

Abstract
The CHD1 gene, encoding the chromo‐domain helicase DNA‐binding protein‐1, is one of the most frequently deleted genes in prostate cancer. Here, we examined the role of CHD1 in DNA double‐strand break (DSB) repair in prostate cancer cells. We show that CHD1 is required for the recruitment of CtIP to chromatin and subsequent end resection during DNA DSB repair. Our data support a role for CHD1 in opening the chromatin around the DSB to facilitate the recruitment of homologous recombination (HR) proteins. Consequently, depletion of CHD1 specifically affects HR‐mediated DNA repair but not non‐homologous end joining. Together, we provide evidence for a previously unknown role of CHD1 in DNA DSB repair via HR and show that CHD1 depletion sensitizes cells to PARP inhibitors, which has potential therapeutic relevance. Our findings suggest that CHD1 deletion, like BRCA1/2 mutation in ovarian cancer, may serve as a marker for prostate cancer patient stratification and the utilization of targeted therapies such as PARP inhibitors, which specifically target tumors with HR defects.
Keywords: CHD1, chromatin, DNA repair, PARP inhibitor, prostate cancer
Subject Categories: Cancer; DNA Replication, Repair & Recombination
Introduction
Recent cancer genome sequencing efforts have uncovered frequent mutations in genes encoding chromatin remodelers and proteins which add, remove, or recognize histone modifications in many types of cancer. The proteins encoded by these genes play central roles in controlling the accessibility of DNA to enzymes and proteins involved in regulating gene transcription, DNA replication, and repair. Notably, the CHD1 gene encoding the chromo‐domain helicase DNA‐binding protein‐1 is the second most frequently deleted or mutated (15–27%) gene in prostate cancer 1, 2, 3. Loss of CHD1 in tumors is correlated with chromosomal instability and poor prognosis 4, 5. However, the importance of CHD1 deletion for tumor cell phenotype, patient stratification, and therapeutic responsiveness remains unknown.
The eukaryotic genome is compacted into chromatin composed of DNA, histones, and other proteins that regulate DNA‐associated processes 6. Notably, most of these processes require physical repositioning, sliding, or removal of nucleosomal histones from the DNA. This regulatory step is enabled by various post‐translational histone modifications catalyzed by histone modifying enzymes and is carried out by histone chaperones and ATP‐dependent chromatin remodeling complexes 7, 8. CHD1 belongs to the family of ATP‐dependent chromatin remodeling factors containing a SNF2‐like helicase domain, where the human CHD1 protein was shown to bind to histone 3 di‐ or trimethylated at lysine 4 (H3K4me2/3) through its two chromo‐domains 9, 10, 11. Studies in Drosophila, yeast, and humans have shown that CHD1 is associated with de‐condensed chromatin 12 and interacts with other proteins that alter chromatin structure 13, 14. CHD1 was also shown to maintain chromatin structure during transcription 15 and be important for the proper positioning of nucleosomes and correct initiation of transcription in yeast 16, 17, 18. Another study also implicated CHD1 in maintaining an open chromatin state in mouse embryonic stem cells where down‐regulation of CHD1 led to increased chromatin condensation and a loss of pluripotency 19.
In addition to roles in transcription, chromatin remodeling is essential for the successful repair of DNA damage 20. Eukaryotic cells have evolved specific mechanisms to repair various types of DNA damage during different stages of the cell cycle 21, 22. Among the different types of DNA damage, DNA double‐strand breaks (DSBs) are the most common cause of genomic instability and tumor formation and are also the product of various anti‐tumor therapies including radio‐ and some chemotherapies. Mammalian cells repair DSBs by two major repair pathways referred to as non‐homologous end joining (NHEJ) and homologous recombination (HR) 23. NHEJ is a fast process and represents the major DSB repair pathway in mammalian cells, repairing DSBs in all cell cycle phases but predominately in G1 24. HR is a comparatively slow repair process which is restricted to S/G2 phase when the intact sister chromatid is available as a template to allow error‐free repair. HR is initiated by processing of DSB ends by nucleolytic enzymes to generate long stretches of single stranded DNA (ssDNA), a mechanism referred to as DNA end resection 25, 26. In mammalian cells, end resection is initiated by the concerted action of MRE11 and CtBP‐interacting protein (CtIP), and the generated ssDNA is coated with replication protein A (RPA) to prevent the formation of secondary structures 27. Subsequently, BRCA2 promotes the replacement of RPA by RAD51, to form a RAD51 nucleofilament which then mediates later steps of HR such as homology search and strand invasion. We have previously described a functional hierarchy in DSB repair 28 which regulates the choice between DSB repair pathways. Deregulation of this hierarchy leads to deficient repair and genetic aberrations. Therefore, this hierarchy is tightly regulated by multiple factors 29, 30, including the DNA damage response (DDR), which ensures appropriate recruitment of DSB repair proteins. The DDR is needed for efficient DSB repair and involves the post‐translational modification of histones which promotes the recruitment of specific repair‐related proteins. Moreover, ATP‐dependent chromatin remodeling complexes including SWI/SNF, INO80, ISWI, and SRCAP, which were previously described to be involved in the repair of DSBs, function to physically alter chromatin structure and direct DNA repair 31, 32, 33.
In this study, we identify a new role for CHD1 in DNA repair and show that it is recruited to DNA damage sites. The loss of CHD1 abrogates the end resection process by resulting in decreased recruitment of CtIP, thereby leading to decreased generation of single stranded DNA (ssDNA) as indicated by decreased binding of RPA1 and RAD51 to chromatin. As a consequence, depletion of CHD1 specifically affects the homologous recombination‐mediated DNA DSB repair process but not the non‐homologous end joining pathway. Consistently, CHD1‐depleted cells are hypersensitive to mitomycin C (MMC) treatment, which generates inter‐strand cross‐links and thereby requires the HR pathway to repair broken DNA, as well as to irinotecan and PARP inhibition. These data provide important mechanistic insight into the tumor suppressor function of CHD1 and may provide a molecular rationale to specifically target the DNA repair defects present in CHD1‐deleted prostate cancers via the use of targeted therapies such as PARP inhibitors.
Results
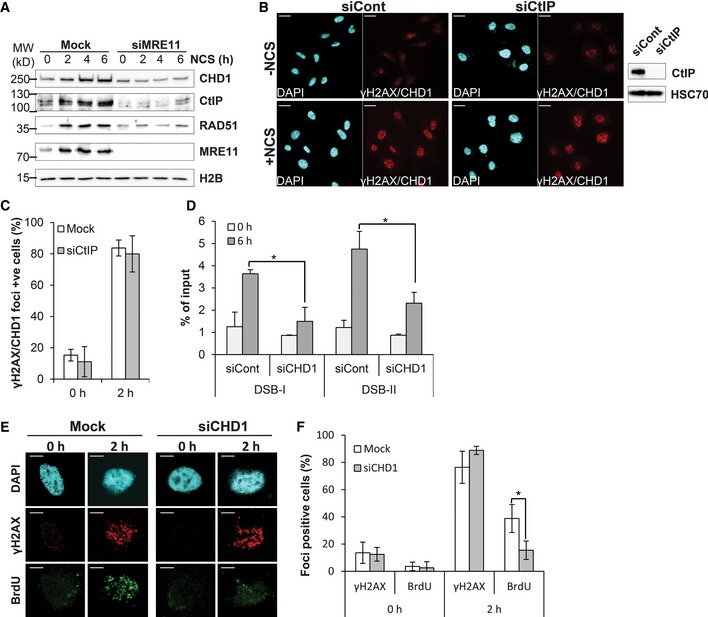
Prostate cancer is one of the most common cancers in men. Like other cancers, prostate cancer is associated with frequently recurring chromosomal abnormalities. It was previously reported that the CHD1 gene is mutated or deleted in 15–27% of prostate cancers. In order to verify these findings, we examined the frequency of CHD1 alteration from various published genome sequencing studies. Consistent with previous reports, most studies displayed CHD1 genetic alterations (mutation or deep deletion) in at least 7% and as high as 21% of patients (Fig 1A).
Figure 1. CHD1 accumulates at the DNA damage sites in proximity to γH2AX.
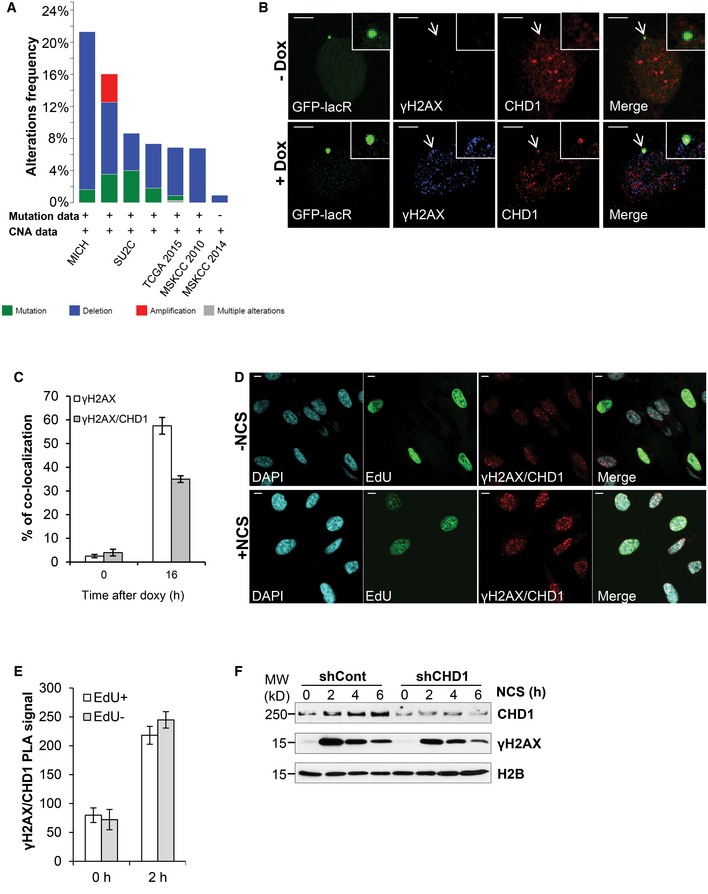
- Frequency of CHD1 gene mutation (green), deep deletion (blue), or amplification (red) in prostate cancer patients.
- CHD1 is recruited to an I‐SceI‐induced DSB site and is co‐localized with γH2AX. Immunofluorescence studies using U2OS19 ptight13 GFP‐LacR cells containing a stably integrated I‐SceI cleavage site flanked by 256 copies of lac operator (lacO) on one side and 96 copies of the tetracycline response element on the other side (tetO). The localization of the GFP‐lac repressor protein (GFP‐LacR) at the lac‐operator DNA sequences in the nucleus before (− I‐SceI) and 16 h after I‐SceI‐induced (+ I‐SceI) DSB is indicated by white arrows. After 16 h of doxy treatment, CHD1 and γH2AX co‐localized at I‐SceI cleavage site, along with DNA‐bound GFP‐LacR but not in uninduced cells (− I‐SceI). Scale bar, 10 μm.
