ωs. The intimate relationship between ωs and the magnetic structure is discussed in terms of geometrical frustration related to the Mn6N octahedron and magnetic stress concept. The results presented herein also show that ωs depends on the number of d electrons in the A atom, suggesting the important role of the d orbitals of the A atom. Not all the dopants in the A site, but the elements that disturb the cubic triangular antiferromagnetic state, are effective in broadening the volume change. This fact suggests that instability neighboring the phase boundary is related to the broadening. The relation between the gradual volume change and the local structure anomaly is suggested by recent microprobe studies.
Keywords: Magnetovolume effect, Magnetism, Frustration, Antiperovskite, sNegative thermal expansion
Introduction
An antiperovskite compound with the formula M3AX (M, transitional metal; A, metal or semiconducting element and X, interstitial light element) is a treasury of functionalities. Various beneficial properties including magnetovolume effects [1, 2], magnetostriction [3, 4], magnetocaloric effects [5, 6], magnetoresistance [7, 8], low-temperature coefficient of resistance [9–11], superconductivity [12, 13] and functional mechanical properties [14, 15] have been obtained in antiperovskites. The wide diversity of physical properties of antiperovskites reflects an equally wide range of chemical-bonding interactions consisting of the d orbital in M and the p orbital in X. Intermetallic compounds containing ordered interstitial light elements, as typified by antiperovskites, form a major class of materials that have different physical backgrounds and different parameters from those of oxides. Their value as a reservoir of smart materials becomes still higher.
Among these compounds, antiperovskite manganese nitrides Mn3AN characterized by their peculiar magnetostructural correlations exhibit giant negative thermal expansion (NTE) [16–19]. The coefficient of linear thermal expansion α is over −30 ppm K−1 for Mn3AN, which is more than ten times as large as that of the NTE materials known before the discovery of this compound. This gigantic, isotropic and non-hysteretic NTE attracts great interest from practical and fundamental viewpoints. The NTE of the manganese antiperovskites was achieved by broadening the sharp volume contraction upon heating from low-temperature (low-T) large-lattice antiferromagnetic (AF) to high-T small-lattice paramagnetic (PM) phases using partial replacement of constituent elements. Such control of electronic phase change is now a key paradigm for the development of smart materials [20].
The gigantic NTE of Mn3AN strongly affected NTE research, leading to the discovery of many NTE materials such as Cd(CN)2·CCl4 [21], ReO3 [22], ScF3 [23], ZnF2 [24], SrCu3Fe4O12 [25], (Bi, La)NiO3 [26] and La(Fe, Si, Co)13 [27]. Particularly, NTE of SrCu3Fe4O12, (Bi, La)NiO3 and La(Fe, Si, Co)13 is achieved using phase control similar to Mn3AN. It is noteworthy that α reaches −80 ppm K−1 in (Bi, La)NiO3. In addition, Ca2Ru1−xCrxO4 [28] was discovered as one NTE material realized by phase control, in which ordering of orbitals as well as spins is relevant to the NTE [29].
Unresolved problems related to the giant NTE in Mn3AN are the origin of the large volume change associated with the magnetic phase transition, defined as spontaneous volume magnetostriction ωs, and the mechanism of how this volume change becomes gradual with T. They reflect the peculiar electronic states of Mn3AN and the central issues to elucidate the physics of the magnetostructural correlations in this class of nitrides. Furthermore, they are dominant factors for the practical functionalities of NTE. Therefore, understanding them is desired as a scientific basis for improvement and control of NTE properties.
This report describes systematic explorations of the magnetism and the crystal structure for stoichiometric and solid solution Mn3Cu1−xAxN (A = Co, Ni, Zn, Ga, Ge, Rh, Pd, Ag, In, Sn or Sb), including typical NTE compositions. Here, we summarize the compositional requirements for large ωs and gradual volume change with T, and discuss their physical background.
Experimental procedures
All measurements were taken using sintered polycrystalline samples [30]. First we obtained Mn3AN using a solid-state reaction. Powders of Mn2N and pure element A (purity: 99.9% or higher), in total about 2 g, were mixed in a bag filled with nitrogen gas and then sealed in a quartz tube (9 mm diameter and 20 cm long) under vacuum (< 10−3 Torr). The sealed quartz tube was heated at 500–760 °C for 40–70 h. To obtain the solid solution, we mixed powders of the stoichiometric antiperovskites in an appropriate molar ratio, pressed it into a pellet and sealed it in a quartz tube under vacuum. The final sintering was conducted at 800 °C for 60 h. The crystal structure was investigated by powder x-ray diffraction (XRD). The nitrogen deficiency δ was confirmed by the method of thermal conductivity detection of the emitted gas. Here, we define δ as the general formula Mn3AN1−δ. The results of δ measurements are presented in tables 1, 2, 4 and 5. In the text, we use the notation Mn3AN for simplicity. The term ‘stoichiometric’ means in this paper that the A site is occupied by a single element, and does not mean δ = 0.
Table 1.
Physical properties of cubic antiperovskite manganese nitrides Mn3AN1−δ showing magnetovolume effects.
| Lattice constant (Å) | Fitting parameters | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mn3AN1−δ A | 1−δ | TN (K) | Magnetic properties | 295 K | 400 K | 10 K | a0 (Å) | γ | θD (K) | ωs (10−3) 10 K |
| Co | 0.74 | 252 | 3.872 | 3.879 | 3.867 | 3.859 | 2.14 | 452.0 | 5.64 | |
| Ni | 0.87 | 256 | 3.888 | 3.893 | 3.886 | 3.875 | 2.11 | 525.9 | 8.18 | |
| Zn | 0.96 | 170 | 3.904 | 3.913 | 3.890 | 3.887 | 2.57 | 344.5 | 20.44 | |
| Ga | 0.92 | 288 | 3.888 | 3.897 | 3.898 | 3.873 | 2.54 | 429.2 | 19.10 | |
| Rh | 0.75 | 226 | 3.930 | 3.936 | 3.918 | 3.916 | 2.19 | 419.2 | 2.07 | |
| Pd | 0.89 | 316 | 3.993 | 3.997 | 3.982 | 3.977 | 2.42 | 484.2 | 3.60 | |
| Ag | 0.88 | 276 | Weak FM | 4.022 | 4.029 | 4.013 | 4.006 | 2.42 | 336.8 | 5.79 |
| In | 0.89 | 366 | Weak FM | 4.012 | 4.012 | 4.000 | 3.983 | 2.65 | 394.8 | 9.24 |
Table 2.
Physical properties of antiperovskite manganese nitrides Mn3AN1−δ without magnetovolume effects: C, cubic; T1 and T4, tetragonal and a∗ represents the cubic root of the pseudo-cubic-cell volume.
| Mn3AN1−δ | Lattice constant Å | |||||||
|---|---|---|---|---|---|---|---|---|
| A | 1−δ | T>T∗1 | T∗1 (K) | T∗1>T>T∗2 | T∗2 (K) | T∗2>T | 295 K | 10 K |
| a=3.907 | ||||||||
| Cu | 0.95 | PM, C | 143 | FM, T−1 | − | − | a=3.907 | c=3.846 |
| (a∗=3.887) | ||||||||
| a=5.431 | a=5.424 | |||||||
| Ge | 0.75 | PM, C | 600 | AF, T4 | 380 | FM, T4 | c=8.119 | c=8.035 |
| (a∗=3.912) | (a∗=3.895) | |||||||
| Sn | 0.76 | PM, C | 550 | AF, C | 45 | FM, C | a=4.061 | a=4.049 |
| a=4.183 | a=4.157 | |||||||
| Sb | 0.91 | PM, C | 360 | FM, T+1 | − | − | c=4.289 | c=4.273 |
| (a∗=4.218) | (a∗=4.195) | |||||||
Table 4.
