

Abstract
SNAREs fuse membranes in several steps. Trans‐SNARE complexes juxtapose membranes, induce hemifused stalk structures, and open the fusion pore. A recent penetration model of fusion proposed that SNAREs force the hydrophilic C‐termini of their transmembrane domains through the hydrophobic core of the membrane(s). In contrast, the indentation model suggests that the C‐termini open the pore by locally compressing and deforming the stalk. Here we test these models in the context of yeast vacuole fusion. Addition of small hydrophilic tags renders bilayer penetration by the C‐termini energetically unlikely. It preserves fusion activity, however, arguing against the penetration model. Addition of large protein tags to the C‐termini permits SNARE activation, trans‐SNARE pairing, and hemifusion but abolishes pore opening. Fusion proceeds if the tags are detached from the membrane by a hydrophilic spacer or if only one side of the trans‐SNARE complex carries a protein tag. Thus, both sides of a trans‐SNARE complex can drive pore opening. Our results are consistent with an indentation model in which multiple SNARE C‐termini cooperate in opening the fusion pore by locally deforming the inner leaflets.
Keywords: exocytosis, lysosome, membrane fusion, SNAREs, vacuole
Subject Categories: Membrane & Intracellular Transport
Introduction
Membrane fusion requires that two bilayers be brought into close proximity and disrupted in a controlled fashion. Fusion of yeast vacuoles occurs through a series of phases that have been experimentally defined 1. It requires the SNAREs Nyv1 (R), Vam3 (Qa), Vti1 (Qb), and Vam7 (Qc) 2, 3, 4. The AAA‐ATPase Sec18 [N‐ethylmaleimide‐sensitive factor (NSF)] and Sec17 (α‐SNAP) disrupts inactive cis‐SNARE complexes leading to the dissociation of Sec17 and of the soluble SNARE Vam7 4, 5, 6. Vacuoles become reversibly docked through HOPS and the Rab‐GTPase Ypt7 7, 8, 9, 10, 11, 12, 13, which is controlled by nucleotide exchange factors and a GTPase activating protein 14, 15, 16, 17. A vertex ring around the zone of membrane contact enriches most regulatory lipids and proteins required for fusion 18, 19, 20. Trans‐SNARE pairing triggers a hemifusion intermediate in which lipids transit between the outer leaflets, but content mixing does not occur 21, 22. Hemifusion requires full SNARE zippering 23 and the V0 sector of the V‐ATPase 24. The reaction culminates in the fusion of inner leaflets and the opening and expansion of a fusion pore, which is driven by SNARE complexes and the SM protein Vps33 25. In this process, the SNARE transmembrane domains (TMDs) appear to act as simple membrane anchors which transduce force but do not undergo sequence‐specific interactions with themselves or other proteins 26.
How the energy provided by SNARE zippering helps to overcome the energy barrier for membrane fusion is an area of active research. SNARE zippering juxtaposes the TMDs of Q‐ and R‐SNAREs, which should exert force on the membrane in order to disturb bilayer structure and induce fusion 23, 27, 28, 29, 30, 31. Theoretical and simulation approaches suggested that the energy barrier for hemifusion should be lower than the one for pore opening 29, 32. In line with this, pore opening can be rate‐limiting for the fusion process also in vacuole fusion 21, 22. In most models of SNARE‐mediated fusion, SNARE TMDs play an important role 33, 34. Truncating or mutating the proteinaceous TMDs of viral fusion proteins or of SNAREs, or replacing them by lipid anchors, can arrest the reaction at hemifusion 33, 35, 36, 37. An exchange of SNARE TMDs against unrelated transmembrane domains can, however, be tolerated without loss of activity 26.
Recent models ascribe a critical role to mechanical forces that zipped SNARE domains transduce to the C‐terminal portion of the SNARE TMDs 38, 39. For example, the addition of only two charged residues to the intra‐vesicular C‐terminus of the synaptobrevin II TMD in chromaffin cells inhibits fusion 40. It was proposed that progression of the fusion reaction likely involves a critical perforation of the membrane in which the C‐terminus is pulled into the hydrophobic core of the membrane(s), and that the addition of hydrophilic or charged residues impairs this process. In contrast, recent molecular dynamics simulations suggested that the C‐termini promote progression of fusion by exerting point‐like forces on the membrane, facilitating strong and highly localized deformations of the inner leaflets rather than involving penetration or perforation by the C‐termini 38. An example of such a process is illustrated in Fig 1.
Figure 1. Active remodeling of the membrane by trans‐SNARE complexes.
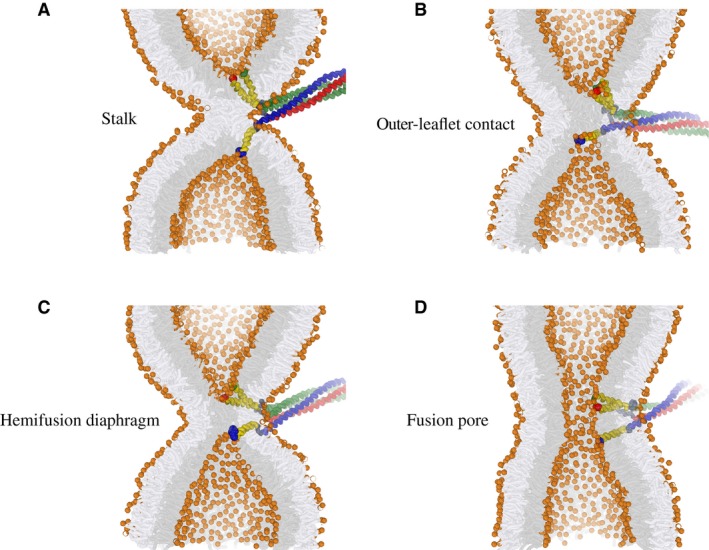
-
A–DExample of a simulated hemifusion‐to‐fusion pore transition in the presence of wild‐type vacuolar SNARE complexes. This example represents a scenario where fusion occurs between two highly curved membrane dimples (1/8 nm−1). Shown are the (A) stalk, (B) inner‐leaflet contact, (C) hemifusion diaphragm, and (D) fusion pore. Nyv1 is shown in blue, Vam3 in red, Vti1/Vam7 in green, the linkers in dark gray, and the TMDs in yellow. The enlarged highlighted ends of the TMDs represent the C‐termini. Lipid head groups are depicted in orange, the inner leaflets of the membrane(s) in gray, and outer leaflets in white.
In order to test these two models in yeast vacuole fusion, we added various tags to the TMDs of yeast vacuolar SNAREs and characterized their effects on membrane penetration, lipid, and content mixing.
Results
SNARE TMDs are expected to experience pulling and/or bending forces and transmit them to the fusion stalk. It has been proposed that these forces promote fusion pore formation by allowing the hydrophilic C‐termini of the SNARE TMDs to either penetrate (we refer to this as penetration regime) or squeeze (termed indentation regime) the inner leaflet, thereby thinning and widening the stalk 32, 38, 39. In order to explore whether these regimes are feasible mechanisms for SNARE‐mediated vacuole fusion, we created a near‐atomic (coarse‐grained) simulation model of the vacuolar SNARE complex in a lipid environment. Fig 2A sketches a scenario were the vacuolar SNARE complex exerts force on a formed (initial) hemifusion structure such as, for example, a hemifusion stalk or a “double membrane”. At this stage, the distance between the C‐termini of Nyv1 and Vam3 is about 8 nm (roughly the thickness of two membranes). Subsequent progression of hemifusion decreases the bending of the SNARE helices, and thus, the mechanical energy stored in the SNARE complex (Fig 1). Therefore, the force exerted on the membranes is expected to reach its maximum value, that is, during the progression of hemifusion, shortly after the initial stalk is formed. We can estimate these forces from the mechanical work that the vacuolar SNARE complex must provide to induce a small indentation (Fig 2A). In order to do this, we exploited the inability of unstructured, flexible TMD linkers to perform mechanical work, which allowed us to estimate the true work required to induce the membrane indentation by artificially pulling the C‐termini (unbiased work). The actual mechanical work performed by the SNARE complex is this unbiased work minus the work performed in the presence of structured linkers (Fig 2A). The slope of the mechanical work profile estimates the force that the C‐termini exert on the membrane. These forces were around 18 pN if the transmembrane‐ and coiled‐coil domains were linked by a fully structured, continuous α‐helix. If the linker of Vti1 was unstructured, this force dropped to 9 pN. These magnitudes are similar to the experimentally estimated force of about 17 pN that is required to unzip the coiled‐coil domain of the neuronal SNARE complex 31. Therefore, forces of 10–20 pN seem a reasonable estimate of the force that the C‐termini exert on the membrane. Do these forces suffice to facilitate membrane perforation of the wild‐type C‐terminus, and does the addition of peptide tags impair such a behavior? Evidently, exerting force on a stalk intermediate leads to indentation and progression of the fusion pore in our simulations (Fig 1). Therefore, analogous to the work of Lindau et al 39, we studied this scenario in a normal, planar model (POPC) membrane. We do not expect a strong dependence of the penetration force on either membrane geometry or topology because such a force is mostly determined by the chemical nature of the lipid head groups. Fig 2B shows the work that the C‐termini require to indent and eventually penetrate the membrane. The plateau region that the wild‐type C‐termini display (red arrow) corresponds to the regime where the membrane gives ups its restoring elastic response and the C‐termini become nearly “free‐floating”. Adding short charged or hydrophilic peptides to the C‐termini does not significantly alter the free energy of membrane indentation up to the penetration regime. Beyond this point, however, the hydrophilic tags substantially increase the free energy of subsequent “indentation” with respect to the wild type. The effects of small peptide tags and large protein domain tags are similar in this scheme. Based on the estimated forces required to allow wild‐type C‐termini to penetrate the bilayer, about 80 pN (Fig 2B), it is thus unlikely that the SNARE complex is able to generate forces sufficient for penetration. The addition of peptide or protein tags to the C‐termini prevents membrane penetration by the C‐termini entirely.
Figure 2. SNARE forces.
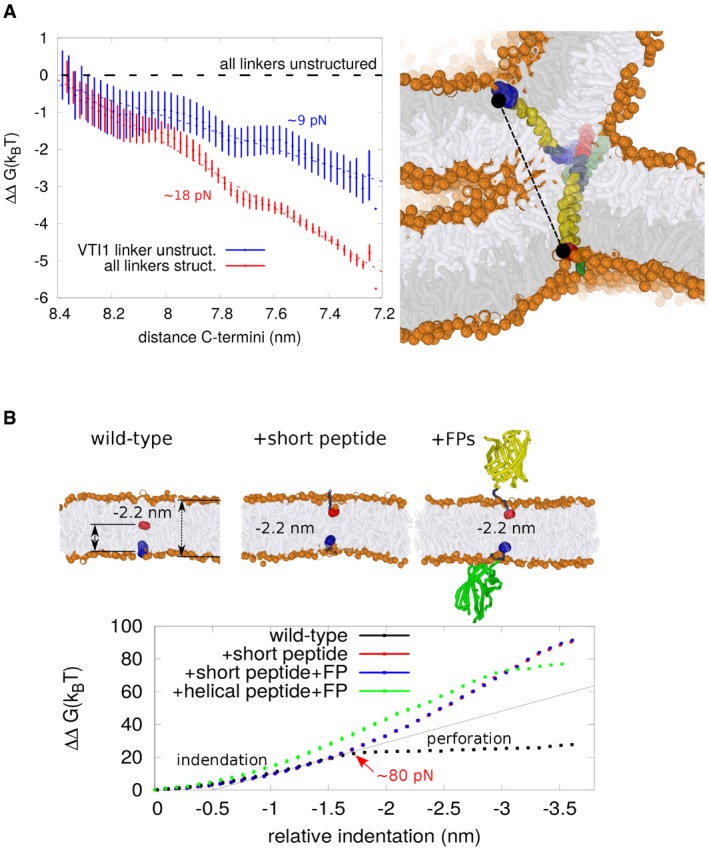
- Estimation of the intrinsic force that the vacuolar SNARE complex can exert on a stalk‐like fusion intermediate (shown in the right panel; coloring as in Fig 1) via its TMD C‐termini (Nyv1 and Vam3). At this stage, the distance between the C‐termini (the two connected black dots) is about 8 nm. The force that the C‐termini exert on the membrane, about 9 pN, is estimated from the difference in work required to induce a small indentation in the presence of structured versus unstructured Nyv1 and Vam3 linker regions (the slope of this profile defines the force). The additional presence of a structured Vti1 linker doubles the exerted force from 9 to 18 pN.