- Quantification of co‐localization of CHD1 with the lac array, mean values ± SD of three independent experiments counting at least 100 cells are represented in the graph.
- PC3 cells were treated with NCS (100 ng/ml) for 2 h and with EdU for 45 min. Cells were stained with EdU first, and then, proximity ligation assay (PLA) was performed using γH2AX and CHD1 antibodies. Scale bar, 10 μm.
- Quantification of PLA signal from (D) in EdU‐positive and EdU‐negative cells using ImageJ. For quantification, more than 100 cells were analyzed for each condition and represented as mean value ± SD (n = 3).
- CHD1 is recruited to chromatin upon DNA double‐strand break induction. PC3 cells with stable control (shCont) or CHD1 shRNA (shCHD1) expression were treated with NCS for the indicated times, and chromatin fractions were immunoblotted with CHD1 and γH2AX antibodies. H2B was used as a loading control.
CHD1 is recruited to chromatin and is required for the DSB repair
Given the frequency of CHD1 genetic aberrations, we sought to determine whether CHD1 may play a role in DNA repair. Upon DNA damage, proteins involved in the DNA damage response and repair are recruited to the chromatin and accumulate at the DNA damage site where they form foci in the nucleus. In order to test whether CHD1 plays a role in DSB repair, we used different methods to examine whether CHD1 is recruited to chromatin and forms foci at the site of DNA damage following DSB induction. We initially treated PC3 cells with the radiomimetic neocarzinostatin (NCS) and co‐stained for CHD1 and γH2AX. We observed that CHD1 is partially co‐localized with γH2AX (Fig EV1A). To further validate CHD1 recruitment at the DSB site, we used U2OS19 ptight13 GFP‐LacR cells harboring a stably integrated I‐SceI cleavage site which is flanked by repeats of the lac operator (lacO) 34, 35. In addition, U2OS19 ptight13 GFP‐LacR cells constitutively express a GFP‐lac repressor fusion protein as well as a doxycycline (doxy)‐inducible I‐SceI endonuclease. The array containing the I‐SceI site can be constitutively visualized by the binding of the GFP‐lac repressor to lacO as a green focus. Upon doxy treatment, a DSB is generated by the I‐SceI enzyme. After generation of a DSB by I‐SceI, the surrounding region is rapidly marked by the phosphorylation of the histone variant H2AX at ser139 (γH2AX) and becomes bound by 53BP1, which can both be visualized by immunofluorescence staining (Fig EV1B). While the genome of these cells contains the single integrated I‐SceI site, we did observe some background staining of γH2AX upon doxycycline treatment, suggesting there may be some minimal off‐target activity of the enzyme. Importantly, CHD1 is co‐localized with both GFP‐LacR and γH2AX specifically in doxy‐treated cells, but not in untreated cells (−I‐SceI) where the I‐SceI site is not cleaved (Fig 1B and C). In addition, we confirmed the close proximity between γH2AX and CHD1 via proximity ligation assay (PLA). The PLA method is used to study the interaction between two proteins in fixed cells based on the utilization of two secondary antibodies from different species which are conjugated to DNA oligonucleotides. When the two antibodies are in close proximity to one another, they can be bridged by two additional circle‐forming oligonucleotides, joined via ligation, amplified by rolling circle amplification, and visualized as foci using a complementary fluorescently labeled oligonucleotide probe. The specificity of the interaction between CHD1 and γH2AX in NCS‐treated samples was further confirmed by the depletion of CHD1 by small interfering RNA (siRNA) which completely abolished the PLA signal (Fig EV1C). Notably, CHD1 depletion did affect the interaction between γH2AX and 53BP1 (Fig EV1C), further confirming the specificity of the PLA signal. Importantly, co‐staining with EdU (to detect cells which are in S phase) and PLA for CHD1 and γH2AX revealed that the recruitment of CHD1 to DSB is not cell cycle‐dependent (Fig 1D and E). We next performed chromatin fractionation to examine the recruitment of CHD1 to chromatin after NCS treatment. Our results revealed that CHD1 is more strongly recruited to chromatin after DSB induction in all cell lines studied including PC3 (Fig 1F) and VCaP (Fig EV1D) prostate cancer cells as well as in the U2OS osteosarcoma cell line (Fig EV1E). Indeed, increased recruitment of CHD1 is observed within 30 min of NCS treatment and is further increased over time. This enrichment is significantly decreased by RNA interference‐mediated CHD1‐depletion either by stable (PC3 and VCaP) or transient (U2OS) knockdown. This increased CHD1 recruitment is not due to overall increased CHD1 protein levels as such (Fig EV1F). Overall, these data indicate that CHD1 is recruited to chromatin in response to DSB induction.
Figure EV1. CHD1 is co‐localized with DNA damage site.
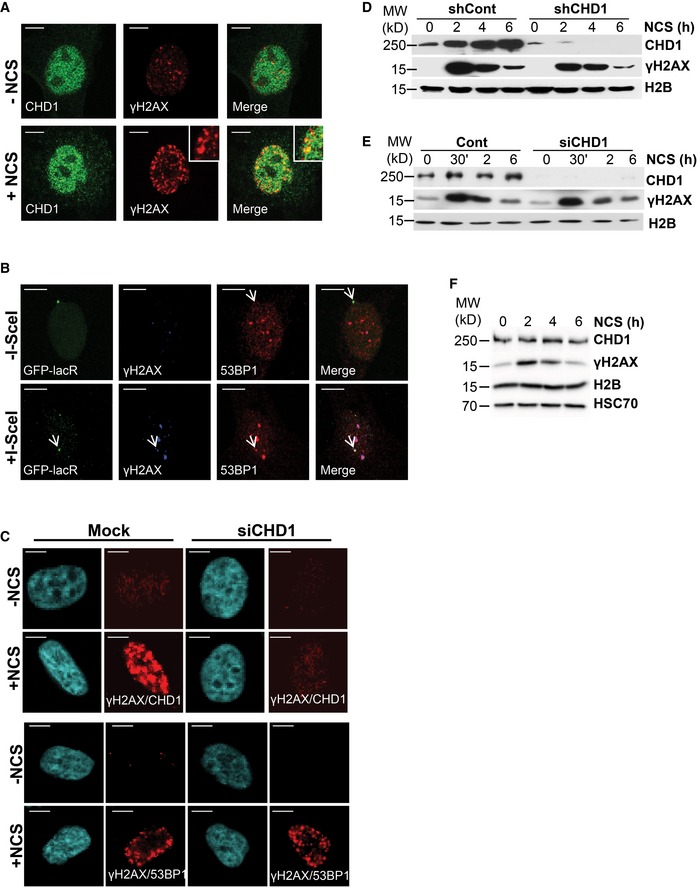
-
ACHD1 is partially co‐localized with γH2AX. PC3 cells were treated with NCS (100 ng/ml) for 2 h and co‐immunostained with γH2AX and CHD1 antibodies. Scale bar, 10 μm.
-
B53BP1 recruitment to I‐SceI‐induced DNA damage sites in U2OS19 ptight13 GFP‐LacR cells indicated by white arrows. Scale bar, 10 μm.
-
CPLA with γH2AX/CHD1 or γH2AX/53BP1 in mock or siCHD1 (SmartPool)‐transfected PC3 cells after 2 h NCS treatment. Scale bar, 10 μm.
-
D, ECHD1 is recruited to the chromatin upon DNA double‐strand break induction. VCaP cells stably expressing either control (shCont) or CHD1 shRNA (shCHD1) (D) and U2OS cells following transient transfection with either mock or CHD1 siRNA (E) were treated with NCS for the indicated times, and chromatin fractions were immunoblotted with CHD1 and γH2AX antibodies, respectively. H2B serves as a loading control.
-
FPC3 cells were treated with NCS (100 ng/ml) for indicated times, and the total protein lysates were analyzed by Western blotting using the indicated antibodies.
Source data are available online for this figure.
Based on its recruitment to chromatin in response to DSBs, we next tested whether CHD1 is involved in DSB repair. To this end, unsynchronized CHD1‐stably‐depleted BHP1 and PC3 cells were irradiated with 3 Gy and γH2AX foci were counted at 1 h and 24 h after IR as a marker for DSB (Fig 2A). Importantly, CHD1‐depleted cells showed a similar number of γH2AX foci at 1 h compared to control cells but significantly more residual foci at the 24‐h time point indicating a DSB repair deficiency (Fig 2B). Further, the prolonged γH2AX signal was confirmed by Western blot analysis after 24 h of IR treatment in CHD1‐depleted cells (Fig 2C). Moreover, neutral comet assay showed increased comet length in CHD1‐depleted cells compared to control cells 6 h after DSB induction with NCS treatment (Fig EV2A). Consistently, CHD1‐depleted cells displayed moderately increased sensitivity to ionizing radiation (IR) compared to control cells (Fig 2D). Similar results were obtained using both PC3 and BHP1 cells (Fig 2D).
Figure 2. Loss of CHD1 leads to DNA damage response defects.
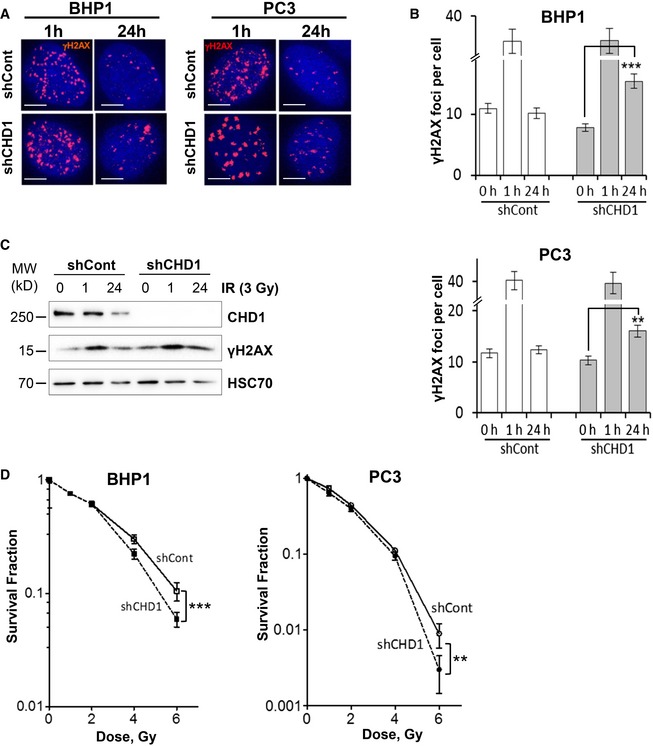
-
A, BBHP1 or PC3 cells with stable expression of either control (shCont) or CHD1 shRNA (shCHD1) were treated with γ‐radiation (3 Gy), and after 1 and 24 h, cells were immunostained for γH2AX (A) and the number of γH2AX foci per cell were determined for each time point. More than 50 cells were counted in each condition (B). Scale bar, 5 μm. The mean values ± SD of three independent experiments are shown. P‐values (**P = 0.006, ***P = 0.0008) were calculated using ANOVA.
-
CCHD1‐depleted cells show prolonged γH2AX accumulation. PC3 cells with shCont or shCHD1 were treated with γ‐radiation as in (A) and total protein was analyzed by Western blotting.
