

Abstract
The pathologic process of chronic phase traumatic brain injury is associated with spreading inflammation, cell death, and neural dysfunction. It is thought that sequestration of inflammatory mediators can facilitate recovery and promote an environment that fosters cellular regeneration. Studies have targeted post-traumatic brain injury inflammation with the use of pharmacotherapy and cell therapy. These therapeutic options are aimed at reducing the edematous and neurodegenerative inflammation that have been associated with compromising the integrity of the blood-brain barrier. Although studies have yielded positive results from anti-inflammatory pharmacotherapy and cell therapy individually, emerging research has begun to target inflammation using combination therapy. The joint use of anti-inflammatory drugs alongside stem cell transplantation may provide better clinical outcomes for traumatic brain injury patients. Despite the promising results in this field of research, it is important to note that most of the studies mentioned in this review have completed their studies using animal models. Translation of this research into a clinical setting will require additional laboratory experiments and larger preclinical trials.
Keywords: stem cells, drugs, neuroinflammation, trauma, neuroprotection, regeneration
Traumatic Brain Injury and Inflammation
Traumatic brain injury (TBI) is characterized by intracranial damage resulting from an external force and can be the product of physical insults such as puncture, blunt impact, or blast (Maas et al., 2008). TBI affects 1.7 million people annually, presenting a significant economic burden, and is particularly prevalent in military casualties, where an increase in explosive warfare has led to a parallel rise in TBI occurrence (Okie, 2005; Acosta et al., 2015b). TBI can be classified as mild, moderate, or severe (Lozano et al., 2015). Determining the severity of TBI is commonly accomplished in humans with the Glasgow Coma Scale (GCS), a simple questionnaire used to evaluate the patient's degree of consciousness in conjunction with medical imaging techniques (Lozano et al., 2015). Associated symptoms such as headache, dizziness, fatigue, or nausea may be short-lived in the case of mild TBI, while in more severe instances cognitive symptoms may progress chronically to resemble neurodegenerative diseases such as Alzheimer's disease (AD) and Parkinson's disease (PD) (Lozano et al., 2015). Epidemiological data also suggest that sufferers of TBI are at an increased risk for developing AD and PD later in life through poorly understood mechanisms (Lozano et al., 2015).
The pathology of TBI is divided into two phases, acute and chronic. The acute phase consists of the immediate damage produced from the insult, and the chronic phase may extend for years and is defined by spreading inflammation, cell death, and neural dysfunction that is triggered by the primary injury (Werner and Engelhard, 2007; Acosta et al., 2015b). The expanded distribution of pro-inflammatory molecules, reactive species, and other damaging byproducts from the primary injury site causes a progressive wave of cell damage, forming a region of dead and endangered cells called the penumbra (Zhao et al., 2005; Acosta et al., 2015b). This region poses the greatest threat of chronic symptom development, but also points to a significant opportunity for cell rescue (Zhao et al., 2005; Acosta et al., 2015b). Once considered to be an acute injury only, our understanding of TBI as possessing this chronic aspect has revealed the opportunity of a greater time frame for therapeutic intervention (Acosta et al., 2015b).
The chronic phase of TBI is marked by a multitude of complex metabolic, immune, and cellular responses which are seen primarily in tissue adjacent to the injury site, but can also spread to distal regions of the brain (Kumar and Loane, 2012; Lozano et al., 2015). While these responses are seen universally in TBI, researchers have shown that the specific pathology and intricacies of the injury response are heterogeneous, and vary depending on the nature and location of the TBI (Pabon et al., 2016). The immediate damage that occurs during a TBI includes necrosis, excitotoxicity, and mutilation of local neurons, microglial activation, and vascular cells - none of which present a significant opportunity for intervention (Kumar and Loane, 2012). Instead, recent research has focused on the multiple aspects of chronic TBI as a means of reducing the secondary cell death that ensues after the initial acute phase (Lozano et al., 2015). In particular, the neuroinflammatory aspect of TBI physiopathology has been explored as a target for preventing secondary cell death and symptom progression (Lozano et al., 2015). Inflammation plays a dual role in the brain after a TBI; in the acute phase, inflammation has been shown to be neuroprotective, while aberrant inflammation throughout the chronic phase is a key factor in perpetuating cell death (Lozano et al., 2015). Sequestration of the chronic inflammation has been proven to be neuroprotective (Lozano et al., 2015). The transition from protective inflammation to degenerative inflammation - as well as the large number of competing pro-inflammatory molecules (i.e., tumor necrosis factor (TNF-α) and interleukin (IL-1β) and anti-inflammatory molecules (i.e., IL-10 and TGF-β) - make for a complex pathology (Lozano et al., 2015).