Physical properties of Mn3Cu0.85A0.15N1−δ showing magnetovolume effects: C, cubic and T1 and T4, tetragonal.
| Mn3Cu0.85A0.15N1−δ | Lattice constant (Å) | ωs (10−3) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A | 1−δ | T>T∗1 | T∗1 (K) | T∗1>T>T∗2 | T∗2 (K) | T∗2>T | 295 K | 10 K | 10 K | |
| Co | 0.93 | C | 152 | C | − | − | 3.901 | a=3.906 | 8.39 | |
| Ni | 0.90 | C | 165 | C | 50 | C(PS) | 3.906 | a1=3.908 | a2=3.888 | 7.92 |
| Zn | 0.90 | C | 95 | T−1 | − | − | 3.903 | a=3.915 | c=3.860 | 2.66 |
| Ga | 0.91 | C | 75 | T−1 | − | − | 3.906 | a=3.905 | c=3.856 | 0.38 |
| Ge | 0.90 | C | 105 | C | − | − | 3.903 | a=3.904 | 12.34 | |
| Rh | 0.92 | C | 248 | (Not measured) | 3.915 | 7.89 | ||||
| Pd | 0.91 | C | 212 | C(PS) | − | − | 3.920 | a1=3.929 | a2=3.903 | 6.93 |
| Ag | 0.94 | C | 145 | C | 70 | T−1 | 3.922 | a=3.936 | c=3.878 | 1.88 |
| In | 0.87 | C | 100 | (Not measured) | 3.916 | 10.99 | ||||
| Sn | 0.86 | C | 95 | C | − | − | 3.917 | a=3.915 | 9.97 | |
Table 5.
Physical properties of Mn3Cu0.5A0.5N1−δ showing magnetovolume effects.
| Mn3Cu0.5A0.5N1−δ | Lattice constant (Å) | ωs (10−3) | |||||
|---|---|---|---|---|---|---|---|
| A | 1−δ | T∗ (K) | ΔT (K) | 295 K | 400 K | 10 K | 10 K |
| Co | 0.88 | 238 | 46 | 3.889 | 3.896 | 3.889 | 10.10 |
| Ni | 0.93 | 232 | 5 | 3.901 | 3.906 | 3.903 | 12.03 |
| Zn | 0.95 | 43 | <1 | 3.901 | 3.910 | 3.900 | 12.20 |
| Ga | 0.91 | 140 | <1 | 3.895 | 3.904 | 3.899 | 14.43 |
| Ge | 0.86 | 365 | 85 | 3.911 | 3.911 | 3.906 | 14.90 |
| Rh | 0.87 | 292 | 10 | 3.934 | 3.939 | 3.926 | 7.82 |
| Pd | 0.92 | 276 | 30 | 3.956 | 3.961 | 3.953 | 8.92 |
| Ag | 0.91 | 212 | 8 | 3.959 | 3.967 | 3.956 | 9.25 |
| In | 0.83 | 243 | 5 | 3.950 | 3.959 | 3.948 | 10.00 |
| Sn | 0.86 | 332 | 36 | 3.966 | 3.967 | 3.958 | 11.03 |
Linear thermal expansion ΔL(T)/L was measured using a strain gage (types KFL and KFH; Kyowa Electronic Instruments Co. Ltd) at temperatures of 4–500 K. This method is simple but requires a material having a known degree of expansion. We used copper (purity: 99.99%) and the corresponding thermal expansion data of pure copper [31, 32]. Measurements of ΔL/L were partly conducted using a laser-interference dilatometer. The T dependence of the lattice constant obtained from XRD data was reported for A = Ni [33], Cu [9], Zn [34], Ga [35] and Ag [36]. Our ΔL/L data for stoichiometric Mn3AN are in quantitative agreement with the previous XRD data presented above, thereby validating our thermal expansion measurements. Temperature-dependent magnetization M(T) was measured at 5000 Oe using a superconducting quantum interference device magnetometer.
Results
Stoichiometric Mn3AN
Results for stoichiometric Mn3AN are presented in tables 1 and 2. The previously reported crystal structure and lattice parameters of the stoichiometric Mn3AX [2, 9, 33–51] are listed in table 3. The inset of figure 2(b) shows a crystal structure of an antiperovskite M3AX.
Table 3.
Literature review of lattice constants at ambient temperature for Mn3AN. Data for carbide counterpart Mn3AC are also listed for comparison: C, cubic and T1 and T4, tetragonal.
| Mn3AN | ||||
|---|---|---|---|---|
| A | Structure | Lattice constant (Å) | References | |
| Al | C | a=3.8545 | [48] | |
| Mn | C | a=3.872 | [39] | |
| Ni | C | a=3.886 | [33] | |
| Cu | C | a=3.906 | [37] | |
| a=3.908 | [41] | |||
| a=3.905 | [9] | |||
| Zn | C | a=3.902 | [42] | |
| a=3.8951 | [34] | |||
| a=3.90082 | [49] | |||
| Ga | C | a=3.888 | [39] | |
| a=3.898 | [40] | |||
| a=3.885 | [35] | |||
| Ge | T4 | a=5.422, | c=8.134 | [41] |
| As | T4 | a=5.782, | c=8.370 | [41] |
| a=5.786, | c=8.378 | [45] | ||
| Rh | C | a=3.9280 | [38] | |
| Pd | C | a=3.9796 | [38] | |
| Ag | C | a=4.0195 | [37] | |
| a=4.025 | [36] | |||
| In | C | a=4.0291 | [50] | |
| Sn | C | a=4.0585 | [38] | |
| a=4.060 | [43] | |||
| a=4.058 | [46] | |||
| Sb | T+1 | a=4.181, | c=4.280 | [41] |
| a=4.172, | c=4.282 | [45] | ||
| Pt | C | a=3.9685 | [38] | |
| a=3.972 | [44] | |||
| Au | C | a=4.0235 | [38] | |
| Hg | C | a=4.0720 | [38] | |
| Mn3ZnC | C | a=3.930 | [2] | |
| Mn3GaC | C | a=3.896 | [39] | |
| a=3.895 | [35] | |||
| a=3.894 | [2] | |||
| Mn3SnC | C | a=3.994 | [47] | |
| a=3.989 | [2] | |||
| a=3.984 | [51] | |||
Figure 2.
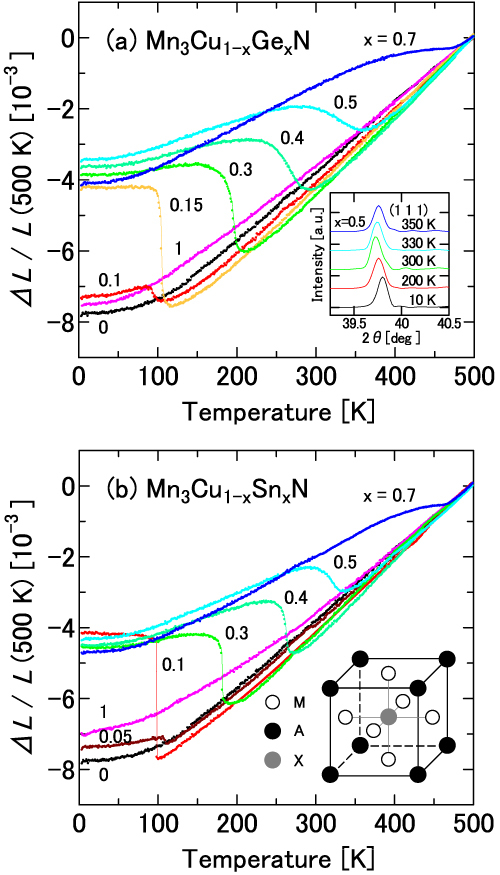
Linear thermal expansion Δ(T)/L of Mn3Cu1−xAxN: A = Ge (a) and Sn (b). The inset in (a) shows the (111) peak of XRD for x = 0.5. The inset in (b) shows the crystal structure of an antiperovskite M3AX.