-
DCHD1‐depleted cells show increased sensitivity to γ‐radiation. For colony formation assay, both BHP1 and PC3 cells with shCont or shCHD1 were treated with the indicated doses of γ‐radiation and surviving fractions were measured by counting colonies after 3 weeks. Mean values ± SD are represented in the plot (n = 3). Data were normalized to the plating efficiency. P‐values (**P = 0.002, ***P = 0.0009) were calculated using ANOVA.
Figure EV2. CHD1 is required for DSB repair.
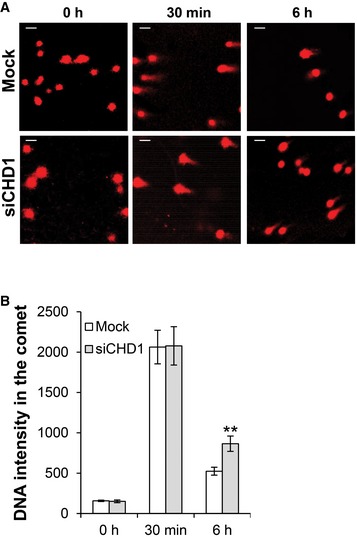
- CHD1‐depleted cells show decreased DNA DSB repair efficiency. PC3 cells were either mock transfected or transfected with siCHD1 (SmartPool). After 48 h of transfection, cells were treated with NCS. Neutral comet assay was performed after the indicated times following NCS treatment, and images were taken. Scale bar, 50 μm.
- Quantification of comet tail (DNA content) from (A) in control and CHD1‐depleted cells is shown. More than 50 cells were counted, and results represent the mean value ± SD (n = 3). P‐values (**P = 0.005) were calculated using ANOVA.
CHD1 is specifically required for homologous recombination DNA repair
In order to determine whether CHD1 plays a role in controlling a specific DSB repair pathway, we depleted CHD1 (Fig EV3A) in HeLa cells harboring a single copy of either HR or NHEJ repair substrates (pGC and pEJ, respectively). The repair efficiency was calculated by flow cytometry based on the fraction of GFP‐positive (GFP+) cells 48 h after transfection with an I‐SceI expression vector. Strikingly, depletion of CHD1 led to an 11‐fold decrease in HR‐mediated repair, but showed no effect on NHEJ. XRCC4 and RAD51 depletion was used as positive controls for NHEJ and HR assays, respectively (Figs 3A and B, and EV3B). We further confirmed that the effects seen on HR in CHD1‐depleted cells were not due to changes in cell cycle regulation (Fig EV3C). Moreover, the inhibition of MRE11 nuclease activity by mirin did not show any further effect on the HR efficiency caused by CHD1 depletion, suggesting that these proteins function epistatically in DNA repair (Fig EV3D). In order to reinforce this data, foci displaying co‐localization of RAD51 and γH2AX were counted as an indicator for HR factories in CHD1‐depleted cells 3 and 24 h after irradiation with 2 Gy. As expected, control cells demonstrated an increase in the number of these foci, which returned to normal levels after 24 h. However, CHD1‐depleted PC3 (Fig 3C and D) and BHP1 (Fig EV3E) cells showed only a moderate increase early and almost no decline in RAD51 foci over time, further confirming a reduced HR capacity.
Figure EV3. CHD1 is required for homologous recombination‐mediated DNA repair.
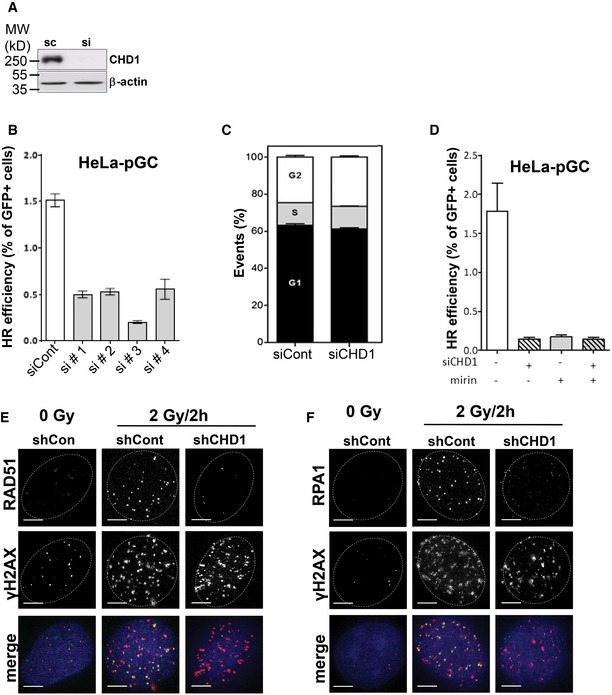
-
AHeLa cells were transfected with control or CHD1 siRNA for 48 h, and protein samples were analyzed by Western blotting for CHD1 expression.
-
BHeLa cells containing single copy HR (pGC) repair substrates were transfected with either control siRNA or four individual siRNAs targeting CHD1. After 24 h of transfection, DSB was induced by transfecting cells with I‐SceI‐expressing vector (pCMV‐I‐SceI‐3xNLS). The GFP‐positive cells were measured by flow cytometry. Data are represented as mean ± SD (n = 3).
-
CCHD1 depletion did not change the cell cycle profile of HeLa cells. Cells were transfected with control or CHD1 siRNA and 48 h after transfection processed for FACS analysis. Mean values ± SD are presented (n = 3).
-
DHeLa cells containing single copy HR (pGC) repair substrates were either mock transfected or with siRNA targeting CHD1. After 24 h of transfection, cells were treated with mirin and DSB was induced by transfecting cells with I‐SceI‐expressing vector (pCMV‐I‐SceI‐3xNLS). The GFP‐positive cells were measured by flow cytometry. Data are represented as mean ± SD (n = 3). For flow cytometry, 50,000 cells were counted for each condition.
-
E, FCHD1‐depleted BHP1 cells show decreased RAD51 and RPA1 foci after DNA damage induction. shCont‐ or shCHD1‐expressing BHP1 cells were irradiated and co‐immunostained with γH2AX and RAD51 or RPA1 antibodies at the indicated time points. Scale bar, 5 μm.
Figure 3. CHD1 is required for homologous recombination (HR) repair.
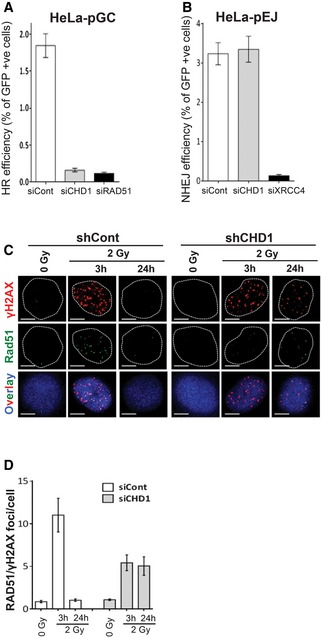
-
A, BHeLa cells harboring single copies of HR (pGC) or NHEJ (pEJ) repair substrates were transfected with either negative control (siCont) or siRNAs targeting CHD1, RAD51, or XRCC4. After 24 h of transfection, DSB was induced by transfecting cells with I‐SceI‐expressing vector (pCMV‐I‐SceI‐3xNLS). After 48 h of transfection, GFP‐positive cells were measured by flow cytometry. HR (A) or NHEJ (B) efficiency was calculated based on the fraction of GFP‐positive cells and represented as mean values ± SD from three independent experiments, and 50,000 cells were counted for each condition.
-
CCHD1‐depleted cells show decreased RAD51 foci after DNA damage induction. The shCont or shCHD1 PC3 cells were irradiated and co‐immunostained with γH2AX and RAD51 antibodies after the indicated times. Scale bar, 5 μm.
-
DThe number of RAD51 foci co‐localized with γH2AX per cell (from C) was counted (n = 50) and represented in a graph. The data are represented as mean value ± SD of three independent experiments. More than 50 cells were counted for each condition.
CHD1 is required for CtIP recruitment to chromatin and end resection
The impaired RAD51 recruitment to DSBs observed in CHD1‐depleted cells (Fig 3C) suggested that CHD1 is required for early steps of HR. Therefore, we tested whether CHD1 may be required for DSB end resection, a key step in the HR process which is mediated by the MRN complex and leads to the recruitment of the C‐terminal binding protein (CtBP)‐interacting protein (CtIP) 27, 36, 37. The resulting ssDNA is subsequently bound by the ssDNA binding proteins RAD51 and RPA1 38. Our results using PLA demonstrate that upon DSB induction following NCS treatment, CHD1 co‐localizes with CtIP, indicating that CHD1 regulates an essential early step of HR (Figs 4A and EV4A). To further test whether CHD1 is required for CtIP recruitment to DSBs, PC3, and VCaP cells stably expressing CHD1 or control shRNAs were treated with NCS for different time points and analyzed for CtIP recruitment to chromatin by chromatin fractionation. As expected, CtIP recruitment to chromatin increased over time following treatment in control cells, but not in CHD1‐depleted cells (Figs 4B and EV4B). Consistent with the chromatin fractionation data, immunofluorescence studies using U2OS19 ptight13 GFP‐LacR cells showed that CHD1 depletion (Fig EV4E) significantly decreased the recruitment of CtIP and subsequent loading of RPA1 to I‐SceI‐induced DSB (Fig 4C and D), further confirming a role for CHD1 in the end resection step of HR. Consistent with these findings, while the kinetics varied slightly between cell lines, CHD1 depletion resulted in impaired recruitment of the ssDNA binding proteins RAD51 and RPA1 to chromatin in PC3, VCaP, and U2OS cells treated with NCS compared to control cells (Figs 4B and EV4B and C). These results were further validated with two individual siRNAs targeting CHD1 in PC3 cells using chromatin fractionation (Fig EV4G). Notably, the expression levels of the aforementioned proteins were not affected by CHD1 depletion (Fig EV4D and F). The impaired recruitment of RPA1 and RAD51 was further confirmed at IR‐induced DSBs (Fig EV3E and F). We also confirmed that the decreased CtIP and RPA1 foci in CHD1‐depleted cells are specifically in S/G2‐phase cells (Fig EV4H–K). Together, our data suggest that CHD1 controls HR‐mediated DSB repair by regulating the end resection step by facilitating CtIP recruitment to DSB sites.
Figure 4. Depletion of CHD1 affects end resection by decreasing binding of CtIP, RPA1, and RAD51 to chromatin in response to DNA DSB induction.
