

See Charidimou (doi:10.1093/aww253) for a scientific commentary on this article.
Retinal Vasculopathy with Cerebral Leukoencephalopathy and Systemic Manifestations is a newly recognised and often misdiagnosed neurovascular syndrome caused by mutations in TREX1. Stam et al. provide the first comprehensive clinical, pathological and radiological characterization of RVCL-S, and highlight cerebral mass lesions, impaired liver and kidney function, and premature death.
Keywords: small vessel disease, molecular genetics, migraine, neuropathology, neuro-ophthalmology

See Charidimou (doi:10.1093/aww253) for a scientific commentary on this article.
Retinal Vasculopathy with Cerebral Leukoencephalopathy and Systemic Manifestations is a newly recognised and often misdiagnosed neurovascular syndrome caused by mutations in TREX1. Stam et al. provide the first comprehensive clinical, pathological and radiological characterization of RVCL-S, and highlight cerebral mass lesions, impaired liver and kidney function, and premature death.
Abstract
See Charidimou (doi:10.1093/aww253) for a scientific commentary on this article.
Cerebroretinal vasculopathy, hereditary vascular retinopathy, and hereditary endotheliopathy, retinopathy, nephropathy and stroke are neurovascular syndromes initially described as distinct entities. Recently they were shown to be one disease caused by C-terminal frame-shift mutations in TREX1, which was termed ‘retinal vasculopathy with cerebral leukodystrophy’. Here we defined the genetic and clinicopathologic spectrum of this clinically and pathophysiologically poorly characterized and frequently misdiagnosed fatal neurovascular disorder. We identified five different TREX1 mutations in 78 members from 11 unrelated families and by using a standardized study protocol we retrospectively reviewed and aggregated the associated clinical, neuroimaging, and pathology data. Findings were similar across mutations and families. Sixty-four mutation carriers had vascular retinopathy. Neuroimaging revealed (i) punctate, hyperintense, white matter lesions with or without nodular enhancement in 97% of them; (ii) rim-enhancing mass lesions in 84%; and (iii) calcifications in the white matter in 52%. Ninety per cent had clinical manifestations of brain disease, including focal neurological deficits (68%), migraine (59%), cognitive impairment (56%), psychiatric disturbances (42%), and seizures (17%). One mutation carrier had enhancing brain lesions and neurological features but unknown retinopathy status. Additional systemic features included liver disease (78%), anaemia (74%), nephropathy (61%), hypertension (60%), mild Raynaud’s phenomenon (40%), and gastro-intestinal bleeding (27%). Mean (± standard deviation) age at diagnosis was 42.9 ± 8.3 years and at death 53.1 ± 9.6 years. Pathological examination revealed systemic vasculopathy with luminal narrowing and multi-laminated basement membranes. The 13 mutation carriers without retinopathy or brain lesions were on average 8 years younger (mean age: 35.1 ± 10.6 years). Of them, 54% had mild Raynaud’s phenomenon, 42% had migraine, and 23% had psychiatric disturbances. Retinal vasculopathy with cerebral leukodystrophy is an autosomal dominant systemic small-vessel disease due to specific TREX1 mutations and clinically primarily characterized by (i) visual impairment from vascular retinopathy; and (ii) neurological decline and premature death due to progressive enhancing cerebral white matter lesions. Impaired liver and kidney function, anaemia sometimes associated with gastrointestinal bleeding, hypertension, migraine, and Raynaud’s phenomenon appear to be part of the clinical spectrum as well. Penetrance seems high. Because of the pathogenetic basis and the emerging clinical picture with systemic manifestations and conspicuous absence of leukodystrophy, we renamed the disease ‘retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations’. We propose diagnostic criteria to facilitate clinical recognition and future studies.
Introduction
Cerebroretinal vasculopathy (CRV) (Grand et al., 1988), hereditary vascular retinopathy (HVR) (Storimans et al., 1991; Terwindt et al., 1998), and hereditary endotheliopathy, retinopathy, nephropathy and stroke (HERNS) (Jen et al., 1997) are devastating fatal syndromes initially described in single families as distinct entities. Comparison of the published data, however, suggested at least some clinical overlap. By concentrating on vascular retinopathy, the most consistently shared feature, our consortium mapped a common locus to chromosome 3p21.1-p21.3 (Ophoff et al., 2001) and subsequently identified pathogenic heterozygous C-terminal frameshift mutations in TREX1 (Richards et al., 2007). This gene encodes a 3′–5′ DNA exonuclease involved in clearing cytosolic nucleic acids (Richards et al., 2007). Pathogenically, the three syndromes were thus united as a single neurovascular disorder, which was termed ‘retinal vasculopathy with cerebral leukodystrophy’ (RVCL) by Online Mendelian Inheritance in Man (OMIM; www.ncbi.nlm.nih.gov/omim).