- The inherent elastic response of a flat lipid membrane (POPC) was studied by enforcing indentations via the isolated SNARE C‐termini in the presence or absence of a short linker peptide, or of the linker plus a fluorescent protein tag (FP). We define “relative indentation” as the change in C‐terminus (Nyv1)—C‐terminus (Vam3) distance (NYV1: blue colored beads, VAM3: red colored beads) that results when the SNARE complex performs mechanical work. It reflects the depth of the formed well upon squeezing. The slope of the obtained work profile estimates the required force (thin black line). Perforation (red arrow) requires a force of 80 pN for the wild‐type C‐termini. The plateau within the work profile indicates that the membrane gives up its elastic response (perforation transition). C‐terminal tags oppose such a perforation transition but do not significantly affect the forces observed within the indentation regime. Furthermore, if the unstructured peptide spacer would be α‐helical—and thus much shorter—indentation forces would still only be slightly affected.
Small peptide tags at the SNARE C‐termini do not impair vacuole fusion
Since it has been reported that additions of two hydrophilic amino acids to the C‐terminus of only one of the SNAREs in the neuronal SNARE complex sufficed to interfere with exocytic membrane fusion 40, we tested the sensitivity of vacuole fusion to various manipulations of the C‐terminus (Fig 3A). First, Nyv1 and Vam3 were fused with small hydrophilic peptide tags, a His6‐(HA)3‐tag or a His6‐(myc)2‐tag, respectively. The tags were separated from the TMDs by a hydrophilic spacer of nine amino acids (S9). Western blotting confirmed that the tagged SNAREs were present on purified vacuoles in similar quantities as their non‐tagged counterparts (Fig 3B). FM4‐64 staining of vacuolar membranes in vivo revealed that individual presence of a small tag on Vam3‐S9‐2myc or Nyv1‐S9‐3HA did not influence vacuole structure at all (Fig 3C and D). Cells that simultaneously expressed both SNAREs with peptide tags tended to show more numerous and smaller vacuoles, but this effect was weak.
Figure 3. Effect of small peptide tags on Vam3 and Nyv1 on vacuole morphology and fusion activity.
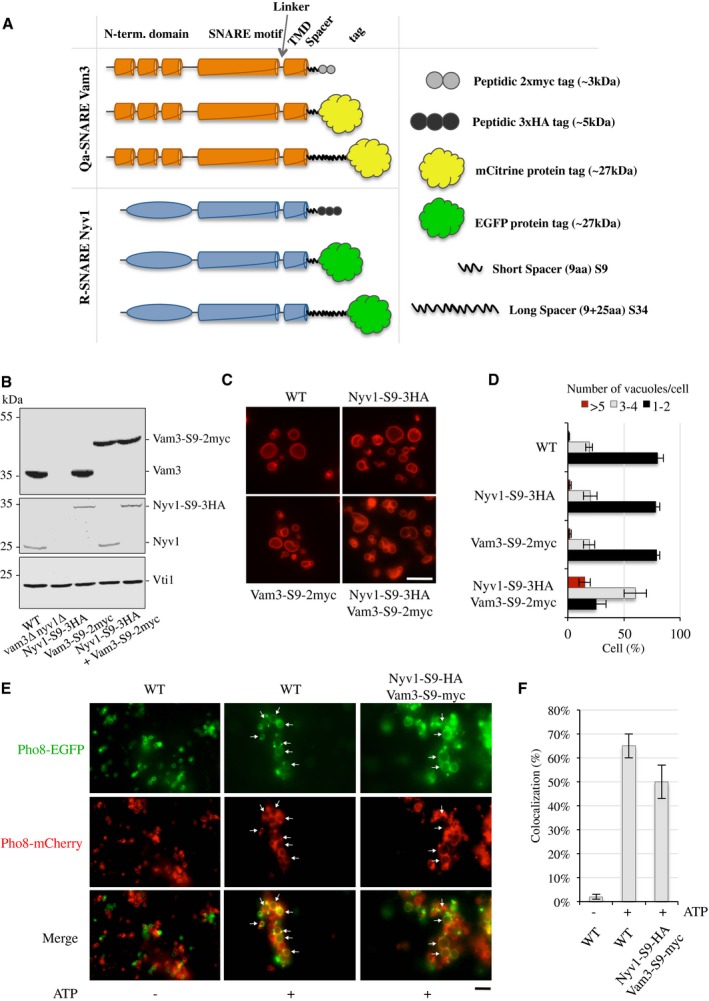
- Schematic view of the constructs for genomically tagging Vam3 and Nyv1 at their C‐termini.
- Vacuoles were isolated from the indicated yeast strains and 30 μg of proteins analyzed by SDS–PAGE and Western blotting.
- In vivo vacuole morphology was assayed by FM4‐64 staining. Scale bar: 5 μm.
- The cells were grouped into three categories according to the number of vacuoles visible per 100 cells. Values represent the means and s.d. from three independent experiments.
- Vacuoles were isolated from BJ3505 strains expressing the indicated tagged versions of Vam3 and Nyv1 and Pho8‐EGFP or Pho8‐mCherry. Ten micrograms of vacuoles was incubated in standard fusion reactions in the presence or absence of ATP and analyzed by confocal microscopy. Arrows indicate examples of fusion products. Scale bar: 5 μm.
- Fusion activity: vacuole fusion was assayed by measuring the percentage of colocalization of the Pho8‐EGFP and Pho8‐mCherry signals. Means ± s.d. are shown for at least 100 stained vacuoles from three experiments.
The in vitro fusion activity of SNAREs carrying the small tags could not be assayed using the alkaline phosphatase (ALP)‐based content mixing assay because this assay requires proteolytically competent vacuoles of the DKY6281 strain, in which the myc tag was efficiently removed from Vam3. Therefore, we resorted to a microscopic in vitro fusion assay that requires only BJ3505 cells, in which the major vacuolar proteases are inactivated and the tags of Nyv1‐S9‐3HA and Vam3‐S9‐2myc remain stable. We generated tagged BJ3505 cells expressing fluorescent versions of the vacuolar transmembrane protein alkaline phosphatase (Pho8) fused to either EGFP or mCherry. Vacuoles were prepared from these two strains, mixed and incubated in fusion reactions in the absence or presence of ATP. After 60 min, up to which timepoint the reaction proceeds in a fairly linear fashion under these conditions 2, 5, fusion was analyzed by fluorescence microscopy. We quantified fusion by determining the percentage of colocalization between EGFP and mCherry. Signals from this colocalization assay reproduce the signals from the ALP‐based content mixing assay very well (Fig EV1). In the absence of ATP, vacuoles appeared dispersed and only 2% of colocalization of EGFP and mCherry was observed (Fig 3E and F). In the presence of ATP, wild‐type vacuoles became bigger and labeled with both fluorescent variants of Pho8, bringing colocalization to > 60%, indicating that these vacuoles had fused. Vacuoles carrying Nyv1‐S9‐3HA and Vam3‐S9‐2myc showed 50% colocalization of EGFP and mCherry, suggesting that their fusion activity was close to that of the wild type. Thus, simultaneous tagging of Nyv1 and Vam3 with small peptide tags maintained the fusogenic activity of these SNAREs.
Figure EV1. Assay of fusion by microscopy and content mixing.
- Distribution of vacuole diameters. Vacuoles were isolated from the indicated strains and used in in vitro fusion reactions. At the end of the reaction, the organelles were analyzed by fluorescence microscopy and vacuole diameters were measured. Only structures that were identifiable as vesicular entities were analyzed. Data are represented as a box‐and‐whisker plot. The boxes provide the median, the upper (blue) and lower (orange) quartile. Whiskers represent the maximal and minimal values observed, respectively. 20 vacuoles have been analyzed for each condition in three independent experiments each.
- Microscopy: Vacuoles were isolated from BJ3505 cells expressing Pho8‐EGFP or Pho8‐mCherry. Ten micrograms of vacuoles was incubated in standard fusion reactions in the absence or presence of ATP with increasing amounts of anti‐Vam3 antibody and analyzed by confocal microscopy. Arrows indicate the products of vacuole fusion. Scale bar: 5 μm.
- Comparison of fusion activity measured through content mixing or the degree of colocalization of vacuolar fluorescent marker proteins, using BJ3505 vacuoles and DKY6281 vacuoles that did not carry tags on their SNARE proteins. Vacuole fusion was assessed by measuring, from parallel fusion reactions, either content mixing by the standard Pho8/Pep4 maturation assay or the colocalization between Pho8‐EGFP and Pho8‐mCherry signals. Reactions were performed in the absence or presence of ATP and with increasing amounts of anti‐Vam3 antibody used as a potent inhibitor of fusion reaction. Means ± s.d. are shown from three independent experiments.
Bulky protein domains at the C‐termini of Q‐ and R‐SNAREs allow SNARE activation
Next, we fused large fluorescent protein domains to the 3′ end of the endogenous genes coding for the Qa‐SNARE Vam3 and the R‐SNARE Nyv1 (Fig 3A). We attached them using the same nine amino acid spacer as for the small peptide tags described above, such that the peptide sequence adjacent to the membrane surface is the same in both cases. The fusion proteins were expressed at levels that were at or slightly above the levels of the non‐tagged versions of these SNAREs, as shown by SDS–PAGE and Western blotting of purified vacuoles (Fig 4A). The addition of fluorescent proteins to the C‐terminus of Nyv1 and Vam3 did not affect the localization of these proteins in the cells. Confocal microscopy confirmed that Vam3‐S9‐mCitrine and Nyv1‐S9‐EGFP localized to the vacuolar membrane (Fig 4B). Nyv1‐S9‐EGFP stained the vacuolar membrane homogeneously, whereas Vam3‐S9‐mCitrine was concentrated in numerous foci along the vacuolar rim. In cells that simultaneously expressed both SNARE proteins with their tags, vacuoles appeared as clusters of fluorescent material. Staining these cells with the vacuole‐specific fluorescent vital dye FM4‐64 revealed that the vacuoles in these cells formed numerous small vesicles that adhered together in grape‐like fashion (Fig 5A). By contrast, vacuolar morphology remained normal if only one of the two SNAREs carried a fluorescent protein tag. Vacuole fragmentation is often due to deficiencies in vacuole fusion 41, 42. Therefore, simultaneous tagging of the transmembrane domains of Qa and R‐SNAREs may interfere with vacuole fusion in vivo.
Figure 4. C‐terminal tagging of the SNAREs Vam3 and Nyv1 with large tags.