The neurodegenerative inflammation and edema seen after TBI is linked to the dysfunction of the blood-brain barrier (BBB) that occurs after the insult (Shlosberg et al., 2010; Neuwelt et al., 2011). The protective effects of the BBB are contingent on the presence of uninterrupted, selective endothelial cells which moderate the entrance of blood-borne particles (Neuwelt et al., 2011). Additionally, the health of these endothelial cells and the fidelity of the BBB is intimately connected to the health of supportive astrocytes. The mechanical force from the TBI compromises this fidelity, allowing a host of exogenous proteins such as albumin, fibrinogen, and thrombin, as well as peripheral immune cells, to enter the brain parenchyma (Shlosberg et al., 2010; Neuwelt et al., 2011; Lozano et al., 2015). These invading substances trigger the activation of microglia within the injured brain region, causing an immune response that persists for as long as the BBB remains compromised (Shlosberg et al., 2010; Neuwelt et al., 2011). Initially, the microglial activation is a protective response, but the activation can become excessive and self-perpetuating over time (Lozano et al., 2015). Additionally, the invading peripheral cells have been shown to release pro-inflammatory mediators such as cytokines, chemokines, prostaglandins, and free radicals, serving to progressively exacerbate the inflammatory process as well as increasing the permeability of the BBB (Lozano et al., 2015). This elevated permeability to large molecules and cells shifts the osmotic pressure within the brain, causing edema and an increase in intracranial pressure (Neuwelt et al., 2011; Lozano et al., 2015). Damage to the BBB is a continuing issue, as increased levels of molecules which damage tight junctions such as matrix metallopeptidase-9 (MMP-9) and vascular endothelial growth factor (VEGF) occur post-TBI and impede the recovery of the BBB (Guo et al., 1989; Lozano et al., 2015).
The immune response is further complicated by differentiation of environment-sensitive microglia into multiple phenotypes (Lozano et al., 2015). Depending on the microenvironment that the microglia is exposed to, the cell may assume an M1, pro-inflammatory, phenotype or an M2, anti-inflammatory phenotype that attenuates the pro-inflammatory M1 microglia (Lozano et al., 2015). Possible therapeutic techniques aim to minimize harmful chronic inflammation, while not preventing acute-phase neuroprotective inflammation, by developing means of downregulating pro-inflammatory molecules, upregulating anti-inflammatory molecules, and aiding in BBB repair (Lozano et al., 2015).
The only defense against the primary injury caused by a TBI is prevention with the use of safety equipment such as helmets and seatbelts (Hirschenfang et al., 1968; Lozano et al., 2015). The window of opportunity for potential therapies lies within the period of secondary cell death which may last for days, months, or years (Iida et al., 1987). Of the different secondary death mechanisms, neuro-inflammation provides the greatest potential for intervention due to its delayed onset. Glutamate excitotoxicity for example has a much narrower range for intervention as it is an immediate response and may normalize within 120 hours after insult (Matsumoto et al., 1986). Based on the length of time required for enough inflammatory cells to migrate and induce secondary cell death, the onset of damaging neuroinflammmation is a slower process (Offit et al., 1986). Neuroinflammation may also persist as a chronic consequence of TBI and this secondary effect is closely linked to many neurodegenerative diseases such as dementia pugilistica (DP), AD, PD and other pathologies (Colmano and Gross, 1971).
Anti-inflammation Pharmacotherapy
While targeting neuroinflammation is a reasonable approach to sequestering secondary cell death in TBI, it should be noted that neuroinflammation also has beneficial effects. Previous studies have shown that high doses of anti-inflammatory drugs administered after TBI led to worse outcomes due to the loss of neuroprotective effects in addition to the existing harmful effects (Ziebell and Morganti-Kossmann, 2010).
Several stand-alone anti-inflammatory drugs have been tested for efficacy in TBI treatment. Minocycline is a tetracycline derivative known to have both neuroprotective and anti-inflammatory properties (Kumar and Loane, 2012). Minocycline is a prime candidate for clinical trials due to its ability to cross the BBB in addition to its safety for human use as declared by the US Food and Drug Administration (Saatman et al., 2008). This drug minimizes the release of proinflammatory cytokines and chemokines, in conjunction with other mediators of inflammation, decreases nitric oxide by directly inhibiting the overactivation and proliferation of microglia cells (Homsi et al., 2009; Kovesdi et al., 2012). Reducing and inactivating microglial cells is key as it attributes to the decline in cytokines IL-1β, IL-6, and matrix metallopeptidase 9 (MMP-9), all of which facilitate the proinflammatory response (Homsi et al., 2009; Ziebell and Morganti-Kossmann, 2010; Guo et al., 2011). The use of minocycline in animal models has illustrated a significant reduction in inflammation and tissue damage, thereby improving outcomes (Homsi et al., 2009; Kovesdi et al., 2012). However, results of other studies contradict this, suggesting there is no beneficial effects of minocycline on TBI. These opposing outcomes are a predicted result of deviations in dosage and administration intervals, calling for further investigation (Homsi et al., 2009; Kelso et al., 2011).
