

Traumatic brain injury (TBI) is an acquired injury to the brain that occurs with sudden trauma that can range from mild (concussive) to severe. TBI is considered a leading cause of death in children and young adults, with the Centers for Disease Control and Prevention estimating that approximately 1.7 million cases of TBI occur in the United States annually (Faul et al., 2010). Further, since the beginning of the global war on terrorism, the Department of Defense has reported over 344,000 U.S. Service Members have been diagnosed with traumatic brain injury from penetrating injuries to mild forms of TBI. TBI, caused by a sudden impact, penetration, or rapid movement of the brain, interrupts the normal functioning of the brain. While the intracranial location and severity of injury contribute to the extent of functional deficits, there are also contributions from physiological changes that occur during hours to days after injury. The progression of molecular changes following TBI leads to a number of secondary injuries including: mitochondrial dysfunction, neuronal degeneration, inflammation, blood-brain barrier (BBB) dysfunction, and edema formation. These changes in the cellular microenvironment contribute to the secondary injury phase when neuronal loss either permanently disables victims, decreasing their quality of life, or results in death. While many preclinical trials have shown positive results in treating TBI, none have resulted in effective neuroprotective treatments in the clinical setting. This likely reflects the multifaceted nature of TBI and incomplete understanding of the contributing mechanisms underlying the progression of events during the secondary injury phase that results in functional deficits. Due to the inability to reverse the damage to the brain during the initial injury, clinicians focus on stabilizing the patient and try to prevent further injury. The need to understand the pathophysiology and subsequently develop and test novel neuroprotective treatments following brain injury has been the driving force in our laboratory.
Mitochondria are considered essential organelles for cell survival as they generate the majority of the cells’ energy production by oxidative phosphorylation. Mitochondria are sensitive to changes in the physiological state of a cell, and their dysfunction has been implicated in numerous neuropathological diseases, including TBI. Increasing evidence suggests that TBI leads to both direct mechanical damage and functional disturbances in mitochondria, and plays a key role in contributing to apoptotic and necrotic cell death. As the brain is a highly aerobic, energy-demanding tissue, an imbalance in energy demand for repair of cellular damage and concomitant decrease in energy generation caused by mitochondrial dysfunction leads to increased cellular damage. Excitotoxicity, production of reactive oxygen species (ROS), Ca2+ overload, apoptosis-inducing factor, and caspases are some of the primary candidates that induce mitochondrial damage following TBI (Vink et al., 1990; Singh et al., 2006). During injury, the surge in ROS facilitates a vicious cycle that accelerates mitochondrial damage, excitotoxicity, lipid peroxidation, and inflammation. Mitochondrial membrane potential is a key indicator of damage to the mitochondria and may be linked to early-stage apoptosis. Thus, mitochondrial targeting strategies in TBI have been increasingly studied, as their maintenance could potentially preserve brain function. Determining the link between mitochondrial dysfunction and vascular physiology may also provide additional potential therapeutic targets following brain injury.
One of our primary goals was to determine whether improving mitochondrial function would reduce lesion volume following TBI and result in improved functional outcomes. To accomplish this we focused on the use of methylene blue (MB), as it is a U.S. Food and Drug Administration-grandfathered drug that has shown promise in reducing neurobehavioral impairment in other disease states such as Parkinson's disease, Alzheimer's disease and ischemic stroke, where mitochondria may also play a critical role in disease progression (Rojas et al., 2012). In addition, low doses of MB have been shown in vivo to increase global glucose uptake, oxygen consumption, cerebral blood flow (Lin et al., 2012) and evoked responses (Huang et al., 2013) in rats. Consistent with these beneficial effects, MB has been shown to be effective in salvaging at-risk tissue (i.e., perfusion-diffusion mismatch which approximates the ischemic penumbra), ultimately reducing infarct size and behavioral deficits in models of ischemic stroke compared to vehicle-treated groups (Shen et al., 2013).
The proposed mechanism of action of MB in promoting cell survival is as an electron cycler that allows the redirection of electrons to the mitochondrial electron transport chain (ETC), resulting in enhanced ATP production. Due to MB's ability to bypass complex I–III activity it also effectively reduces ROS production from the mitochondrial ETC, which has the potential to minimize injury following TBI. MB's dual mechanism of increasing energy (ATP) production and decreasing ROS makes it a unique drug for TBI and makes it a highly promising tool to study the impact of mitochondrial function on TBI injury progression.
