

Abstract
The structure and the toughening mechanism of nacre have been the subject of intensive research over the last 30 years. This interest originates from nacre’s excellent combination of strength, stiffness and toughness, despite its high, for a biological material, volume fraction of inorganic phase, typically 95%. Owing to the improvement of nanoscale measurement and observation techniques, significant progress has been made during the last decade in understanding the mechanical properties of nacre. The structure, microscopic deformation behavior and toughening mechanism on the order of nanometers have been investigated, and the importance of hierarchical structure in nacre has been recognized. This research has led to the fabrication of multilayer composites and films inspired by nacre with a layer thickness below 1 μm. Some of these materials reproduce the inorganic/organic interaction and hierarchical structure beyond mere morphology mimicking. In the first part of this review, we focus on the hierarchical architecture, macroscopic and microscopic deformation and fracture behavior, as well as toughening mechanisms in nacre. Then we summarize recent progress in the fabrication of materials inspired by nacre taking into consideration its mechanical properties.
Keywords: nacre, mechanical properties, toughening mechanism, multilayers, composites
Introduction
Biological structures are constructed under ambient conditions from common materials, yet they often exhibit superior performance achieved through the complex hierarchical utilization of composition and architecture. In particular, materials such as bone, shells and tooth enamel are composite structures of ceramics and organic polymers and have high strength and toughness. The design principles found in these materials present promising opportunities for the development of new nature-inspired materials [1–4].
A well-known example is nacre, which forms the iridescent inner surface of certain molluscan shells; it is commonly called the ‘mother of pearl’ and is traditionally used for ornamentation. Apart from decorative usage, the structure of nacre has been studied for many decades owing to its excellent mechanical properties. The basic structure is a two-phase composite material, with polygonal platelets of aragonite (a polymorph of CaCO3) within layers of an organic polymer matrix. In its natural environment, the main role of nacre is to keep the shell from breaking catastrophically by absorbing fracture energy during attacks by predators [1, 5]; remarkably, the work of fracture has been reported to be up to three orders of magnitude greater than the pure mineral, despite being almost 95% aragonite by volume [5].
The excellent bulk mechanical performance of nacre in seashells has been well reported in the literature and is attributed to the combination of hard minerals and soft biopolymers in the structure of nacre [6–12]. Compared with other seashells such as conch and clam shells that can have several hierarchical orders of structure [13–15], nacre has a relatively low-order structure, which has made it a more attractive material to study the fundamental structural principles of biological materials.
In this article, we review the nacre structure, deformation mechanisms and toughening mechanism, as well as materials designed to mimic nacre and their fabrication methods; we also outline directions for further research. We first introduce the well-studied basic structure, together with brief discussions on biomineralization, nanostructures and the organic matrix, to describe the formation of structures in shells. The bulk of this article is devoted to current research on the mechanical behavior of nacre, followed by a review of methods for fabricating synthetic materials which have been inspired by nacre.
Nacre structure
Hierarchical architecture
The photographs in figure 1 show the shell of the abalone Haliotis gigantea, with nacre on the inner surface. This figure also illustrates the characteristic composite ‘brick and mortar’ structure of nacre [1, 5–7]. It consists predominantly (over 95% by volume) of uniformly oriented, ordered layers of aragonite-rich platelets separated by a thin organic polymer matrix. Single platelets are approximately 5 μm wide and 200–900 nm thick and are uniformly aligned along the vertical c-axis [001] direction. The organic sheets between the platelets have an insoluble chitin sheet sandwiched by soluble proteins [16, 17], which controls the biomineralization process as described in section 2.2.
Figure 1.
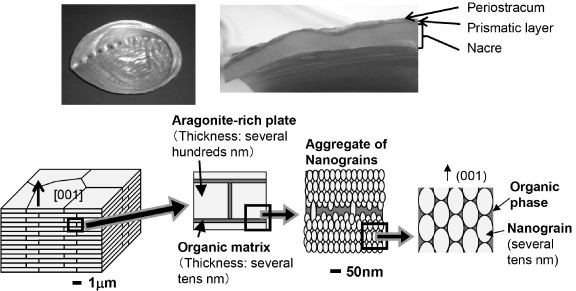
Photographs of the abalone H. gigantea: bottom view (top left) and cross-section (top right) showing nacre at the inner surface. The bottom row presents a schematic diagram of the nacre structure, sequentially magnified from left to right. It shows the ‘brick and mortar’ cross-sectional structure of nacre, consisting of horizontal layers of polygonal aragonite plates within a matrix of organic polymers. Single plates are uniformly aligned vertically in the c-axis [001] direction, and are approximately 5 μm wide and 500 nm thick. The platelet consists of ‘nanograins’, aragonite particles of the order of several tens of nanometers and the organic phase surrounding the nanograins.
Some studies have focused on the internal structure of the platelets, which can provide details of the biomineralization processes and mechanical behavior. Nanoscale structures within individual platelets were reported in the 1960s with transmission electron microscopy (TEM) observation of aragonite nanoparticles called ‘blocks’ surrounded by an ‘intracrystalline’ matrix [18, 19]. More recently, aragonite nanoparticles (called ‘nanograins’) surrounded by organic material within individual nacre platelets were observed using scanning electron microscopy (SEM), atomic force microscopy (AFM) and TEM [20–27] (figure 1). While the nacre platelets consist of nanostructures, TEM diffraction patterns reported in the literature generally showed uniform crystal orientation within the nacre layers. This led to the common belief that the platelet structure was a single crystal rather than a group of similarly oriented crystals. This unusual phenomenon was described as the pseudo-single-crystal effect [28].
Biomineralization
The biomineralization process involved in nacre formation has also been well reported. Amorphous CaCO3 is the precursor material, and biomolecules in the organic matrix control the nucleation and arrangement of aragonite particles, which are then constructed into the uniformly oriented platelets of the nacre structure [3, 16, 29–36]. This process is repeated many times, as thousands of these layers are typically formed over the life cycle of the mollusc to make up the millimeter-order thickness of the shell. Exploring these processes is a significant part of research into nature-inspired materials, since they involve important concepts on how the hierarchical structures are built within the shell.
Figure 2 illustrates the growth of columnar nacre, which occurs, for example, in abalone shells. The extrapallial space between the outer epithelium of the animal and the shell inner surface contains proteins and amorphous CaCO3 precursor [30]. Initially, the animal deposits organic sheets consisting of protein and chitin layers [16, 17] to form a template, upon which aragonite crystals sequentially nucleate and grow from the colloidal amorphous CaCO3 precursor [29, 37] via the assembly of nanoparticles [20]. Subsequent layers form within the organic sheets, initiating from the one below via mineral bridges, growing to full thickness and then expanding laterally. Hence the growth surface is made of many conical structures. The preferential growth in the c-axis [001] direction and uniform crystallographic orientation are reportedly controlled by charge interactions and specific proteins [16, 20, 29, 38–44].
Figure 2.
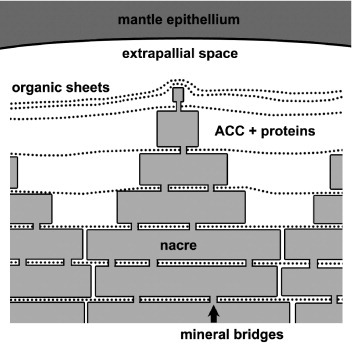
Illustration of the growth surface of columnar nacre. Amorphous CaCO3 precursor is sequentially deposited as layers of aragonite platelets to form nacre.
Recent studies on the growth of larval shells have provided additional information on the shell biomineralization. The larval shells were found to consist of a series of different aragonite and organic structures with distinct crystal orientations, much like mature shell structures. This result demonstrates that ordered structures begin to form in the early life stage [37, 45, 46].
Organic material
The organic layer of nacre has been investigated using various methods such as x-ray diffraction (XRD), AFM and TEM. It has a laminar structure consisting of a fibrous chitin core [16, 17, 35, 47, 48] between layers of macromolecules. The outer layers contain proteins that are important in biomineralization, controlling the nucleation and growth of aragonite [29–33, 49, 50].
The proteins have modular structures [51] resembling the silks of spiders and silkworm cocoons, as revealed by electron diffraction [52] and amino acid sequencing [53]. In both types of silk, the fundamental protein structures with modular repeating hydrophilic or hydrophobic domains adopt various conformations depending on the presence of water [54–56]; these structures account for mechanical characteristics such as strength, elasticity and toughness [57–60]. The presence of water has been detected in the nacre matrix using Fourier transform infrared spectroscopy [21].