Melatonin is a hormone produced in the pineal gland and is currently being explored as a stand-alone drug due to its neuroprotective characteristics (Lozano et al., 2015). It is an enzyme that easily passes through cell membranes due to it lipophilic properties. The mechanism of action for its anti-inflammatory effects are by way of inhibiting microglial activation and reducing pro-inflammatory cytokine secretion, such as IL-1β and TNF-α (Wang et al., 2013; Ding et al., 2014a). The effects of melatonin as a TBI therapeutic drug vary, similar to the results of minocycline administration. Lower brain edema and reduced cortical neural degeneration was displayed in successful trials, implying improvement of cognitive deficits (Ding et al., 2014b). However, there was no significant cognitive enhancements in experimental models, the likely cause being attributed to dosage (Kelso et al., 2011). While melatonin is a prospective therapy to sequester secondary damage caused by inflammation, additional studies are necessary to address the long-term feasibility, safety, and efficacy of melatonin in multiple models of TBI (Hirschenfang et al., 1968; Lozano et al., 2015).
Statins are a well-known group of drugs used to treat high cholesterol that have additionally shown neuroprotective and anti-inflammatory effects in a mouse model of subarachnoid hemorrhage (Uekawa et al., 2014). The proposed mode of action is through interaction with microglia and astrocytes. Statins inhibits the signaling pathways of toll-like receptor 4, nuclear factor κB (NF-κB) activation, and some small G-proteins, all of which contribute to the reduction of microglia activation (Loane and Faden, 2010; Wang et al., 2014). By additionally inhibiting epidermal growth factor receptors that play a part in astrogliosis, statins also decrease astrocyte activation (Wu et al., 2010). Without microglia and astrocytes contributing their proinflammatory effects, there is a reduction in the expression of proinflammatory cytokines IL-1β and TNF-α, as well as intracellular and intercellular adhesion molecules, and consequently an overall sequestration of the neuroinflammatory process (Loane and Faden, 2010; Wu et al., 2010; Uekawa et al., 2014). In TBI models accompanied by statin administration, results have displayed an increase in neuronal survival, growth, and differentiation and a lessening of functional impairment (Loane and Faden, 2010). Minor improvements in amnesia and disorientation (as assessed by the Galveston Orientation Amnesia Test) were achieved after rosuvastatin was given over a 10 day period to TBI patients within a clinical trial (Tapia-Perez et al., 2008). Statins are an already well established class of drugs that are tolerated by patients, with occasional mild side effects that are distinct and can be monitored without difficulty. Statins show promise in preclinical for other neurological deficits such as stroke, intracerebral hemorrhage, but more extensive laboratory studies in TBI models are needed to improve clinical outcomes (Hirschenfang et al., 1968; Lozano et al., 2015).
Currently, there remain safety and efficacy optimization hurdles about the use of anti-inflammatory pharmacotherapy for the treatment of TBI. The use of these drugs has demonstrated a notable reduction in the inflammatory response; however, it is crucial to further investigate the applicability of these treatment options in a clinical setting. As previously mentioned, concerns regarding dosage and long-term outcomes warrant further investigation of these drugs prior to their clinical use.
Stem Cell Therapy for Sequestration of Inflammation
An alternative approach to treating TBI is the concept of stem cell therapy. Stem cells are undifferentiated cells that have the potential to regenerate damaged tissue secondary to their ability to proliferate numerously, differentiate into multiple cell lines, and provide restorative resources to surviving cells (Antonucci et al., 2014; Tajiri et al., 2014c; Tajiri et al., 2014a). Preclinical studies have demonstrated the remarkable regenerative ability of stem cells to transform into newly differentiated neurons following TBI (Antonucci et al., 2012; Rodrigues et al., 2012; Liu et al., 2013; Acosta et al., 2014; Tajiri et al., 2014b, c). Following brain injury, stem cell therapy shows notable potential in sequestering neural cell death and a prolonged inflammatory response, which results in increased recovery in both cognitive and motor function (Acosta et al., 2014; de la Pena et al., 2014).