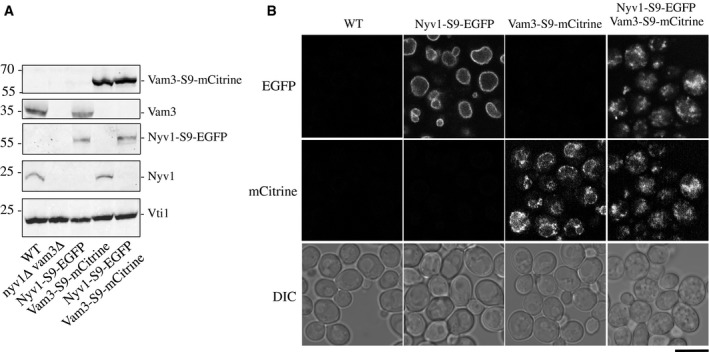
- Vacuoles were isolated from BJ3505 cells expressing the indicated versions of Vam3 and/or Nyv1. 10 μg of the organelles were analyzed by SDS–PAGE and Western blotting using antibodies to Vti1, Vam3, and Nyv1.
- Localization in vivo. The indicated strains were grown logarithmically in synthetic complete (SC) medium. After 16 h at 30°C, when the final OD600 of each culture was about 1, the cells were observed by confocal fluorescence and differential interference (DIC) microscopy. Scale bar: 5 μm.
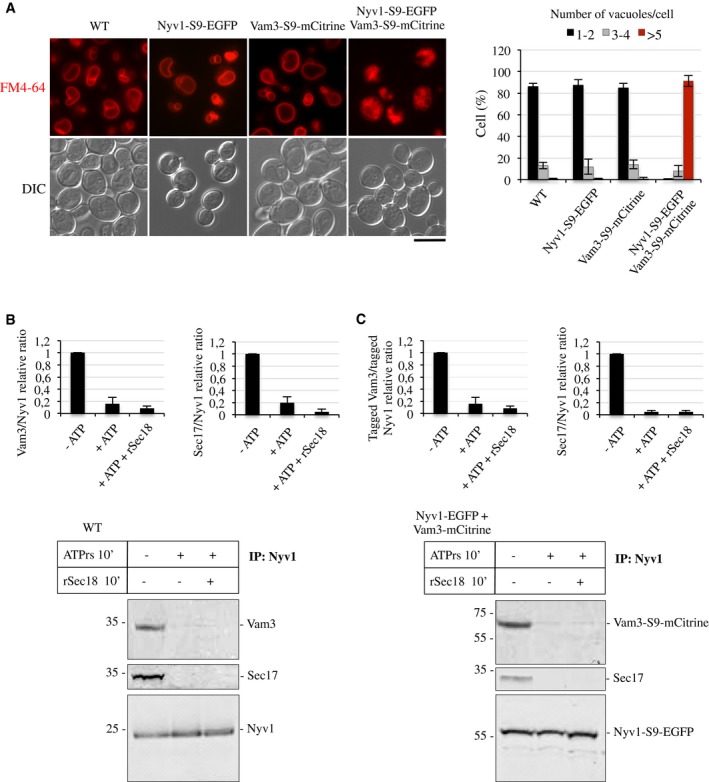
Figure 5. Effect of tags on vacuole morphology and cis‐SNARE activation.
-
AIn vivo morphology. Cells expressing the indicated SNARE variants were grown in SC medium (16 h, 30°C), stained with FM4‐64, and their relative morphology was assessed by fluorescence and DIC microscopy. Scale bar: 5 μm. The cells were grouped into three categories according to the number of vacuoles visible per 100 cells. Values represent the means and s.d. from three independent experiments.
-
B, CSNARE activation on isolated vacuoles. Vacuoles were isolated from a strain co‐expressing NYV1‐S9‐EGFP and VAM3‐S9‐mCitrine and from an isogenic wild type. One hundred and fifty micrograms of the organelles was incubated in fusion reactions in the presence or absence of an ATP‐regenerating system and recombinant, purified Sec18/NSF (rSec18, 50 μg/ml). After 10 min of incubation at 27°C, the vacuoles were solubilized and immunoprecipitated with antibodies to Nyv1. Co‐immunoprecipitated proteins were analyzed by SDS–PAGE and Western blotting. The histograms provide quantifications of the band intensities as the means ± s.d. from three independent experiments.
We hypothesized that the simultaneous presence of two C‐terminal tags might cause steric problems in a cis‐SNARE complex, distort the arrangement of the TMDs, and interfere with SNARE activation. This hypothesis was supported by earlier observations that replacing the TMD of Vam3 by a palmitoyl anchor interferes with the disruption and activation of cis‐SNARE complexes 43. We used the in vitro fusion of purified vacuoles to test this aspect. ATP‐dependent SNARE activation by Sec18/NSF disrupts cis‐SNARE complexes and coincides with the release of Sec17/α‐SNAP from the membrane 4. We incubated isolated vacuoles in the presence or absence of an ATP‐regenerating system, solubilized them in detergent and immuno‐precipitated Nyv1. The amount of co‐precipitated Vam3 and Sec17/α‐SNAP was determined by Western blotting. Upon incubation in the absence of ATP, Vam3, Nyv1, and Sec17/α‐SNAP were found in association (Fig 5B). Incubation with ATP diminished this association by > 75% and addition of purified recombinant Sec18/NSF enhanced dissociation to around 90%. Vacuoles carrying Vam3‐S9‐mCitrine and Nyv1‐S9‐EGFP behaved like wild‐type organelles in these assays (Fig 5C). Thus, simultaneous presence of two protein tags on Vam3 and Nyv1 does not interfere with cis‐SNARE activation.
Simultaneous C‐terminal tagging of Q‐ and R‐SNAREs in trans blocks the hemifusion‐to‐fusion transition
Next, we tested whether the tags inhibited fusion at the level of trans‐SNARE complexes. This could be expected if the luminal C‐termini of SNAREs had a role in lipid mixing or in the opening of the fusion pore. In order to generate a situation where only trans‐SNARE complexes could be affected by simultaneous tagging of Vam3 and Nyv1, we created strain in which the NYV1 gene was deleted and VAM3 was fused to mCitrine (nyv1∆ VAM3‐S9‐mCitrine). These cells expressed VAM3‐S9‐mCitrine at the same levels as seen for VAM3 in wild‐type cells (Fig 6A), and they showed wild‐type‐like vacuole morphology (Fig 6B). Vacuoles from this strain were incubated under fusion conditions, either with vacuoles from a NYV1‐S9‐EGFP strain (NYV1‐S9‐EGFP VAM3) or with non‐tagged “wild‐type” vacuoles (NYV1 VAM3). Content mixing was measured via the maturation of pro‐alkaline phosphatase, which was contained in one fusion partner, by the peptidase Pep4 that was contained in the other fusion partner. Vacuoles carrying NYV1‐S9‐EGFP fused efficiently with non‐tagged wild‐type vacuoles, as did vacuoles from nyv1∆ VAM3‐S9‐mCitrine cells (Fig 6C). However, fusion between vacuoles carrying NYV1‐S9‐EGFP and vacuoles carrying VAM3‐S9‐mCitrine was reduced to background levels, giving signals comparable to those of vacuoles that lack both SNAREs, or of wild‐type vacuoles incubated on ice or without ATP. Vacuoles carrying both NYV1‐S9‐EGFP and VAM3‐S9‐mCitrine in the same membrane fused efficiently with wild‐type organelles. Therefore, tagging of Vam3 and Nyv1 interferes with content mixing only if these tags are present on both sides of a trans‐SNARE complex.
Figure 6. Effect of large protein tags on lipid and content mixing.
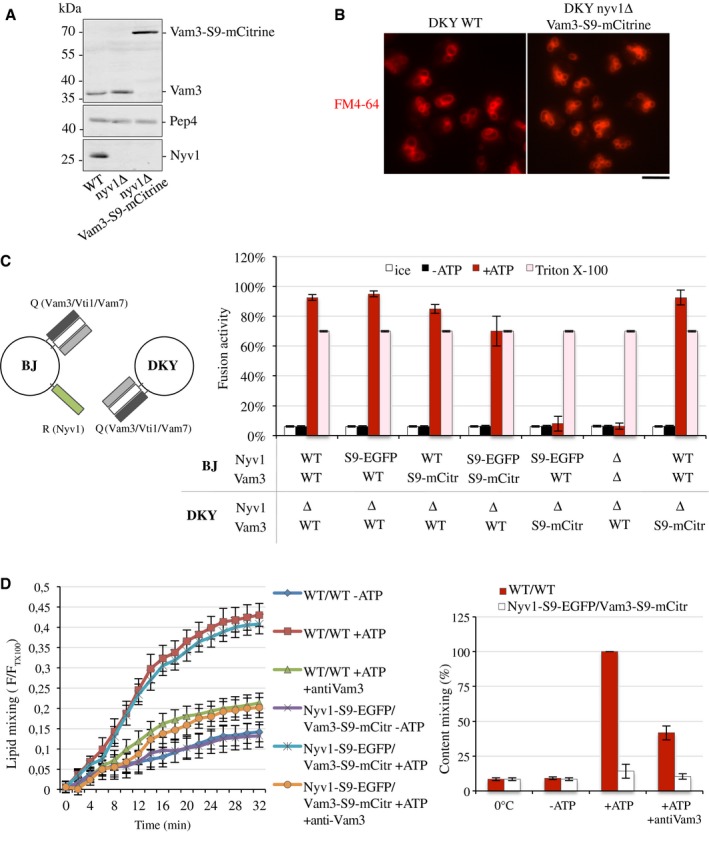
- Vacuoles were isolated from the indicated yeast strains and 30 μg of proteins analyzed by SDS–PAGE and Western blotting.
- Vacuole morphology was assessed for the indicated cells as in Fig 3C. Scale bar: 5 μm.
- Vacuoles were isolated from BJ3505 and DKY6281 cells with SNAREs tagged or deleted as indicated. Note that the presence of Nyv1 on only one fusion partner still permits efficient fusion 64 but ensures that trans‐SNARE complexes can only form in one orientation, between the Q‐SNARE in DKY6281 and the R‐SNARE in BJ3505. The vacuoles were used in standard fusion reactions, and content mixing was measured via the alkaline phosphatase assay. In parallel, identical samples were incubated either on ice, which prevents fusion, or in the presence of 0.5% Triton X‐100, which allows fusion‐independent access of the maturase Pep4 to pro‐ALP and controls for the levels of these two reporter enzymes in the samples. Means ± s.d. are shown from three independent experiments.
- Using strains from (C), lipid and content mixing (left and right panel, respectively) were performed in parallel in the presence or absence of ATP. Anti‐Vam3 antibody (3 μg) was added to some reactions in order to inhibit trans‐SNARE pairing and fusion. Means ± s.d. are shown from three independent experiments.
Lipid mixing was assessed for vacuoles from the same strains by incorporating rhodamine–phosphatidyl ethanolamine (Rh‐PE) into one of the fusion partners at a self‐quenching concentration 21. These donor vacuoles were mixed in fusion reactions with an excess of acceptor vacuoles that were free of Rh‐PE. Lipid mixing between donors and acceptors results in fluorescence dequenching. Lipid mixing between vacuoles carrying NYV1‐S9‐EGFP and vacuoles carrying VAM3‐S9‐mCitrine occurred with very similar kinetics and efficiency as between wild‐type vacuoles (Fig 6D). In both cases, lipid mixing depended on the presence of ATP and was sensitive to the addition of antibodies to Vam3, a potent inhibitor of fusion. Thus, the presence of large tags on the Qa and R‐SNAREs of a trans‐SNARE complex permits normal lipid mixing but prevents content mixing. This suggests that the reaction proceeds up to a hemifusion state, which also requires trans‐SNARE pairing 21, 22, 23, 44, but that the opening of the fusion pore is inhibited.