Proteins have been identified that bind to chitin or control the nucleation and growth of the aragonite on organic sheets [61–63]. A recent analysis of green sheets in growth lines (see section 2.4) has shown that these sheets are made of proteins and polysaccharides with a chitin core, similar to the nacre matrix structure. One side of the sheet is thought to provide adhesion to the formed aragonite shell, while the other plays a role in further shell growth [45].
Growth lines
Growth lines are a distinct structure that occurs periodically within the shell between the nacre regions. These layers have been given various terms in the literature, such as ‘mesolayers’ or ‘heterolayers’, and studies of their structures revealed aragonite and organic material, but with a different morphology from that of nacre. The different growth layers observed in the shell structure of abalone are illustrated in figure 3. The outer layer of the shell is made of prismatic calcite, while the bulk of the shell consists of nacreous aragonite. Also observed at irregular intervals are growth lines, made up of block-like aragonite, organic layers and spherulitic aragonite. The sequence of the formation of growth lines is well reported; it involves complex control of the architecture including the crystal structure and morphologies of aragonite in the shell [29, 34, 37, 45, 46, 64, 65]. First, nacre growth is interrupted by the appearance of prismatic or block-like aragonite structures, followed by a layer of granular aragonite. An organic green sheet then forms that prepares for further nacre growth. A layer of spherulitic aragonite is deposited next, from which nacreous aragonite is nucleated. The crystal orientation of the aragonite layers is uniform, demonstrating that their formation is closely controlled [34, 45, 64].
Figure 3.
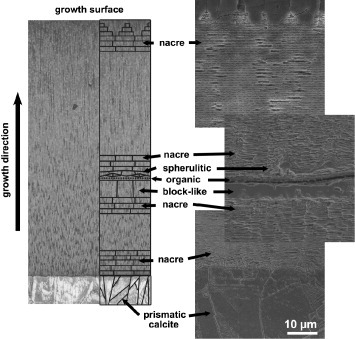
Cross-section showing the different growth layers in an abalone shell. A schematic diagram is superimposed onto an optical micrograph (left) and the schematic layers have been magnified for clarity. SEM images of the different growth layers (right).
Growth lines are important for the study of biomineralization processes, as they are transition structures during shell growth [66]. Although when and why they occur are still not completely understood, endogenous processes as well as diet and environmental factors are thought to influence the formation of growth lines [34, 39, 67–72].
Deformation and fracture behavior
The foundation of studies of the macroscopic mechanical properties of nacre was established by Currey in the 1970s. Since then, properties such as tensile strength, bending strength, Young’s modulus and fracture toughness have been reported, as summarized in table 1 [5, 6, 73–76]. Although their individual values are lower than those of engineering ceramics, the balance of strength and toughness in nacre is comparable to that of metals and polymers. No artificial material has achieved a comparable level of overall performance with a ceramic composition approaching 95 vol.% [77–79]. Current artificial ceramics composites usually sacrifice the intrinsic high strength and stiffness of ceramics in order to reduce brittleness. Understanding the mechanism underlying the good balance of properties in nacre will help in developing a new design concept of composite materials combining toughness, strength, stiffness and hardness.
Table 1.
| Fracture toughness | Work of fracture | Bending strength | Tensile strength | Young’s modulus | |
|---|---|---|---|---|---|
| (MPa m−1/2) | (J m−2) | (MPa) | (MPa) | (GPa) | |
| Wet (across) | 3.7–4.5 | 1034–1650 | 223–309 | 78–130 | 58–70 |
| Wet (along) | 4.2–5.0 | 553–800 | 194–275 | ||
| Dry (across) | 3.3–4.6 | 437 | 280–289 | 90–167 | 68–90 |
| Dry (along) | 3.7–5.0 | 250 | 322 |
Deformation behavior
Figure 4 shows a typical example of the stress–strain curve in a tensile test of nacre. Nacre has a different mechanical response depending on its hydration condition. Hydrated nacre deforms elastically at first, followed by continuous yielding at ∼50 MPa, and then shows ‘work hardening’ behavior like that of metals, with a tensile strength of ∼100 MPa and a total elongation of over 1%. Observation of the specimen during loading reveals ‘whitening’ of the surface after the yielding, as is observed in polymers under deformation, spreading over the specimen. Microscopic observation suggests that the color change can be attributed to the accumulation of pull-out of the platelets (figure 4(b)). The fracture surface shows that fracture occurs at the boundaries of the platelets, as a result of pull-out (figure 4(c)). Thus the deformation and fracture mechanism of the nacre in tension is as follows (see the illustration in figure 4(a)): the platelets are interlocked firmly during the linear deformation and then start to slide along the interface between the platelets at the yielding point. The sliding proceeds with loading that results in the macroscopic elongation of nacre. Finally, the platelets are pulled out completely, causing a macroscopic fracture.
Figure 4.
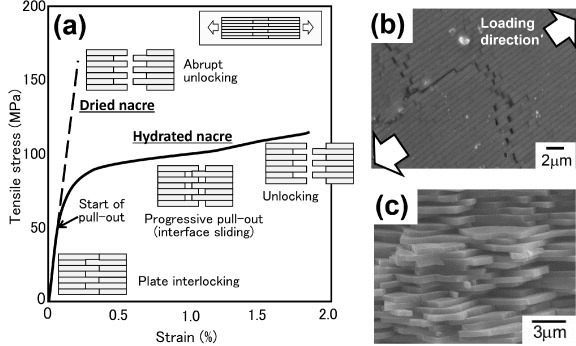
(a) Stress–strain curves measured in a tensile test of dried and hydrated nacre. The insets illustrate the deformation behavior during loading. (b) Deformation behavior in the ‘work-hardening’ stage in the stress–strain curve (SEM image). Platelets are progressively pulled out in an accumulative manner. (c) SEM image of the fracture surface after tensile testing. Fracture occurs at the boundaries of a platelet as a result of pull-out.
To affect such a gradual pull-out and macroscopic work hardening behavior, the interface sliding resistance must increase as the sliding proceeds; otherwise, only the weakest interface which slides first will be pulled out, and the accumulation of the pull-out cannot occur. As the frictional resistance at a sliding interface becomes larger than the resistance for another interface, the sliding interface stops and the other interface starts to slide instead. This mechanism causes macroscopic work hardening, and more and more load is needed for further deformation. This mechanism is unique as in conventional laminar composites, the sliding resistance decreases monotonically due to the decrease of contact area by pull-out and the decrease of frictional coefficient by wear.
Dried nacre is quite brittle. It shows linear deformation almost up to the maximum stress of ∼150 MPa with little yielding and then fractures catastrophically. The apparent fracture behavior is similar to that of ceramics, but the fracture surface shows that the fracture is not the unstable crack propagation through the platelets from a defect, but rather the pull-out of the platelets, as in hydrated nacre. Thus the substantial fracture of the dried nacre is a consequence of abrupt unlocking of the platelets at the maximum load after firm interlocking during the linear deformation (figure 4(a)).
The mechanism of the local sliding at the interface is not yet fully understood. Several origins of the interlocking and frictional resistance have been reported (figure 5), including nanoasperity on the platelet surface [8, 9, 80], mineral bridging [38, 80–83] and waviness of the platelet [73, 80, 83, 84]. Nanoasperity and mineral bridging may be effective at the initial interlocking and may also generate some frictional resistance during the sliding. However, these mechanisms by themselves cannot account for an interface where the sliding resistance increases as the sliding proceeds. Some researchers ascribe this situation to the variation of individual platelets, i.e. waviness of the platelet surface. When the edges of the platelets are thickened and interlocked with each other (figure 5(c)), the load for pulling out the platelet would increase as the pull-out proceeds. Simulation studies show that this hypothesis can explain the actual deformation behavior of nacre. Recently, local stress distribution in a single platelet during loading was measured. The result indicates that high stress is concentrated at the curved boundaries of the platelets, showing the locking effect caused by the waviness [85].
Figure 5.

Three models of the origin of interface sliding resistance: (a) the mineral bridging model, (b) the nanoasperity model and (c) the surface waviness model.
The morphology of the interface alone cannot explain the dependence of the mechanical response of nacre on its hydrated condition, however. The organic phase between the platelets is believed to play an important role in controlling the interface sliding behavior. The gel-like organic phase is susceptible to water content, as mentioned in section 3.2.2, so the interface properties are changed with nacre’s hydrated condition, thereby affecting the macroscopic mechanical response of the nacre. The hydrated organic phase acts as a lubricant at the interface and controls interface shear sliding [5, 9, 73, 74]. When the organic phase is dehydrated, it stiffens and loses flexibility and fluidity [73, 74]. This results in strong bonding of the interface that brings about higher macroscopic strengthening but causes brittleness.