The hallmark of stem cells dampening the TBI-induced inflammatory response lies within their mechanism of action. Mesenchymal stem cells (MSCs) possess the ability to migrate to the site of injury and activate cellular effectors of the inflammatory response such as microglia, T lymphocytes, and neutrophils (Borlongan, 2011; Borlongan et al., 2011; Zhang et al., 2013). Key inflammatory signals, such as TNF-α and IL-1, induce MSCs secrete an anti-inflammatory protein called TNF-α-stimulated gene/protein 6 (TSG-6) (Watanabe et al., 2013; Zhang et al., 2013). The anti-inflammatory role of TSG-6 is carried out by disrupting the inflammatory signaling pathways of both toll-like receptors (TLRs) and NF-κB (Watanabe et al., 2013; Zhang et al., 2013). The inflammatory NF-κB signaling cascade is characterized by the production of proinflammatory cytokines by T cells, such as interferon γ (Russo et al., 2011). Interestingly, TSG-6 is able to modulate the activity of T cells to instead produce anti-inflammatory cytokines, such as IL-4 (Russo et al., 2011).
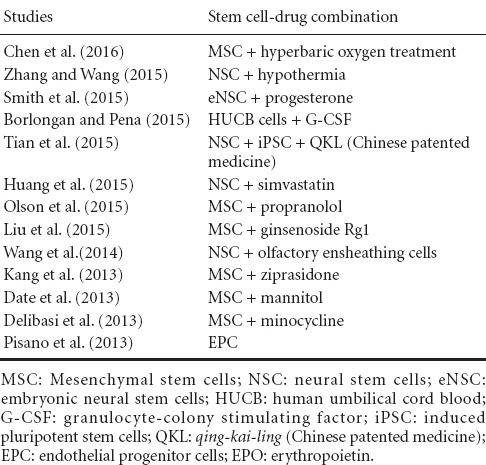
Once transplanted, stem cells are faced with the struggle of surviving in the hostile environment at the site of injury (Dela Pena et al., 2014). Certain factors, such as granulocyte-colony stimulating factor (G-CSF), can be introduced alongside stem cells to promote the neuroprotection of the transplanted stem cells (Acosta et al., 2014). Combining stem cells with factors such as G-CSF provide a significant improvement in neurogenesis and a reduction in cell death, when compared to being administered alone (Acosta et al., 2014). G-CSF, a cytokine, has the inherent capability to reduce brain edema, enable recovery of motor function, and to improve control of glutamate levels (Acosta et al., 2014). The mechanism of G-CSF utilizes receptor-mediated transport to recruit endogenous stem cells from the bone marrow into the peripheral blood. These mobilized stem cells can then migrate to the site of injury, where they can synthesize and release growth factors, chemokines, and cytokines that aid in the process of brain tissue repair (Acosta et al., 2014). Recent studies demonstrate that transplanted stem cells preferentially migrate to the spleen, rather than the brain (Acosta et al., 2015a). The concept of increased stem cell survival in the spleen over the brain suggests that direct, targeted therapy may not always be the best treatment option (Acosta et al., 2015a). Indeed, studies are investigating the efficacy of systemic delivery of stem cells in TBI and other disease models, such as stroke (Acosta et al., 2015a). Several types of transplantable cells have been tested in the laboratory, with a few reaching clinical trials, for cell therapy in stroke, including fetal cells, NT2N cells, CTX0E3, embryonic stem cells, neural stem/progenitor cells, umbilical cord blood, amnion, adipose, and induced pluripotent stem cells (Hara et al., 2008; Li et al., 2008; Stroemer et al., 2009; Kaneko et al., 2011; Liu et al., 2014; De La Pena et al., 2015). Primarily due to solid safety profile in other disease indications, preclinical studies and on-going clinical trials have given special attention to bone marrow and its cellular derivatives (Borlongan et al., 2011; Steinberg et al., 2016). Direct intracerebral implantation and peripheral transplantation, such as intravenous, intra-arterial, and intranasal, have documented the functional benefits of bone marrow-derived stem cells (Borlongan et al., 2004; Borlongan, 2011; Borlongan et al., 2011; Prasad et al., 2014; Acosta et al., 2015a). Clinical trials have been initiated, and preliminary reports have demonstrated safety, although efficacy warrants additional investigations (Steinberg et al., 2016). This concept highlights the need for investigations of combination therapy (Figure 1 and Table 1), in order to improve the outcomes of drugs and stem transplantation in TBI and other related brain disorders.
Figure 1.
Combination therapies for traumatic brain injury (TBI).
Stand-alone treatments may not be optimal for conferring brain repair. Combination of drugs and stem cell therapy may synergistically enhance the therapeutic outcomes. Indeed, combining drugs, such as granulocyte-colony stimulating factor (G-CSF), erythropoietin (EPO), and statins may improve the functional benefits of stem cells towards brain repair in TBI and other related disorders.
Table 1.