Based on these observations we investigated acute and subacute mild TBI in rats using a double-blind, randomized, vehicle-controlled design to avoid unintentional bias. TBI was induced using the cortical impact model in rats over the somatosensory cortex with MB (1 mg/kg) administered intravenously 1 and 3 hours after TBI. MRI and behavioral assessments of motor function using the foot fault and cylinder tests were assessed from pre-injury to 14 days post TBI. In the vehicle group, the lesion volume (measured using MRI) peaked at 2 days, and decreased at 14 days post TBI (Talley Watts et al., 2014). In the MB group, by contrast, the lesion volume did not change substantially with time and was significantly smaller than that in the vehicle group at all time points studied. The MB-treated animals also exhibited improved functional outcomes compared to the vehicle-treated animals at all time points studied. We also found decreased dark-stained Nissl cells and Fluro Jade positive cells in the MB-treated group compared to vehicle-treated animals 14 days post TBI (Talley Watts et al., 2014). These results indicated that MB markedly reduces TBI lesion volume, vasogenic edema, functional deficits and neuronal degeneration.
In many instances there is a substantial delay in treatment time following a TBI, therefore we wanted to extend the treatment time to test whether delayed MB treatment would also show efficacy in reducing lesion and behavioral deficits following TBI using the same injury model. In this study, MB was applied 24 hours post injury and comparisons were made between vehicle and acute (1 and 3 hours) MB treated animals. Multimodal MRI and behavioral studies were also performed at 1 and 3 hours, 2, 7, and 14 days following an impact which was centered over the primary forelimb somatosensory cortex. The results from this study demonstrated that delayed MB treatment was also effective in reducing lesion volumes and behavioral deficits on days 2, 7 and 14 when compared to vehicle treatment (Talley Watts et al., 2016). We also performed a more extensive MRI analysis of T2, apparent diffusion coefficient (ADC), cerebral blood flow (CBF) values within the lesion for this study. Multimodal MRI offers the unique means to non-invasively image anatomical, physiological, and functional changes associated with TBI longitudinally in the same animal. Using MRI we found that the T2 determined lesion volume peaked on day 2 in vehicle treated animals and acute MB treated animals while delayed MB lesion volume peaked at 3 hours. Further, the T2 values increased at 3 hours after TBI, and significantly decreased by 3 hours in acutely treated MB animals and by day 2 in delayed MB animals, with all groups gradually returning toward normal by day 14. ADC values increased initially but significantly reduced by acute and delayed MB treatment on days 2 and 7 and then gradually returned toward normal on day 14. CBF measures demonstrated severe hypoperfusion at 3 hours, marked hyperperfusion peaking on day 2, hypoperfusion again on day 7, and a return towards normal on day 14 in all groups with only a significant increase with acute and delayed MB found on day 14 (Talley Watts et al., 2016). This data provided further evidence that MB acts as a neuroprotective agent by decreasing lesion volume, reducing cytotoxic edema formation, improving MRI defined parameters, and improving functional outcome even when administered 24 hours after the impact. This has substantial implications in providing neuroprotection when treatment is not readily available.
In addition to MBs ability to reduce lesion volume and functional deficits, we also found that in the same rat model of mild TBI there were severe and widespread disruptions in CBF and vasoreactivity (VR) beyond the impact area that change dynamically during the acute phase (Figure 1A; adapted from Long et al., 2015). These initial disruptions in CBF and VR surrounding the impact area appeared to correlate with poor tissue and functional outcomes (Long et al., 2014). Based on this data we hypothesized that administration of low doses of MB could normalize hemodynamic and metabolic function, thereby reducing lesion volume and functional deficits based on MBs known ability to increase CBF and brain metabolism and is an area we are currently investigating (Figure 1B).
Figure 1.
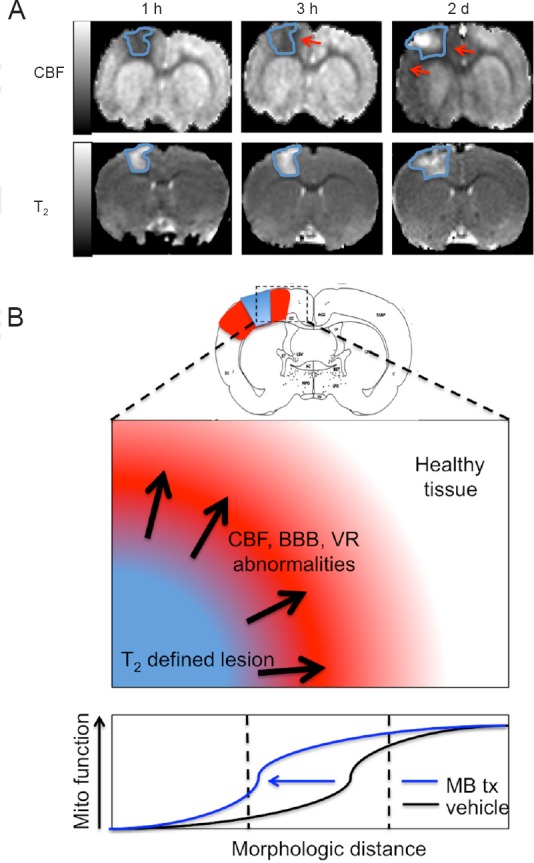
Cerebral blood flow (CBF) abnormality exceeds T2 defined lesion volume.