Figure 1 displays ΔL/L measured for Mn3Cu1−xAxN for eight dopants A = Co, Ni, Zn, Ga, Rh, Pd, Ag and In. Each of the eight elements forms the stoichiometric Mn3AN that exhibits magnetovolume effects in the cubic AF ground state. Cadmium, which is also categorized in the same group, is omitted from the present study. Figure 2 displays ΔL/L of typical NTE compounds Mn3Cu1−xAxN with A = Ge and Sn. A comparison with the magnetization M(T) depicted in figure 3 shows that the anomaly temperature in ΔL/L, T∗, corresponds to the magnetic transition temperature (the Curie temperature TC for the ferromagnetic (FM) transition; the Neel temperature TN for the AF transition).
Figure 1.
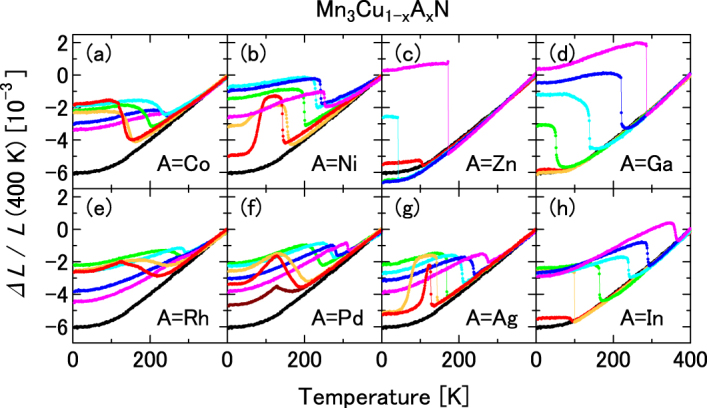
Linear thermal expansion Δ(T)/L of Mn3Cu1−xAxN for x = 0 (black), 0.1 (red), 0.15 (orange), 0.3 (green), 0.5 (light blue), 0.7 (blue) and 1 (magenta): A = Co (a), Ni (b), Zn (c), Ga (d), Rh (e), Pd (f), Ag (g) and In (h).
Figure 3.
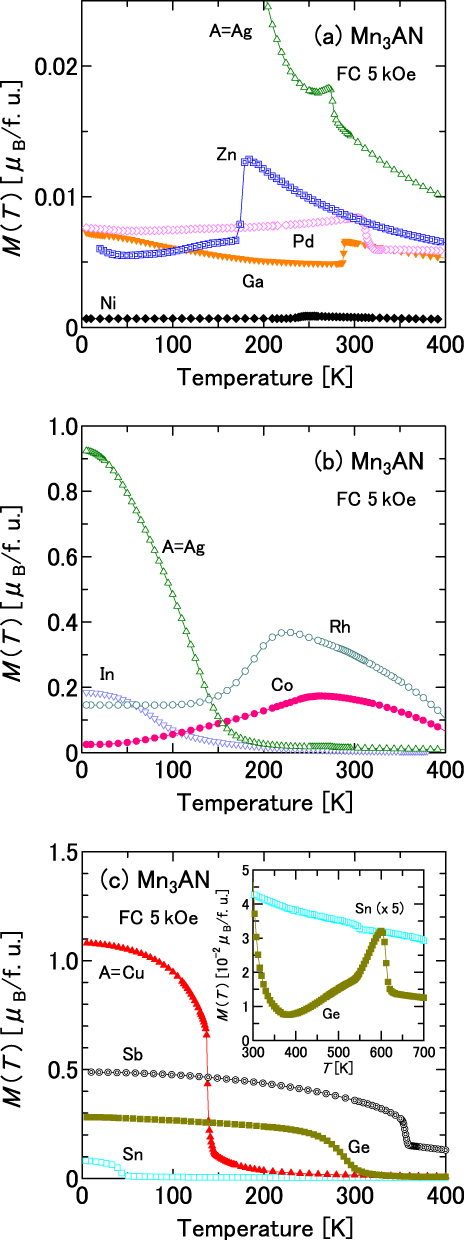
Temperature-dependent magnetization M(T) of stoichiometric Mn3AN measured during a cooling process in an applied field of 5 kOe: (a) A = Ni, Zn, Ga, Pd and Ag; (b) A = Co, Rh, Ag and In and (c) A = Cu, Ge, Sn and Sb. The inset in (c) shows magnified magnetization for A = Ge and Sn at the high-temperature region of T = 300–700 K.
The PM–FM transition temperature (T∗ = TC) of Mn3CuN is 143 K, which is the lowest one among the stoichiometric Mn3AN. Mn3CuN exhibits negligibly small magnetovolume effects (figure 4). At TC, the structural deformation occurs simultaneously from high-T cubic C to low-T tetragonal T−1 (c/a < 1) symmetry. The magnetic unit cell of the FM phase is enlarged two times within the ab plane (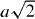 ,
, 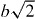 , c), containing six manganese atoms or magnetic moments; four moments are canceled out within the ab plane, whereas two moments are aligned ferromagnetically. The study of magnetostriction indicates that the magnetic easy axis is the a-axis (ab plane) [3]. A similar ferrimagnetic-like spin configuration is also realized in Mn3SbN, in which the magnetic unit cell is twice as long as the c-axis (a, b, 2c) [1], but the direction of tetragonal distortion is opposite. That is, T+1 (c/a > 1) [4].
, c), containing six manganese atoms or magnetic moments; four moments are canceled out within the ab plane, whereas two moments are aligned ferromagnetically. The study of magnetostriction indicates that the magnetic easy axis is the a-axis (ab plane) [3]. A similar ferrimagnetic-like spin configuration is also realized in Mn3SbN, in which the magnetic unit cell is twice as long as the c-axis (a, b, 2c) [1], but the direction of tetragonal distortion is opposite. That is, T+1 (c/a > 1) [4].
Figure 4.
Linear thermal expansion Δ(T)/L of Mn3CuN (open circle) and the ideal lattice thermal expansion determined by fitting analysis using equation (1) (red line). The fitting parameters are a0 = 3.891 Å, θD = 320.3 K and γ = 2.33.
The eight stoichiometric cubic Mn3AN compounds presented in figure 1, respectively, exhibit anomalies in ΔL/L at T∗ and contract upon heating. For quantitative evaluation of the magnetovolume effects, we estimate the magnetic contribution to the volume, i.e. spontaneous volume magnetostriction ωs, defined as the difference between the hypothetical phononic thermal expansion (ΔL/L)ph and the measured thermal expansion. As presented in figure 1, ΔL/L of Mn3Cu1−xAxN at the PM phase is apparently universal irrespective of x. Consequently, the hypothetical phononic expansion is determined by extrapolating this envelope using the following equation of the lattice constant a at T due to the anharmonic phonon [52]:
(ϕ, Thacher's approximate function [53]; a0, the lattice constant at absolute zero temperature; V 0, the volume of the unit cell at absolute zero temperature; r, the number of atoms in the unit cell; K, the bulk modulus; θD, the Debye temperature; γ, the Grüneisen parameter and kB, the Boltzmann constant). Because the cubic crystal structure is maintained over the T range, we assume that V =a3. Based on the previous results on the Young modulus of the antiperovskites [54], we fixed K at 130 GPa. For an antiperovskite structure, r is five. Consequently, the fitting parameters were reduced to three in the present analysis: a0, γ and θD. The obtained parameters are listed in table 1. The result for Mn3CuN is portrayed in figure 4. These fitting parameters are physically reasonable because they are comparable to those of typical transition metal-based intermetallic compounds [55–57].
For A = Ni, it might be difficult to define the envelope because ΔL/L in the PM phase varies according to x. If we estimate (ΔL/L)ph by extrapolating the measured ΔL/L at the PM phase of Mn3NiN, then we would obtain an unphysically large Debye temperature (a0 = 3.883 Å, γ = 3.23 and θD = 1441 K). However, ΔL/L at the PM phase is apparently universal for x = 0.1–0.5. We estimated (ΔL/L)ph for Mn3NiN using this common part of ΔL/L for x = 0.1–0.5, instead of the measured Mn3NiN data. In this analysis, the fitting parameters are in the reasonable range (a0 = 3.875 Å, γ = 2.11 and θD = 525.9 K). Assuming this (ΔL/L)ph, ωs does not disappear even at the PM phase for Mn3NiN. Empirically, the finite ωs originates from the rather weak T dependent ΔL/L of Mn3NiN at the PM phase. This flat ΔL/L is also the origin of an unphysically large θD in the fitting analysis of (ΔL/L)ph. The finite ωs suggests spin fluctuations at the PM phase [58].