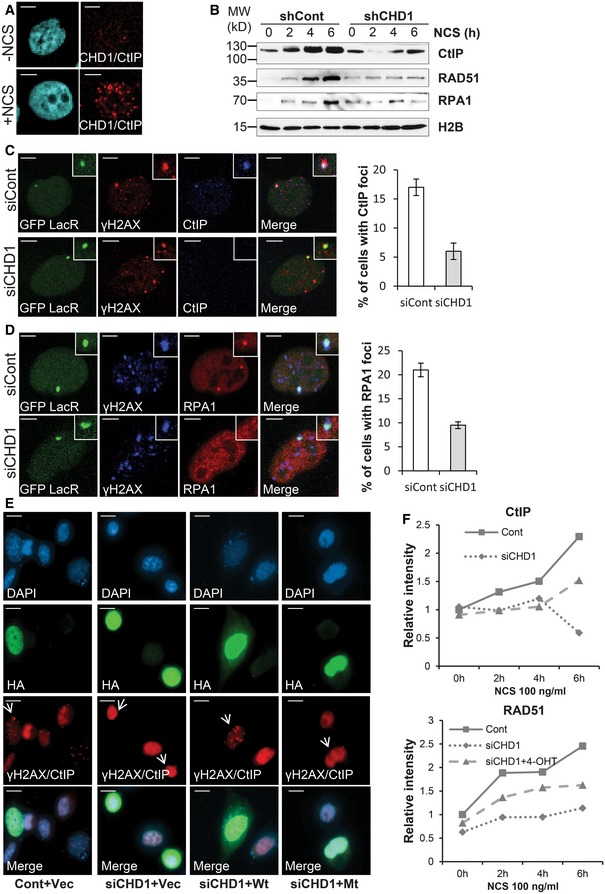
-
APLA assay with CtIP and CHD1 in PC3 cells after 2 h of NCS treatment. Scale bar, 5 μm.
-
BshCont and shCHD1 PC3 cells were treated with NCS for the indicated times, and chromatin fractions were immunoblotted for CtIP, RPA1, and RAD51. H2B is shown as a loading control.
-
C, DCHD1 depletion leads to decreased CtIP (C) and RPA1 (D) recruitment to I‐SceI‐induced DSB in U2OS19 ptight13 GFP‐LacR cells. CHD1 was depleted in U2OS19 ptight13 GFP‐LacR cells by siRNA and 48 h after transfection cells were treated with doxy for 16 h and co‐immunostained for γH2AX and CtIP or RPA1. The number of cells with CtIP or RPA1 foci were counted and represented in the graphs as mean percentage ± SD of foci‐positive cells (n = 3). More than 50 cells were counted. Scale bar, 5 μm.
-
EPC3 cells were transfected with either mock or siCHD1 followed by empty vector, wt mChd1 (Wt) or ATPase‐mutant (Mt) mChd1. After 48 h of transfection, cells were treated with 4‐OH tamoxifen (4‐OHT) for 24 h and processed for PLA with γH2AX and CtIP antibodies. Cells demonstrating positive focal interactions indicative of DNA repair hubs (punctate staining) are present only in control and wt CHD1‐rescued cells indicated by white arrows but not following knockdown or reconstitution of an ATPase‐mutated CHD1 (Mt). Scale bar, 20 μm.
-
FPC3 cells which stably express HA‐mChd1‐ERT2 were transfected with either mock or siCHD1. After 24 h of transfection, cells were treated with 4‐OHT for 24 h to induce HA‐mChd1‐ERT2 nuclear translocation. Western blot analysis of chromatin fractions for CHD1, ERT2 (mChd1), CtIP, and RAD51 shown in Fig EV4O was analyzed by densitometry using ImageJ. The relative quantification for the indicated proteins is shown in the graph.
Figure EV4. CHD1 is required for DNA end resection.
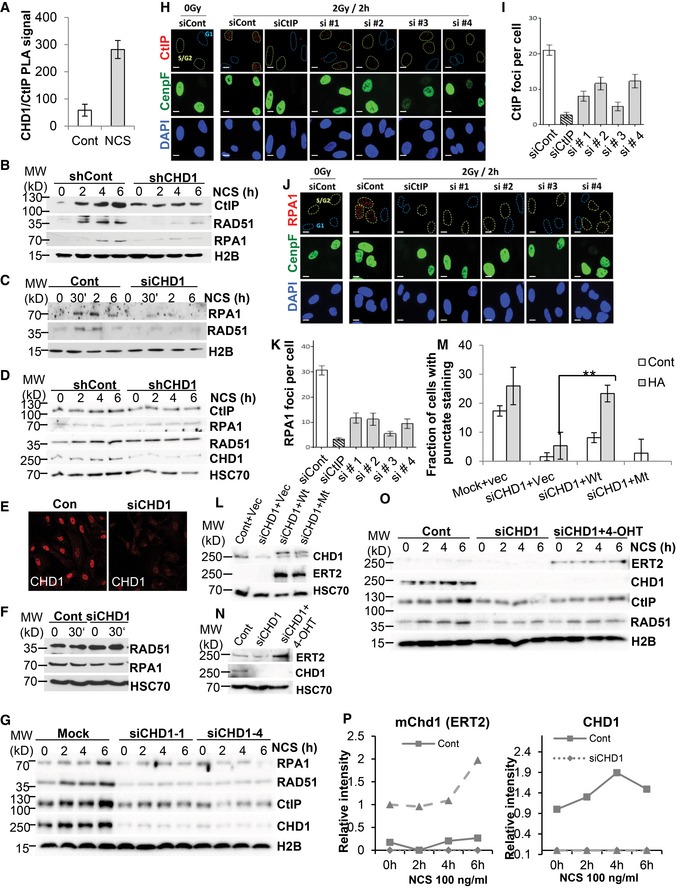
-
AQuantification of CHD1/CtIP PLA signal from Fig 4A in control and NCS‐treated PC3 cells, mean values ± SD are presented (n = 3). More than 50 cells were counted for each condition.
-
BSimilar to Fig EV1D, VCaP cells with shCont or shCHD1 were treated with NCS for different times and chromatin fractions were analyzed by Western blotting.
-
CSimilar to Fig EV1E, U2OS cells were transfected and treated with NCS for different times and chromatin fractions were analyzed by Western blot analysis.
-
DTotal protein lysates from shCont and shCHD1 PC3 cells after NCS treatment were analyzed by Western blotting using the indicated antibodies.
-
EU2OS19 ptight13 GFP‐LacR cells were transfected with either mock or siCHD1. After 48 h of transfection, cells were immunostained for CHD1. Scale bar, 50 μm.
-
FTotal protein lysate from mock or siCHD1 U2OS cells from (E) were analyzed by Western blotting.
-
GPC3 cells were transfected with either mock, siCHD1‐1, or siCHD1‐4. After 48 h of transfection, cells were treated with NCS for the indicated times and chromatin fractions were analyzed by Western blotting.
-
HPC3 cells were transfected with either control or CtIP or with four individual CHD1 siRNAs. After 48 h of transfection, cells were irradiated and processed for immunostaining for CenpF and CtIP. Scale bar, 10 μm.
-
IQuantification of CtIP foci from (H), data are presented as mean ± SD (n = 3). More than 50 cells were counted per condition.
-
J, KSimilar to (H) PC3 cells were transfected and immunostained with RPA1 and CenpF antibodies, RPA1 foci were quantified, and mean values ± SD are presented in the graph (K) (n = 3). More than 50 cells were counted per condition. Scale bar, 10 μm.
-
LPC3 cells were transfected with either mock or siCHD1 (1 and 4) followed by transfection with empty vector or plasmids expressing ERT2‐fusions of Wt‐mChd1 or ATPase Mt‐mChd1. After 48 h of transfection, cells were treated with 300 ng/ml of 4‐OH tamoxifen for 24 h. Whole‐cell protein lysates were analyzed by Western blotting using antibodies that recognize mouse and human CHD1, the ERT2 fusion protein, or HSC70 (loading control).
-
MQuantification of the fraction of cells with punctate staining after PLA assay for γH2AX/CtIP from Fig 4E, mean values ± SD are presented (n = 30, **P = 0.007, ANOVA).
-
NPC3 cells stably expressing ERT2‐HA‐mChd1 were transfected with either mock or siCHD1 (1 and 4). After 24 h of transfection, cells were treated with either vehicle or 4‐OHT for 24 h and whole‐cell lysates were analyzed by Western blotting using antibodies specific for hCHD1, the ERT2 fusion protein or HSC70 (loading control).
-
OChromatin fractions from (N) after NCS treatment for the indicated times were analyzed by Western blotting.
-
PQuantification of ERT2 (mChd1) and hCHD1 intensity from (O).
To further validate the role of CHD1 in promoting CtIP recruitment to DSBs, we generated expression vectors for wild‐type or ATPase‐mutant (Mt) HA‐tagged murine CHD1 proteins (mChd1; 98% identical to the hCHD1 protein) which were fused to a tamoxifen‐inducible mutant form of the estrogen receptor ligand binding domain (ERT2). Due to variation in codon usage, the transgenes are resistant to the siRNAs against human CHD1. The PLA assay for γH2AX/CtIP demonstrated that the CHD1‐depleted cells show decreased PLA signals, indicating that the CtIP recruitment is affected but can be rescued by re‐expressing wild‐type (Wt) mChd1 but not an ATPase‐mutant (Mt) form (Figs 4E and EV4L–M). In addition, PC3 cells which stably express HA‐mChd1‐ERT2 (Wt) were depleted with siRNA targeting endogenous hCHD1 and treated with NCS (Fig EV4N). Chromatin fractionation analysis confirmed that the decreased recruitment of CtIP and RAD51 to chromatin in CHD1‐depleted cells could be partially rescued by re‐expressing mChd1 (Figs 4F and EV4O and P).
In order to determine whether CHD1 recruitment to chromatin is dependent on MRN complex activity, we depleted MRE11 or inhibited its activity by mirin in PC3 cells (Figs 5A and EV5A). Chromatin fractionation analysis from NCS‐treated cells showed that MRE11 activity is required for the stabile recruitment of both CHD1 and CtIP (Figs 5A and EV5B). In contrast, PARP inhibitor (PARPi) treatment did not affect the recruitment of CHD1 (Fig EV6B). Importantly, CtIP depletion did not affect the recruitment of CHD1 (PLA assay for γH2AX/CHD1) to DSB, further supporting a role for CHD1 upstream of CtIP (Fig 5B and C). Consistent with this, decreased CtIP expression in CHD1‐depleted cells did not show any synergistic effect on sensitivity to IR in either PC3 or BHP1 cells, suggesting these proteins function epistatically in DSB repair (Fig EV5C and D).
Figure 5. CHD1 facilitates chromatin opening at HR DSB sites.
- CHD1 recruitment is dependent on MRE11 activity. PC3 cells were transfected with mock or siMRE11 (SmartPool), and 48 h after transfection, cells were treated with NCS, and chromatin fractions were isolated and analyzed by Western blotting.
- CHD1 is upstream of CtIP. PLA with γH2AX and CHD1 antibodies in mock or CtIP‐depleted cells after 2 h of NCS. Verification of knockdown efficiency is shown by Western blot on the right. HSC70 is shown as a loading control. Scale bar, 20 μm.
- Quantification of PLA signal from (B) represented as mean values ± SD from three independent experiments. More than 100 cells were counted per condition.
- qPCR analyses for chromatin accessibility at two HR‐repaired sites (DSB‐I and DSB‐II) were analyzed by FAIRE in AsiSI–ER‐U20S cells transfected with either mock or CHD1 siRNA. After 48 h of transfection, cells were treated with 4‐OHT for the indicated times and processed for FAIRE. The data are represented as mean ± SD (n = 3). P‐values were calculated using ANOVA (*P = 0.02 for DSB‐I and *P = 0.03 for DSB‐II).