Hemifusion arrest depends on physical proximity of the protein tag to the TMD
If the large protein tags interfered sterically with the movement and local force transmission by TMDs, an elongated spacer between the tags and the TMDs might rescue fusion. We added an additional 25 amino acid to extend the previous short spacer (S34) between the TMDs and the protein tags (Fig 3A), which is predicted to adopt an unstructured extended conformation. SNAREs with this long spacer were present in the vacuolar fraction at similar levels as their counterparts with the short spacers (Fig 7A). In order to avoid proteolytic digestion of the spacers, these constructs were expressed in BJ3505 cells that lacked the vacuolar proteinases A and B. Vacuolar morphology was analyzed in vivo by FM4‐64 staining (Fig 7B and C). As described above, NYV1‐S9‐EGFP and VAM3‐S9‐mCitrine showed strong vacuolar fragmentation. Extension of the spacer in VAM3‐S34‐mCitrine gave a pronounced rescue of this phenotype. NYV1‐S34‐EGFP also rescued vacuolar morphology, albeit less profoundly. Simultaneous extension of both spacers rescued vacuolar morphology to wild‐type‐like appearance. This effect suggested that spacer extension could rescue the fusion activity of SNAREs carrying a bulky FP in vivo. Since S34 was not stable in the proteolytically competent vacuoles of the DKY6281 strain, which are required for the alkaline phosphatase‐based content mixing assay, we again resorted to the fusion assay by microscopy described above (Fig 3E) for analyzing fusion activity in vitro. To this end, the vacuolar proteins Pho8‐mCFP or Pho8‐mCherry were expressed in the strains carrying different combinations of tagged Nyv1 and Vam3. After incubation in the absence of ATP, vacuoles of all combinations appeared dispersed and mCFP did not colocalize with mCherry (Fig 7D and E). Upon incubation in the presence of ATP, wild‐type vacuoles as well as vacuoles that simultaneously carried Nyv1‐S34‐EGFP and Vam3‐S34‐mCitrine became bigger and labeled with both fluorescent variants of Pho8, indicating that these vacuoles had fused. Hardly any colocalization was observed in reactions with vacuoles that simultaneously carried Nyv1‐S9‐EGFP and Vam3‐S9‐mCitrine. These results are consistent with the notion that the protein tags inhibit fusion by a steric effect on the hemifusion zone.
Figure 7. Effect of extending the spacers between the SNARE C‐termini and the fluorescent protein tags.
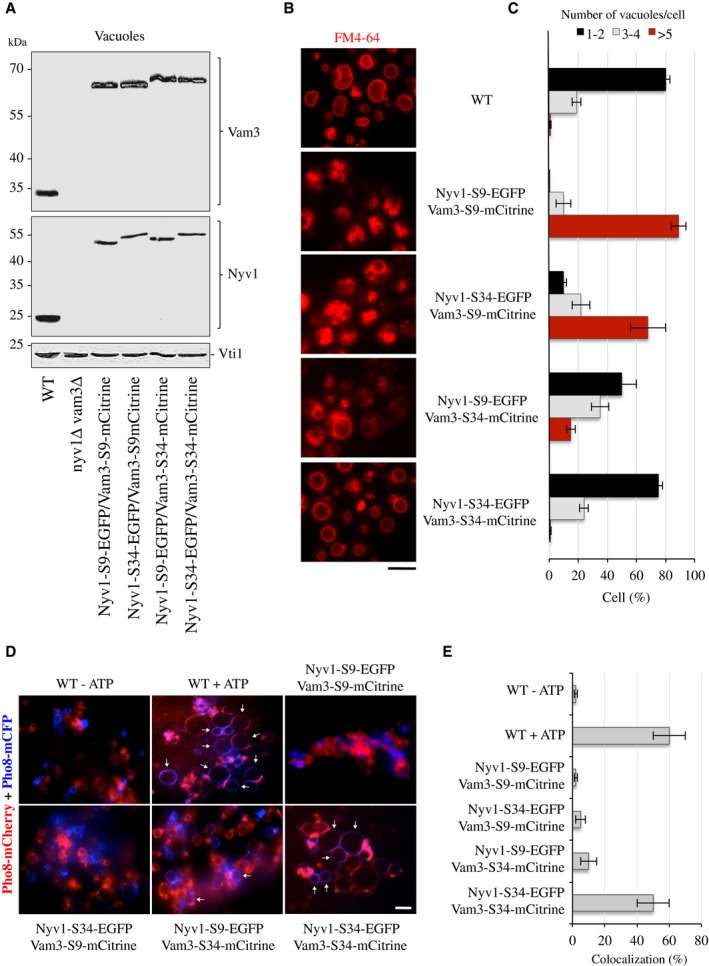
- Protein levels on isolated vacuoles from these strains were compared as in Fig 3B.
- Vacuole morphology was assessed as in Fig 3C. Scale bar: 5 μm.
- The cells were grouped into three categories according to the number of vacuoles visible per 100 cells. Values represent the means and s.d. from three independent experiments.
- Fusion activity: Vacuoles were isolated from BJ3505 strains expressing the indicated versions of Vam3 and Nyv1 and Pho8‐mCFP or Pho8‐mCherry. Ten micrograms of vacuoles was incubated in standard fusion reactions in the presence or absence of ATP and analyzed by confocal microscopy. Arrows indicate examples of fusion products. Scale bar: 5 μm.
- Vacuole fusion was assayed by measuring the percentage of colocalization of the Pho8‐mCFP and Pho8‐mCherry signals. Means ± s.d. are shown for at least 100 vacuoles from three experiments.
Chlorpromazine rescues fusion pore opening
Membrane curvature modulates the efficiency of SNARE‐mediated fusion. Chlorpromazine (CPZ) is an amphipathic molecule that inserts into membranes, perturbs bilayer structure, and increases membrane fluidity 45. It changes spontaneous membrane curvature and promotes the hemifusion‐to‐fusion transition 46, 47. Since vacuole fusion depends on non‐bilayer lipids 48 and CPZ can promote vacuole fusion in vitro 49, we tested whether CPZ might suffice to lift the fusion block imposed by SNAREs with bulky protein tags in vivo. In order to verify that CPZ reached the vacuoles in living yeast cells, we measured its effect on vacuolar membrane fluidity in wild‐type cells, using the diffusion of the vacuolar transmembrane protein Vph1‐GFP as a readout. Vph1‐GFP was bleached in a small region of the vacuolar membrane by a laser pulse. Fluorescence recovery after photobleaching (FRAP) was recorded in this area over the next seconds. CPZ reduced the half‐time for fluorescence recovery from 4 to 1.8 s (Fig 8). Thus, CPZ reaches the vacuolar membrane.
Figure 8. Effect of chlorpromazine on fusion activity and morphology of double‐tagged vacuoles.
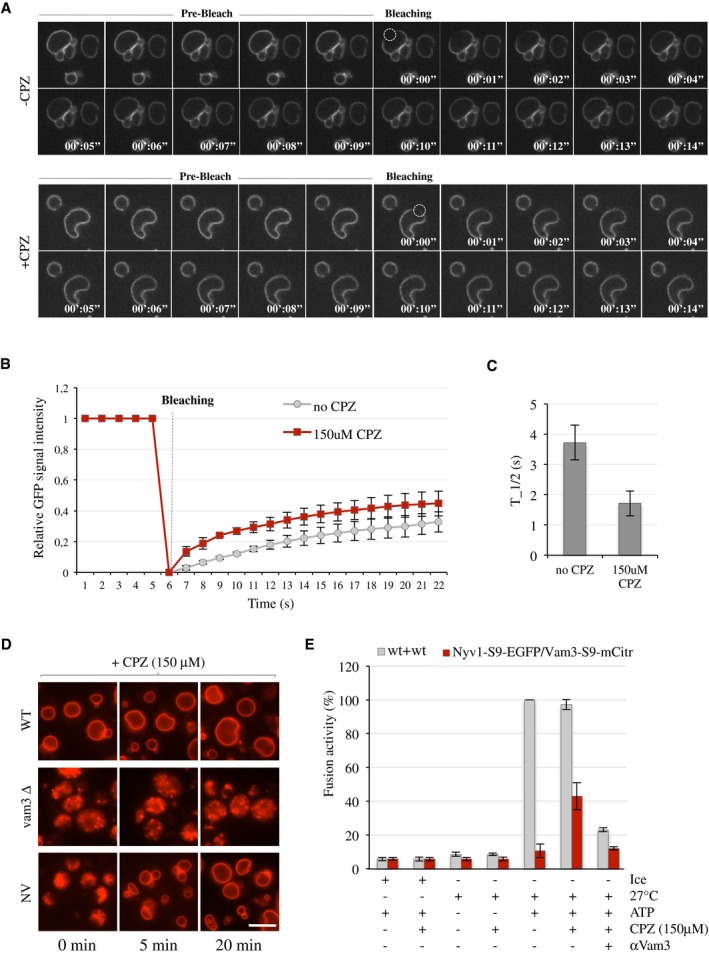
- Yeast strain expressing a plasmid with Vph1‐GFP was grown in SC‐URA medium for 16 h at 30°C. The mobility of Vph1‐GFP in the absence (upper panels) or presence (lower panel) of CPZ (150 μM) was assessed by FRAP.
- The histogram shows the GFP signals in the bleached areas during the FRAP procedure. Means ± s.d. are shown for 20 cells of three independent experiments.
- The half‐time is shown. Means and s.d. are shown for 20 cells of three independent experiments.
- Yeast cells were grown in SC medium for 16 h at 30°C. After staining with FM4‐64, cells were treated with CPZ (150 μM). Vacuole morphology was assessed by confocal microscopy before and after 5 and 20 min of CPZ addition. Scale bar: 5 μm.
- Fusion activity: Vacuoles were isolated from BJ cells carrying NyV1‐S9‐EGFP and DKY cells deleted for Nyv1 and carrying VaM3‐S9‐mCitrine or from wild‐type cells and incubated in standard fusion reactions in the presence or absence of chlorpromazine (CPZ, 150 μM) and anti‐Vam3 (3 μg). Content mixing was assayed via alkaline phosphatase activity. Means and s.d. are shown for three independent experiments.
Chlorpromazine rescued the fragmented vacuolar morphology of cells carrying VAM3‐S9‐mCitrine and NYV1‐S9‐EGFP. Within 5 min after the addition of CPZ, the vacuoles began to fuse and formed one large organelle within 20 min of incubation (Fig 8D). CPZ treatment did not induce fusion in cells lacking Vam3, suggesting that in the absence of a hemifusion structure, CPZ is not sufficient to facilitate vacuole fusion in living cells. In line with these in vivo observations, CPZ also rescued the in vitro fusion activity between Nyv1‐S9‐EGFP and Vam3‐S9‐mCitrine vacuoles to ≈40% of the wild‐type value (Fig 8E). Simultaneous addition of antibodies to the SNARE Vam3, which prevent hemifusion 26, abrogated this rescue. Thus, pharmacological perturbation of vacuolar lipid structure in vivo can partially overcome the inhibition of fusion pore opening imposed by bulky luminal protein tags at the C‐termini of vacuolar SNAREs.
Discussion
The vacuole system provides good tools to assay the abundance of the tagged SNAREs on the isolated organelle, dissect their molecular interactions, and to identify hemifusion intermediates. In contrast to previous studies on tagged synaptobrevin II in chromaffin cells, in which these molecular properties are hard to access 40, this allowed us to demonstrate that the effect of large luminal tags was restricted to content mixing, whereas lipid mixing was essentially unaffected. This suggests that mixing of the outer leaflets may be less dependent on a collective perturbation of lipid structure by SNARE TMDs than the rearrangement of the inner leaflets. It is consistent with theory and simulations on the energetics of SNARE‐driven fusion, which suggested that fusion pore opening is limited by a larger free energy barrier than the induction of hemifusion 38. In line with this, opening of the fusion pore has been found to be rate‐limiting for vacuole fusion 22.