Recent studies combining stem cell therapy and drugs
Future Directions
In this review, the outlined studies detailed treatment options for TBI that focus on either stand-alone pharmacotherapy or cell therapy. A single therapeutic approach may not be optimal in reducing inflammation and in initiating the brain repair process after TBI. It is more likely that combination therapy of anti-inflammatory drugs alongside stem cell transplantation will yield enhanced results that can be translated to the clinical setting. In addition, most preclinical studies are limited by their use of small animal models for TBI, which may not accurately mimic the clinical scenario in humans. Thus, limited clinical trials of stand-alone therapies, alongside more advanced laboratory studies incorporating optimization of safety and efficacy on dosages and long-term functional assessments of combined pharmacotherapy and stem cell transplantation in large animal models, are required to allow these treatment options to become available at the bedside.
Acknowledgments
We thank Ms. Donna Morrison and Ms. Inger Mills for technical assistance in processing this manuscript.
Footnotes
Funding: This work was funded by NIH R01NS071956, NIH R01 NS090962, NIH R21NS089851, NIH R21 NS094087, DOD W81XWH-11-1-0634, and VA Merit Review I01 BX001407 to CVB.
Conflicts of interest: None declared.
References
- Acosta SA, Tajiri N, Hoover J, Kaneko Y, Borlongan CV. Intravenous bone marrow stem cell grafts preferentially migrate to spleen and abrogate chronic inflammation in stroke. Stroke. 2015a;46:2616–2627. doi: 10.1161/STROKEAHA.115.009854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta SA, Tajiri N, de la Pena I, Bastawrous M, Sanberg PR, Kaneko Y, Borlongan CV. Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson's disease. J Cell Physiol. 2015b;230:1024–1032. doi: 10.1002/jcp.24830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta SA, Tajiri N, Shinozuka K, Ishikawa H, Sanberg PR, Sanchez-Ramos J, Song S, Kaneko Y, Borlongan CV. Combination therapy of human umbilical cord blood cells and granulocyte colony stimulating factor reduces histopathological and motor impairments in an experimental model of chronic traumatic brain injury. PLoS One. 2014;9:e90953. doi: 10.1371/journal.pone.0090953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonucci I, Pantalone A, Tete S, Salini V, Borlongan CV, Hess D, Stuppia L. Amniotic fluid stem cells: a promising therapeutic resource for cell-based regenerative therapy. Curr Pharm Des. 2012;18:1846–1863. doi: 10.2174/138161212799859602. [DOI] [PubMed] [Google Scholar]
- Antonucci I, Di Pietro R, Alfonsi M, Centurione MA, Centurione L, Sancilio S, Pelagatti F, D’Amico MA, Di Baldassarre A, Piattelli A, Tete S, Palka G, Borlongan CV, Stuppia L. Human second trimester amniotic fluid cells are able to create embryoid body-like structures in vitro and to show typical expression profiles of embryonic and primordial germ cells. Cell Transplant. 2014;23:1501–1515. doi: 10.3727/096368914X678553. [DOI] [PubMed] [Google Scholar]
- Borlongan CV. Bone marrow stem cell mobilization in stroke: a ‘bonehead’ may be good after all! Leukemia. 2011;25:1674–1686. doi: 10.1038/leu.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlongan CV, Glover LE, Tajiri N, Kaneko Y, Freeman TB. The great migration of bone marrow-derived stem cells toward the ischemic brain: therapeutic implications for stroke and other neurological disorders. Prog Neurobiol. 2011;95:213–228. doi: 10.1016/j.pneurobio.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlongan CV, Lind JG, Dillon-Carter O, Yu G, Hadman M, Cheng C, Carroll J, Hess DC. Intracerebral xenografts of mouse bone marrow cells in adult rats facilitate restoration of cerebral blood flow and blood-brain barrier. Brain Res. 2004;1009:26–33. doi: 10.1016/j.brainres.2004.02.050. [DOI] [PubMed] [Google Scholar]
- Colmano G, Gross WB. Effect of metyrapone and DDD on infectious diseases. Poult Sci. 1971;50:850–854. doi: 10.3382/ps.0500850. [DOI] [PubMed] [Google Scholar]
- de la Pena I, Sanberg PR, Acosta S, Lin SZ, Borlongan CV. Umbilical cord blood cell and granulocyte-colony stimulating factor: combination therapy for traumatic brain injury. Regen Med. 2014;9:409–412. doi: 10.2217/rme.14.32. [DOI] [PubMed] [Google Scholar]