- Native BrdU staining of PC3 cells transfected with either mock or siCHD1 (SmartPool) were grown for 48 h and then treated with NCS for 2 h prior to staining with anti‐BrdU and γH2AX antibodies. Scale bar, 5 μm.
- Quantification of BrdU‐positive cells from (E). The data are represented as mean ± SD from three independent experiments. More than 50 cells were counted for each condition. P‐values (*P = 0.03) were calculated using ANOVA.
Figure EV5. CHD1 facilitates chromatin opening during HR‐mediated repair.
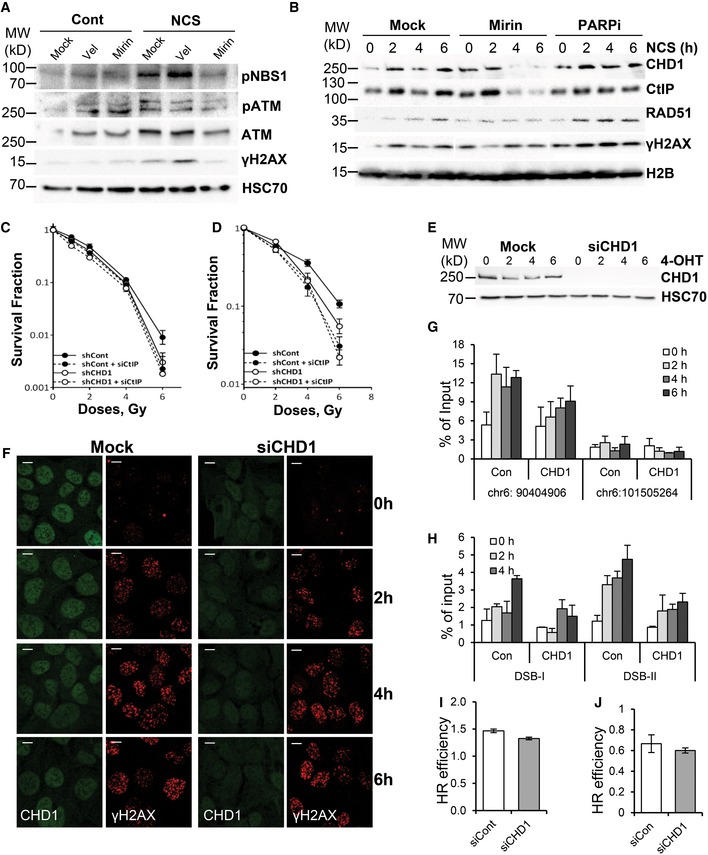
-
ATotal protein lysate from cells treated with DMSO, mirin, or veliparib (PARPi) was analyzed by Western blotting.
-
BPC3 cells were pretreated with DMSO, mirin (50 μM), or veliparib (20 μM, PARPi) for 1 h prior to NCS treatment for 2, 4, or 6 h. Chromatin fractions were analyzed by Western blotting for the recruitment of CHD1, CtIP, RAD51, and γH2AX.
-
C, DPC3 (C) and BHP1 (D) cells with stable shCont or shCHD1 expression were transfected with either siCont or siCtIP for 24 h before irradiation with the indicated doses. Surviving fractions were measured by counting colonies after 3 weeks, and mean values ± SD are presented (n = 3).
-
EAsiSI–ER‐U20S cells were transfected either control or CHD1 siRNA. After 48 h of transfection, cells were treated with 4‐OHT for the indicated times and the whole‐cell protein lysates were immunoblotted with CHD1 and HSC70 antibodies.
-
FCells from (E) were processed for immunostaining with γH2AX and CHD1. Scale bar, 20 μm.
-
G, HqPCR analysis from FAIRE DNA from AsiSI–ER‐U20S cells from (E) at sites repaired (H) or not repaired (G) by HR. Mean values ± SD are presented in the graphs (n = 3).
-
I, JCHD1 is required only for chromatin‐associated homologous recombination (HR) repair. PC3 cells or BHP1 cells with stable expression of either control or CHD1 shRNA were transfected with I‐SceI‐linearized pGC pGC repair substrate. After 24 h of transfection, HR efficiency was calculated based on the fraction of GFP‐positive cells plotted in the graph, and 50,000 cells were counted per condition. Data are represented as mean ± SD from three independent experiments.
Figure EV6. CHD1 loss increases chemotherapeutic repressiveness.
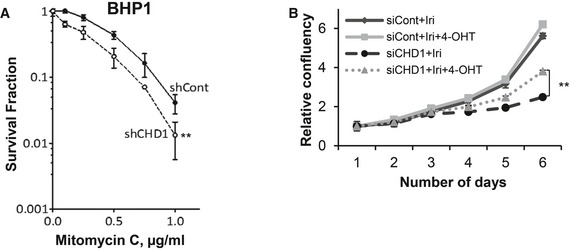
- CHD1‐depleted BHP1 cells show hypersensitivity to mitomycin C (MMC) treatment. Control and CHD1‐depleted BHP1 cells were treated with the indicated doses of MMC for 4 h, and surviving fractions were measured by counting colonies after 3 weeks. Data are represented as mean ± SD (n = 3), and P‐value was calculated using ANOVA (**P = 0.005).
- Similar to Fig EV4O, rescue experiments were performed with cells expressing mChd1‐ERT2. Cells were treated as indicated with 0.5 μM of irinotecan and/or 4‐OHT, proliferation was measured by Celigo, and the relative confluency was plotted in the graph. Data are represented as mean ± SD (n = 3). Data were normalized to the plating efficiency. P‐value (**P = 0.006) was calculated using ANOVA.
We next hypothesized that CHD1 may control the early steps of HR by physically opening chromatin structure near the DSB. To test this hypothesis, we performed formaldehyde‐assisted isolation of regulatory elements (FAIRE), which enables the assessment of an open chromatin status or nucleosome depletion 39. For this, we used AsiSI‐ER‐U2OS cells, where DSBs are induced at specific sites in the genome by the AsiSI restriction enzyme upon 4‐OHT treatment 40. FAIRE data using AsiSI‐ER‐U2OS cells treated with 4‐OHT for different time points demonstrate an opening of chromatin near the DSBs site within 2 h of DSB induction, which remained open up to 6 h (Fig EV5G). Notably, the opening of chromatin was observed only at the positive site (chr6:90404906), where AsiSI is known induce DSBs, but not at a negative site (chr6:101505264) (Fig EV5G). Interestingly, the opening of chromatin at DSB sites (DSB‐I and DSB‐II) previously shown to recruit RAD51 and be repaired by the HR pathway 41 was significantly decreased in CHD1‐depleted cells (Figs 5D and EV5H). Moreover, CHD1 depletion only affected the opening of chromatin at the positive site known to result in DSB formation, but not at the negative site, thereby confirming a role of CHD1 in the opening of chromatin at the break site. As a control, we also confirmed that there were no substantial differences in the induction of DSBs by AsiSI in control and CHD1‐depleted cells (Fig EV5F). To further validate that CHD1 is important for the end resection process and mediates the ssDNA generation, we performed native BrdU assay in PC3 cells where the depletion of CHD1 led to decreased ssDNA generation (Fig 5E and F).
Given its established role as a regulator of chromatin structure, we hypothesized that the requirement of CHD1 for HR‐mediated DNA repair is likely dependent upon DNA packaging into a chromosomal chromatin context. Thus, we transiently transfected control and CHD1‐depleted PC3 and BHP1 cells with the linearized HR repair substrate vector and measured HR efficiency. In this case, DNA repair does not occur in a fully genomic chromatin context. Interestingly, in this non‐chromosomal context of transfected linear plasmid DNA, CHD1 depletion did not affect HR efficiency (Fig EV5I and J), indicating that CHD1 likely functions to promote the end resection process of HR DNA repair via a chromatin remodeling mechanism and also confirms that the HR machinery is otherwise intact following CHD1 depletion.
Loss of CHD1 increases sensitivity to DNA damaging agents and PARP inhibition
Given the evidence for the role of CHD1 in the HR pathway, we further tested the sensitivity of CHD1‐depleted cells to mitomycin C (MMC), which induces S phase‐specific DSBs which are predominantly repaired by HR. Stable depletion of CHD1 in PC3 and BHP1 cells resulted in increased sensitivity to MMC (Figs 6A and EV6A). Moreover, CHD1‐depleted cells (PC3) also showed increased sensitivity to the clinically used topoisomerase I inhibitor irinotecan (Iri), which also induces secondary DSBs in S phase that largely require homologous recombination for repair (Fig 6B). Notably, the increased sensitivity of CHD1‐depleted cells to irinotecan can be partially rescued by expression of mChd1 (Fig EV6B), further confirming the importance of CHD1 for therapeutic responsiveness of prostate cancer cells.
Figure 6. CHD1‐depleted cells show hypersensitivity to mitomycin C, irinotecan, and PARP inhibition.
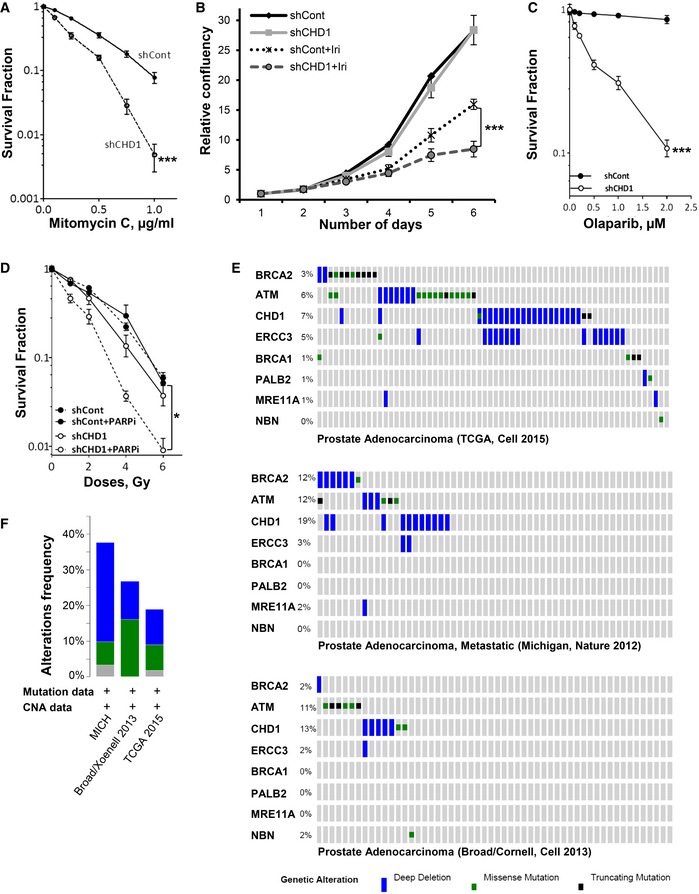
- CHD1‐depleted cells show hypersensitivity to mitomycin C (MMC) treatment. For colony formation assay, shCont and shCHD1 PC3 cells were treated with the indicated MMC concentrations for 4 h and surviving fractions were measured by counting colonies after 3 weeks and mean values ± SD are represented (n = 3, ***P = 0.0002, ANOVA).