The observation that even bulky protein tags do not inhibit vacuole fusion if they are attached to only one side of the trans‐SNARE complex suggests that fusion pore opening can be induced by both the Q‐ and R‐SNAREs. There is apparently not a gross differentiation of the fusogenic potential between Q‐ and R‐SNARE, despite the fact that the sequences of their TMDs are different. This does not support a sequence‐specific contribution of the SNARE TMDs and argues in favor of local mechanical force transduction as a key factor for the action of SNARE TMDs in vacuole fusion 26. It remains, however, in stark contrast to the situation found in regulated exocytosis, where tagging of the R‐SNARE synaptobrevin II alone inhibited exocytosis 39 and where sequence changes in the SNARE TMDs can influence fusion pore dynamics 50, 51. This difference may reflect the highly specialized nature of the machinery for regulated exocytosis, which is tuned for speed and tight temporal control 52. It also operates with a number of accessory factors which seem not to play a role for other SNARE‐driven fusion reactions, such as complexin and synaptotagmin. Another factor might be that in exocytosis the two fusing membranes are quite different in composition and curvature. Therefore, one may speculate that the plasma membrane could be less easily perturbed by pulling or bending forces transmitted by its resident Q‐SNAREs than the vesicle membrane. In vacuole fusion, by contrast, both bilayers are identical. This excludes effects of differences in bilayer properties and allows to directly compare the potential of Q‐ and R‐SNARE TMDs for fusion pore opening.
The force that the C‐termini exert on the membrane ultimately depends on the ability of the SNARE complex to store mechanical energy; that is, the SNARE molecules must be able to fold into a partly zipped state and simultaneously adopt or conserve a sufficiently stiff helical structure to permit force transduction to their TMDs. Other vacuolar fusion factors, such as the SM protein containing HOPS complex, assist fusion by tethering the membranes but, in addition, they may promote pore opening by enhancing SNARE zipping 25, 53, 54. Upon SNARE complex zippering, the TMDs of trans‐complexes should experience bending and pulling forces 23, 28, 55, 56, 57, 58. These forces might provoke hemifusion by inducing local lipid disorder in the outer leaflets and subsequently promote thinning and widening of the stalk to open a fusion pore. Coarse‐grained simulations and biophysical and electron microscopic studies elucidated a number of different fusion pathways and led to different suggestions concerning the behavior of the TMDs during the fusion process 32, 38, 39, 59. It has been proposed that the C‐terminus of synaptobrevin II perforates the hydrophobic core of the bilayer as a result of SNARE zippering 40. The proposal was based on the observation that the addition of only two charged residues to the C‐terminus of synaptobrevin II interferes with exocytosis in chromaffin cells. At first sight, this model appears compatible with the effects of large C‐terminal tags in vacuole fusion, but there are arguments that stand against it. We could attach small HA or myc tags to vacuolar SNAREs, thereby adding up to five charged residues to the C‐terminus, without causing significant effects on fusion. Unlike in chromaffin cells, where attachment of a tag to only a single SNARE had profound effects, vacuole fusion remains almost unaffected if a tag is attached to only one side of the trans‐SNARE complex. And even if both sides are tagged, the tag must include a large hydrophilic protein that is attached very close to the C‐terminus in order to arrest pore opening.
Our simulations suggest that the addition of small peptide tags renders membrane perforation by the C‐termini energetically virtually impossible. This is in agreement with the simulations of Lindau et al 39. However, in order to provide an argument for the perforation model, two requirements would have to be met: (i) The force at the perforation threshold must be less than the force required to remodel the membrane during progression from hemifusion to pore expansion (Figs 1 and 9A), and (ii) the C‐termini of a SNARE complex must be able to exert a substantial force of about 80 pN. Based on our simulations, we estimated that the force that a vacuolar SNARE complex can exert via its C‐termini on, for example, a stalk‐like intermediate is approximately 9–18 pN (Fig 2A). This force is of similar magnitude as the experimentally estimated force of about 17 pN that is required to unzip the coiled‐coil domain of the neuronal SNARE complex 31. Thus, it is very unlikely that forces of 80 pN, which are required to perforate the membrane, are accessible for the wild‐type SNARE complex.
Figure 9. Model for the movement of SNARE TMDs during vacuole fusion.
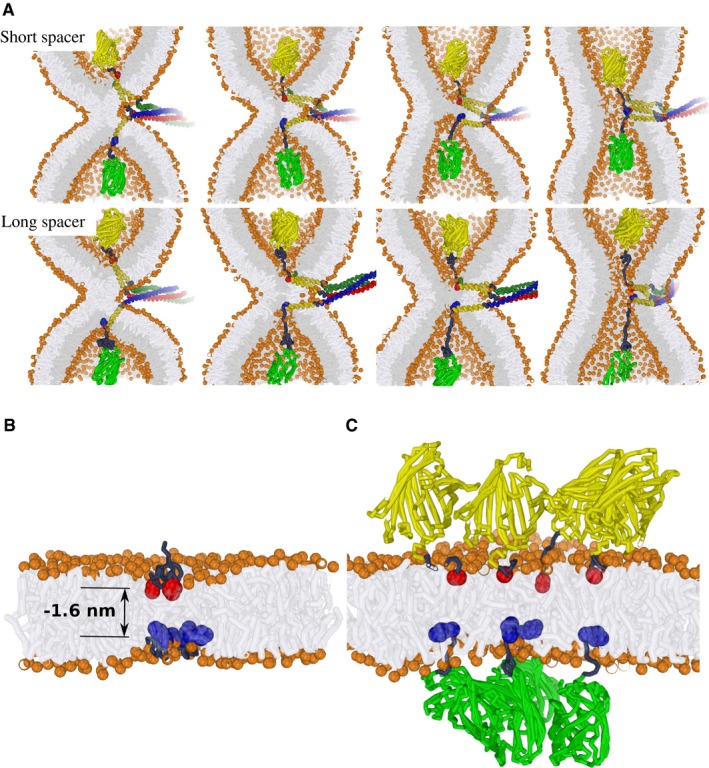
-
AExamples of simulated hemifusion‐to‐fusion transitions in the presence of modified vacuolar SNARE complexes. Upper panel: Short spacer. Lower panel: Long spacer. These stirred molecular dynamics simulations illustrate that the progression of fusion is sterically achievable for both of the fluorescent protein‐tagged SNARE complexes, even when the geometry of the fusion zone is extremely confined, that is, the fusion between two highly curved membrane dimples (1/8 nm−1). This scenario suggests that the fusion‐inhibiting effect of a fluorescent protein added via a short peptide spacer is unlikely to result from a direct “steric clash” with the membrane. Vam3 in red/yellow; Nyv1 in blue/yellow; Vti1/Vam7 in dark green and yellow; bulky fluorescent protein tags in light green and yellow, respectively.
-
B, CA scenario simulating the presence of four SNARE complexes near the fusion site. The modeled indentation corresponds to a relative indentation of −1.6 nm (see Fig 2B). This corresponds to an exaggerated indentation force of about 80 pN which we exploit to demonstrate the clustering between “indentations” within our limited simulation time. The C‐terminal amino acid of each Vam3 and Nyv1 molecule is represented by a red or blue bead, respectively; bulky fluorescent protein tags in light green and yellow, respectively. (B) Isolated SNARE C‐termini with short peptide tag. The C‐termini concentrate in a small zone (similar membrane perturbations attract each other). (C) Steric hindrance by the addition of bulky proteins prevents collective, localized force transduction by multiple SNARE complexes.
Could multiple SNARE complexes collaborate to overcome the threshold for perforation? We consider this as unlikely because “perforation” relates more to local pressure—force divided by the effective cross‐sectional area of the C‐terminus—than to force itself. For example, when multiple SNARE complexes collectively exert a force of about 80 pN on the membrane, the pressure directly “under” each individual C‐terminus is still similar to that of a single, isolated SNARE complex (even when the C‐termini cluster) and will thus remain below the perforation threshold. Although additional hydrophilic residues markedly increase the perforation threshold of the C‐termini, this does not affect the force required to indent the membrane. Our observation that C‐termini with small tags preserve vacuole fusion activity therefore suggests that progression of fusion does not involve active perforation. It is consistent with the hypothesis that the TMDs drive an active remodeling of the membrane via indentation. In order to facilitate such a remodeling mechanism, the SNAREs must be able to exert sufficient force on the membrane via their C‐termini without perforating it.
Why does the addition of bulky 30 kDa fluorescent protein domains to the C‐termini of SNAREs arrest the reaction at the hemifusion stage only when the domain is attached via a short peptide spacer but not when it is attached via a long spacer? Our simulations suggest that, even in case of a sterically confined (highly curved) fusion site, adding this bulky protein to the short peptide spacer is unlikely to hinder membrane indentation (Fig 9A). This intuition is confirmed by the corresponding fusion energetics which revealed that the SNARE complex required the same work to open the fusion pore (~20 KBT, see Fig EV2). Thus, the bulky tags seem not to interfere with force transduction of a single SNARE complex. Therefore, we stipulate that attachment of the bulky fluorescent protein very likely interferes with the local organization of multiple SNARE complexes at the fusion site. To this aim, we studied the self‐assembly between four membrane “indentations” in both the absence and presence of the bulky fluorescent protein. These “indentations” mimic the squeezing of the membrane by the C‐termini of Nyv1 and Vam3. Such a “squeezing” facilitates strong lateral clustering of the present SNARE C‐termini (Fig 9B). The observed clustering is driven by a favorable reduction of membrane elastic energy—isolated membrane indentations are energetically unfavorable (this effect is similar to the hydrophobic mismatch‐driven clustering of transmembrane proteins). Notably, clustering facilitates a collective transduction of the point‐like forces that the C‐termini exert on the membrane, and which can thereby facilitate an indentation‐mediated opening of the fusion pore.
Figure EV2. Energetics of SNARE‐mediated fusion.
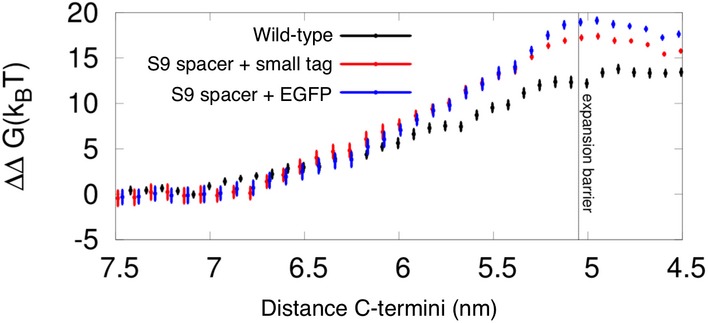
Shown is the work required to open the fusion pore in both the absence and presence of small or large tags at the C‐termini. The system corresponds to the setup presented in Fig 9, that is, the SNARE‐mediated fusion between two highly curved (POPC) dimples (1/8 nm−1). The plateau (stalk expansion barrier) marks the distance at which fusion proceeds spontaneously and without the need for mechanical work performed by the SNARE complex. Adding the large tags does not significantly alter the work required to open the fusion pore. Note that this method relies on an irreversible, nucleated transition. Therefore, an inaccuracy of about ± 5 KbT should be taken into account within the obtained work profile. For further technical details, see 38.