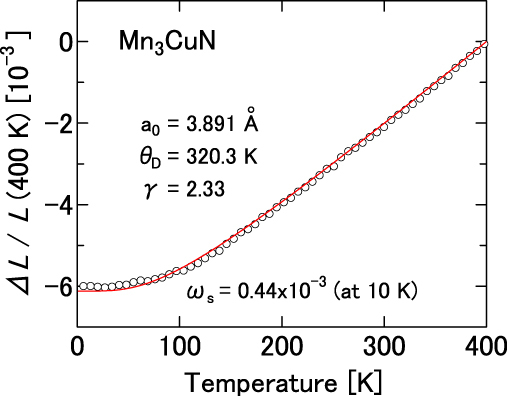
For a cubic system, ωs is defined as 3[ΔL/L − (ΔL/L)ph]. The obtained ωs values are presented in figure 5 and table 1. For Mn3GeN and Mn3SnN, we do not obtain finite values of ωs. We infer that they are zero. Furthermore, for Mn3CuN (figure 4), ωs is negligibly small (ωs is less than 0.44 × 10−3). However, slight dopants, typically x ∼ 0.1, produce large ωs. Therefore, we plot in figure 5 the value for A = Cu0.85Ge0.15 and ωs = 12.59 × 10−3, as a potential ωs of Mn3CuN (see table 4). The presented result clearly exhibits a good agreement between ωs and the number of d electrons in the A atom. ωs is larger for the 3d series than for the 4d series. It increases concomitantly with increasing atomic number in the same series. For Mn3ZnN and Mn3GaN, ωs becomes prominently large, more than twice the size of the second largest group, A = In, Ni or Ag. In previous arguments, the number of valence (outer s and p) electrons in A, nv, was regarded as the dominant factor for the electronic states, based on the clear relationship between T∗ and nv [1]. However, ωs is unrelated to T∗. Therefore, it is unrelated to nv in the presented results. In terms of the electronic structure, this fact implies that ωs increases as the d level of the A atom moves away from the Fermi level, suggesting an important role of the d level of the A atom, although it has been disregarded to date.
Figure 5.
Spontaneous volume magnetostriction ωs for stoichiometric Mn3AN at 10 K. For A = Cu, the value of Mn3Cu0.85Ge0.15N is shown as a representative value of potential ωs because Mn3CuN-based antiperovskites exhibit pronounced ωs when the cubic triangular AF state is recovered by slight dopants on the A site.
From the viewpoint of nv, the cubic AF phase showing the magnetovolume effects appears for nv = 2 and 3. When nv becomes smaller (nv = 1: A = Cu) or larger (nv = 4: A = Ge, Sn; nv = 5: A = As, Sb), the system undergoes structural deformation, possesses different magnetic structure and does not exhibit magnetovolume effects (table 2). The structural deformation is interpreted as a consequence of the band Jahn–Teller effects. The band filling varies according to nv. At both ends of small and large nv its condition is satisfied, i.e. the Fermi level approaches singularity in the density of states [59].
3.2. Solid solution Mn3Cu1−xAxN
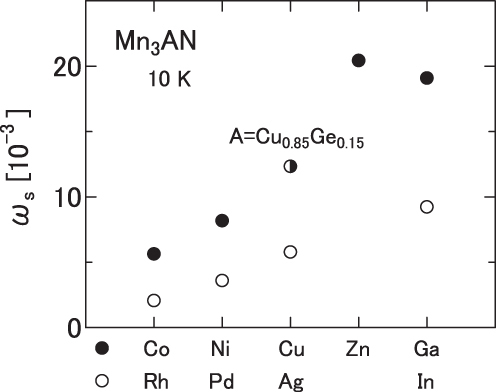
The results are presented in tables 4 (x = 0.15) and 5 (x = 0.5). The giant NTE of the antiperovskites was first discovered in the Mn3Cu1−xGexN solid solution. Surprisingly, neither end material Mn3CuN nor Mn3GeN exhibits magnetovolume effects or NTE. The drastic change in physical properties of Mn3Cu1−xGexN exemplifies the diverse physical properties of Mn3AN.
First, we examine the doping effects of two representative dopants: Ge and Sn. The linear thermal expansion and the magnetization are shown, respectively, in figures 2 and 6. Doping effects of Ge and Sn are fundamentally the same. The sequence starting from the low-doping level is the following: (i) Mn3CuN and slightly doped (x < 0.1) compounds possess the tetragonal FM ground state, (ii) cubic and AF structures are recovered and magnetovolume effects appear (x = 0.1–0.15), (iii) TN increases concomitantly with increasing x, (iv) the volume change at TN becomes broader as the doping proceeds and shows the giant NTE (x ∼ 0.5), (v) further doping decreases ωs and (vi) eventually the phase transition occurs and the crystal structure and the magnetism become the same as those of the end material Mn3GeN or Mn3SnN, without showing magnetovolume effects (x ∼ 0.9). For the other eight dopants presented in figure 1, the sequence is similar to that of the Ge- or Sn-doped compounds, except that the cubic AF state with magnetovolume effects survives the doping to the end. The change in T∗ is interpreted as a consequence of the change in nv by doping. For A = Ni and Ag, the re-entrant behavior was clearly observed for the low-doping region of x = 0.1–0.15. Upon cooling, the volume expands at the onset of the AF state and shrinks at the reentrant (figures 1(b) and (g)).
Figure 6.
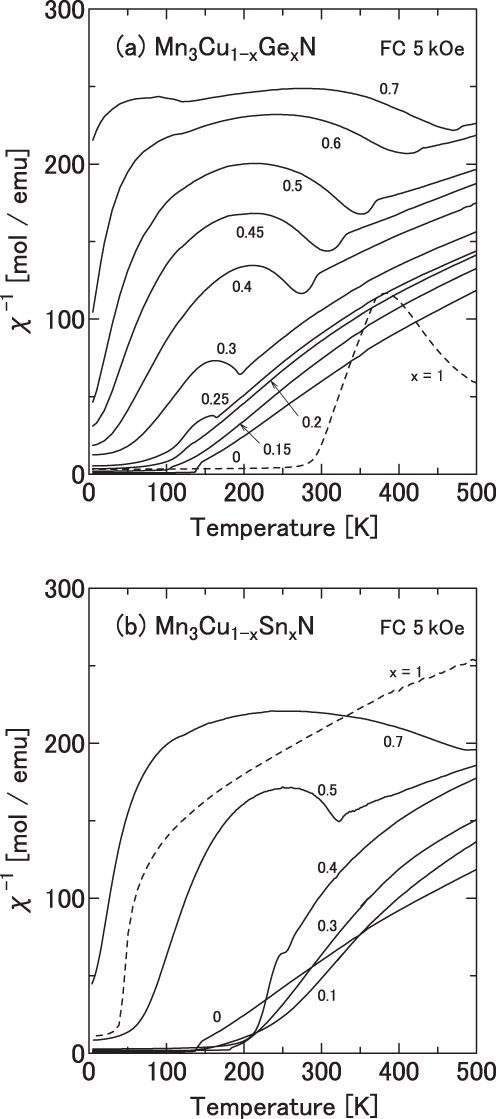
Inverse susceptibility χ−1(T) of Mn3Cu1−xAxN measured during a cooling process in an applied field of 5 kOe. A = Ge (a) and Sn (b).
Although the minimum x required for recovery of the cubic AF state varies between 0.1 and 0.3 according to the A dopant, we find a remarkable relation between the recovery of cubic structure and the appearance of magnetovolume effects. This is illustrated nicely in the series of Mn3Cu0.85A0.15N. Temperature-dependent XRD measurements were conducted for all the compounds except for A = Rh and In. Table 4 shows the crystal structure and the lattice parameter determined by the XRD measurements and ωs estimated using a procedure similar to that of the case of stoichiometric Mn3AN.