Toughening mechanism
The most attractive characteristic of nacre, extremely high toughness, has been investigated by many researchers. The toughening of shells is attributed to at least ten mechanisms [1] and most of them are applicable to nacre. Mechanisms acting at the submicrometer scale within the brick and mortar structure were reported by the 1990s, and since the work of Smith et al in 1999 [86], there have been many studies of the toughening mechanism on the nanoscopic level. Most of them only mention the qualitative phenomena with little discussion of quantitative findings and reference to macroscopic behavior. Recent works confirm that the main mechanism contributing to significant toughening of nacre is energy-dissipating sliding of the platelets at the submicrometer scale [73, 74, 83]. Nano-order mechanisms are, however, important in controlling the platelet sliding behavior. Here the mechanisms are classified according to the components involved and the dimensions at which the mechanism operates, as summarized in table 2 and figure 6.
Table 2.
.Classification of toughening mechanisms in nacre.
| References | |
|---|---|
| (1) Submicrometer interaction of platelets | |
| • Surface creation by interface delamination and microcrack accumulation | [5, 6, 8–12, 73–76, 80–85, 87, 88] |
| • Crack deflection | |
| • Plate interlocking | |
| • Mineral bridging | |
| • Pull-out of plates | |
| (2) Energy-dissipating mechanism of the organic phase | |
| • Organic bridging between plates | [5, 6, 8–12, 84, 86–88, 91] |
| • Ligament formation in the organic phase with viscoelastic property | |
| • Unfolding of domains in chain molecules and breaking of cross-links | |
| (3) Nanostructural toughening mechanisms | |
| • Rotation and sliding of nanograins | [96, 97] |
| • Organic bridging between nanograins |
Figure 6.
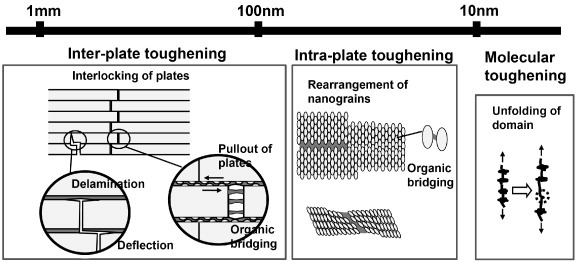
Hierarchical toughening mechanism of nacre. Toughening can be classified according to the operating dimension, from an inter-platelet mechanism operating at the submicrometer scale, to an intra-platelet mechanism of the order of several tens of nanometers, down to individual organic molecules (nm scale).
Submicrometer interaction of platelets
Mechanisms categorized in group 1 in table 2 [5, 6, 8–12, 73–76, 80–85, 87, 88] are well known in conventional laminar composites and fiber-reinforced composites; these microfracture-accumulating mechanisms occur at two orders of magnitude lower in nacre compared with artificial composites. For example, in laminar composites, when the layer thickness is decreased 100 times, a hundredfold increase in the interface is introduced within the material. This is a great advantage in accumulating microcracks, delamination and crack deflection as well as energy dissipation during platelet sliding mentioned below. The advantages of segmented, i.e. brick and mortar structure over the continuous layer structure have been demonstrated in a large-scale model composite [89].
Figure 7 shows the single-edged notch specimen of a hydrated nacre sample in tension. A color change area has formed at the notch tip and spread through the ligament, indicating that the stress intensity at the notch tip causes multiple pull-out actions of the platelet [9, 74]. The pull-out results in yielding and work hardening behavior, as described in section 3.1, so this ‘damage zone’ can behave as a yielding zone in metals or a ‘whitening zone’ in polymers that relaxes the stress concentration and dissipates the energy for crack propagation. Nacre exhibits ‘R-curve’ behavior, i.e. increased crack growth resistance with crack extension. The energy dissipation in the damage zone is estimated at 750 J m−2, using the displacement distribution in the damage zone and the corresponding sliding resistance to the measured displacement [74]. The value agrees well with the value measured by the fracture test, 1200 J m−2, suggesting that interlocking and accumulative pull-out of the platelets is the dominant toughening mechanism.
Figure 7.

The formation of a damage zone in front of the notch tip in the tensile fracture test.
Dried nacre exhibits brittle fracture and has attracted less attention than hydrated nacre. It has a much higher critical stress intensity factor, KIC, of 3–5 MPa·m−1/2 [5, 76] compared to that of monolithic aragonite, 0.25 MPa·m−1/2 [90]. A mechanical model in which interlocking in front of the notch tip gives a closure force to prevent crack extension was proposed and showed that the primary mechanism of the dried nacre is the platelet interlocking [76]. This mechanism may provide a simpler and more practical composite design principle than the complex multistage mechanisms of the hydrated nacre.
Energy-dissipating mechanism of the organic phase
The mechanisms listed in group 2 in table 2 are related to the unique organic phase of nacre [5, 6, 8–12, 84, 86–88, 91]. The organic phase itself dissipates energy by viscoelastic deformation or redistributes the stress near the crack tip. The mechanical characteristics of the matrix at the molecular level are attributed to proteins with modular structures [51, 86]. The protein mechanical behavior has been investigated using AFM [86], in situ TEM observation [91] and steered molecular dynamics simulations [92, 93]. Figure 8 shows a schematic diagram of a saw-tooth load–displacement curve of the molecule due to unfolding of the domains which increases energy dissipation during the deformation. The unfolding behavior is affected by the presence of water that changes bonding and other molecular interactions [93].
Figure 8.
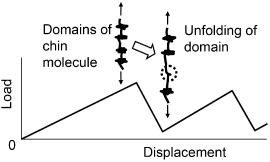
Schematic diagram of the deformation behavior of proteins with modular structures.
The mechanical properties of the organic matrix have been investigated in situ using a TEM pulling holder [91]. Figure 9 shows the corresponding TEM images where organic matrix ligaments are stretched between separating platelets and then subsequently fail and recoil. Viscoelastic reversible behavior was observed and attributed to unfolding and refolding protein domains similar to those in silkworm and spider silks. A large-scale model composite confirmed the importance of the viscoelastic and resilient phase between the platelets [89]. The deformation of the sheets of the organic matrix between growing nacre platelets was studied by nanoindentation measurements [47]. Deformation strengthening of the stretched organic strand in bending has also been reported [94], confirming the energy dissipation by the modular structure in the macromolecule.
Figure 9.

In situ deformation of organic matrix between platelets in nacre. (a) Schematic diagram showing the top view of nacre platelets as they separate and the deformation of the matrix between the interfaces. (b) TEM video still image sequence showing matrix ligament bridging between platelets and the subsequent deformation, failure and recoiling of the ligaments (adapted with permission from [91] © 2008 Cambridge University Press).
Nanostructural toughening mechanisms
The platelet has been considered as a single crystal in the discussions of the toughening mechanism because the crystal orientation of nacre as a whole has been generally observed as being uniform; however, nacre is composed of nanograins, as described in section 2.1. To understand the macroscopic mechanical properties of nacre it is adequate to treat platelets as single crystals because the fracture occurs at the boundaries of the platelets, leaving the platelets intact in macroscopic mechanical tests. However, fracture resistance against compressive local loading such as indentation and drilling, which are likely to be induced in the natural habitat by predators, has not been understood. Considering that biological structures are constructed bottom-up following a hierarchical architecture [11, 22, 95], further study of the nanoscale behavior remains a key step in understanding the bulk mechanical properties. AFM has been used in conjunction with nanoindentation of single platelets to show ductility and viscoelastic behavior, suggesting deformation by the interaction of nanograins and organic material [21, 23, 25]. In situ tension and bending tests revealed rotation and deformation of nanograins [96]. Deformation induced within single platelets using ultramicrotomy or micro-indentation was investigated by TEM [97]. The platelets sheared during ultramicrotomy showed deformation; toughening mechanisms such as nanograin separation and shear, and bridging by organic material within the platelets (figure 10), are thought to contribute to overall damage tolerance of nacre.
Figure 10.

TEM images of nacre platelet cross-sections prepared using ultramicrotomy. (a) A crack through platelets (numbered 1–3) and columnar structures approximately 50 nm across within the platelets are observed. Regions marked b and c are magnified in the right images. (b) White arrows indicate ligaments of organic matrix stretched between the columns. (c) Crack deflection around a nanoparticle within a column (arrow) (adapted with permission from [97] © 2008 Cambridge University Press).