- De La Pena I, Sanberg PR, Acosta S, Lin SZ, Borlongan CV. G-CSF as an adjunctive therapy with umbilical cord blood cell transplantation for traumatic brain injury. Cell Transplant. 2015;24:447–457. doi: 10.3727/096368915X686913. [DOI] [PubMed] [Google Scholar]
- Dela Pena I, Sanberg PR, Acosta S, Tajiri N, Lin SZ, Borlongan CV. Stem cells and G-CSF for treating neuroinflammation in traumatic brain injury: aging as a comorbidity factor. J Neurosurg Sci. 2014;58:145–149. [PMC free article] [PubMed] [Google Scholar]
- Ding K, Wang H, Xu J, Lu X, Zhang L, Zhu L. Melatonin reduced microglial activation and alleviated neuroinflammation induced neuron degeneration in experimental traumatic brain injury: possible involvement of mTOR pathway. Neurochem Int. 2014a;76:23–31. doi: 10.1016/j.neuint.2014.06.015. [DOI] [PubMed] [Google Scholar]
- Ding K, Wang H, Xu J, Li T, Zhang L, Ding Y, Zhu L, He J, Zhou M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2-ARE signaling pathway as a potential mechanism. Free Radic Biol Med. 2014b;73:1–11. doi: 10.1016/j.freeradbiomed.2014.04.031. [DOI] [PubMed] [Google Scholar]
- Guo YS, Singh P, Draviam E, Greeley GH, Jr, Thompson JC. Peptide YY inhibits the insulinotropic action of gastric inhibitory polypeptide. Gastroenterology. 1989;96:690–694. [PubMed] [Google Scholar]
- Guo ZD, Wu HT, Sun XC, Zhang XD, Zhang JH. Protection of minocycline on early brain injury after subarachnoid hemorrhage in rats. Acta Neurochir Suppl. 2011;110:71–74. doi: 10.1007/978-3-7091-0353-1_13. [DOI] [PubMed] [Google Scholar]
- Hara K, Yasuhara T, Maki M, Matsukawa N, Masuda T, Yu SJ, Ali M, Yu G, Xu L, Kim SU, Hess DC, Borlongan CV. Neural progenitor NT2N cell lines from teratocarcinoma for transplantation therapy in stroke. Prog Neurobiol. 2008;85:318–334. doi: 10.1016/j.pneurobio.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Hirschenfang S, Silber M, Benton JG. Personality patterns in peripheral neuropathy. Dis Nerv Syst. 1968;29:46–50. [PubMed] [Google Scholar]
- Homsi S, Federico F, Croci N, Palmier B, Plotkine M, Marchand-Leroux C, Jafarian-Tehrani M. Minocycline effects on cerebral edema: relations with inflammatory and oxidative stress markers following traumatic brain injury in mice. Brain Res. 2009;1291:122–132. doi: 10.1016/j.brainres.2009.07.031. [DOI] [PubMed] [Google Scholar]
- Iida K, Mitomo K, Fujita T, Tamura N. Characterization of three monoclonal antibodies against C3 with selective specificities. Immunology. 1987;62:413–417. [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y, Hayashi T, Yu S, Tajiri N, Bae EC, Solomita MA, Chheda SH, Weinbren NL, Parolini O, Borlongan CV. Human amniotic epithelial cells express melatonin receptor MT1, but not melatonin receptor MT2: a new perspective to neuroprotection. J Pineal Res. 2011;50:272–280. doi: 10.1111/j.1600-079X.2010.00837.x. [DOI] [PubMed] [Google Scholar]
- Kelso ML, Scheff NN, Scheff SW, Pauly JR. Melatonin and minocycline for combinatorial therapy to improve functional and histopathological deficits following traumatic brain injury. Neurosci Lett. 2011;488:60–64. doi: 10.1016/j.neulet.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovesdi E, Kamnaksh A, Wingo D, Ahmed F, Grunberg NE, Long JB, Kasper CE, Agoston DV. Acute minocycline treatment mitigates the symptoms of mild blast-induced traumatic brain injury. Front Neurol. 2012;3:111. doi: 10.3389/fneur.2012.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain Behav Immun. 2012;26:1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- Li Z, McKercher SR, Cui J, Nie Z, Soussou W, Roberts AJ, Sallmen T, Lipton JH, Talantova M, Okamoto S, Lipton SA. Myocyte enhancer factor 2C as a neurogenic and antiapoptotic transcription factor in murine embryonic stem cells. J Neurosci. 2008;28:6557–6568. doi: 10.1523/JNEUROSCI.0134-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Zhang Z, Lu Z, Borlongan C, Pan J, Chen J, Qian L, Liu Z, Zhu L, Zhang J, Xu Y. Human umbilical cord stem cells ameliorate experimental autoimmune encephalomyelitis by regulating immunoinflammation and remyelination. Stem Cells Dev. 2013;22:1053–1062. doi: 10.1089/scd.2012.0463. [DOI] [PubMed] [Google Scholar]