(A) Representative CBF and T2 maps (150 × 150 × 1,000 μm) of 1 slice at 1 and 3 hours and 2 days after TBI from one animal. The T2 lesion is outlined in blue and is overlaid in the CBF map to demonstrate the extent of abnormality between the two measures. Red arrows indicate the areas of CBF abnormality that extend beyond the T2 lesion. Hypoperfusion is apparent at 1 and 3 hours. On day 2, hyperperfusion was detected in the center of impact and widespread hypoperfusion remained peripheral to the impact zone and exceeds the T2 defined lesion volume (adapted from Long et al., 2015). (B) Hypothesized alteration of stimulating mitochondrial metabolism with methylene blue following TBI. The extent of CBF, BBB, and VR abnormalities may be mitigated by stimulating mitochondrial metabolism thereby reducing the extent of these alterations and reducing lesion volume and functional deficits. TBI: Traumatic brain injury; BBB: blood-brain barrier; VR: vasoreactivity. h: hour(s); d: days.
The significance of this work is that it addresses one of the critical gaps in knowledge identified by the CDC that there is a compelling need to better understand injury mechanisms of TBI and their effect on functional outcomes in the hopes of developing novel neuroprotective agents for brain injury. Improving our understanding of the impact of physiological changes associated with TBI will allow for improved clinical assessment and could provide novel assessment tools to better predict long-term functional outcomes. A more complete understanding of the underlying injury mechanisms will improve clinical management, responses to therapy, and quality of life in patients with TBI and will allow clinicians to better tailor treatments to an individual's injury.
References
- Faul M, Xu L, Wald MM, Coronado VG. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002-2006. Atlanta, GA: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2010. [Google Scholar]
- Huang S, Du F, Shih YY, Shen Q, Gonzalez-Lima F, Duong TQ. Methylene blue potentiates stimulus-evoked fMRI responses and cerebral oxygen consumption during normoxia and hypoxia. Neuroimage. 2013;72:237–242. doi: 10.1016/j.neuroimage.2013.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AL, Poteet E, Du F, Gourav RC, Liu R, Wen Y, Bresnen A, Huang S, Fox PT, Yang SH, Duong TQ. Methylene blue as a cerebral metabolic and hemodynamic enhancer. PLoS One. 2012;7:e46585. doi: 10.1371/journal.pone.0046585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JA, Watts LT, Chemello J, Shen Q, Duong TQ. MRI reveals widespread disruptions in CBF and vascular reactivity following focal TBI. J Neurotrauma. 2014;31:A27. [Google Scholar]
- Long JA, Watts LT, Li W, Shen Q, Muir ER, Huang S, Boggs RC, Suri A, Duong TQ. The effects of perturbed cerebral blood flow and cerebrovascular reactivity on structural MRI and behavioral readouts in mild traumatic brain injury. J Cereb Blood Flow Metab. 2015;35:1852–1861. doi: 10.1038/jcbfm.2015.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas JC, Bruchey AK, Gonzalez-Lima F. Neurometabolic mechanisms for memory enhancement and neuroprotection of methylene blue. Prog Neurobiol. 2012;96:32–45. doi: 10.1016/j.pneurobio.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Q, Du F, Huang S, Rodriguez P, Watts LT, Duong TQ. Neuroprotective efficacy of methylene blue in ischemic stroke: an MRI study. PLoS One. 2013;8:e79833. doi: 10.1371/journal.pone.0079833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh IN, Sullivan PG, Deng Y, Mbye LH, Hall ED. Time course of post-traumatic mitochondrial oxidative damage and dysfunction in a mouse model of focal traumatic brain injury: implications for neuroprotective therapy. J Cereb Blood Flow Metab. 2006;26:1407–1418. doi: 10.1038/sj.jcbfm.9600297. [DOI] [PubMed] [Google Scholar]
- Talley Watts L, Long JA, Boggs RC, Manga H, Huang S, Shen Q, Duong TQ. Delayed methylene blue improves lesion volume, multi-parametric quantitative magnetic resonance imaging measurements, and behavioral outcome after traumatic brain injury. J Neurotrauma. 2016;33:194–202. doi: 10.1089/neu.2015.3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talley Watts L, Long JA, Chemello J, Van Koughnet S, Fernandez A, Huang S, Shen Q, Duong TQ. Methylene blue is neuroprotective against mild traumatic brain injury. J Neurotrauma. 2014;31:1063–1071. doi: 10.1089/neu.2013.3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink R, Head VA, Rogers PJ, McIntosh TK, Faden AI. Mitochondrial metabolism following traumatic brain injury in rats. J Neurotrauma. 1990;7:21–27. doi: 10.1089/neu.1990.7.21. [DOI] [PubMed] [Google Scholar]