Effect of hierarchical structure
Hierarchical structures responsible for mechanical protection of shells occur at various scales. In the largest dimension (from millimeters down to tens of micrometers), the whole shell itself has a hybrid structure constructed from several layers, such as prismatic, cross-lamellar, foliated and nacreous layers. Each layer has a different role against external mechanical actions, e.g. a columnar layer providing a hard protective layer on the outer side of the shell and nacre providing a tough structural component on the inner side [78, 98]. In the smaller dimension (from tens of micrometers down to nanometers), the cross-lamellar layer consists of three or four order lamella [90, 99–101], which provide damage tolerance behavior by accumulating multi-scale fractures.
Nacre also exhibits hierarchical toughening mechanisms at submicrometer to nanometer scales. Nanoscale mechanisms by the interaction of nanograins and organic phase in the platelet can cause resistance against crack initiation induced by local damage. The laminar structure of the platelet prevents crack propagation by deflection at the crack tip; it also provides mechanisms of energy dissipation and stress redistribution in front of the crack by multiple pull-out behavior. The energy dissipation mechanism at the sliding interface involves a structural change at the macromolecular level within the organic phase. Thus, multi-scale mechanisms operate in stages according to various magnitudes and types of loading. Quantitative studies of the effect of each mechanism are being carried out; however, mechanical conditions for activating each mechanism and their synergistic effect are not still fully understood.
Multilayer composites inspired by nacre and their mechanical properties
Although the toughening mechanism of nacre is not yet fully understood, nanolaminar composites inspired by nacre have been fabricated in a variety of ways [102–105]. In this section, we briefly introduce the classification and principles of the fabrication methods and provide typical examples, paying particular attention to works in which the mechanical properties of the fabricated composites have been evaluated.
Self-organized growth of minerals from solution
Methods categorized in this group are bottom-up approaches in which mineral formation is controlled in a supersaturated solution using an organic material as a template. Depending on the organic material’s molecular weight, concentration, functional group, functional group density, the solution’s temperature and pH, the orientation of crystal growth is accelerated or inhibited. Consequently, various shapes of the mineral such as plate, helicoid and rhombohedron can be formed. K2SO4 formed by a self-assembly technique using poly(acrylic acid) has a hierarchical structure containing nanocrystals that resemble nacre’s nanograins, columnar units such as nacre platelets and macroscopic crystals with various shapes [106, 107]. In another study, poly(acrylic acid) was used for the synthesis of CaCO3. The formation of an inorganic connection between the nanocrystals (such as ‘mineral bridging’ in nacre) was observed, confirming that the oriented architectures are generated from bridged nanocrystals with incorporated organic polymers [108]. A schematic diagram of the mineral-bridged nanocrystal growth controlled by the organic material is shown in figure 11.
Figure 11.
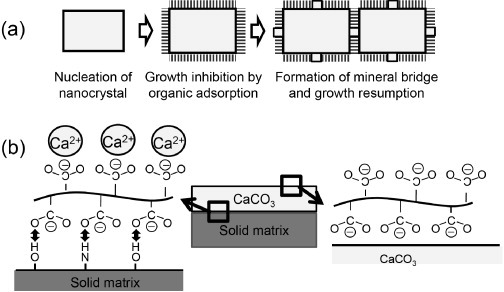
(a) Schematic diagram of sequential growth to the oriented structure via a mineral bridge. A nanocrystal is nucleated, and its growth is inhibited because of the adsorption of polymers. The growth restarts by the formation of a mineral bridge, leading to the growth of the adjacent nanocrystals. Thus the nanoscale-orientated architecture continues from nanocrystal to nanocrystal, resulting in an architecture with a specific macroscopic shape. (b) The formation of submicron-thick film is illustrated as follows: (1) acidic macromolecules are adsorbed onto the surface of the solid matrix due to the interaction of the carboxyl group of the acidic macromolecule and the hydroxy group or amino group of the matrix, (2) the macromolecule binds Ca2+ and causes local concentration of Ca2+ on the surface and nucleation of CaCO3, (3) the acidic macromolecules are adsorbed on the growing CaCO3 and inhibit growth in the thickness direction [108, 109].
Self-organization techniques are often used to form a mineral film or an inorganic/organic layered film, by anchoring the organic material on a flat substrate. For example, thin-film crystals of CaCO3 are formed on chitosan (as a solid organic matrix) coated on a glass substrate in the presence of acidic macromolecule (e.g. poly(acrylic acid), poly(aspartic acid), poly(glycolic acid)) as a soluble additive [109]. An inorganic/organic multilayer can be fabricated by repeating the process. For example, a layered polymer/CaCO3 film was fabricated by alternating spin coating of the polysaccharides and mineralization of the CaCO3 layer with a thickness of 0.8 μm [110].
Self-organization methods are quite effective in forming thin, well-structured mineral crystals for electrical or optical applications. They also have the advantages of a mild processing route, using aqueous solution and popular molecules in ambient air at room temperature. Although the fabrication of bulk or film multilayer materials for use as structural components is quite difficult, some works combined self-organization and other coating techniques to obtain multilayers with submicrometer-thick layers in which a smaller nanoparticle structure is built, as introduced below [111–113]. The spontaneous formation of a hierarchical structure has a great advantage over any other method.
The layer-by-layer (LBL) method
The LBL method is a bottom-up technique that involves alternately immersing a substrate into two solutions of oppositely charged electrolytes. The method was applied to nanostructured organic–inorganic films using anionic montmorillonite (MMT) clay platelets and poly(diallydimethylammonium) chloride (PDDA) polycation [114]. Repeating the adsorption of cationic PDDA and anionic MMT clay platelets resulted in alternating coating of MMT and PDDA owing to attractive electrostatic and van der Waals interactions between them. Intercalation of the PDDA into MMT also occurred during the process, so the dimension of the layer structure was much finer than that of nacre; the typical single-layer thickness was several nanometers. Repeating the process 200 times, a freestanding multilayer with a thickness of 4.9 μm, a tensile strength of 109 MPa, an elongation of 10% and a Young’s modulus of 13 GPa was obtained. The tensile behavior was like that of a polymer film rather than that of other bulk structural materials. Later, in the work by the same research group, the organic material was changed from PDDA to poly(vinyl alcohol) (PVA), which gives stronger covalent and hydrogen bonding between MMT and PVA [115]. The strength and Young’s modulus were improved to 400 MPa and 106 GPa, respectively; however, the material was brittle with an elongation of 0.33%. They investigated the effect of the organic materials further, comparing PDDA with chitosan, which is one of the strongest natural polymers, and found that the mechanical properties of the MMT/chitosan composite were inferior to those of MMT/PDDA. They attributed this fact to the high rigidity of the polymer, resulting in poor interfacial adhesion with the clay [116].
In other works, multilayers of TiO2 and several kinds of polyelectrolytes (PEs) were fabricated by the combination of the LBL method and chemical bath deposition (CBD) [111, 112]. CBD is a kind of self-mineralization process involving nanoparticle growth. In this process, nanoparticulate structures akin to nacre’s nanograin structure within the inorganic layer can be produced as well as organic/inorganic laminar structure. Positively charged PEs were adsorbed onto the negatively charged TiO2 layer produced by CBD, and a multilayer with the same dimension as that of nacre was produced, consisting of a ∼100-nm-thick inorganic layer and a 10-nm-thick organic layer. CBD and adsorption of PEs were repeated a maximum of eight times. Mechanical properties were evaluated with indentation techniques. The evaluation showed an enhancement of fracture toughness by a factor of four in comparison with that of the single TiO2 layer, whereas hardness and Young’s modulus were preserved.
The LBL method has the advantage of easily producing thick freestanding film composites, although it requires time-consuming, repeated steps. It also has the advantages of a wide range of material selection and mild processing conditions. It can easily be combined with other film fabrication methods, but it is difficult to obtain bulk materials or materials with a high volume fraction of inorganic phase.