The XRD results at 10 K for Mn3Cu0.85A0.15N are classified into three categories: (i) single-phase cubic (A = Co, Ge and Sn), (ii) tetragonal T−1 (A = Zn, Ga and Ag) and (iii) phase-separated cubic (A = Ni and Pd) states. The XRD patterns are portrayed in figure 7 for A = Ge (a), Ga (b), Ag (c) and Ni (d) as representatives. Here, the contribution from the Kα2 was subtracted. For A = Ge, the (200) peak keeps a single component down to the lowest temperature, implying the cubic structure over a whole T range. For A = Ga, the (200) peak is a single component at 300 K, but it splits into two parts at 10 K. The intensity ratio of the lower-angle peak to that of the higher-angle one is 2:1. Therefore, the lower and higher peaks are assigned, respectively, to (200) and (002) in the tetragonal notation. The cubic unit cell is deformed into the tetragonal one with the shorter c-axis, namely T−1, which is the same as Mn3CuN [3, 9]. For A = Ag, the (200) peak keeps a single component down to 100 K, but it splits into two parts at 10 K similar to Mn3CuN and Mn3Cu0.85Ga0.15N, which indicates that the crystal structure is cubic in the intermediate state (70 < T < 145 K) and it is deformed into T−1 in the ground state (T < 70 K). For A = Ni, the (111) peak splits into two parts. However, this (111) peak does not split by tetragonal deformation in principle. Similar splitting occurs for all XRD peaks for A = Ni and Pd. These profiles are reasonably explained by assuming the co-existence of two cubic phases with different lattice parameters.
Figure 7.
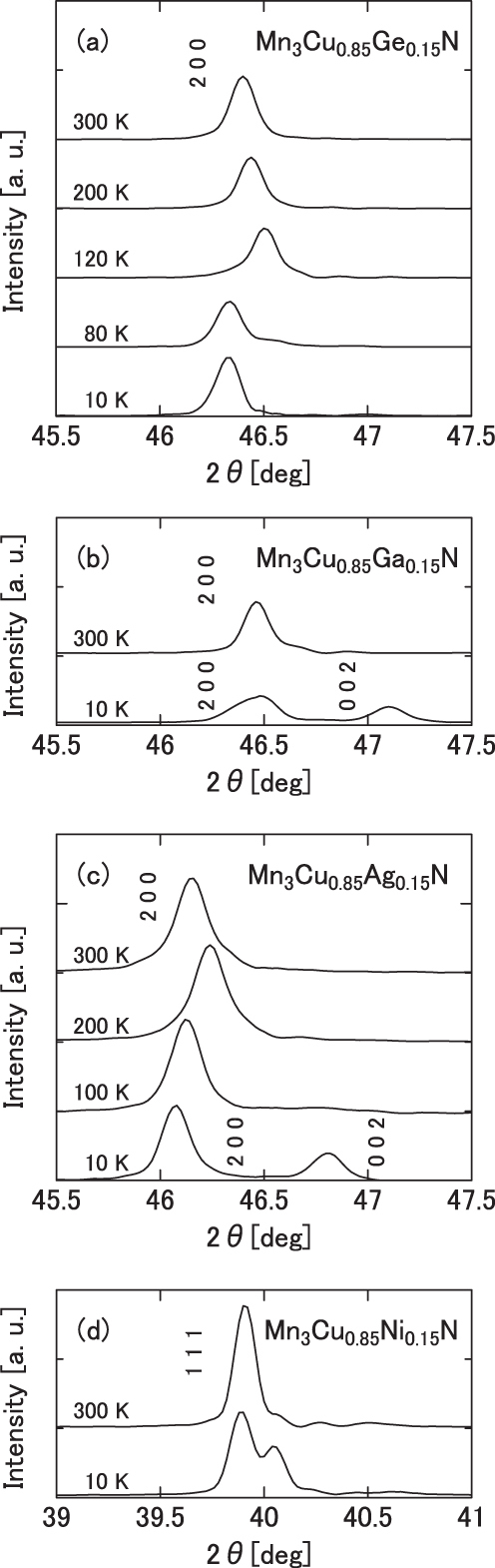
Temperature dependence of XRD profiles of Mn3Cu0.85A0.15N: A = Ge (a), Ga (b), Ag (c) and Ni (d).
Good agreement exists between the cubic crystal symmetry and the large ωs, as shown in table 4. ωs is large for (8–10) × 10−3, at largest 12.34 × 10−3, for cubic compounds including phase-separated ones, although it is at its largest 2.66 × 10−3 for A = Zn and becomes much smaller for A = Ga and Ag, where the lattice is tetragonally distorted. For A = Ag, however, ωs becomes large (10.51 × 10−3) in the cubic intermediate state (70 < T < 145 K), although it is small (1.88 × 10−3) in the T−1 ground state. The presented result on A = Ga is in agreement with the previous result [43].
Even though 15% doping is small for magnetovolume effects, such as A = Ga or Zn, further doping achieves the cubic AF state showing magnetovolume effects. This is nicely presented in the series of Mn3Cu0.5A0.5N (table 5). The experiments here reveal that all the systems listed in table 5 maintain a cubic structure over the whole T range and possess the AF ordered state. ωs is (7.82–14.90) × 10−3. An important difference is not the magnitude of volume change ωs, but the temperature window ΔT in which the volume gradually contracts on heating. The broadening of the volume change that causes NTE at room temperature does not appear for all dopants. In this study, broadening is remarkable in the order of A = Ge (ΔT = 85 K), Co (46 K), Sn (36 K) and Pd (30 K). For the other dopants, broadening is limited and the volume change remains sharp. Particularly, ΔT is less than 1 K for A = Zn and Ga. Such a sharp volume change is a characteristic feature of the end materials Mn3ZnN and Mn3GaN. The difference in atomic size between Cu and the dopant A is unrelated to the broadening.
Discussion
Physical background of magnetovolume effects
Requisites for large ωs.
From the results, we might conclude that the cubic crystal structure is a requisite for large ωs in Mn3AN. In addition, the AF state, particularly, the triangular (Γ5g and Γ4g) magnetic structure, is apparently required for large ωs. Although the dc magnetization measurements do not provide us with information related to the magnetic structure, this study shows that large ωs is incompatible with FM states. A comparison between the previous neutron diffraction study and the presented results offers direct evidence for the relation between the triangular AF structure and the large ωs.
The stoichiometric compounds, Mn3NiN, Mn3ZnN and Mn3GaN, which were confirmed to have the triangular AF configuration by neutron diffraction study [1], exhibit a large ωs in this study, 8.18 × 10−3, 20.44 × 10−3 and 19.10 × 10−3, respectively. However, for Mn3RhN, which is confirmed to have a different AF configuration by the neutron study [1], ωs is small (2.07 × 10−3). The neutron study [1] also indicates that the Γ5g AF structure is realized just below TN in Mn3AgN, but the Γ4g FM component is developed as T decreases. The small ωs of Mn3AgN at 10 K (5.79 × 10−3) might be partly explained by this mixing of the FM components.
A recent neutron study [60] revealed good agreement between the Γ5g AF structure and the appearance of ωs for the solid solution Mn3Cu1−xGexN. In addition, the Γ5g AF state with large ωs of Mn3GaN is found to be unstable against slight Fe dopants on the Mn site. Moreover, the FM state is induced at lower temperatures [61]. Impressively, the ωs disappears at the impurity-induced FM state. The detailed magnetic structure is yet to be confirmed.
An exception is Mn3SnN. An earlier neutron study [1] revealed that it possesses the cubic Γ5g AF state at 237 < T < 357 K, although no magnetovolume effects are confirmed in previous [46] studies or the present study. It must be considered that Mn3SnN easily loses nitrogen (1 − δ = 0.76 in this study). The nitrogen deficiency drastically alters the physical properties of this class of nitrides [62]. Indeed, the physical properties of Mn3SnN studied here differs from those of the previous one [1]: the former undergoes two successive transitions at 550 and 45 K (figure 3(c)), whereas the latter undergoes four transitions at 475, 357, 237 and 186 K.