- CHD1‐depleted cells show increased hypersensitivity to irinotecan. For cell proliferation analysis, shCont and shCHD1 cells were treated with 1 μM irinotecan and proliferation was measured by Celigo and the relative confluency (mean ± SD) are plotted in the graph (n = 3, ***P = 0.0004, ANOVA).
- Loss of CHD1 leads to increased sensitivity to PARP inhibition. Control or CHD1‐depleted BPH1 cells were treated with the indicated concentrations of the PARP inhibitor olaparib, and surviving fractions were measured by counting colonies after 3 weeks. Data are represented as mean values ± SD (n = 3, ***P = 0.0003, ANOVA).
- CHD1 depletion render cells sensitive to PARP inhibition in combination with irradiation. shCont‐ or shCHD1‐expressing BHP1 cells were treated with 1 μM of PARP inhibitor olaparib for 2 h before irradiation with the indicated doses of X‐rays, and surviving fractions (normalized to the unirradiated condition) were measured by counting colonies after 3 weeks. Data are represented as mean ± SD (n = 3, *P = 0.07, ANOVA).
- Frequency of CHD1 gene alterations in comparison with DSB repair genes in prostate cancer patients. Data obtained from cBioPortal for cancer genomics.
- Frequency of CHD1 gene alterations from the indicated data sets.
It is well established that cells which lack HR efficiency, such as BRCA1‐ and ‐2‐deficient ovarian cancer, are particularly sensitive to PARP inhibition 42, 43. This has led to numerous clinical trials with various PARP inhibitors (PARPi) and the approval of olaparib as a therapy for BRCA‐mutated ovarian cancers. Therefore, we tested whether CHD1‐depleted cells display synthetic lethality for PARP inhibition (PARPi). Indeed, not only were CHD1‐depleted cells sensitive to PARPi alone (Fig 6C), but PARPi also significantly increased cell responsiveness to irradiation (Fig 6D; Tables EV3 and EV4). Together, these data further confirm that CHD1 is required for HR efficiency and reveal a previously unknown synthetic lethal relationship between CHD1‐deficiency and PARP inhibition.
Discussion
In this study, we provide the first evidence that the chromo‐domain helicase DNA‐binding protein 1 (CHD1) is recruited to chromatin upon DSB induction and is required for DSB repair. Specifically, CHD1 is required for the recruitment of CtIP to chromatin in DNA damage‐induced cells, indicating a role for CHD1 in the end resection process during HR‐mediated DSB repair. To date, CHD1 was primarily implicated in transcription and the maintenance of an open chromatin status 19, 44. Studies from yeast and Drosophila described an association of CHD1 with promoters and the transcribed regions of active genes 45, 46. In vitro studies demonstrated that CHD1 functions in the assembly, remodeling, and spacing of nucleosomes 10, 47. CHD1 may also work in cooperation with histone modifications. For example, one study suggested a dependence of H2B monoubiquitination (H2Bub1) on CHD1 48. Consistent with this finding, we and others previously demonstrated that the H2B ubiquitin ligase complex containing RNF20 and RNF40 is important for activation of the DNA damage response and DSB repair in an ATM‐dependent manner 49, 50, 51. We further uncovered an intimate connection between H2Bub1 and the histone chaperone complex FACT. Specifically, the FACT subunit SUPT16H was required for DNA DSB repair and its depletion elicited a similar phenotype as RNF40 knockdown 51. Interestingly, CHD1 was also reported to interact with the FACT subunit SSRP1 13, lending further support for the functional interaction of CHD1, FACT, and H2Bub1 in DNA repair.
Our current results indicate that CHD1 is recruited to chromatin upon induction of DSB and co‐localizes with γH2AX, similar to many proteins involved in the DNA damage response and repair. Moreover, the co‐localization of CHD1 with γH2AX is not dependent on cell cycle phase. Chromatin fractionation showed that CHD1 recruitment to chromatin gradually increases in response to DSB induction. It appears that CHD1 is associated with both fast and slow repair kinetics, possibly indicating that CHD1 recruitment is not restricted to euchromatic regions. In mammalian cells, DSB are predominantly repaired by NHEJ and HR. The balance between these two pathways is controlled by a tightly regulated hierarchy 28 which is further controlled by other repair proteins such as KU 52, RAD51, or ATM 30. In the absence of one of these factors, a deregulated repair hierarchy may result in the inappropriate switching to another repair mechanism. One decisive regulatory step controlling HR is the DNA end resection process, which commits the repair to HR. DSB end resection must be appropriately regulated in order to avoid inappropriate initiation of HR, which may otherwise lead to genomic aberrations. We now extend our understanding of this process and show that CHD1 is required for end resection. Consistent with a mechanical function in opening chromatin structure, we showed that the ATPase domain of CHD1 is important for the recruitment of CtIP to DSBs. While CHD1 was shown to interact with methylated H3 (H3K4me3) during transcriptional regulation 11, it remains unclear whether this mechanism is required for CHD1 recruitment during DNA repair. Notably, a recent study showed that the chromo‐domains of CHD1 are more important for its enzymatic activity than chromatin localization 53.
Stably integrated plasmid‐based HR and NHEJ reporter assays showed that CHD1 depletion specifically affects HR but not NHEJ, suggesting that the ATP‐dependent chromatin remodeling activity of CHD1 is important for HR‐mediated DSB repair. One key step in HR is the generation of ssDNA through the end resection process, which requires the binding of CtIP to the damage site. Subsequently, RPA1 binds to ssDNA generated by the end resection process and later replaced by RAD51 for the formation of the presynaptic filament 54. We show that CHD1 depletion leads to reduced recruitment of CtIP to chromatin upon DSB induction. CtIP was initially identified as a CtBP‐interacting protein and interacts with the MRN complex and BRCA1 27, 37. CtIP promotes ATR recruitment in S/G2 phase and the end resection process 27. The defects we observed on ssDNA binding protein (RPA1 and RAD51) recruitment in CHD1‐depleted cells are likely due to decreased recruitment of CtIP to chromatin in response to DSB. Experiments with MRE11 inhibition or CtIP depletion indicate that the recruitment of CHD1 to DSB is dependent on MRE11 activity, but not on CtIP, thereby placing CHD1 downstream of the MRN complex and upstream of CtIP in the repair process.
Data from FAIRE analyses support a role for CHD1 in the opening of chromatin at the DSB site. Based on the data presented here, we suggest a model for an early function of CHD1 downstream of ATM‐mediated H2AX phosphorylation (Fig 7). In this model, the chromatin remodeling function of CHD1 is required for the repositioning or the ejection of nucleosomes adjacent the DSB site. In this manner, the exposed DNA may serve as a substrate for CtIP‐dependent DNA end resection and subsequent HR events. In contrast, NHEJ does not require end resection and can therefore function independent of CHD1. The regulation of chromatin structure during the DSB repair process has been described in the “access–repair–restore” model 55, where chromatin modifiers and regulators are the key components. This study provides evidence that CHD1 might function during the “access” step to open chromatin structure to enable DSB repair via the HR pathway. Other CHD family members including CHD3, CHD4, and CHD1L have also been shown to be important for the repair of DSB 32, 56. Studies on the CHD1‐like protein (CHD1L) have shown that it interacts with PARP and is involved in the PARP‐mediated nucleosome sliding. Similar to CHD1 depletion, CHD1L depletion leads to DNA damage sensitivity 56. Many other ATP‐dependent chromatin remodelers including the SWI/SNF complex, INO80, ISWI, and SRCAP also play roles in DSB repair. These complexes appear to have diverse, but specific, functions in the DNA repair process. For example, the ARID1A subunit of the BAF complex was recently shown to be important for NHEJ and cell survival. Similar to CHD1, ARID1A is mutated in many cancers 57, 58. Due to the diverse cell systems and contexts investigated for the various remodelers, the degree of redundancy and epistasis between the various chromatin remodeling complexes is not well established.
Figure 7.
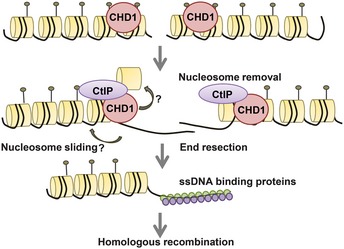
Model for the role of CHD1 in the HR repair pathway.
Importantly, the CHD1 gene is frequently deleted or mutated in prostate cancer where these aberrations are associated with a poorer prognosis 1, 2, 3. Our data suggest that these effects may be caused at least in part by impaired HR. Importantly, CHD1 deletion or mutation, like BRCA1 or ‐2 mutation in breast and ovarian cancer, which also show defects in HR, may provide an Achilles' heel for targeted treatment of this subgroup of tumors using small molecules such as PARP inhibitors, which display selectivity for HR‐deficient cells. Notably, PARP inhibitors are also already being tested in prostate cancer. For example, a single metastatic prostate cancer patient with BRCA2 alterations was reported to show a complete response to veliparib 59. More recently, a phase II clinical trial (NCT01682772) demonstrated the utility of the PARP inhibitor olaparib for the treatment of metastatic prostate cancer, where 33% of patients demonstrated a positive clinical response 59, 60. Notably, patients with mutations in known DNA repair‐associated genes (e.g., BRCA2 and ATM) displayed a better progression‐free and overall survival compared to the patients without DNA repair defects where 14 out of 16 (88%) of PARPi responders were biomarker‐positive for DNA repair defects. A retrospective analysis of CHD1 status in these samples may reveal its potential utility as a biomarker for predicting PARPi responsiveness. Interestingly, when we combine the frequency of gene alterations of CHD1 in prostate cancer with other DNA repair genes, it is striking that the percentage of alterations is similar to the response rate observed by Mateo, et al (Fig 6E) 1, 4, 61. Thus, our data demonstrating a central role of CHD1 in HR‐mediated DSB repair provide a foundation for further exploration of CHD1 status as a diagnostic marker for prostate cancer patient stratification and the development of individualized tumor therapies.
Materials and Methods
Cell culture and siRNA transfection
PC3, VCaP, and BHP1 cells which have stable integration of shControl or shCHD1 3 were grown in RPMI medium containing 10% bovine growth serum (BGS; HyClone, USA), 1× penicillin–streptomycin (Sigma, St. Louis, USA), and 1 μg/ml of puromycin (Sigma). U2OS cells were grown in high‐glucose DMEM (Invitrogen) containing 10% bovine growth serum (HyClone), 1× sodium pyruvate (Invitrogen), and 1× penicillin–streptomycin (Sigma). HeLa cells harboring pEJ or pGC substrates were grown in DMEM medium containing 800 μg/ml G418 or 1 μg/ml puromycin, respectively. U2OS19 ptight13 GFP‐lacR cells were grown in DMEM high glucose, 10% BGS, 1× penicillin–streptomycin (Sigma), 800 mg/ml of G418, and 2 mM IPTG containing medium for maintenance 34, 35. To induce DNA damage, cells were treated with doxycycline (doxy) for 16 h without IPTG and processed for immunostaining. AsiSI‐U2OS cells were grown in medium containing 1 μg/ml puromycin and treated with 300 ng/ml of 4‐OH tamoxifen (4‐OHT) (T176, Sigma) to induce DSB as previously described 40. To transiently knockdown CHD1, siRNA transfections were performed using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. Cells were treated either with neocarzinostatin 100 ng/ml (NCS; Sigma), doxy, 4‐OHT, veliparib (ABT‐888, Selleckchem), mirin (M9948, Sigma), or irinotecan (I1406, Sigma) as indicated. Cloning of mChd1 and siRNAs used in this study is described and listed in Table EV1.