Electrophoretic deposition
Electrophoretic deposition is the deposition of charged nanoparticles in suspension by the application of an external electric field. In this process, charged nanoparticles are forced to migrate and are deposited onto an oppositely charged electrode. MMT/poly(amic acid) layered composite was fabricated by this method. An emulsion of poly(amic acid), which was synthesized from pyromellitic dianhydride and 4,4′-dianminodiphenyl ether (ODA), containing various loadings of ODA-modified MMT was deposited on a stainless steel electrode [117]. The ODA molecules were either intercalated into the interlayer of the silicates or adsorbed onto the surface of MMT, and thus the ordered layered assembly of the MMT/ poly(amic acid) composite films was accomplished. The composite films exhibited a tensile strength of 63.25 MPa and a Young’s modulus of 3.67 GPa, with an increase of 155% in modulus and 40% in strength over the values for a pure poly(amic acid) film. MMT/polyacrylamide was also fabricated by the same research group [118]. The resulting material had a significantly high volume fraction of inorganic phase of 95.3% because an aqueous suspension of acrylamide-modified MMT without excess organic content was used for electrophoretic deposition. The film combined flexibility and rigidity, due to its brick and mortar structure, and had a hardness of 0.95 GPa and a modulus of 16.9 GPa as measured by the nanoindentation test. In another work, MMT was hydrothermally intercalated with acrylic anodic electrophoretic resin (AAER) and deposited on the electrode [119]. It had a nanoindentation hardness of 5 GPa, showing an increase of 2.1 GPa compared to that without intercalation. In other works, gibbsite/ethoxylated trimethylolpropane triacrylate (ETPTA) with a thickness of ∼30 μm with a gibbsite volume fraction of 50% was fabricated by infiltrating ETPTA into a nanocomposite in which surface-modified gibbsite nanoplates were aligned by electrophoresis. A tensile strength of ∼57 MPa was obtained [120].
Electrophoretic deposition can easily obtain a freestanding film with a thickness of several tens of micrometers within a few minutes’ electrophoresis. Like other bottom-up approaches, it has the advantages of a wide range of material selection and mild processing conditions, but it is usually difficult to obtain bulk materials or materials with a high volume fraction of inorganic phase with this process.
Sputtering
Alternate sputtering of different materials, usually amorphous or crystallized ceramics and polymer, on a substrate results in a multilayer. Multilayers of ceramics (TiC, Si3Ni4 or B4C) and Teflon were produced with an individual layer thickness of several tens of nanometers, and their mechanical properties were evaluated using Vickers indentation [121].
Multilayer ceramics composed of fully crystalline alternate Al2O3 and TiO2 layers with an individual layer thickness of ∼65–70 nm were fabricated by reactive pulse magnetron sputtering [122]. The multilayer ceramics was composed of columnar grains and elongated voids with their longitudinal axes parallel to the deposition direction. Spherical indentation was done on the multilayer; cross-sectional observation of the area below the indentation revealed that the layers near the surface endured a localized bending without breaking (figures 12(a) and (b)). This damage tolerance behavior was attributed to reorientation/relocation of the columnar grains via grain boundary sliding within the layers, analogous to the nanograins’ behavior in nacre [91, 96] (figures 12(c) and (d)). These results confirmed the microscopic non-brittle fracture upon localized loading in multilayer ceramics, occurring due to their nanoscale structure, and suggest that the organic phase (or soft phase) may not be necessary for a microdamage-tolerant behavior.
Figure 12.

(a) Cross-sections of residual impressions formed by spherical indentation at 300 mN. (b) Magnified view of the central region in (a), showing details of the deformation behavior. (c) Cross-sectional TEM images taken from the bent site of a residual impression, revealing the movement of columnar grains along the intercolumnar boundaries. (d) Schematic diagram of the microscopic mechanism, showing the distortion in the alignment of the columnar grains and columnar unit movement in the form of reorientation/relocation via sliding along intercolumnar boundaries (adapted with permission from [122] © 2009 Cambridge University Press).
Sputtering usually needs a controlled atmosphere, and it is time consuming and expensive to form a multilayer. However, it can obtain a dense layer with a highly uniform, controlled thickness, and is an attractive method of depositing a nacre-like multilayer structure for surface protection in structural applications. In this sense, the process should be distinguished from other bottom-up techniques which usually produce polymer-based films reinforced by inorganic platelets.
Liquid coating (spin coating and dipping)
Alternate coating on a substrate is performed using a liquid suspension where ceramic flakes or ceramics sol is used for inorganic layers, and a low-viscosity resin, polymer emulsion or polymer solution provides the organic phase. Surface-modified submicrometer-thick Al2O3 platelets were spread over the water surface and transferred to a glass substrate by dip coating. Then the dip-coated substrate was spin-coated with an organic layer from chitosan solution. Repetition of these steps led to multilayered inorganic–organic films with a total thickness of a few tens of micrometers [123]. The platelet fraction was 20% at maximum. The fabricated film had a tensile strength of 315 MPa, a Young’s modulus of 9.6 GPa and a total elongation of 21%. The same procedure of dipping and spin coating was used to synthesize layered double hydroxides (Cu–NO3, Co–Al–CO3 and Eu–Cl)/chitosan multilayers [124]. The obtained films were transparent, with a tensile strength of 160 MPa and a Young’s modulus of 12.7 GPa.
Methods for bulk, ceramics-based materials
The methods introduced above all have the potential to form thin multilayers; however, they are basically suited for the fabrication of coating or film and are not suited for the fabrication of bulk materials for structural use. Even when such processes are repeated thousands of times, only a thin plate with less than 1 mm thickness can be obtained. Also, in most cases, the multilayer composites are polymer-based materials, unlike nacre, which has a high volume fraction of inorganic phase. In this section, other methodologies that attempt to fabricate bulk, inorganic-based material for structural use are summarized.
A unique technique to utilize the dendrite formation during freezing of ice as a template was proposed [125, 126]. When aqueous slurry of ceramic powder was frozen and then removed from the mixture with dendrites, a layered scaffold of ceramic powders concentrated at the spacing of the dendrites was formed. Bulk layered materials can be obtained by infiltrating the space of the scaffold, i.e. the site where the dendrites existed originally. Al2O3/Al–Si [125] and Al2O3/polymethyl methacrylate (PMMA) [126] composites were fabricated using this method. Al2O3/Al–Si showed a nonlinear load–displacement curve in a bending test and had a critical stress intensity factor of 10 MPa m−1/2. The work also demonstrated the superiority of the segmented brick and mortar structure over the continuous layer structure [126].
Several works utilized flake ceramics powder or layered clay as a reinforcing inorganic platelet material and mixed it with matrix using a variety of ways of aligning the inorganic platelets [127–131]. Thin Al2O3 flake powder was aligned by slip casting and then hot-pressed in an epoxy matrix, yielding a flake volume fraction of 60% [127]. The fabricated material showed inelastic fracture behavior in a bending test rather than brittle fracture. In another work using the Al2O3 flake powder, the powder was trapped in gel, aligned during drying and then hot-pressed [128]. The composites with platelet volume fractions up to 50% were fabricated. The maximal achieved yield strength and elastic modulus of the composites were 82% and 13 times higher, respectively, than the values of the polymer alone.
Glass flake powder coated with ductile metal was aligned by a roller using a 3D printing machine to form a green sample and then hot-pressed [129]. The coating layer became the matrix phase between the flake powders after hot-pressing; a well-organized brick and mortar structure with a high flake volume fraction of 87% was obtained. The material showed crack propagation resistance by interface delamination, crack deflection, platelet bridging and pull-out in indentation and bending tests. Those mechanisms brought a work of fracture of 300 J m−2, which is two orders larger than that of monolithic glass.
An attempt to align talc tablets by centrifugation, shearing, sedimentation, dipping and other processes was made, and the effect of these aligning processes on the resultant structure was assessed by XRD, SEM and bending tests [130]. The results indicated that physical methods inducing shear to the tablet suspension enhanced the alignment. Recently, another study was carried out to determine how to align the platelet powder by testing three aligning methods of paper making, using a doctor blade and simple painting using PVA-coated MMT, and the results showed the importance of alignment as well as cross-linking agents in the organic matrix phase [131].
Summary
Understanding of the deformation and toughening mechanisms has advanced significantly in the past decade, supported by the spread of nanoscale measurement and observation tools. Although some important details remain unknown, the principal guidelines for the design of new composites can be summarized as follows:
Segmented brick and mortar structure.
Control of sliding resistance at the interface between the laminar platelets: appropriate shear strength that allows crack deflection and the following interface sliding without breaking the platelets; sliding shear stress that increases with the progress of the sliding.
Hierarchical architecture ranging from the nanometer to the micrometer order.
The effectiveness of the segmented structure and the importance of controlling the interface were recognized in composite design for a long time; however, we note that the idea of an interface becoming increasingly resistant to slide did not exist in conventional design concepts to the best of our knowledge.