These arguments enable us to predict the low-T crystal structure of Mn3Cu0.85Rh0.15N and Mn3Cu0.85In0.15N, which were not measured in this study. Rather, the large ωs, estimated as 7.89 × 10−3 and 10.99 × 10−3 for the Rh- and In-doped compounds, respectively, predicts the cubic crystal structure for these solid solutions.
Frustration.
An intimate relation between the triangular AF structure and the large ωs suggests that geometrical frustration plays a role in the magnetovolume effects. An Mn6N octahedron has three-dimensional geometrical frustration because the nearest-neighbor (Mn–Mn) magnetic interaction J1 is AF [63]. As a result, non-collinear long-range order such as Γ5g or Γ4g AF structure is stabilized, keeping the cubic structure only by a very narrow margin of energy with the assistance of the strong FM next-nearest-neighbor interaction J2 [40]. The pronounced magnetovolume effects in the cubic triangular AF phase imply that lattice expansion with cubic symmetry brings larger energy gain than removal of degeneracy by lattice deformation. An idea incorporating the frustration into the magnetovolume effects is that the strong frustration might assist lattice contraction at the PM state because the volume contraction increases the bandwidth and hence reduces the electronic energy. This is energetically favorable, although the amplitude of a magnetic moment is decreased, when the short-range magnetic ordering, or spin fluctuation, is suppressed by the strong frustration.
This study produces a result supporting the idea presented above. Figure 8 displays the x dependence of the lattice parameter deduced from XRD and thermal expansion measurements. As presented in figure 8(a), Vegard's law is not fulfilled for Mn3Cu1−xGexN. The deviation from Vegard's law at 10 K is reasonably ascribed to the large ωs. At this temperature, the lattice parameter is fairly scaled to ΔL/L. That is, it increases suddenly at x ∼ 0.1 and possesses a broad peak around x = 0.5. Then, it decreases slowly with increasing x. What is important is the deviation from Vegard's law at 500 K, at which the magnetovolume effects cannot affect the lattice parameter. The a − x curve at 500 K has a dip around x = 0.5, in contrast to that at 10 K. Similar deviation from Vegard's law is also observed for Mn3Cu1−xSnxN at 500 K (figure 8(b)), although it is less clear because the atomic size of Sn is much greater than that of Cu. The large ωs might be partly attributable to the small volume at the PM state because of the frustration. A similar effect might play an important role also for the huge ωs up to 5% in YMn2 [64], in which strong frustration is regarded as suppressing spin fluctuations and hence the volume immediately above TN. We refer to an important difference: TN of NTE manganese nitrides (∼ 300 K or higher) is much higher than that of YMn2 (∼ 100 K).
Figure 8.
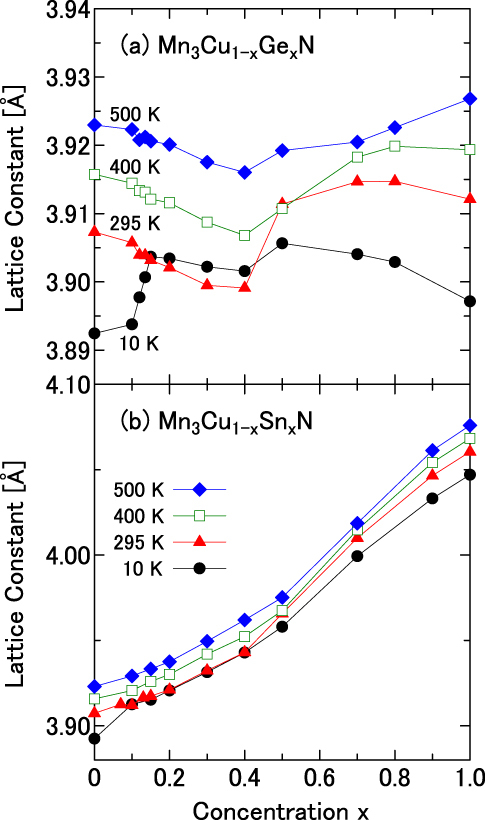
Deviation from Vegard's law in the lattice constant of solid solutions Mn3Cu1−xAxN: A = Ge (a) and Sn (b).
Magnetic structure.
The magnetovolume effect is a change in volume attributable to a variation in the amplitude of the magnetic moment m in a magnetic metal. It was first discovered in Ni–Fe Invar, showing low thermal expansion of α = 0.5 − 1 ppm K−1 below TC = 500 K [65]. It appears as a huge volume change up to 5% in YMn2 [64] as described above. A general explanation of NTE caused by the magnetovolume effect is that a larger volume favors the appearance of a magnetic moment in a metal. The electronic theory of solids provides a microscopic picture: an increase in volume suppresses the overlap of electronic orbitals and therefore reduces the electronic bandwidth. Narrowing of the bandwidth can increase the density of states ρ(ε) at Fermi energy εF, which favors magnetism. The magnetovolume effect is related directly to a longstanding problem, the origin of magnetism in a metal, and has long been regarded as a fundamental topic of physics3 [66, 67].
Magnetovolume effects are formulated in terms of itinerant-electron magnetism, or the Stoner–Edwards–Wohlfarth concept [68], ascribed to singularity in ρ(ε). In this framework, ωs is related to the amplitude of the magnetic moment m in a manner: ωs = Cmvm2/K. Here, Cmv is the magnetovolume coupling constant. In the spin fluctuation theory [69], this relationship is modified as ωs = Cmv[m2 + ξ2]/K (ξ: the amplitude of spin fluctuations). In these frameworks, the physics related to the magnetic structure would be incorporated via the coupling constant Cmv. However, magnetovolume effects related to the magnetic structure are apparently not fully discussed in the framework explained above.
In an itinerant-electron magnet, the magnetic and electronic structures are intimately related. Therefore, ωs is naively related to the magnetic structure. However, the physics of the electronic structure is too rich to incorporate only through the unique constant Cmv or singularity in εF. One attempt to take the physics omitted from the arguments presented above into consideration is the concept of magnetic stress [70]. Magnetic stress is related to the strain tensor 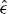 derivative of the energy exchange interaction parameter J, which determines the magnetic structure. In fact, J and its strain tensor derivative depend strongly on the origin of the magnetic interactions, and consequently on the distance between the sites. They provide us with information related to the anisotropy or strains. We can also examine volume effects by considering the trace of the stress tensor.
derivative of the energy exchange interaction parameter J, which determines the magnetic structure. In fact, J and its strain tensor derivative depend strongly on the origin of the magnetic interactions, and consequently on the distance between the sites. They provide us with information related to the anisotropy or strains. We can also examine volume effects by considering the trace of the stress tensor.
For CrN [70], the nearest-neighbor AF Cr–Cr coupling J1 is mediated by direct t2g orbital interactions. The shorter atomic distance enhances overlapping of the orbitals. Therefore, the magnitude of J1 becomes larger. In that case, the AF nearest-neighbor Cr–Cr bonds tend to decrease, although the FM nearest-neighbor Cr–Cr bonds tend to increase. This scheme explains the observed AF ordering and lattice distortion in CrN at TN to an excellent degree. Consequently, the magnetostructural correlations of the antiperovskites also might be discussed comprehensively using the magnetic-stress concept.
Mechanism of broadening in volume change
Chemical disorder is not a primary cause for broadening of the volume change. The XRD measurement of typical broadened composition Mn3Cu0.5Ge0.5N (the inset of figure 2(a)) clearly contra-indicates compositional inhomogeneity: the width of the (111) peak does not widen in the operating temperature window of NTE, compared with that above and below it. Moreover, the neutron and NMR measurements [71] reveal that the volume expands as the Γ5g AF ordered moment gradually develops. Instead, the broadening seems to result from dopants that disturb the cubic triangular AF state because Mn3GeN and Mn3SnN have different electronic states from it [72]4.