Our simulations further suggest that bulky fluorescent protein tags at the C‐termini sterically oppose a close lateral assembly of multiple SNARE complexes around the fusion site (Fig 9C). The SNARE complexes must therefore distribute the point‐like forces exerted by the TMDs over a larger membrane area, preventing the respective indentations caused by them from self‐assembling and reducing the capacity of the TMDs to deform the inner leaflet. In this sense, the large luminal tags can counteract the collective force transduction of SNARE domains to the inner leaflets in a scenario were several SNARE complexes cooperate in opening the fusion pore.
Overall, our results are quite compatible with the results and conclusions from recent simulations on SNARE‐driven membrane fusion 38. They provide experimental support for the model that the TMDs from several SNARE complexes must exert highly localized force on the lipids in the inner leaflet in order to open and expand the fusion pore, for example, by thinning the stalk and promoting lipid reorientation.
Materials and Methods
Strains and culture conditions
All strains were grown in either in YPD (yeast extract, peptone, dextrose) containing 2% glucose in the presence or absence of G418, or in SC (synthetic complete) medium containing 2% glucose in the presence or absence of the appropriate supplements to select for auxotrophies.
All strains used in this study can be found in Table 1. Primers used can be found in Table 2. For the generation of Nyv1‐S9‐EGFP, Nyv1‐S34‐EGFP and Nyv1‐S9‐HA strains we used pKT209 and pUG6H3HA plasmids as a template, while for the generation of Vam3‐S9‐mCitrine, Vam3‐S34‐mCitrine, and Vam3‐S9‐HA strains, we used pKT140 and pU6H3HA plasmids, respectively.
Table 1.
Strains used
| Strain | Genotype | References |
|---|---|---|
| BJ3505 | MATa pep4::HIS3 prb1‐Δ1.6R lys2‐208 trp1‐Δ101 ura3‐52 gal2 can | 65 |
| DKY6281 | MATα pho8::TRP1 leu2‐3 leu2‐112 lys2‐ 801 suc2‐Δ9 trp1‐Δ901 ura3‐52 | 65 |
| BJ Nyv1‐S9‐EGFP | BJ3505; Nyv1‐S9‐EGFP (URA) | This study |
| BJ Vam3‐S9‐mCitrine | BJ3505; Vam3‐S9‐mCitrine (G418) | This study |
| BJ Nyv1‐S9‐EGFP + Vam3‐S9‐mCitrine | BJ3505; Nyv1‐S9‐EGFP (URA), Vam3‐S9‐mCitrine (G418) | This study |
| BJ Nyv1‐S9‐HA | BJ3505; Nyv1‐S9‐His6(HA)3 (G418) | 25 |
| BJ Vam3‐S9‐myc | BJ3505; Vam3‐S9‐His6(myc)2 (URA) | 25 |
| BJ Nyv1‐S9‐HA + Vam3‐S9‐myc | BJ3505; Nyv1‐S9‐His6(HA)3 (G418), Vam3‐S9‐His6(myc)2 (URA) | This study |
| DKY ∆nyv1 | DKY6281; Nyv1::Nat | This study |
| DKY ∆nyv1 + Vam3‐S9‐mCitrine | DKY6281; Nyv1::Nat, Vam3‐S9‐mCitrine (G418) | This study |
| BJ Nyv1‐S34‐EGFP | BJ3505; Nyv1‐S34‐EGFP (URA) | This study |
| BJ Vam3‐S34‐mCitrine | BJ3505; Vam3‐S34‐mCitrine (G418) | This study |
| BJ Nyv1‐S34‐EGFP + Vam3‐S9‐mCitrine | BJ3505; Nyv1‐S34‐EGFP (URA), Vam3‐S9‐mCitrine (G418) | This study |
| BJ Nyv1‐S9‐EGFP + Vam3‐S34‐mCitrine | BJ3505; Nyv1‐S9‐EGFP (URA), Vam3‐S34‐mCitrine (G418) | This study |
| BJ Nyv1‐S34‐EGFP + Vam3‐S34‐mCitrine | BJ3505; Nyv1‐S34‐EGFP (URA), Vam3‐S34‐mCitrine (G418) | This study |
| BJ Nyv1‐HA + Vam3‐myc + Pho8‐GFP | BJ3505; Nyv1‐His6(HA)3 (G418), Vam3‐His6(myc)2 (URA) Pho8‐GFP (TRP) | This study |
| BJ Nyv1‐HA + Vam3‐myc + Pho8‐mCherry | BJ3505; Nyv1‐His6(HA)3 (G418), Vam3‐His6(myc)2 (URA) Pho8‐mCherry (TRP) | This study |
| BJ Pho8‐mCFP | BJ3505; Pho8‐mCFP (LYS) | This study |
| BJ Pho8‐mCherry | BJ3505; Pho8‐mCherry (TRP) | This study |
| BJ Nyv1‐S34‐EGFP + Vam3‐S34‐mCitrine + Pho8‐mCFP | BJ3505; Nyv1‐S34‐EGFP (URA), Vam3‐S34‐mCitrine (G418) Pho8‐mCFP (LYS) | This study |
| BJ Nyv1‐S34‐EGFP + Vam3‐S34‐mCitrine + Pho8‐mCherry | BJ3505; Nyv1‐S34‐EGFP (URA), Vam3‐S34‐mCitrine (G418) Pho8‐mCherry (TRP) | This study |
| BJ Nyv1‐S34‐EGFP + Vam3‐S9‐mCitrine + Pho8‐mCFP | BJ3505; Nyv1‐S34‐EGFP (URA), Vam3‐S9‐mCitrine (G418) Pho8‐mCFP (LYS) | This study |
| BJ Nyv1‐S34‐EGFP + Vam3‐S9‐mCitrine + Pho8‐mCherry | BJ3505; Nyv1‐S34‐EGFP (URA), Vam3‐S9‐mCitrine (G418) Pho8‐mCherry (TRP) | This study |
| BJ Nyv1‐S9‐EGFP + Vam3‐S34‐mCitrine + Pho8‐mCFP | BJ3505; Nyv1‐S9‐EGFP (URA), Vam3‐S34‐mCitrine (G418) Pho8‐mCFP (LYS) | This study |
| BJ Nyv1‐S9‐EGFP + Vam3‐S34‐mCitrine + Pho8‐mCherry | BJ3505; Nyv1‐S9‐EGFP (URA), Vam3‐S34‐mCitrine (G418) Pho8‐mCherry (TRP) | This study |
Table 2.
Primers used
| Strain | Primers |
|---|---|
| Nyv1‐S9‐EGFP | Fw: 5′‐CTATTTGTAAGTGCTGCTTTCATGTTTTTCTATCTGTGGGGTGACGGTGCTGGTTTA‐3′ |
| Rv: 5′‐GTTATTGTCGTGGGACAGCTCCCCTTTTTTTTTATTTACTCGATGAATTCGAGCTCG‐3′ | |
| Nyv1‐S34‐EGFP | Fw: 5′‐CTATTTGTAAGTGCTGCTTTCATGTTTTTCTATCTGTGGAGTCTAAGTGGTGGTGGTGGTTCTGGAGGAGGAGGATCCGGTGGTGGAGGAAGTGGAGGTGGAGGTGCTGCCGCAGGTGACGGTGCTGGTTTA‐3′ |
| Rv: 5′ ‐GTTATTGTCGTGGGACAGCTCCCCTTTTTTTTTATTTACTCGATGAATTCGAGCTCG‐3′ | |
| Vam3‐S9‐mCitrine | Fw: 5′‐AATAGTTGTGTGCATGGTGGTATTGCTTGCTGTATTAAGTGGTGACGGTGCTGGTTTA‐3′ |
| Rv: 5′‐TACCAGAAAGTCTGTGCTCAATGCGCGTTTAAGGAGATTATCGATGAATTCGAGCTCG‐3′ | |
| Vam3‐S34‐mCitrine |
Fw: 5′‐TAATCATTATAATAGTTGTGTGCATGGTGGTATTGCTTGCTGTATTAAGTCTAAGTGGTGGTGGTGGTTCTGGAGGAGGAGGATCCGGTGGTGGAGGA AGTGGAGGTGGAGGTGCTGCCGCAGGTGACGGTGCTGGTTTAATTAAC‐3′ |
| Rv: 5′‐CGAGCTCGAATTCATCGATAATCTCCTTAAACGCGCATTGAGCACAGACTTTCTGGTAGACCCAATCTTATCTATTTA‐3′ | |
| Nyv1‐HA | Fw: 5′‐ATTATACTATTTGTAAGTGCTGCTTTCATGTTTTTCTATCTGTGGTCCCACCACCATCATCATCAC‐3′ |
| Rv: 5′‐GTAAATAAAAAAAAAGGGGAGCTGTCCCACGACAATAACATTAATACTATAGGGAGACCGGCAGATC‐3′ |
Vacuole isolation
BJ3505 and DKY6281 strains carrying tagged SNAREs were grown in YPD at (30°C, 225 rpm) to OD600 = 1 and harvested (3 min, 5,000 g). Vacuoles were isolated through hydrolyzing yeast cell walls using lyticase, recombinantly expressed in E. coli RSB805 (provided by Dr. Randy Schekman, Berkeley) and prepared from a periplasmic supernatant 21. Harvested cells were resuspended in reduction buffer (30 mM Tris/Cl pH 8.9, 10 mM DTT) and incubated for 5 min at 30°C. After harvesting as described above, cells were resuspended in 15 ml digestion buffer (600 mM sorbitol, 50 mM K‐phosphate pH 7.5 in YP medium with 0.2% glucose and 0.1 mg/ml lyticase preparation). After 25 min at 30°C, cells were centrifuged (2 min, 2,500 g, JLA25.5 rotor). The spheroplasts were resuspended in 2 ml 15% Ficoll‐400 in PS buffer (10 mM PIPES/KOH pH 6.8, 200 mM sorbitol) and 250 μl DEAE dextran (0.4 mg/ml in PS). After 2 min of incubation at 30°C, the cells were transferred to SW41 tubes and overlaid with steps of 8, 4, and 0% Ficoll‐400 in PS. Cells were centrifuged for 90 min at 4°C and 150,000 g in a SW41 rotor.
Vacuole fusion
DKY6281 and BJ3505 vacuoles were adjusted to a protein concentration of 0.5 mg/ml and incubated in a volume of 30 μl PS buffer (10 mM PIPES/KOH pH 6.8, 200 mM sorbitol) with 125 mM KCl, 0.5 mM MnCl2, 1 mM DTT. Inhibitors were added before starting the fusion reaction by addition of the ATP‐regenerating system (0.25 mg/ml creatine kinase, 20 mM creatine phosphate, 500 μM ATP, 500 μM MgCl2). After 60 min at 27°C, or on ice, 1 ml of PS buffer was added, vacuoles were centrifuged (2 min, 20,000 g, 4°C) and resuspended in 500 μl developing buffer (10 mM MgCl2, 0.2% Triton X‐100, 250 mM Tris–HCl pH 8.9, 1 mM p‐nitrophenyl phosphate). After 5 min at 27°C, the reactions were stopped with 500 μl 1 M glycine pH 11.5 and the OD was measured at 405 nm.
Microscopic fusion assay
Vacuoles were prepared from BJ3505 cells carrying different tagged variants of SNAREs and different fluorescently tagged alkaline phosphatase (Pho8‐EGFP, Pho8‐mCherry, or Pho8‐mCFP). Three micrograms each of vacuoles carrying a single fluorescently tagged Pho8 were mixed in fusion reactions with three micrograms of vacuoles carrying a Pho8 with a different fluorescent tag. At the end of the 60‐min incubation period, the samples were analyzed by spinning disc fluorescence microscopy. Fusion efficiency was determined by measuring the percentage of colocalization of two fluorescently tagged Pho8 variants by using the ImageJ colocalization plugin.