When we adopt the wisdom of nature, we should note the importance of ‘re-designing’. Nacre realizes a unique interface by a combination of the organic matrix material and the surface morphology of inorganic platelets; however, this combination may not need to be faithfully copied. It is essential to reproduce its function, e.g. the function of the interface to increase the sliding resistance, rather than just copy the structure (such a copy would be impractical, inefficient and expensive). Revealing the principles of mechanisms and re-designing them to suit our current technology are important subjects for engineering.
Individual mechanical properties of nacre are not exceptional; they can be exceeded by the use of advanced engineering ceramics and polymers as component materials even when they are simply laminated without constructing the complicated architecture of nacre. However, we are still far from being able to reproduce the complex combination of mechanisms found in nacre. Continuing improvements in both technical approaches and the refinement of re-design concepts are expected to lead to the fusion of advanced engineering and the wisdom of nature present in nacre.
References
- Mayer G. Science. 2005;310:1144. doi: 10.1126/science.1116994. [DOI] [PubMed] [Google Scholar]
- Sarikaya M, Tamerler C, Jen A K Y, Schulten K. and Baneyx F. Nat. Mater. 2003;2:577. doi: 10.1038/nmat964. [DOI] [PubMed] [Google Scholar]
- Meyers M A, Chen P-Y, Lin A Y-M. and Seki Y. Prog. Mater. Sci. 2008;53:1. doi: 10.1016/j.pmatsci.2007.05.002. [DOI] [Google Scholar]
- Weiner S. and Addadi L. J. Mater. Chem. 1997;7:689. doi: 10.1039/a604512j. [DOI] [Google Scholar]
- Jackson A P, Vincent J F V. and Turner R M. Proc. R. Soc. 1988;234(B):415. doi: 10.1098/rspb.1988.0056. [DOI] [Google Scholar]
- Currey J D. Proc. R. Soc. 1977;196(B):443. doi: 10.1098/rspb.1977.0050. [DOI] [Google Scholar]
- Sarikaya M. Microsc. Res. Tech. 1994;27:360. doi: 10.1002/(ISSN)1097-0029. [DOI] [PubMed] [Google Scholar]
- Evans A G, Suo Z, Wang R Z, Aksay I A, He M Y. and Hutchinson J W. J. Mater. Res. 2001;16:2475. doi: 10.1557/JMR.2001.0339. [DOI] [Google Scholar]
- Wang R Z, Suo Z, Evans A G, Yao N. and Aksay I A. J. Mater. Res. 2001;16:2485. doi: 10.1557/JMR.2001.0340. [DOI] [Google Scholar]
- Menig R, Meyers M H, Meyers M A. and Vecchio K S. Acta Mater. 2000;48:2383. doi: 10.1016/S1359-6454(99)00443-7. [DOI] [Google Scholar]
- Ji B. and Gao H. J. Mech. Phys. Solids. 2004;52:1963. doi: 10.1016/j.jmps.2004.03.006. [DOI] [Google Scholar]
- Lin A Y-M. and Meyers M A. J. Mech. Behav. Biomed. 2009;2:607. doi: 10.1016/j.jmbbm.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Menig R, Meyers M H, Meyers M A. and Vecchio K S. Mater. Sci. Eng. 2001;297(A):203. doi: 10.1016/S0921-5093(00)01228-4. [DOI] [Google Scholar]
- Zhu Z, Tong H, Ren Y. and Hu J. Micron. 2006;37:35. doi: 10.1016/j.micron.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Lin A Y M, Meyers M A. and Vecchio K S. Mater. Sci. Eng. 2006;26(C):1380. doi: 10.1016/j.msec.2005.08.016. [DOI] [Google Scholar]
- Weiner S. and Traub W. Phil. Trans. R. Soc. 1984;304(B):425. doi: 10.1098/rstb.1984.0036. [DOI] [Google Scholar]
- Nakahara H, Bevelander G. and Kakei M. Venus Japan J. Malacol. 1982;41:33. [Google Scholar]
- Watabe N. J. Cell Biol. 1963;18:70. doi: 10.1083/jcb.18.3.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe N. J. Ultra Res. 1965;12:351. doi: 10.1016/S0022-5320(65)80104-6. [DOI] [PubMed] [Google Scholar]
- Oaki Y, Kotachi A, Miura T. and Imai H. Adv. Funct. Mater. 2006;16:1633. doi: 10.1002/(ISSN)1616-3028. [DOI] [Google Scholar]
- Mohanty B, Katti K S, Katti D R. and Verma D. J. Mater. Res. 2006;21:2045. doi: 10.1557/jmr.2006.0247. [DOI] [Google Scholar]
- Oaki Y. and Imai H. Angew. Chem. Int. Ed. 2005;44:6571. doi: 10.1002/(ISSN)1521-3773. [DOI] [PubMed] [Google Scholar]
- Bruet B J F, Qi H J, Boyce M C, Panas R, Tai K, Frick L. and Ortiz C. J. Mater. Res. 2005;20:2400. doi: 10.1557/jmr.2005.0273. [DOI] [Google Scholar]
- Rousseau M, Lopez E, Stempflé P, Bendlé M, Franke L, Guette A, Naslain R. and Bourrat X. Biomaterials. 2005;26:6254. doi: 10.1016/j.biomaterials.2005.03.028. [DOI] [PubMed] [Google Scholar]
- Li X, Chang W-C, Chao Y J, Wang R. and Chang M. Nano Lett. 2004;4:613. doi: 10.1021/nl049962k. [DOI] [Google Scholar]
- Stempflé P. and Brendlé M. Tribol. Int. 2006;39:1485. doi: 10.1016/j.triboint.2006.01.011. [DOI] [Google Scholar]
- Takahashi K, Yamamoto H, Onoda A, Doi M, Inaba T, Chiba M, Kobayashi A, Taguchi T, Okamura T-A. and Ueyama N. Chem. Commun. 2004;996 doi: 10.1039/b315478e. [DOI] [PubMed] [Google Scholar]
- Li X. and Huang Z. Phys. Rev. Lett. 2009;102:075502. doi: 10.1103/PhysRevLett.102.075502. [DOI] [PubMed] [Google Scholar]
- Addadi L, Joester D, Nudelman F. and Weiner S. Chem. Eur. J. 2006;12:980. doi: 10.1002/(ISSN)1521-3765. [DOI] [PubMed] [Google Scholar]
- Bevelander G. and Nakahara H. Calcif. Tissue Res. 1969;3:84. doi: 10.1007/BF02058648. [DOI] [PubMed] [Google Scholar]
- Belcher A M, Wu X H, Christensen R J, Hansma P K, Stucky G D. and Morse D E. Nature. 1996;381:56. doi: 10.1038/381056a0. [DOI] [Google Scholar]
- Feng Q L, Pu G, Pei Y, Cui F Z, Li H D. and Kim T N. J. Cryst. Growth. 2000;216:459. doi: 10.1016/S0022-0248(00)00396-1. [DOI] [Google Scholar]
- Thompson J B, Paloczi G T, Kindt J H, Michenfelder M, Smith B L, Stucky G, Morse D E. and Hansma P K. Biophys. J. 2000;79:3307. doi: 10.1016/S0006-3495(00)76562-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Belcher A M, Zaremba C M, Morse D E, Stucky G D. and Heuer A H. Chem. Mater. 2002;14:3106. doi: 10.1021/cm011739q. [DOI] [Google Scholar]
- Cartwright J H E. and Checa A G. J. R. Soc. Interface. 2007;4:491. doi: 10.1098/rsif.2006.0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saruwatari K, Matsui T, Mukai H, Nagasawa H. and Kogure T. Biomaterials. 2009;30:3028. doi: 10.1016/j.biomaterials.2009.03.011. [DOI] [PubMed] [Google Scholar]
- Weiss I M, Tuross N, Addadi L. and Weiner S. J. Exp. Zool. 2002;293:478. doi: 10.1002/(ISSN)1097-010X. [DOI] [PubMed] [Google Scholar]