- Liu SP, Fu RH, Wu DC, Hsu CY, Chang CH, Lee W, Lee YD, Liu CH, Chien YJ, Lin SZ, Shyu WC. Mouse-induced pluripotent stem cells generated under hypoxic conditions in the absence of viral infection and oncogenic factors and used for ischemic stroke therapy. Stem Cells Dev. 2014;23:421–433. doi: 10.1089/scd.2013.0182. [DOI] [PubMed] [Google Scholar]
- Loane DJ, Faden AI. Neuroprotection for traumatic brain injury: translational challenges and emerging therapeutic strategies. Trends Pharmacol Sci. 2010;31:596–604. doi: 10.1016/j.tips.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano D, Gonzales-Portillo GS, Acosta S, de la Pena I, Tajiri N, Kaneko Y, Borlongan CV. Neuroinflammatory responses to traumatic brain injury: etiology, clinical consequences, and therapeutic opportunities. Neuropsychiatr Dis Treat. 2015;11:97–106. doi: 10.2147/NDT.S65815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas AI, Stocchetti N, Bullock R. Moderate and severe traumatic brain injury in adults. Lancet Neurol. 2008;7:728–741. doi: 10.1016/S1474-4422(08)70164-9. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Kitazono S, Ohtsuru Y, Himeno R. Orthodontic treatment for 3 impacted upper anterior teeth. Fukuoka Shika Daigaku Gakkai Zasshi. 1986;13:60–66. [PubMed] [Google Scholar]
- Neuwelt EA, Bauer B, Fahlke C, Fricker G, Iadecola C, Janigro D, Leybaert L, Molnar Z, O’Donnell ME, Povlishock JT, Saunders NR, Sharp F, Stanimirovic D, Watts RJ, Drewes LR. Engaging neuroscience to advance translational research in brain barrier biology. Nat Rev Neurosci. 2011;12:169–182. doi: 10.1038/nrn2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offit PA, Shaw RD, Greenberg HB. Passive protection against rotavirus-induced diarrhea by monoclonal antibodies to surface proteins vp3 and vp7. J Virol. 1986;58:700–703. doi: 10.1128/jvi.58.2.700-703.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okie S. Traumatic brain injury in the war zone. N Engl J Med. 2005;352:2043–2047. doi: 10.1056/NEJMp058102. [DOI] [PubMed] [Google Scholar]
- Pabon MM, Acosta S, Guedes VA, Tajiri N, Kaneko Y, Borlongan CV. Brain region-specific histopathological effects of varying trajectories of controlled cortical impact injury model of traumatic brain injury. CNS Neurosci Ther. 2016;22:200–211. doi: 10.1111/cns.12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad K, Sharma A, Garg A, Mohanty S, Bhatnagar S, Johri S, Singh KK, Nair V, Sarkar RS, Gorthi SP, Hassan KM, Prabhakar S, Marwaha N, Khandelwal N, Misra UK, Kalita J, Nityanand S, Inve STSG. Intravenous autologous bone marrow mononuclear stem cell therapy for ischemic stroke: a multicentric, randomized trial. Stroke. 2014;45:3618–3624. doi: 10.1161/STROKEAHA.114.007028. [DOI] [PubMed] [Google Scholar]
- Rodrigues MC, Voltarelli J, Sanberg PR, Allickson JG, Kuzmin-Nichols N, Garbuzova-Davis S, Borlongan CV. Recent progress in cell therapy for basal ganglia disorders with emphasis on menstrual blood transplantation in stroke. Neurosci Biobehav Rev. 2012;36:177–190. doi: 10.1016/j.neubiorev.2011.05.010. [DOI] [PubMed] [Google Scholar]
- Russo I, Barlati S, Bosetti F. Effects of neuroinflammation on the regenerative capacity of brain stem cells. J Neurochem. 2011;116:947–956. doi: 10.1111/j.1471-4159.2010.07168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatman KE, Duhaime AC, Bullock R, Maas AI, Valadka A, Manley GT Workshop Scientific T, Advisory Panel M. Classification of traumatic brain injury for targeted therapies. J Neurotrauma. 2008;25:719–738. doi: 10.1089/neu.2008.0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlosberg D, Benifla M, Kaufer D, Friedman A. Blood-brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat Rev Neurol. 2010;6:393–403. doi: 10.1038/nrneurol.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg GK, Kondziolka D, Wechsler LR, Lunsford LD, Coburn ML, Billigen JB, Kim AS, Johnson JN, Bates D, King B, Case C, McGrogan M, Yankee EW, Schwartz NE. Clinical outcomes of transplanted modified bone marrow-derived mesenchymal stem cells in stroke: a phase 1/2a Study. Stroke. 2016;47:1817–1824. doi: 10.1161/STROKEAHA.116.012995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroemer P, Patel S, Hope A, Oliveira C, Pollock K, Sinden J. The neural stem cell line CTX0E03 promotes behavioral recovery and endogenous neurogenesis after experimental stroke in a dose-dependent fashion. Neurorehabil Neural Repair. 2009;23:895–909. doi: 10.1177/1545968309335978. [DOI] [PubMed] [Google Scholar]