Lipid and content mixing assay
Lipid and content mixing were assayed as described 21. In brief, 30 μg of unlabeled BJ3505 vacuoles and 6 μg of rhodamine‐labeled phosphoethanolamine DKY6281 vacuoles were mixed in 190 μl of 0.3 mM MnCl2, 75 mM KCl in PS buffer. Inhibitors were pre‐warmed to 37°C before adding to the tubes. Anti‐Vam3 was used at 0.13 μM. Fusion reactions were started by adding 9.5 μl of 20× ATP‐regeneration system, yielding 0.125 mg/ml creatine kinase, 20 mM creatine phosphate, 0.5 mM ATP, 0.5 mM MgCl2. One hundred microliters was used to assay lipid mixing in a fluorescent plate reader at 27°C for 32 min. 80 μl were incubated separately for 60 min prior to adding alkaline phosphatase developing buffer for 5 min.
Immunoprecipitations
Vacuoles from a 1 ml of fusion reaction were pelleted (5 min, 6,000 g, 4°C), solubilized for 10 min in lysis buffer (0.5% Triton X‐100, 0.5 mM MnCl2, 100 mM CaCl2, 1 mM PMSF), and centrifuged for 10 min at 12,000 g and 4°C. The supernatant was supplemented with 30 μg of anti‐Nyv1 antibody and 25 μl of protein G Sepharose and shaken for 60 min at 4°C. The beads were washed three times with lysis buffer and suspended in SDS sample buffer.
Gel electrophoresis and Western blot
Protein samples were dissolved in reducing sample buffer and heated to 95°C for 5 min. The samples were run on either 10 or 12.5% polyacrylamide gels. The stacking gels were prepared as follows: 6% acrylamide, 0.16% bis‐acrylamide, 0.1 M Tris pH 6.8, 0.1% SDS, 0.1% TEMED, 0.05% ammonium persulfate. Running gels were as follows: 10 or 12.5% acrylamide, 0.27 or 0.34% bis‐acrylamide, 0.38 M Tris pH 8.8, 0.1% SDS, 0.06% TEMED, 0.06% APS. The gels were run at constant current (25–35 mA). Proteins were blotted on nitrocellulose membrane by the semidry method for 80 min at 400 mA. After incubation with the primary antibody overnight, the signals were detected by secondary antibodies coupled to IR dyes.
Western blot images
The Western blot images were taken on a LI‐COR Odyssey Infrared Imager. The files were exported as TIFF and processed in Adobe Illustrator CS3. The band intensity was quantified using densitometry software supplied with the Odyssey Infrared Imager.
FM4‐64 staining
Cells were inoculated from a pre‐culture in stationary phase and grown overnight to logarithmic phase (OD600 between 0.2 and 0.8). After dilution to an OD600 of 0.2 in 1 ml of culture, FM4‐64 in DMSO was added to a final concentration of 10 μM. Cells are stained for one hour, followed by three washing steps in medium without stain (2 min, 3,000 g) and a subsequent chase of 1–2 h in medium without stain, depending on the endocytotic capacity of the strain. The cells for microscopy were grown at 30°C. The temperature was kept constant during staining and visualization.
FRAP analysis
FRAP experiments were performed with the Photokinesis unit on the Ultra‐VIEW Vox confocal system. The full‐size non‐constricting ring was selected, and the middle slice from the z‐stacks was used for bleaching. After collecting two pre‐bleaching images, a selected region of interest (ROI) was bleached to > 80% of the original signal by 20 ms of a 100% 546 nm or 488 nm laser pulse. Post‐bleaching images were collected each second for a total period of 10 s. After subtracting the background and correcting for the photobleaching during image acquisition (using the intensity of unbleached cells), the ROI intensity was normalized with the mean pre‐bleached intensity set to 100%.
Simulation model and settings
The molecular dynamics simulations were performed with the GROMACS simulation package 60, version 4.5.7. We used the MARTINI coarse‐grained model 61, 62 to simulate the lipids, amino acids and solvent. In all simulations, the system was coupled to a constant temperature bath using the “vscale” algorithm with a relaxation time of 1.0 ps. We performed our simulations at a temperature of 293 K. Periodic boundary conditions were applied to simulate bulk behavior. The time step used in the simulation was 20 fs. The dielectric constant in the simulations was εr = 15. The neighbor list was updated every 10 simulation steps. The pressure was weakly coupled to one bar with a relaxation time of 1.0 ps.
Modeling the modified vacuolar SNARE complex
The vacuole SNARE complex was modeled using the MARTINI model for proteins 62, which qualitatively captures the chemical nature of each individual amino acid and includes the secondary structure.
For NYV1, the modeled sequence is “IGDATEDQIK DVIQIMNDNI DKFLERQERV SLLVDKTSQL NSSSNKFRRK AVNIKEIMWW [QKVKN]ITLLT FTIILFVSAAF MFFYLW”; for VAM3, “TIIHQERSQQ IGRIHTAVQE VNAIFHQLGS LVKEQGEQVT TIDENISHLH DNMQNANKQL TRA[DQHQRDRNK] CGKVTLIIII VVCMVVLLAV LS”; for VTI1, “IDDDQRQQLL SNHAILQKSG DRLKDASRIA NETEGIGSQI MMDLRSQRET LENARQTLFQ ADSYVDKSIK TLKTMTR [RLVANK]FISY AIIAVLILLI LLVLFSKFK”; and for VAM7, “MQMVRDQEQE LVALHRIIQA QRGLALEMNE ELQTQNELLT ALEDDVDNTG RRLQIANKKA RHF”. Here, the brackets [] depict the defined linker regions. The resolved and earlier simulated structure 63 of the neuronal SNARE complex was used as a template structure for the vacuolar SNARE complex. To this aim, we applied an external field, using a self‐modified version of Gromacs, to drive the structure of the vacuole SNARE complex toward the known structure of the neuronal SNARE complex based on the known alignment. All residues are defined α‐helical except for the defined SNARE linkers (random coil “~” or α‐helical “H”). The linkers which connected the Nyv1 and Vam3 C‐termini with the fluorescent proteins (EGFP and mCitrine) were modeled as a random coil (“~”). We conserved the resolved ternary structure of the fluorescent proteins (PDB: 1GFL and 1HUY) in our simulations. To this aim, we included an additional elastic network between backbone residues; that is, we connected all backbone beads within a lower cutoff of 0.5 nm and higher cutoff of 0.9 nm with an elastic bond (Kforce = 500 kJ nm−2 mol−1). This network included all “structured” backbone beads starting from LYS3 in EGFP, and SER2 in mCitrine.
Membrane simulation setups
To study the effect of the fluorescent proteins, we performed simulations of the isolated fluorescent protein+spacer+C‐termini in POPC membranes consisting of 512 lipids. Membrane indentations are obtained by bringing the C‐termini in closer proximity and thereby squeezing the membrane. The hydrophobic thickness of the membrane is about 4.0 nm (defined as the distance between the two phosphate planes). However, “squeezing” also requires work when the distance between the C‐termini is about 5.0 nm (albeit minor). The latter is an artifact introduced by excessive rotational entropy at large separation distances (an additional rotational axis is introduced because of excessive in‐membrane “tilting” of the vector which connects the C‐termini). To this aim, we define a separation distance of 4.1 nm as the zero point (i.e., a relative indentation of 0 nm) within our work (free energy) profile. Note that the force, that is, the slope of the work profile, is completely independent of the choice of such an offset.
To derive the free energy, we applied umbrella simulations techniques in combination with the weighted histogram method (see 38 for technical details). The isolated C‐terminus representing the wild type is charged in these simulation, whereas the C‐termini representing the modified SNAREs are modeled electrically neutral. The presented “relative spacer effects” are obtained by extracting the data of the wild type from the two other data sets using the gnuplot software. As a result, the line y = 0 now represents the effect of the wild type (curvilinear coordinate system).
The “dimple” or “vesicle” fusion setup consisted of 2217 POPC lipids. This setup is described in detail in 5. We artificially pulled the Nyv1 and Vam3 C‐termini in these stirred simulations with a rate of 5 × 10−5 nm/ps (Kforce = 1000 kJ nm−2mol−1).
Statistics
For all the experiments not involving simulation, data points with error bars represent the mean of at least three independent experiments. Error bars represent the standard deviation. The significance of differences was evaluated by an unpaired Student's t‐test. Differences are interpreted only if P < 0.005.
Author contributions
MDA and HJR performed the experiments and assembled the figures. MDA, HJR, and AM conceived the experiments, interpreted the data, and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Acknowledgements
We thank the Swiss National Science Foundation and the ERC for funding AM and the State of Lower Saxony (life@nano initiative) for supporting HJR.