- Schäffer T E. et al Chem. Mater. 1997;9:1731. doi: 10.1021/cm960429i. [DOI] [Google Scholar]
- Lin A. and Meyers M A. Mater. Sci. Eng. 2005;390(A):27. doi: 10.1016/j.msea.2004.06.072. [DOI] [Google Scholar]
- Nakahara H. Venus Japan J. Malacol. 1979;38:205. [Google Scholar]
- Rousseau M, Lopez E, Couté A, Mascarel G, Smith D C, Naslain R. and Bourrat X. J. Struct. Biol. 2005;149:149. doi: 10.1016/j.jsb.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Nassif N, Pinna N, Gehrke N, Antonietti M, Jäger C. and Cölfen H. Proc. Natl Acad. Sci. USA. 2005;102:12653. doi: 10.1073/pnas.0502577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C-S. and Kim Y-W. Biomaterials. 2000;21:213. doi: 10.1016/S0142-9612(99)00120-9. [DOI] [PubMed] [Google Scholar]
- Falini G, Albeck S, Weiner S. and Addadi L. Science. 1996;271:67. doi: 10.1126/science.271.5245.67. [DOI] [Google Scholar]
- Falini G, Sartor G, Fabbri D, Vergni P, Fermani S, Belcher A M, Stucky G D. and Morse D E. J. Struct. Biol. 2011;173:128. doi: 10.1016/j.jsb.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Auzoux-Bordenave S, Badou A, Gaume B, Berland S, Helléouet M-N, Milet C. and Huchette S. J. Struct. Biol. 2010;171:277. doi: 10.1016/j.jsb.2010.05.012. [DOI] [PubMed] [Google Scholar]
- Meyers M A, Lim C T, Li A, Hairul Nizam, Tan E P S, Seki Y. and McKittrick J. Mater. Sci. Eng. 2009;29(C):2398. doi: 10.1016/j.msec.2009.07.005. [DOI] [Google Scholar]
- Levi-Kalisman Y, Falini G, Addadi L. and Weiner S. J. Struct. Biol. 2001;135:8. doi: 10.1006/jsbi.2001.4372. [DOI] [PubMed] [Google Scholar]
- Williams A. Nature. 1966;211:1146. doi: 10.1038/2111146a0. [DOI] [Google Scholar]
- Wheeler A P, George J W. and Evans C A. Science. 1981;212:1397. doi: 10.1126/science.212.4501.1397. [DOI] [PubMed] [Google Scholar]
- Shen X, Belcher A M, Hansma P K, Stucky G D. and Morse D E. J. Biol. Chem. 1997;272:32472. doi: 10.1074/jbc.272.51.32472. [DOI] [PubMed] [Google Scholar]
- Weiner S. and Traub W. FEBS Lett. 1980;111:311. doi: 10.1016/0014-5793(80)80817-9. [DOI] [Google Scholar]
- Sudo S, Fujikawa T, Nagakura T, Ohkubo T, Sakaguchi K, Tanaka M, Nakashima K. and Takahashi T. Nature. 1997;387:563. doi: 10.1038/42391. [DOI] [PubMed] [Google Scholar]
- Bini E, Knight D P. and Kaplan D L. J. Mol. Biol. 2004;335:27. doi: 10.1016/j.jmb.2003.10.043. [DOI] [PubMed] [Google Scholar]
- Dicko C, Knight D P, Kennedy J M. and Vollrath F. Int. J. Biol. Macromol. 2005;36:215. doi: 10.1016/j.ijbiomac.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Jin H-J. and Kaplan D L. Nature. 2003;424:1057. doi: 10.1038/nature01809. [DOI] [PubMed] [Google Scholar]
- Gosline J M, Denny M W. and DeMont M E. Nature. 1984;309:551. doi: 10.1038/309551a0. [DOI] [Google Scholar]
- Oroudjev E, Soares J, Arcidiacono S, Thompson J B, Fossey S A. and Hansma H G. Proc. Natl Acad. Sci. USA. 2002;99:6460. doi: 10.1073/pnas.082526499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulha H, Foo C W P, Kaplan D L. and Tsukruk V V. Polymer. 2006;47:5821. doi: 10.1016/j.polymer.2006.06.002. [DOI] [Google Scholar]
- Xu M. and Lewis R V. Proc. Natl Acad. Sci. USA. 1990;87:7120. doi: 10.1073/pnas.87.18.7120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Saruwatari K, Kogure T, Yamamoto Y, Nishimura T, Kato T. and Nagasawa H. Science. 2009;325:1388. doi: 10.1126/science.1173793. [DOI] [PubMed] [Google Scholar]
- Blank S, Arnoldi M, Khoshnavaz S, Treccani L, Kuntz M, Mann K, Grathwohl G. and Fritz M. J. Microsc. 2003;212:280. doi: 10.1111/j.1365-2818.2003.01263.x. [DOI] [PubMed] [Google Scholar]
- Fu G, Valiyaveettil S, Wopenka B. and Morse D E. Biomacromolecules. 2005;6:1289. doi: 10.1021/bm049314v. [DOI] [PubMed] [Google Scholar]
- Zaremba C M, Belcher A M, Fritz M, Li Y, Mann S, Hansma P K, Morse D E, Speck J S. and Stucky G D. Chem. Mater. 1996;8:679. doi: 10.1021/cm9503285. [DOI] [Google Scholar]
- Lin A Y-M, Chen P-Y. and Meyers M A. Acta Biomater. 2008;4:131. doi: 10.1016/j.actbio.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Sumitomo T, Kakisawa H. and Kagawa Y. J. Struct. Biol. 2011;174:31. doi: 10.1016/j.jsb.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Shepherd S A, Al-Wahaibi D. and Al-Azri A R. Mar. Freshwater Res. 1995;46:575. doi: 10.1071/MF9950575. [DOI] [Google Scholar]
- Shepherd S A, Avalos-Borja M. and Quintanilla M O. Mar. Freshwater Res. 1995;46:607. doi: 10.1071/MF9950607. [DOI] [Google Scholar]
- Erasmus J, Cook P A. and Sweijd N. J. Shellfish Res. 1994;13:493. [Google Scholar]
- Richardson C A. J. Exp. Mar. Biol. Ecol. 1987;111:77. doi: 10.1016/0022-0981(87)90021-9. [DOI] [Google Scholar]
- Richardson C A. J. Exp. Mar. Biol. Ecol. 1988;122:105. doi: 10.1016/0022-0981(88)90179-7. [DOI] [Google Scholar]
- Lopez M I, Chen P Y, McKittrick J. and Meyers M A. Mater. Sci. Eng. 2011;31(C):238. doi: 10.1016/j.msec.2010.09.003. [DOI] [Google Scholar]
- Barthelat F, Tang H, Zavattieri P D, Li C-M. and Espinosa H D. J. Mech. Phys. Solids. 2007;55:306. doi: 10.1016/j.jmps.2006.07.007. [DOI] [Google Scholar]
- Barthelat F. and Espinosa H D. Exp. Mech. 2007;47:311. doi: 10.1007/s11340-007-9040-1. [DOI] [Google Scholar]
- Jackson A P, Vincent J F V. and Turner R M. J. Mater. Sci. 1990;25:3173. doi: 10.1007/BF00587670. [DOI] [Google Scholar]
- Inoue R. to be published [Google Scholar]
- Barthelat F. Phil. Trans. R. Soc. 2007;365(A):2907. doi: 10.1098/rsta.2007.0006. [DOI] [PubMed] [Google Scholar]
- Luz M G. and Mano F J. Phil. Trans. R. Soc. 2009;367(A):1587. doi: 10.1098/rsta.2009.0007. [DOI] [Google Scholar]
- Li X. JOM. 2007;59:71. doi: 10.1007/s11837-007-0043-2. [DOI] [Google Scholar]
- Barthelat F, Li C-M, Comi C. and Espinosa H D. J. Mater. Res. 2006;21:1977. doi: 10.1557/jmr.2006.0239. [DOI] [Google Scholar]
- Song F, Soh A K. and Bai Y L. Biomaterials. 2003;24:3623. doi: 10.1016/S0142-9612(03)00215-1. [DOI] [PubMed] [Google Scholar]
- Meyers M A, Lin A Y-M, Chen P-Y. and Muyco J. J. Mech. Behav. Biomed. 2008;1:76. doi: 10.1016/j.jmbbm.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Katti K S, Katti D R, Pradhan S M. and Bhosle A. J. Mater. Res. 2005;20:1097. doi: 10.1557/JMR.2005.0171. [DOI] [Google Scholar]