Clinically, however, RVCL remained under-recognized. Diagnostic criteria were lacking and awareness of RVCL and its clinical manifestations was limited and possibly biased as the available clinical characteristics were mainly based on only a few, mostly old and genetically unverified and thus potentially misclassified, case reports (Grand et al., 1988; Storimans et al., 1991; Jen et al., 1997; Terwindt et al., 1998; Ophoff et al., 2001; Richards et al., 2007). As a result, RVCL has been misdiagnosed as brain tumour, multiple sclerosis, vasculitis, or organ disease of unknown origin, leading to unnecessary and often hazardous diagnostic procedures such as biopsies of brain, liver or kidney.
To increase clinical awareness and to facilitate clinical diagnosis, genetic counselling, and future studies we set out to define the genetic, clinical, radiological, and pathological spectrum of RVCL. We here provide a characterization of the genetic and clinicopathological spectrum of RVCL based on the aggregated retrospective data from 78 TREX1 mutation carriers (MC) from 11 unrelated families. In addition, we summarize the clinical features of five additional cases of RVCL, which have been reported in the literature but were not included in our dataset. To aid clinical recognition, we defined the typical clinical picture and formulated operational diagnostic criteria. Finally, after careful analysis of all available data and prompted by the pathogenetic basis and emerging clinical picture with systemic manifestations and conspicuous absence of leukodystrophy, we renamed the disease ‘retinal vasculopathy with cerebral leukoencephalopathy and systemic manifestations’ (RVCL-S). To preclude confusion with the old and pathophysiologically incorrect term RVCL (which incorrectly suggests the presence of leukodystrophy and absence of systemic features), we decided to use the term RVCL-S hereafter.
Material and methods
We genetically identified (Richards et al., 2007) and evaluated the medical records and reviewed, aggregated and correlated the genetic, clinical, neuroimaging, and pathology data of 78 TREX1 MC (35 females; 43 males) from 11 families from The Netherlands (37 MC; three families), USA (32 MC; five families), Australia (five MC; two families), and Germany (four MC; one family) (Supplementary Tables 1, 2 and 3). All subjects were personally interviewed and examined by one or more of the authors. We used a standardized study protocol and generally accepted test limits and formal diagnostic criteria (Supplementary material). Pathological findings were obtained from biopsy and autopsy reports in Families 1, 2, 5 and 7–11. Some clinical and genetic data from Families 1–5 and 7–11 (Grand et al., 1988; Storimans et al., 1991; Jen et al., 1997; Terwindt et al., 1998; Weil et al., 1999; Ophoff et al., 2001; Cohn et al., 2005; Richards et al., 2007; Gruver et al., 2011), and some neuropathological data of Families 1 and 7–9 (Grand et al., 1988; Jen et al., 1997; Weil et al., 1999; Gruver et al., 2011) have previously been published in brief in various independent reports, but never been subjected to a detailed and coherent meta-analysis.
Cerebral lesions were identified by review of images on brain CT or MRI. Sequences available for review were: T1-weighted (fat suppressed and with gadolinium-enhanced), T2-weighted [fat suppressed and fluid attenuation inversion recovery (FLAIR)], and in some patients, diffusion-weighted. No susceptibility weighted sequences were included because most scans were done more than 10 years ago, when this technique was not yet routinely performed.
The following classification was used for the three types of T2 hyperintense white matter lesions (T2-H-WMLs) identified with neuroimaging: (i) punctate lesions without enhancement; (ii) punctate lesions with nodular enhancement; and (iii) larger mass lesions with rim enhancement, mass effect and surrounding oedema.
Descriptive statistics are based on the number of subjects for whom the relevant data were available. As not all data were available for all subjects, the denominator may vary per item. The institutional ethics committee at each participating institution approved the study and all living subjects provided written informed consent.
In addition to the analysis of our own 78 cases, we also identified and reviewed five cases of RVCL-S that have been reported in the literature but were not included in our dataset.