No data presented here contradict the assumption that disturbance to the cubic triangular AF state is relevant to the broadening of the volume change. Because we know the detailed magnetic structure of seven stoichiometric Mn3AN (A = Ni, Zn, Ga, Ge, Rh, Ag and Sn) [1], we first examine these seven dopants. The ground state of Mn3AN (A = Ni, Zn, Ga or Ag) was confirmed to be the cubic triangular AF state, showing remarkable magnetovolume effects. Actually, Mn3AgN is weak FM at low temperatures, but it clearly shows a peak in the M(T) curve (figure 3(a)) and TN is definable. This weak FM behavior is ascribed to the mixing of the Γ4g FM component. However, the ground state of Mn3AN (A = Ge, Rh or Sn) has different magnetic structures from the triangular AF. This study demonstrates that the former four dopants do not induce remarkable broadening of the volume change, although the latter three dopants induce broadening.
Indium also does not induce broadening. For Mn3InN, we have no information related to the magnetic structure, but this study confirmed that the crystal structure is cubic and the magnetic state is suggested to be weak FM, similar to Mn3AgN, which are consistent with a recent report [50]. Therefore, Mn3InN might have the ground state of the triangular AF structure. However, Co and Pd induce broadening of the volume change. Results of this study confirmed that Mn3CoN and Mn3PdN have a cubic structure and AF magnetic properties. Although the magnetic structures of Mn3CoN and Mn3PdN are yet to be confirmed, this study predicts that they have no triangular AF structure.
For Mn3Cu1−xGexN and Mn3Cu1−xSnxN, the cubic triangular AF states spread over a large area up to x ∼ 0.9. The volume change is gradual as T from x ∼ 0.4 to this boundary. The broadening of volume change near the phase boundary suggests that a kind of phase instability plays an important role in this broadening. It is interesting and suggestive that Ni–Fe Invar showing anomaly in thermal expansion lies near the fcc–bcc boundary in the Ni–Fe phase diagram [66]. Microscopic probes such as pair-distribution-function analysis of the neutron diffraction [73], x-ray absorption fine structure [74] and electron microscope observation [75] indicate that local structure anomaly is relevant to the broadening of the volume change in the antiperovskites. The relation to phase instability is yet to be explored.
Concluding remarks
We performed systematic explorations of the crystal structure and magnetism of stoichiometric and solid-solution Mn3Cu1−xAxN (A = Co, Ni, Zn, Ga, Ge, Rh, Pd, Ag, In, Sn or Sb), particularly addressing the origin of pronounced spontaneous volume magnetostriction ωs and the mechanism of broadening of the volume change.
An entire set of linear thermal expansion data enables us to evaluate the magnetic contribution to the thermal expansion, ωs. The results demonstrate that ωs depends highly on crystal and magnetic structures. Particularly, the cubic triangular AF state is apparently a prerequisite for large ωs. In addition, ωs was found to depend on the number of d electrons in A, suggesting an important role of the d orbitals of the A atom. Based on these results, we discussed the effects of the geometrical frustration related to the Mn6N octahedron and a concept of magnetic stress.
Not all dopants in the A site, but the elements that disturb the cubic triangular AF state, are effective in broadening the volume change. The results suggest that chemical disorder is not a primary cause for the broadening, but that a kind of instability in structural and/or magnetic states is related to the broadening. As an important issue for future study, we refer to the relation between local structure anomaly and broadening of the volume change.
Acknowledgments
The authors are grateful to H Ikuta, Y Kakehashi, T Kanomata, H Takagi, K Fukamichi, A Fujita and A Filippetti for their helpful comments. They also would like to thank D Hashizume for his assistance in the XRD study. One of the authors (KT) would like to express his appreciation to his wife Masumi for her devotion and support during the whole research period. This work was supported in part by the Ministry of Education, Culture, Sports, Science and Technology of Japan (grant number 22360291), by NEDO, Japan (grant number 08A19009d) and by the Asahi Glass Foundation.
Footnotes
For a review see [66].
Of course, also in the antiperovskites, chemical inhomogeneity can induce the coexistence of larger-volume and smaller-volume regions and be the origin of broadened volume change, which is realized in the case of a huge NTE material (Bi, La)NiO3 [26]. Nevertheless, what is important is that even without such inhomogeneity, the volume change becomes broadened in the antiperovskites.
References
- Fruchart D. and Bertaut E F. J. Phys. Soc. Japan. 1978;44:781. [Google Scholar]
- Kaneko T, Kanomata T. and Shirakawa K. J. Phys. Soc. Japan. 1987;56:4047. [Google Scholar]
- Asano K, Koyama K. and Takenaka K. Appl. Phys. Lett. 2008;92:161909. [Google Scholar]
- Takenaka K, Shibayama T, Kasugai D. and Shimizu T. Japan. J. Appl. Phys. 2012;51:043001. [Google Scholar]
- Tohei T, Wada H. and Kanomata T. J. Appl. Phys. 2003;94:1800. [Google Scholar]
- Wang B S. Europhys. Lett. 2009;85:47004. [Google Scholar]
- Kamishima K, Goto T, Nakagawa H, Miura N, Ohashi M, Mori N, Sasaki T. and Kanomata T. Phys. Rev. B. 2001;63:024426. [Google Scholar]
- Wang B S, Tong P, Sun Y P, Li L J, Tang W, Lu W J, Zhu X B, Yang Z R. and Song W H. Appl. Phys. Lett. 2009;95:222509. [Google Scholar]
- Chi E O, Kim W S. and Hur N H. Solid State Commun. 2001;120:307. [Google Scholar]
- Sun Y, Wang C, Chu L H, Wen Y C, Nie M. and Liu F S. Scr. Mater. 2010;62:686. [Google Scholar]
- Takenaka K, Ozawa A, Shibayama T, Kaneko N, Oe T. and Urano C. Appl. Phys. Lett. 2011;98:022103. [Google Scholar]
- He T. Nature. 2001;411:54. [Google Scholar]
- Uehara M, Uehara A, Kozawa K. and Kimishima Y. J. Phys. Soc. Japan. 2009;78:033702. [Google Scholar]
- Kimura Y, Sakai K, Wei F-G. and Mishima Y. Intermetallics. 2006;14:1262. [Google Scholar]
- Zhang X W, Wang X H, Li F Z. and Zhou Y C. J. Am. Ceram. Soc. 2009;92:2698. [Google Scholar]
- Takenaka K. and Takagi H. Appl. Phys. Lett. 2005;87:261902. [Google Scholar]
- Sun Y, Wang C, Wen Y C, Zhu K G. and Zhao J T. Appl. Phys. Lett. 2007;91:231913. [Google Scholar]
- Huang R J, Li L F, Cai F S, Xu X D. and Qian L H. Appl. Phys. Lett. 2008;93:081902. [Google Scholar]