- Song F. and Bai Y L. J. Mater. Res. 2003;18:1741. doi: 10.1557/JMR.2003.0239. [DOI] [Google Scholar]
- Tanaka Y, Kakisawa H. and Kagawa Y. Proc. 7th Asian-Australasian Conf. on Composite Materials. (in CD-ROM) 2010 [Google Scholar]
- Smith B L. et al Nature. 1999;399:761. doi: 10.1038/21607. [DOI] [Google Scholar]
- Wang R Z, Wen H B, Cui F Z, Zhang H B. and Li H D. J. Mater. Sci. 1995;30:2299. doi: 10.1007/BF01184577. [DOI] [Google Scholar]
- Nukala P K V V. and Simunovic S. Biomaterials. 2005;26:6087. doi: 10.1016/j.biomaterials.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Mayer G. Mater. Sci. Eng. 2006;26(C):1261. doi: 10.1016/j.msec.2005.08.031. [DOI] [Google Scholar]
- Kamat S, Su X, Ballarini R. and Heuer A H. Nature. 2000;405:1036. doi: 10.1038/35016535. [DOI] [PubMed] [Google Scholar]
- Sumitomo T, Kakisawa H, Owaki Y. and Kagawa Y. J. Mater. Res. 2008;23:1466. doi: 10.1557/JMR.2008.0184. [DOI] [Google Scholar]
- Ghosh P, Katti D R. and Katti K S. Mater. Manuf. Process. 2006;21:676. doi: 10.1080/10426910600611706. [DOI] [Google Scholar]
- Ghosh P, Katti D R. and Katti K S. Biomacromolecules. 2007;8:851. doi: 10.1021/bm060942h. [DOI] [PubMed] [Google Scholar]
- Xu Z H. and Li X. Adv. Funct. Mater. 2011;21:3883. doi: 10.1002/adfm.v21.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H. Int. J. Fract. 2006;138:101. doi: 10.1007/s10704-006-7156-4. [DOI] [Google Scholar]
- Li X, Xu Z. and Wang R. Nano Lett. 2006;6:2301. doi: 10.1021/nl061775u. [DOI] [PubMed] [Google Scholar]
- Sumitomo T, Kakisawa H, Owaki Y. and Kagawa Y. J. Mater. Res. 2008;23:3213. doi: 10.1557/JMR.2008.0389. [DOI] [Google Scholar]
- Sarikaya M. Proc. Natl Acad. Sci. USA. 1999;96:14183. doi: 10.1073/pnas.96.25.14183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. and Nardi P. Nanotechnology. 2004;15:211. doi: 10.1088/0957-4484/15/1/038. [DOI] [Google Scholar]
- Pokroy B. and Zolotoyabko E. J. Mater. Chem. 2003;13:682. doi: 10.1039/b300167a. [DOI] [Google Scholar]
- Kessler H, Ballarini R, Mullen R L, Kuhn L T. and Heuer A H. Comput. Mater. Sci. 1996;5:157. doi: 10.1016/0927-0256(95)00067-4. [DOI] [Google Scholar]
- Kato T, Sugawara A. and Hosoda N. Adv. Mater. 2002;14:869. doi: 10.1002/1521-4095(20020618)14:12<869::AID-ADMA869>3.0.CO;2-E. [DOI] [Google Scholar]
- Lobmann P. Curr. Nanosci. 2007;3:306. doi: 10.2174/157341307782418603. [DOI] [Google Scholar]
- Ruiz-Hitzky E R, Darder M. and Aranda P. J. Mater. Chem. 2005;15:3650. doi: 10.1039/b505640n. [DOI] [PubMed] [Google Scholar]
- Luz G M. and Mano J F. Phil. Trans. R. Soc. 2009;367(A):1587. doi: 10.1098/rsta.2009.0007. [DOI] [PubMed] [Google Scholar]
- Oaki Y. and Imai H. Adv. Funct. Mater. 2005;15:1407. doi: 10.1002/(ISSN)1616-3028. [DOI] [Google Scholar]
- Oaki Y. and Imai H. Angew. Chem. Int. Ed. 2005;44:6571. doi: 10.1002/(ISSN)1521-3773. [DOI] [PubMed] [Google Scholar]
- Oaki Y, Kotachi A, Miura T. and Imai H. Adv. Funct. Mater. 2006;16:1633. doi: 10.1002/(ISSN)1616-3028. [DOI] [Google Scholar]
- Kato T. Adv. Mater. 2000;12:1543. doi: 10.1002/(ISSN)1521-4095. [DOI] [Google Scholar]
- Kato T S, Suzuki T. and Irie T. Chem. Lett. 2000;2:186. doi: 10.1246/cl.2000.186. [DOI] [Google Scholar]
- Burghard Z, Tucic A, Jeurgens L P H, Hoffmann R C, Bill J. and Aldinger F. Adv. Mater. 2007;19:970. doi: 10.1002/(ISSN)1521-4095. [DOI] [Google Scholar]
- Burghard Z, Zini L, Srot V, Bellina P, van Aken P A. and Bill J. Nano Lett. 2009;94:103. doi: 10.1021/nl902324x. [DOI] [PubMed] [Google Scholar]
- Zlotnikov I, Gotman I, Burghard Z, Bill J. and Gutmanas E Y. Colloids Surf. 2010;361(A):138. doi: 10.1016/j.colsurfa.2010.03.013. [DOI] [Google Scholar]
- Tang Z, Kotov N A, Maganov S. and Ozturk B. Nat. Mater. 2003;2:413. doi: 10.1038/nmat906. [DOI] [PubMed] [Google Scholar]
- Podsiadlo P. et al Science. 2007;318:80. doi: 10.1126/science.1143176. [DOI] [PubMed] [Google Scholar]
- Podsiadlo P, Tang Z, Shim B S. and Kotov N A. Nano Lett. 2007;7:1224. doi: 10.1021/nl0700649. [DOI] [PubMed] [Google Scholar]
- Wang C A, Long B, Lin W, Huang Y. and Sun J. J. Mater. Res. 2008;23:1706. doi: 10.1557/JMR.2008.0209. [DOI] [Google Scholar]
- Long B, Wang C A, Lin W, Huang Y. and Sun J. Compos. Sci. Technol. 2997;67:2770. doi: 10.1016/j.compscitech.2007.02.007. [DOI] [Google Scholar]
- Lin W, Wang C, Le H, Long B. and Huang Y. Mater. Sci. Eng. 2008;28(C):1031. doi: 10.1016/j.msec.2007.04.030. [DOI] [Google Scholar]
- Lin T H, Huang W H, Jun I K. and Jiang P. Chem. Mater. 2009;21:2039. doi: 10.1021/cm802372f. [DOI] [Google Scholar]
- He J L, Wang L D, Li W Z. and Li H D. Mater. Chem. Phys. 1998;54:333. doi: 10.1016/S0254-0584(98)00036-4. [DOI] [Google Scholar]
- Dericioglu A F, Liu Y F. and Kagawa Y. J. Mater. Res. 2009;24:3387. doi: 10.1557/jmr.2009.0418. [DOI] [Google Scholar]
- Bonderer L J, Studart A R. and Gauckler L J. Science. 2008;319:1069. doi: 10.1126/science.1148726. [DOI] [PubMed] [Google Scholar]
- Yao H B, Fang H Y, Tan Z H, Wu L H. and Yu S H. Angew. Chem. Int. Ed. 2010;49:2140. doi: 10.1002/anie.v49:12. [DOI] [PubMed] [Google Scholar]
- Deville S, Saiz E, Nalla R K. and Tomsia A P. Science. 2006;311:515. doi: 10.1126/science.1120937. [DOI] [PubMed] [Google Scholar]
- Munch E, Launey M E, Alsem D H, Saiz E, Tomsia A P. and Ritchie R O. Science. 2008;322:1516. doi: 10.1126/science.1164865. [DOI] [PubMed] [Google Scholar]
- Ekiz O O, Dericioglu A F. and Kakisawa H. Mater. Sci. Eng. 2009;29(C):2050. doi: 10.1016/j.msec.2009.04.001. [DOI] [Google Scholar]
- Bonderer L J, Feldman K. and Gauckler L J. Compos. Sci. Technol. 2010;70:1958. doi: 10.1016/j.compscitech.2010.07.014. [DOI] [Google Scholar]
- Kakisawa H, Sumitomo T, Inoue R. and Kagawa Y. Compos. Sci. Technol. 2010;70:161. doi: 10.1016/j.compscitech.2009.10.003. [DOI] [Google Scholar]
- Almqvist N, Thomson N H, Smith B L, Stucky G D, Morse D E. and Hansma P K. Mater. Sci. Eng. 1999;7(C):37. doi: 10.1016/S0928-4931(98)00072-1. [DOI] [Google Scholar]
- Walther A, Bjurhager I, Malho J M, Pere J, Ruokolainen J, Berglund L A. and Ikkala O. Nano Lett. 2010;10:2742. doi: 10.1021/nl1003224. [DOI] [PubMed] [Google Scholar]