Results
Genetic findings and study population
We identified five distinct pathogenic C-terminal frameshift TREX1 mutations (V235fs, T236fs, T249fs, R284fs, and L287fs) (Table 1 and Supplementary Fig. 1), which were all inherited; none had occurred de novo. The families, most likely, are unrelated. This could be confirmed by haplotype analysis in all but Families 6 and 9. Families 2 and 6 carried a same mutation (V235fs) as did Families 8 and 9 (T249fs) but both mutations are recurrent and also found on different haplotypes in other families. Moreover, Families 8 and 9 are from different ethnic origins, making a common ancestor unlikely.
Table 1.
Manifestations of RVCL-S TREX1 mutation carriers for all mutation carriers and all MC+, which in addition have been split in all living MC+ and all deceased MC+
| All MC | All MC+ | Living MC+ | Deceased MC+ | |
|---|---|---|---|---|
| Demographics | ||||
| Mutation carriers (n) | 78 | 65 | 30 | 35 |
| Age at diagnosis of retinopathy | ||||
| Mean ± SD (years) | 42.5 ± 8.1 | 42.5 ± 8.1 | 41.2 ± 8.4 | 43.7 ± 7.7 |
| Range (yeas) | 25–61 | 25–61 | 25–61 | 30–61 |
| Age at last follow-up | ||||
| Mean ± SD (years) | NA | NA | 47.3 ± 8.1 | NA |
| Range (years) | NA | NA | 34–62 | NA |
| Age at death | ||||
| Mean ± SD (years) | NA | NA | NA | 53.1 ± 9.6 |
| Range (years) | NA | NA | NA | 32–72 |
| Survival time from onset (years) | ||||
| Mean ± SD (years) | NA | NA | NA | 9.1 ± 6.7 |
| Range (years) | NA | NA | NA | <1–26 |
| Major featuresa | ||||
| Retinopathyb | 84 (64/76) | 100 (64/64) | 100 (30/30) | 100 (34/34) |
| Cerebral features | ||||
| Focal brain featuresc | 56 (40/72) | 68 (40/59) | 40 (12/30) | 97 (28/29) |
| Cognitive impairmentd | 47 (33/70) | 56 (32/57) | 38 (11/29) | 75 (21/28) |
| Psychiatric diseasee | 39 (29/75) | 42 (26/62) | 31 (9/29) | 52 (17/33) |
| Seizuresf | 14 (9/66) | 17 (9/54) | 7 (2/27) | 26 (7/27) |
| Neuroimaging evidence of white matter disease | ||||
| Rim-enhancing mass lesionsg | 71 (36/51) | 84 (36/43) | 75 (15/20) | 91 (21/23)i |
| Punctate non-enhancing and/or nodular contrast-enhancing lesionsj | 92 (35/38) | 97 (34/35) | 95 (18/19)h | 100 (16/16) |
| Calcifications on CT | 52 (14/27) | 52 (14/27) | 71 (5/7) | 45 (9/20) |
| Other commonly found features | ||||
| Liver diseasek | 70 (28/40) | 70 (28/40) | 65 (11/17) | 74 (17/23) |
| Kidney diseasek | 49 (22/45) | 50 (22/44) | 50 (9/18) | 50 (13/26) |
| Hypertension | 58 (30/52) | 60 (30/50) | 47 (9/19) | 68 (21/31) |
| Anaemia | 74 (25/34) | 74 (25/34) | 67 (8/12) | 77 (17/22) |
| Gastrointestinal bleedingl | 22 (9/77) | 27 (9/64) | 3 (1/30) | 24 (8/34) |
| Migraine with or without aura | 53 (28/53) | 59 (24/41) | 48 (12/25) | 75 (12/16) |
| Raynaud’s phenomenon (mild) | 42 (31/73) | 40 (24/60) | 52 (14/27) | 30 (10/33) |
aUnless indicated otherwise, the disease manifestations presented in the table are shown as percentage of subjects followed by the number of subjects. The denominator varies according to the number of individuals with available data.
bOne MC+ with unknown retinopathy status.
Cerebral features included:.
cProgressive focal neurological abnormalities such as hemiparesis, facial weakness, aphasia, hemianopia, hemisensory deficit, frontal release signs.
dCognitive impairment with bradyphrenia, apathy, irritability, and impaired memory and judgement.
eDepression, anxiety, and other psychiatric disturbances.
fSeizures.
gRim-enhancing mass lesions are defined as T2 hyperintense mass lesions with rim-enhancement, mass effect and surrounding oedema.
h One subject with no evidence of white matter hyperintensities had only one MRI, which was done within 1 year of diagnosis with retinopathy.
iFive patients were excluded as the last neuroimaging available was more than 5 years before their death.
jBased on MRI scans only.
kBased on laboratory values.
lOne of the patients had progressive gastrointestinal bleeding that led to subtotal colectomy but the others were intermittent. Two patients also had gastric or duodenal ulcers and one had the progressive colonic ulcerations associated with bleeding. Two patients had telangiectasias in the colon.