- Tajiri N, Duncan K, Borlongan MC, Pabon M, Acosta S, de la Pena I, Hernadez-Ontiveros D, Lozano D, Aguirre D, Reyes S, Sanberg PR, Eve DJ, Borlongan CV, Kaneko Y. Adult stem cell transplantation: is gender a factor in stemness. Int J Mol Sci. 2014a;15:15225–15243. doi: 10.3390/ijms150915225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajiri N, Duncan K, Antoine A, Pabon M, Acosta SA, de la Pena I, Hernadez-Ontiveros DG, Shinozuka K, Ishikawa H, Kaneko Y, Yankee E, McGrogan M, Case C, Borlongan CV. Stem cell-paved biobridge facilitates neural repair in traumatic brain injury. Front Syst Neurosci. 2014b;8:116. doi: 10.3389/fnsys.2014.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajiri N, Acosta SA, Shahaduzzaman M, Ishikawa H, Shinozuka K, Pabon M, Hernandez-Ontiveros D, Kim DW, Metcalf C, Staples M, Dailey T, Vasconcellos J, Franyuti G, Gould L, Patel N, Cooper D, Kaneko Y, Borlongan CV, Bickford PC. Intravenous transplants of human adipose-derived stem cell protect the brain from traumatic brain injury-induced neurodegeneration and motor and cognitive impairments: cell graft biodistribution and soluble factors in young and aged rats. J Neurosci. 2014c;34:313–326. doi: 10.1523/JNEUROSCI.2425-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia-Perez J, Sanchez-Aguilar M, Torres-Corzo JG, Gordillo-Moscoso A, Martinez-Perez P, Madeville P, de la Cruz-Mendoza E, Chalita-Williams J. Effect of rosuvastatin on amnesia and disorientation after traumatic brain injury (NCT003229758) J Neurotrauma. 2008;25:1011–1017. doi: 10.1089/neu.2008.0554. [DOI] [PubMed] [Google Scholar]
- Uekawa K, Hasegawa Y, Ma M, Nakagawa T, Katayama T, Sueta D, Toyama K, Kataoka K, Koibuchi N, Kawano T, Kuratsu J, Kim-Mitsuyama S. Rosuvastatin ameliorates early brain injury after subarachnoid hemorrhage via suppression of superoxide formation and nuclear factor-kappa B activation in rats. J Stroke Cerebrovasc Dis. 2014;23:1429–1439. doi: 10.1016/j.jstrokecerebrovasdis.2013.12.004. [DOI] [PubMed] [Google Scholar]
- Wang KW, Chen HJ, Lu K, Liliang PC, Liang CL, Tsai YD, Cho CL. Simvastatin attenuates the cerebral vascular endothelial inflammatory response in a rat traumatic brain injury. Ann Clin Lab Sci. 2014;44:145–150. [PubMed] [Google Scholar]
- Wang Z, Wu L, You W, Ji C, Chen G. Melatonin alleviates secondary brain damage and neurobehavioral dysfunction after experimental subarachnoid hemorrhage: possible involvement of TLR4-mediated inflammatory pathway. J Pineal Res. 2013;55:399–408. doi: 10.1111/jpi.12087. [DOI] [PubMed] [Google Scholar]
- Watanabe J, Shetty AK, Hattiangady B, Kim DK, Foraker JE, Nishida H, Prockop DJ. Administration of TSG-6 improves memory after traumatic brain injury in mice. Neurobiol Dis. 2013;59:86–99. doi: 10.1016/j.nbd.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner C, Engelhard K. Pathophysiology of traumatic brain injury. Br J Anaesth. 2007;99:4–9. doi: 10.1093/bja/aem131. [DOI] [PubMed] [Google Scholar]
- Wu H, Mahmood A, Lu D, Jiang H, Xiong Y, Zhou D, Chopp M. Attenuation of astrogliosis and modulation of endothelial growth factor receptor in lipid rafts by simvastatin after traumatic brain injury. J Neurosurg. 2010;113:591–597. doi: 10.3171/2009.9.JNS09859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Liu Y, Yan K, Chen L, Chen XR, Li P, Chen FF, Jiang XD. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J Neuroinflammation. 2013;10:106. doi: 10.1186/1742-2094-10-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Moore AN, Clifton GL, Dash PK. Sulforaphane enhances aquaporin-4 expression and decreases cerebral edema following traumatic brain injury. J Neurosci Res. 2005;82:499–506. doi: 10.1002/jnr.20649. [DOI] [PubMed] [Google Scholar]
- Ziebell JM, Morganti-Kossmann MC. Involvement of pro- and anti-inflammatory cytokines and chemokines in the pathophysiology of traumatic brain injury. Neurotherapeutics. 2010;7:22–30. doi: 10.1016/j.nurt.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]