EMBO Reports (2016) 17: 1590–1608
References
- 1. Wickner W (2010) Membrane fusion: five lipids, four SNAREs, three chaperones, two nucleotides, and a Rab, all dancing in a ring on yeast vacuoles. Annu Rev Cell Dev Biol 26: 115–136 [DOI] [PubMed] [Google Scholar]
- 2. Alpadi K, Kulkarni A, Comte V, Reinhardt M, Schmidt A, Namjoshi S, Mayer A, Peters C (2012) Sequential analysis of Trans‐SNARE formation in intracellular membrane fusion. PLoS Biol 10: e1001243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dietrich LEP, Peplowska K, LaGrassa TJ, Hou H, Rohde J, Ungermann C (2005) The SNARE Ykt6 is released from yeast vacuoles during an early stage of fusion. EMBO Rep 6: 245–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ungermann C, Nichols BJ, Pelham HR, Wickner W (1998) A vacuolar v‐t‐SNARE complex, the predominant form in vivo and on isolated vacuoles, is disassembled and activated for docking and fusion. J Cell Biol 140: 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mayer A, Wickner W, Haas A (1996) Sec18p (NSF)‐driven release of Sec17p (alpha‐SNAP) can precede docking and fusion of yeast vacuoles. Cell 85: 83–94 [DOI] [PubMed] [Google Scholar]
- 6. Boeddinghaus C, Merz AJ, Laage R, Ungermann C (2002) A cycle of Vam7p release from and PtdIns 3‐P‐dependent rebinding to the yeast vacuole is required for homotypic vacuole fusion. J Cell Biol 157: 79–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mayer A, Wickner W (1997) Docking of yeast vacuoles is catalyzed by the Ras‐like GTPase Ypt7p after symmetric priming by Sec18p (NSF). J Cell Biol 136: 307–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hickey CM, Wickner W (2010) HOPS initiates vacuole docking by tethering membranes before trans‐SNARE complex assembly. Mol Biol Cell 21: 2297–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hickey CM, Stroupe C, Wickner W (2009) The major role of the Rab Ypt7p in vacuole fusion is supporting HOPS membrane association. J Biol Chem 284: 16118–16125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Price A, Seals D, Wickner W, Ungermann C (2000) The docking stage of yeast vacuole fusion requires the transfer of proteins from a cis‐SNARE complex to a Rab/Ypt protein. J Cell Biol 148: 1231–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Seals DF, Eitzen G, Margolis N, Wickner WT, Price A (2000) A Ypt/Rab effector complex containing the Sec1 homolog Vps33p is required for homotypic vacuole fusion. Proc Natl Acad Sci USA 97: 9402–9407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Starai VJ, Hickey CM, Wickner W (2008) HOPS proofreads the trans‐SNARE complex for yeast vacuole fusion. Mol Biol Cell 19: 2500–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ungermann C, Price A, Wickner W (2000) A new role for a SNARE protein as a regulator of the Ypt7/Rab‐dependent stage of docking. Proc Natl Acad Sci USA 97: 8889–8891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brett CL, Plemel RL, Lobinger BT, Vignali M, Fields S, Merz AJ (2008) Efficient termination of vacuolar Rab GTPase signaling requires coordinated action by a GAP and a protein kinase. J Cell Biol 182: 1141–1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cabrera M, Nordmann M, Perz A, Schmedt D, Gerondopoulos A, Barr F, Piehler J, Engelbrecht‐Vandré S, Ungermann C (2014) The Mon1‐Ccz1 GEF activates the Rab7 GTPase Ypt7 via a longin‐fold‐Rab interface and association with PI3P‐positive membranes. J Cell Sci 127: 1043–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lachmann J, Barr FA, Ungermann C (2012) The Msb3 GAP controls the activity of the Rab GTPases Vps21 and Ypt7 at endosomes and vacuoles. Mol Biol Cell 23: 2516–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nordmann M, Cabrera M, Perz A, Bröcker C, Ostrowicz C, Engelbrecht‐Vandré S, Ungermann C (2010) The Mon1‐Ccz1 complex is the GEF of the late endosomal Rab7 homolog Ypt7. Curr Biol 20: 1654–1659 [DOI] [PubMed] [Google Scholar]
- 18. Fratti RA, Jun Y, Merz AJ, Margolis N, Wickner W (2004) Interdependent assembly of specific regulatory lipids and membrane fusion proteins into the vertex ring domain of docked vacuoles. J Cell Biol 167: 1087–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jun Y, Fratti RA, Wickner W (2004) Diacylglycerol and its formation by phospholipase C regulate Rab‐ and SNARE‐dependent yeast vacuole fusion. J Biol Chem 279: 53186–53195 [DOI] [PubMed] [Google Scholar]
- 20. Wang L, Seeley ES, Wickner W, Merz AJ (2002) Vacuole fusion at a ring of vertex docking sites leaves membrane fragments within the organelle. Cell 108: 357–369 [DOI] [PubMed] [Google Scholar]
- 21. Reese C, Heise F, Mayer A (2005) Trans‐SNARE pairing can precede a hemifusion intermediate in intracellular membrane fusion. Nature 436: 410–414 [DOI] [PubMed] [Google Scholar]
- 22. Reese C, Mayer A (2005) Transition from hemifusion to pore opening is rate limiting for vacuole membrane fusion. J Cell Biol 171: 981–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schwartz ML, Merz AJ (2009) Capture and release of partially zipped trans‐SNARE complexes on intact organelles. J Cell Biol 185: 535–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Strasser B, Iwaszkiewicz J, Michielin O, Mayer A (2011) The V‐ATPase proteolipid cylinder promotes the lipid‐mixing stage of SNARE‐dependent fusion of yeast vacuoles. EMBO J 30: 4126–4141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pieren M, Schmidt A, Mayer A (2010) The SM protein Vps33 and the t‐SNARE H(abc) domain promote fusion pore opening. Nat Struct Mol Biol 17: 710–717 [DOI] [PubMed] [Google Scholar]
- 26. Pieren M, Desfougères Y, Michaillat L, Schmidt A, Mayer A (2015) Vacuolar SNARE transmembrane domains serve as non‐specific membrane anchors with unequal roles in lipid mixing. J Biol Chem 290: 12821–12832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen YA, Scales SJ, Patel SM, Doung YC, Scheller RH (1999) SNARE complex formation is triggered by Ca2+ and drives membrane fusion. Cell 97: 165–174 [DOI] [PubMed] [Google Scholar]
- 28. Gao Y, Zorman S, Gundersen G, Xi Z, Ma L, Sirinakis G, Rothman JE, Zhang Y (2012) Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science 337: 1340–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kozlov MM, McMahon HT, Chernomordik LV (2010) Protein‐driven membrane stresses in fusion and fission. Trends Biochem Sci 35: 699–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McNew JA, Weber T, Parlati F, Johnston RJ, Melia TJ, Söllner TH, Rothman JE (2000) Close is not enough: SNARE‐dependent membrane fusion requires an active mechanism that transduces force to membrane anchors. J Cell Biol 150: 105–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Min D, Kim K, Hyeon C, Hoon Cho Y, Shin Y‐K, Yoon T‐Y (2013) Mechanical unzipping and rezipping of a single SNARE complex reveals hysteresis as a force‐generating mechanism. Nat Commun 4: 1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Risselada HJ, Grubmüller H (2012) How SNARE molecules mediate membrane fusion: recent insights from molecular simulations. Curr Opin Struct Biol 22: 187–196 [DOI] [PubMed] [Google Scholar]
- 33. Chernomordik LV, Kozlov MM (2005) Membrane hemifusion: crossing a chasm in two leaps. Cell 123: 375–382 [DOI] [PubMed] [Google Scholar]
- 34. Jahn R, Grubmüller H (2002) Membrane fusion. Curr Opin Cell Biol 14: 488–495 [DOI] [PubMed] [Google Scholar]
- 35. Giraudo CG, Hu C, You D, Slovic AM, Mosharov EV, Sulzer D, Melia TJ, Rothman JE (2005) SNAREs can promote complete fusion and hemifusion as alternative outcomes. J Cell Biol 170: 249–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Grote E, Baba M, Ohsumi Y, Novick PJ (2000) Geranylgeranylated SNAREs are dominant inhibitors of membrane fusion. J Cell Biol 151: 453–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xu Y, Zhang F, Su Z, McNew JA, Shin Y‐K (2005) Hemifusion in SNARE‐mediated membrane fusion. Nat Struct Mol Biol 12: 417–422 [DOI] [PubMed] [Google Scholar]
- 38. Risselada HJ, Bubnis G, Grubmüller H (2014) Expansion of the fusion stalk and its implication for biological membrane fusion. Proc Natl Acad Sci USA 111: 11043–11048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lindau M, Hall BA, Chetwynd A, Beckstein O, Sansom MSP (2012) Coarse‐grain simulations reveal movement of the synaptobrevin C‐terminus in response to piconewton forces. Biophys J 103: 959–969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ngatchou AN, Kisler K, Fang Q, Walter AM, Zhao Y, Bruns D, Sørensen JB, Lindau M (2010) Role of the synaptobrevin C terminus in fusion pore formation. Proc Natl Acad Sci USA 107: 18463–18468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baars TL, Petri S, Peters C, Mayer A (2007) Role of the V‐ATPase in regulation of the vacuolar fission‐fusion equilibrium. Mol Biol Cell 18: 3873–3882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Seeley ES, Kato M, Margolis N, Wickner W, Eitzen G (2002) Genomic analysis of homotypic vacuole fusion. Mol Biol Cell 13: 782–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Laage R, Ungermann C (2001) The N‐terminal domain of the t‐SNARE Vam3p coordinates priming and docking in yeast vacuole fusion. Mol Biol Cell 12: 3375–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mima J, Hickey CM, Xu H, Jun Y, Wickner W (2008) Reconstituted membrane fusion requires regulatory lipids, SNAREs and synergistic SNARE chaperones. EMBO J 27: 2031–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ogiso T, Iwaki M, Mori K (1981) Fluidity of human erythrocyte membrane and effect of chlorpromazine on fluidity and phase separation of membrane. Biochim Biophys Acta 649: 325–335 [DOI] [PubMed] [Google Scholar]
- 46. Melikyan GB, Brener SA, Ok DC, Cohen FS (1997) Inner but not outer membrane leaflets control the transition from glycosylphosphatidylinositol‐anchored influenza hemagglutinin‐induced hemifusion to full fusion. J Cell Biol 136: 995–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chernomordik LV, Frolov VA, Leikina E, Bronk P, Zimmerberg J (1998) The pathway of membrane fusion catalyzed by influenza hemagglutinin: restriction of lipids, hemifusion, and lipidic fusion pore formation. J Cell Biol 140: 1369–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zick M, Stroupe C, Orr A, Douville D, Wickner WT (2014) Membranes linked by trans‐SNARE complexes require lipids prone to non‐bilayer structure for progression to fusion. eLife 3: e01879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Karunakaran S, Fratti RA (2013) The lipid composition and physical properties of the yeast vacuole affect the hemifusion‐fusion transition. Traffic 14: 650–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bao H, Goldschen‐Ohm M, Jeggle P, Chanda B, Edwardson JM, Chapman ER (2016) Exocytotic fusion pores are composed of both lipids and proteins. Nat Struct Mol Biol 23: 67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Han X, Wang C‐T, Bai J, Chapman ER, Jackson MB (2004) Transmembrane segments of syntaxin line the fusion pore of Ca2 + ‐triggered exocytosis. Science 304: 289–292 [DOI] [PubMed] [Google Scholar]
- 52. Südhof TC, Rothman JE (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323: 474–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zick M, Wickner WT (2014) A distinct tethering step is vital for vacuole membrane fusion. eLife 3: e03251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Baker RW, Jeffrey PD, Zick M, Phillips BP, Wickner WT, Hughson FM (2015) A direct role for the Sec1/Munc18‐family protein Vps33 as a template for SNARE assembly. Science 349: 1111–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sutton RB, Fasshauer D, Jahn R, Brunger AT (1998) Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395: 347–353 [DOI] [PubMed] [Google Scholar]
- 56. Stein A, Weber G, Wahl MC, Jahn R (2009) Helical extension of the neuronal SNARE complex into the membrane. Nature 460: 525–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hernandez JM, Stein A, Behrmann E, Riedel D, Cypionka A, Farsi Z, Walla PJ, Raunser S, Jahn R (2012) Membrane fusion intermediates via directional and full assembly of the SNARE complex. Science 336: 1581–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kesavan J, Borisovska M, Bruns D (2007) v‐SNARE actions during Ca(2+)‐triggered exocytosis. Cell 131: 351–363 [DOI] [PubMed] [Google Scholar]
- 59. Diao J, Grob P, Cipriano DJ, Kyoung M, Zhang Y, Shah S, Nguyen A, Padolina M, Srivastava A, Vrljic M et al (2012) Synaptic proteins promote calcium‐triggered fast transition from point contact to full fusion. eLife 1: e00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hess B, Kutzner C, van der Spoel D, Lindahl E (2008) GROMACS 4: algorithms for highly efficient, load‐balanced, and scalable molecular simulation. J Chem Theory Comput 4: 435–447 [DOI] [PubMed] [Google Scholar]
- 61. Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH (2007) The MARTINI force field: coarse grained model for biomolecular simulations. J Phys Chem B 111: 7812–7824 [DOI] [PubMed] [Google Scholar]
- 62. Monticelli L, Kandasamy SK, Periole X, Larson RG, Tieleman DP, Marrink S‐J (2008) The MARTINI coarse‐grained force field: extension to proteins. J Chem Theory Comput 4: 819–834 [DOI] [PubMed] [Google Scholar]
- 63. Risselada HJ, Kutzner C, Grubmüller H (2011) Caught in the act: visualization of SNARE‐mediated fusion events in molecular detail. ChemBioChem 12: 1049–1055 [DOI] [PubMed] [Google Scholar]
- 64. Nichols BJ, Ungermann C, Pelham HR, Wickner WT, Haas A (1997) Homotypic vacuolar fusion mediated by t‐ and v‐SNAREs. Nature 387: 199–202 [DOI] [PubMed] [Google Scholar]
- 65. Haas A, Conradt B, Wickner W (1994) G‐protein ligands inhibit in vitro reactions of vacuole inheritance. J Cell Biol 126: 87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File