- Song X Y, Sun Z G, Huang Q Z, Rettenmayr M, Liu X M, Seyring M, Li G N, Rao G G. and Yin F X. Adv. Mater. 2011;23:4690. doi: 10.1002/adma.201102552. [DOI] [PubMed] [Google Scholar]
- Takagi H. and Hwang H Y. Science. 2010;327:1601. doi: 10.1126/science.1182541. [DOI] [PubMed] [Google Scholar]
- Phillips A E, Goodwin A L, Halder G J, Southon P D. and Kepert C J. Angew. Chem. Int. Edn Engl. 2008;47:1396. doi: 10.1002/anie.200704421. [DOI] [PubMed] [Google Scholar]
- Chatterji T, Hansen T C, Brunelli M. and Henry P F. Appl. Phys. Lett. 2009;94:241902. [Google Scholar]
- Greve B K, Martin K L, Lee P L, Chupas P J, Chapman K W. and Wilkinson A P. J. Am. Chem. Soc. 2010;132:15496. doi: 10.1021/ja106711v. [DOI] [PubMed] [Google Scholar]
- Chatterji T, Zbiri W. and Hansen T C. Appl. Phys. Lett. 2011;98:181911. [Google Scholar]
- Yamada I. Angew. Chem. Int. Edn Engl. 2011;50:6579. doi: 10.1002/anie.201102228. [DOI] [PubMed] [Google Scholar]
- Azuma M. Nature Commun. 2011;2:347. doi: 10.1038/ncomms1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R J, Liu Y Y, Fan W, Tan J, Xiao F R, Qian L H. and Li L F. J. Am. Chem. Soc. 2013;135:11469. doi: 10.1021/ja405161z. [DOI] [PubMed] [Google Scholar]
- Qi T F, Korneta O B, Parkin S, De Long L E, Schlottmann P. and Cao G. Phys. Rev. Lett. 2010;105:177203. doi: 10.1103/PhysRevLett.105.177203. [DOI] [PubMed] [Google Scholar]
- Takenaka K. Sci. Technol. Adv. Mater. 2012;13:013001. doi: 10.1088/1468-6996/13/1/013001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka K. and Takagi H. Mater. Trans. 2006;47:471. [Google Scholar]
- White G K. and Collins J G. J. Low Temp. Phys. 1972;7:43. [Google Scholar]
- White G K. J. Phys. D: Appl. Phys. 1973;6:2070. [Google Scholar]
- Fruchart D, Bertaut E F, Madar R, Lorthioir G. and Fruchart R. Solid State Commun. 1971;9:1793. [Google Scholar]
- Kim W S, Chi E O, Kim J C, Hur N H, Lee K W. and Choi Y N. Phys. Rev. B. 2003;68:172402. [Google Scholar]
- l‘Heritier Ph, Boursier D, Fruchart R. and Fruchart D. Mater. Res. Bull. 1979;14:1203. [Google Scholar]
- Fruchart D. and Bertaut E F. Proc. Int. Conf. Magn. 1974;4:572. [Google Scholar]
- Samson C, Bouchaud J-P. and Fruchart R. C. R. Acad. Sci. Paris. 1964;259:392. [Google Scholar]
- Madar R, Gilles L, Rouault A, Bouchaud J-P, Fruchart E, Lorthioir G. and Fruchart R. C. R. Acad. Sci. Paris. 1967;264:308. [Google Scholar]
- Bouchaud J-P. Ann. Chim. France. 1968;3:81. [Google Scholar]
- Bertaut E F, Fruchart D, Bouchaud J-P. and Fruchart R. Solid State Commun. 1968;6:251. [Google Scholar]
- Barberon M, Madar R, Fruchart E, Lorthioir G. and Fruchart R. Mater. Res. Bull. 1970;5:1. [Google Scholar]
- Fruchart D, Bertaut E F, Madar R. and Fruchart R. J. Phys. (Paris) 1971;32:876. [Google Scholar]
- Fruchart R, Madar R, Barberon M, Fruchart E. and Lorthioir M G. J. Phys. (Paris) 1971;32:982. [Google Scholar]
- Krén E, Zsoldos É, Barberon M. and Fruchart R. Solid State Commun. 1971;9:27. [Google Scholar]
- Barberon M, Fruchart E, Fruchart R, Lorthioir G, Madar R. and Nardin M. Mater. Res. Bull. 1972;7:109. [Google Scholar]
- Fruchart D, Bertaut E F, Senateur J P. and Fruchart R. J. Phys. (Paris) 1977;38:L21. [Google Scholar]
- l‘Heritier Ph, Senateur J P, Fruchart R, Fruchart D. and Bertaut E F. Mater. Res. Bull. 1977;12:533. [Google Scholar]
- Lin J C, Wang B S, Tong P, Li W J, Zhang L, Zhu X B, Yang Z R, Song W H, Dai J M. and Sun Y P. Appl. Phys. Lett. 2011;98:092507. [Google Scholar]
- Sun Y S, Guo Y F, Wang X X, Tsujimoto Y, Matsushita Y, Shi Y G, Wang C, Belik A A. and Yamaura K. Appl. Phys. Lett. 2012;100:161907. [Google Scholar]
- Sun Y S, Guo Y F, Wang X X, Yi W, Li J J, Zhang S B, Sathish C I, Belik A A. and Yamaura K. J. Phys.: Conf. Ser. 2012;400:032094. [Google Scholar]
- Wang B S, Lu W J, Lin S, Lin J C, Tong P, Zhao B C, Song W H. and Sun Y P. J. Magn. Magn. Mater. 2012;324:773. [Google Scholar]
- Sayetat F, Fertey P. and Klessler M. J. Appl. Crystallogr. 1998;31:121. [Google Scholar]
- Thacher A C J. Chem. Phys. 1960;32:638. [Google Scholar]
- Nakamura Y, Takenaka K, Kishimoto A. and Takagi H. J. Am. Ceram. Soc. 2009;92:2999. [Google Scholar]
- Staudenmann J L, DeFacio B, Testardi L R, Werner S A, Flükiger R. and Muller J. Phys. Rev. B. 1981;24:6446. [Google Scholar]
- Siethoff H. Intermetallics. 1997;5:625. [Google Scholar]
- Mayer N, Anton H, Bott E, Methfessel M, Sticht J, Harris J. and Schmidt P C. Intermetallics. 2003;11:23. [Google Scholar]
- Moriya T. 1985. Spin Fluctuations in Itinerant Electron Magnetism (Berlin: Springer; ) [Google Scholar]
- Jardin J P. and Labbe J. J. Solid State Chem. 1983;46:275. [Google Scholar]
- Iikubo S, Kodama K, Takenaka K, Takagi H. and Shamoto S. Phys. Rev. B. 2008;77:020409. doi: 10.1103/PhysRevLett.101.205901. [DOI] [PubMed] [Google Scholar]
- Takenaka K, Inagaki T. and Takagi H. Appl. Phys. Lett. 2009;95:132508. [Google Scholar]
- Takenaka K, Shibayama T, Asano K. and Koyama K. J. Phys. Soc. Japan. 2010;79:073706. [Google Scholar]
- Tahara D, Motome Y. and Imada M. J. Phys. Soc. Japan. 2007;76:013708. [Google Scholar]
- Nakamura H, Wada H, Yoshimura K, Shiga M, Nakamura Y, Sakurai J. and Komura Y. J. Phys. F: Met. Phys. 1988;18:981. [Google Scholar]
- Hayase M, Shiga M. and Nakamura Y. J. Phys. Soc. Japan. 1973;34:925. [Google Scholar]
- Wasserman E F. 1990. Ferromagnetic Materials vol 5 Buschow K H J and Wohlfarth E P. Amsterdam: Elsevier; 237–322 [Google Scholar]
- Takahashi Y. and Nakano H. J. Phys.: Condens. Matter. 2006;18:521. [Google Scholar]
- Wohlfarth E P. Physica B&C. 1977;91:305. [Google Scholar]
- Moriya T. and Usami K. Solid State Commun. 1980;34:95. [Google Scholar]
- Filippetti A. and Hill N A. Phys. Rev. Lett. 2000;85:5166. doi: 10.1103/PhysRevLett.85.5166. [DOI] [PubMed] [Google Scholar]
- Kodama K, Iikubo S, Takenaka K, Takigawa M, Takagi H. and Shamoto S. Phys. Rev. B. 2010;81:224419. doi: 10.1103/PhysRevLett.101.205901. [DOI] [PubMed] [Google Scholar]
- Takenaka K, Asano K, Misawa M. and Takagi H. Appl. Phys. Lett. 2008;92:011927. [Google Scholar]
- Iikubo S, Kodama K, Takenaka K, Takagi H, Takigawa M. and Shamoto S. Phys. Rev. Lett. 2008;101:205901. doi: 10.1103/PhysRevLett.101.205901. [DOI] [PubMed] [Google Scholar]
- Matsuno J, Takenaka K, Takagi H, Matsumura D, Nishihata Y. and Mizuki J. Appl. Phys. Lett. 2008;94:181904. [Google Scholar]
- Mori S. and Kurushima K. private communication. [Google Scholar]