Chromatin fractionation
Chromatin fractionation was performed as previously described 51. Briefly cells were re‐suspended in lysis buffer [10 mM HEPES (pH 7.9), 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.1% Triton X‐100, 1 mM DTT, and protease inhibitors] and centrifuged at 1,300 g for 5 min, and the nuclear pellet was lysed in nuclear lysis buffer [3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, and protease inhibitors] for 30 min on ice. Soluble chromatin fractions were separated by centrifuging at 1,700 g for 5 min. Chromatin fractions were analyzed by SDS–PAGE electrophoresis. Immunoblotting and antibody incubations were performed according to the standard protocol. Antibodies used in this study are listed in Table EV5.
DSB reporter assay
Double‐strand break repair efficiency was measured using DSB reporter assay as previously described 28. Briefly, HeLa cells harboring single copies of an HR (pGC) or NHEJ (pEJ) repair substrate were transfected with control or CHD1 siRNA. Twenty‐four hours after siRNA transfection, cells were transfected with I‐SceI‐expressing vector (pCMV3xnls‐I‐SceI, a kind gift from M. Jasin) to induce DSBs, or with pCMV‐Neo as a control. After 48 h, the percentage of GFP‐positive cells was monitored using flow cytometry (FACScan, BD Bioscience) as an indication for HR and NHEJ efficiency. Data were normalized to transfection efficiency.
Immunofluorescence staining
Cells were grown on coverslips and treated with NCS, doxy, or 4‐OHT for the indicated time points and then fixed with 4% paraformaldehyde for 10 min before permeabilization with 0.5% Triton X‐100 for 10 min at room temperature. After blocking with 3% BSA, cells were incubated with primary antibodies overnight at 4°C and then with fluorescently conjugated secondary antibodies. The nuclei were counterstained with DAPI (10 ng/ml). Images were acquired with a Zeiss LSM 510 Meta confocal microscope using 25× or 63× oil immersion lens. For radiation experiments, cells grown on coverslips were irradiated (RS225 research system, GLUMAY MEDICAL, UK at 200 kV, 15 mA) with the indicated doses. EdU incorporation was performed using the Invitrogen Click‐iT® EdU Alexa Fluor® 488 HCS Assay (C10351) according to the manufacturer's instructions. Proximity ligase assays were performed using the Duolink® In Situ Red Starter Kit Mouse/Rabbit (DUO92101) kit (Sigma) according to the manufacturer's instructions.
For native BrdU assay, cells were pre‐extracted with pre‐extraction buffer (25 mM Hepes 7.4, 50 mM NaCl, 1 mM EDTA, 3 mM MgCl2, 300 mM sucrose, and 0.5% Triton X‐100) and fixed with 4% formaldehyde (w/v). Immunostaining was performed with anti‐BrdU and γH2AX antibodies, and images were taken with Zeiss LSM 510 Meta confocal microscope.
Colony formation assays
Colony formation assay was performed as previously described 52. PC3 and BHP1 cells stably expressing either control or CHD1 shRNA were seeded and allowed to adhere before MMC treatment or irradiation. For PARP inhibition, 1 μM olaparib (SelleckChem) was added 2 h prior to irradiation. Survival fractions were measured by counting colonies after 3 weeks. The mean values of three independent experiments are shown. Data were normalized to the plating efficiency. For proliferation assays, cells were grown in 96‐well plates and the confluency was measured daily using the CeligoTM Cytometer (Cyntellect) with alternative days of indicated treatments.
Formaldehyde‐assisted isolation of regulatory elements
For FAIRE, AsiSI‐ER‐U2OS cells were transfected with mock or siCHD1 and treated with 4‐OHT for different time points. The cells were cross‐linked with 1% formaldehyde in PBS for 10 min and quenched with 125 mM glycine for 5 min. Nuclei was isolated with nuclear preparation buffer [50 mM Tris (pH 7.5), 20 mM EDTA (pH 8.0), 150 mM NaCl, 0.5% (v/v) NP‐40, 1% (v/v) Triton X‐100, and 20 mM NaF with protease inhibitors]. The nuclear pellet was re‐suspended in sonication buffer‐I containing 50 mM Tris–HCl (pH 8.0), 10 mM EDTA, 1% SDS plus with protease inhibitors and incubated for 15 min at 4°C. Equal volumes of sonication buffer‐II [50 mM Tris–HCl (pH 8.0), 20 mM EDTA, 50 mM NaCl, 1% (v/v) NP‐40, 20 mM NaF, and protease inhibitors] was added, and samples were sonicated for 30 cycles using Bioruptor® Plus (Diagenode SA, Liège, Belgium). After clearing the chromatin extracts by centrifugation at high speed, extracts were diluted with 600 μl of dilution buffer containing 50 mM Tris–HCl (pH 8.0), 20 mM EDTA, 150 mM NaCl, 0.5% sodium deoxycholate, 1% (v/v) NP‐40, 20 mM NaF, and protease inhibitors. Accessible DNA was isolated from fragmented chromatin by phenol:chloroform:isoamyl alcohol extraction and de‐cross‐linked overnight at 65°C. The DNA was precipitated with linear polyacrylamide and analyzed using quantitative real time–PCR (qRT–PCR) with specific primers near to the break site. The primer sequences utilized are listed in Table EV2. Data were normalized to the input.
ERT2‐HA‐mChd1 cloning and stable cells lines
For the rescue experiments, we expressed mouse Chd1 (mChd1), which bears 96% identity at the amino acid level to the human CHD1 protein. The coding region of mChd1 (NM_007690.3) was cloned into pSG5‐HA‐MCS‐ERT2‐P2A‐Hygro vector (S.A. Johnsen, unpublished) using cloning primers mChd1‐Cl‐NotI‐For GCTGACGCGGCCGCAATGAATGGACACAGTGATGAAGAAAG and mChd1‐Cl‐NheI‐Rev GCTGACGCTAGCTGTCTTCCGACTACTCCAGGTGTG, and clones were confirmed by sequencing. The resulting construct results in the production of two polypeptides including the HA‐tagged mChd1‐ERT2 fusion protein and the hygromycin resistance gene separated by a viral P2A sequence in a single open reading frame. Stable cell lines were generated by transfecting 2.5 μg of plasmid DNA into PC3 cells using Lipofectamine 2000. Cells were then selected with 100 μg/ml hygromycin. For the ATPase‐mutant mChd1, the amino acid 510 lysine (K) was mutated to arginine (R) (AAA to AGA) using the QuikChange II XL Site‐Directed Mutagenesis Kit (200521, Agilent Technologies), and the mutation was confirmed by sequencing. For the transient transfections, PC3 cells were transfected with Lipofectamine 2000 and treated with 4‐OHT for 24 h before NCS treatment and processed for immunostaining and PLA.
Neutral comet assay
Neutral comet assay cells were mixed with 1% low‐melting agarose and coated on comet slides. The slides were incubated with lysis buffer [2.5 M NaCl, 100 mM EDTA, 10 mM Tris–HCl, 1% N‐lauroylsarcosine, pH 9.5, 0.5% Triton X‐100 and 10% dimethylsulfoxide (DMSO)] for 1 h at 37°C in the dark. After incubation, slides were washed with electrophoresis buffer (300 mM sodium acetate, 100 mM Tris–HCl, pH 8.3) and left in the fresh buffer for 1 h. Then, slides were electrophoresed for 20 min at 20 V, stained with propidium iodide (PI), and images were obtained using Zeiss LSM 510 Meta confocal microscope.
Author contributions
VK, WYM, and SAJ designed the experiments. VK and SAJ wrote the manuscript. VK, WYM, SKR, and SJB performed the experiments. AM and HW provided U2OS19 ptight13 GFP‐lacR cells and helped with the experiments. HS and RS provided the stable shCHD1 prostate cancer cell lines. MD, MG and ED provided helpful guidance in study design. All authors provided intellectual input and edited the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Source Data for Expanded View
Review Process File
Source Data for Figure 2C
Acknowledgements
The authors would like to acknowledge R. Reimer (Microscope and Imaging facility, HPI, Hamburg) for help with the confocal fluorescence microscopy, G. Legube for providing AsiSI‐U2OS cells, and S. Bolte and N. Molitor for technical support. A.M. is supported by a Marie Curie Intra‐European Fellowship for Career Development (Project #627187). S.K.R. is supported by a National Overseas Scholarship, Government of India (11015/49/2010‐SCD‐V). This work was funded by the Deutsche Forschungsgemeinschaft (GRK1034) to S.A.J..
EMBO Reports (2016) 17: 1609–1623
Contributor Information
Vijayalakshmi Kari, Email: vkari1@gwdg.de.
Steven A Johnsen, Email: steven.johnsen@med.uni-goettingen.de.
References
- 1. Grasso CS, Wu Y‐M, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC et al (2012) The mutational landscape of lethal castration‐resistant prostate cancer. Nature 487: 239–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huang S, Gulzar ZG, Salari K, Lapointe J, Brooks JD, Pollack JR (2012) Recurrent deletion of CHD1 in prostate cancer with relevance to cell invasiveness. Oncogene 31: 4164–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burkhardt L, Fuchs S, Krohn A, Masser S, Mader M, Kluth M, Bachmann F, Huland H, Steuber T, Graefen M et al (2013) CHD1 is a 5q21 tumor suppressor required for ERG rearrangement in prostate cancer. Cancer Res 73: 2795–2805 [DOI] [PubMed] [Google Scholar]
- 4. Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M et al (2013) Punctuated evolution of prostate cancer genomes. Cell 153: 666–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rodrigues LU, Rider L, Nieto C, Romero L, Karimpour‐Fard A, Loda M, Lucia MS, Wu M, Shi L, Cimic A et al (2015) Coordinate loss of MAP3K7 and CHD1 promotes aggressive prostate cancer. Cancer Res 75: 1021–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kornberg RD, Lorch Y (1999) Twenty‐five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98: 285–294 [DOI] [PubMed] [Google Scholar]
- 7. Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074–1080 [DOI] [PubMed] [Google Scholar]
- 8. Strahl BD, Allis CD (2000) The language of covalent histone modifications. Nature 403: 41–45 [DOI] [PubMed] [Google Scholar]
- 9. Delmas V, Stokes DG, Perry RP (1993) A mammalian DNA‐binding protein that contains a chromodomain and an SNF2/SWI2‐like helicase domain. Proc Natl Acad Sci USA 90: 2414–2418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lusser A, Urwin DL, Kadonaga JT (2005) Distinct activities of CHD1 and ACF in ATP‐dependent chromatin assembly. Nat Struct Mol Biol 12: 160–166 [DOI] [PubMed] [Google Scholar]
- 11. Sims RJ III, Chen C‐F, Santos‐Rosa H, Kouzarides T, Patel SS, Reinberg D (2005) Human but not yeast CHD1 binds directly and selectively to histone H3 methylated at lysine 4 via its tandem chromodomains. J Biol Chem 280: 41789–41792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stokes DG, Perry RP (1995) DNA‐binding and chromatin localization properties of CHD1. Mol Cell Biol 15: 2745–2753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kelley DE, Stokes DG, Perry RP (1999) CHD1 interacts with SSRP1 and depends on both its chromodomain and its ATPase/helicase‐like domain for proper association with chromatin. Chromosoma 108: 10–25 [DOI] [PubMed] [Google Scholar]
- 14. Simic R, Lindstrom DL, Tran HG, Roinick KL, Costa PJ, Johnson AD, Hartzog GA, Arndt KM (2003) Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. EMBO J 22: 1846–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smolle M, Venkatesh S, Gogol MM, Li H, Zhang Y, Florens L, Washburn MP, Workman JL (2012) Chromatin remodelers Isw1 and Chd1 maintain chromatin structure during transcription by preventing histone exchange. Nat Struct Mol Biol 19: 884–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gkikopoulos T, Schofield P, Singh V, Pinskaya M, Mellor J, Smolle M, Workman JL, Barton GJ, Owen‐Hughes T (2011) A role for Snf2‐related nucleosome‐spacing enzymes in genome‐wide nucleosome organization. Science 333: 1758–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hennig BP, Bendrin K, Zhou Y, Fischer T (2012) Chd1 chromatin remodelers maintain nucleosome organization and repress cryptic transcription. EMBO Rep 13: 997–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pointner J, Persson J, Prasad P, Norman‐Axelsson U, Strålfors A, Khorosjutina O, Krietenstein N, Svensson JP, Ekwall K, Korber P (2012) CHD1 remodelers regulate nucleosome spacing in vitro and align nucleosomal arrays over gene coding regions in S. pombe. EMBO J 31: 4388–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gaspar‐Maia A, Alajem A, Polesso F, Sridharan R, Mason MJ, Heidersbach A, Ramalho‐Santos J, McManus MT, Plath K, Meshorer E et al (2009) Chd1 regulates open chromatin and pluripotency of embryonic stem cells. Nature 460: 863–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Price BD, D'Andrea AD (2013) Chromatin remodeling at DNA double‐strand breaks. Cell 152: 1344–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bont RD, van Larebeke N (2004) Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 19: 169–185 [DOI] [PubMed] [Google Scholar]
- 22. Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155–168 [DOI] [PubMed] [Google Scholar]
- 23. Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40: 179–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Helleday T, Lo J, van Gent DC, Engelward BP (2007) DNA double‐strand break repair: from mechanistic understanding to cancer treatment. DNA Repair 6: 923–935 [DOI] [PubMed] [Google Scholar]
- 25. Chapman JR, Taylor MRG, Boulton SJ (2012) Playing the end game: DNA double‐strand break repair pathway choice. Mol Cell 47: 497–510 [DOI] [PubMed] [Google Scholar]
- 26. Critchlow SE, Jackson SP (1998) DNA end‐joining: from yeast to man. Trends Biochem Sci 23: 394–398 [DOI] [PubMed] [Google Scholar]
- 27. Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP (2007) Human CtIP promotes DNA end resection. Nature 450: 509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mansour WY, Schumacher S, Rosskopf R, Rhein T, Schmidt‐Petersen F, Gatzemeier F, Haag F, Borgmann K, Willers H, Dahm‐Daphi J (2008) Hierarchy of nonhomologous end‐joining, single‐strand annealing and gene conversion at site‐directed DNA double‐strand breaks. Nucleic Acids Res 36: 4088–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Köcher S, Rieckmann T, Rohaly G, Mansour WY, Dikomey E, Dornreiter I, Dahm‐Daphi J (2012) Radiation‐induced double‐strand breaks require ATM but not Artemis for homologous recombination during S‐phase. Nucleic Acids Res 40: 8336–8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mansour WY, Borgmann K, Petersen C, Dikomey E, Dahm‐Daphi J (2013) The absence of Ku but not defects in classical non‐homologous end‐joining is required to trigger PARP1‐dependent end‐joining. DNA Repair 12: 1134–1142 [DOI] [PubMed] [Google Scholar]
- 31. Chai B, Huang J, Cairns BR, Laurent BC (2005) Distinct roles for the RSC and Swi/Snf ATP‐dependent chromatin remodelers in DNA double‐strand break repair. Genes Dev 19: 1656–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lans H, Marteijn JA, Vermeulen W (2012) ATP‐dependent chromatin remodeling in the DNA‐damage response. Epigenetics Chromatin 5: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dong S, Han J, Chen H, Liu T, Huen MSY, Yang Y, Guo C, Huang J (2014) The human SRCAP chromatin remodeling complex promotes DNA‐end resection. Curr Biol 24: 2097–2110 [DOI] [PubMed] [Google Scholar]
- 34. Lemaître C, Fischer B, Kalousi A, Hoffbeck A‐S, Guirouilh‐Barbat J, Shahar OD, Genet D, Goldberg M, Betrand P, Lopez B et al (2012) The nucleoporin 153, a novel factor in double‐strand break repair and DNA damage response. Oncogene 31: 4803–4809 [DOI] [PubMed] [Google Scholar]
- 35. Mund A, Schubert T, Staege H, Kinkley S, Reumann K, Kriegs M, Fritsch L, Battisti V, Ait‐Si‐Ali S, Hoffbeck A‐S et al (2012) SPOC1 modulates DNA repair by regulating key determinants of chromatin compaction and DNA damage response. Nucleic Acids Res 40: 11363–11379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van den Bosch M, Bree RT, Lowndes NF (2003) The MRN complex: coordinating and mediating the response to broken chromosomes. EMBO Rep 4: 844–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu X, Chen J (2004) DNA damage‐induced cell cycle checkpoint control requires CtIP, a phosphorylation‐dependent binding partner of BRCA1 C‐terminal domains. Mol Cell Biol 24: 9478–9486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Symington LS (2002) Role of RAD52 epistasis group genes in homologous recombination and double‐strand break repair. Microbiol Mol Biol Rev 66: 630–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Giresi PG, Kim J, McDaniell RM, Iyer VR, Lieb JD (2007) FAIRE (Formaldehyde‐assisted isolation of regulatory elements) isolates active regulatory elements from human chromatin. Genome Res 17: 877–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, Legube G (2010) High‐resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J 29: 1446–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Aymard F, Bugler B, Schmidt CK, Guillou E, Caron P, Briois S, Iacovoni JS, Daburon V, Miller KM, Jackson SP et al (2014) Transcriptionally active chromatin recruits homologous recombination at DNA double‐strand breaks. Nat Struct Mol Biol 21: 366–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T (2005) Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature 434: 913–917 [DOI] [PubMed] [Google Scholar]
- 43. Patel AG, Sarkaria JN, Kaufmann SH (2011) Nonhomologous end joining drives poly(ADP‐ribose) polymerase (PARP) inhibitor lethality in homologous recombination‐deficient cells. Proc Natl Acad Sci 108: 3406–3411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Marfella CGA, Imbalzano AN (2007) The Chd family of chromatin remodelers. Mutat Res 618: 30–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Walfridsson J, Khorosjutina O, Matikainen P, Gustafsson CM, Ekwall K (2007) A genome‐wide role for CHD remodelling factors and Nap1 in nucleosome disassembly. EMBO J 26: 2868–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. McDaniel IE, Lee JM, Berger MS, Hanagami CK, Armstrong JA (2008) Investigations of CHD1 function in transcription and development of Drosophila melanogaster. Genetics 178: 583–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Stockdale C, Flaus A, Ferreira H, Owen‐Hughes T (2006) Analysis of nucleosome repositioning by yeast ISWI and Chd1 chromatin remodeling complexes. J Biol Chem 281: 16279–16288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee J‐S, Garrett AS, Yen K, Takahashi Y‐H, Hu D, Jackson J, Seidel C, Pugh BF, Shilatifard A (2012) Codependency of H2B monoubiquitination and nucleosome reassembly on Chd1. Genes Dev 26: 914–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nakamura K, Kato A, Kobayashi J, Yanagihara H, Sakamoto S, Oliveira DVNP, Shimada M, Tauchi H, Suzuki H, Tashiro S et al (2011) Regulation of homologous recombination by RNF20‐dependent H2B ubiquitination. Mol Cell 41: 515–528 [DOI] [PubMed] [Google Scholar]
- 50. Moyal L, Lerenthal Y, Gana‐Weisz M, Mass G, So S, Wang S‐Y, Eppink B, Chung YM, Shalev G, Shema E et al (2011) Requirement of ATM‐dependent monoubiquitylation of histone H2B for timely repair of DNA double‐strand breaks. Mol Cell 41: 529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kari V, Shchebet A, Neumann H, Johnsen SA (2011) The H2B ubiquitin ligase RNF40 cooperates with SUPT16H to induce dynamic changes in chromatin structure during DNA double‐strand break repair. Cell Cycle 10: 3495–3504 [DOI] [PubMed] [Google Scholar]
- 52. Mansour WY, Rhein T, Dahm‐Daphi J (2010) The alternative end‐joining pathway for repair of DNA double‐strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res 38: 6065–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hauk G, McKnight JN, Nodelman IM, Bowman GD (2010) The chromodomains of the Chd1 chromatin remodeler regulate DNA access to the ATPase motor. Mol Cell 39: 711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krejci L, Altmannova V, Spirek M, Zhao X (2012) Homologous recombination and its regulation. Nucleic Acids Res 40: 5795–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Soria G, Polo SE, Almouzni G (2012) Prime, repair, restore: the active role of chromatin in the DNA damage response. Mol Cell 46: 722–734 [DOI] [PubMed] [Google Scholar]
- 56. Ahel D, Hořejší Z, Wiechens N, Polo SE, Garcia‐Wilson E, Ahel I, Flynn H, Skehel M, West SC, Jackson SP et al (2009) Poly(ADP‐ribose)–dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 325: 1240–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Watanabe R, Ui A, Kanno S, Ogiwara H, Nagase T, Kohno T, Yasui A (2014) SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res 74: 2465–2475 [DOI] [PubMed] [Google Scholar]
- 58. Wu JN, Roberts CWM (2013) ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov 3: 35–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. VanderWeele DJ, Paner GP, Fleming GF, Szmulewitz RZ (2015) Sustained complete response to cytotoxic therapy and the PARP inhibitor veliparib in metastatic castration‐resistant prostate cancer – a case report. Front Oncol 5: 169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez‐Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N et al (2015) DNA‐repair defects and olaparib in metastatic prostate cancer. N Engl J Med 373: 1697–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cancer Genome Atlas Research Network . (2015) The molecular taxonomy of primary prostate cancer. Cell 163: 1011–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Source Data for Expanded View
Review Process File
Source Data for Figure 2C