NA = not applicable.
Clinical and neuroimaging findings in all 78 mutation carriers
We found a range of retinal, cerebral, and systemic features in 78 mutation carriers, which were similar across mutations and families (Supplementary Table 1). Sixty-four mutation carriers (84%) had vascular retinopathy. Manifestations of brain disease were found in 60/74 (81%) and included focal neurological deficits (56%), migraine (53%), cognitive impairment (47%), psychiatric disturbances (39%), and seizures (14%); neuroimaging revealed (i) punctate, hyperintense, white matter lesions with or without nodular enhancement (92%); (ii) rim-enhancing mass lesions (71%); and (iii) calcifications in the white matter (52%). Systemic features were present in 62/63 (98%) and included liver disease (70%), anaemia (74%), nephropathy (49%), hypertension (58%), mild Raynaud’s phenomenon (42%), and gastro-intestinal bleeding (12%).
Vascular retinopathy, in most cases leading to progressive visual impairment, and contrast-enhancing brain lesions typically associated with progressive neurological features, were the most consistent and diagnostically most striking and disease-specific findings. Both features were particularly prevalent among older mutation carriers in the more advanced stages of the disease and, even more so, among mutation carriers who already had died. Of the 35 deceased mutation carriers with available data, all had been diagnosed with vascular retinopathy and nearly all with contrast-enhancing brain lesions. Of the 43 still-living mutation carriers, 13 did not have retinopathy or brain lesions. These were designated MC− and showed no, or only non-specific, features (see below), which were too general to alert physicians to consider a clinical diagnosis of RVCL-S in the absence of a positive family history. In contrast, the 65 MC+ with retinopathy (64/64; 100%), contrast-enhancing brain lesions (44/48; 92%), or both (44/47; 94%), showed a range of, mostly severe, associated cerebral and systemic features (see below); one MC+ had contrast-enhancing brain lesions but unknown retinopathy status.
MC− were on average 8 years younger than MC+, suggesting that MC− still are in an early non-specific phase of the disease. Lumping of the features of MC− and MC+ across all disease stages would likely obscure the typical clinical picture of RVCL-S, hampering clinical recognition of patients. We therefore decided to present the clinical data in two complementary ways: (i) as an unbiased general review of all 78 mutation carriers (see above and Supplementary Table 1); and (ii) separately for MC+ and MC− (see Table 1 and below) to better highlight the characteristic combination of features.
Mutation carriers with retinopathy and brain lesions
Clinical features
A clinical diagnosis of RVCL-S was made in 65 MC+ at a mean [± standard deviation (SD)] age of 42.9 ± 8.3 years (range 25–61). Reasons for medical screening were: visual (40/65; 61%) or neurological (14/65; 21%) features, family history (5/65; 8%), or unknown (6/65; 9%). At diagnosis, vascular retinopathy was present in 59/64 (92%) mutation carriers, visual disturbances such as decreased visual acuity or field defects in 50/65 (77%), and neurological features in 17/65 (26%); for 6/65 (9%) MC+, information on the clinical features at diagnosis was unavailable. Eventually, 64/64 (100%) MC+ developed vascular retinopathy, 40/59 (68%) focal neurological deficits, 32/57 (56%) cognitive impairment, 26/62 (42%) psychiatric symptoms such as depression and anxiety, and 9/54 (17%) seizures. All but one MC+ showed progressive increase in number and size of characteristic brain lesions on imaging (see below). In the single MC+ without brain lesions, imaging was done only once, less than a year after retinopathy was diagnosed.
Thirty-five MC+ had died, primarily from complications of neurological decline, at a mean age of 53.1 ± 9.6 years (range 32–72) and on average 9.0 ± 6.7 years (range <1–26) after diagnosis. Compared to still living MC+, deceased MC+ more frequently had focal neurological features (28/29; 97% versus 12/30; 40%) and cognitive impairment (21/28; 75% versus 11/29; 38%), indicating progressive neurological decline among all MC+. The single deceased MC+ without cerebral manifestations had died of heart disease 2 years after retinopathy was diagnosed. A typical case history of RVCL-S is described in Fig. 1.
Figure 1.
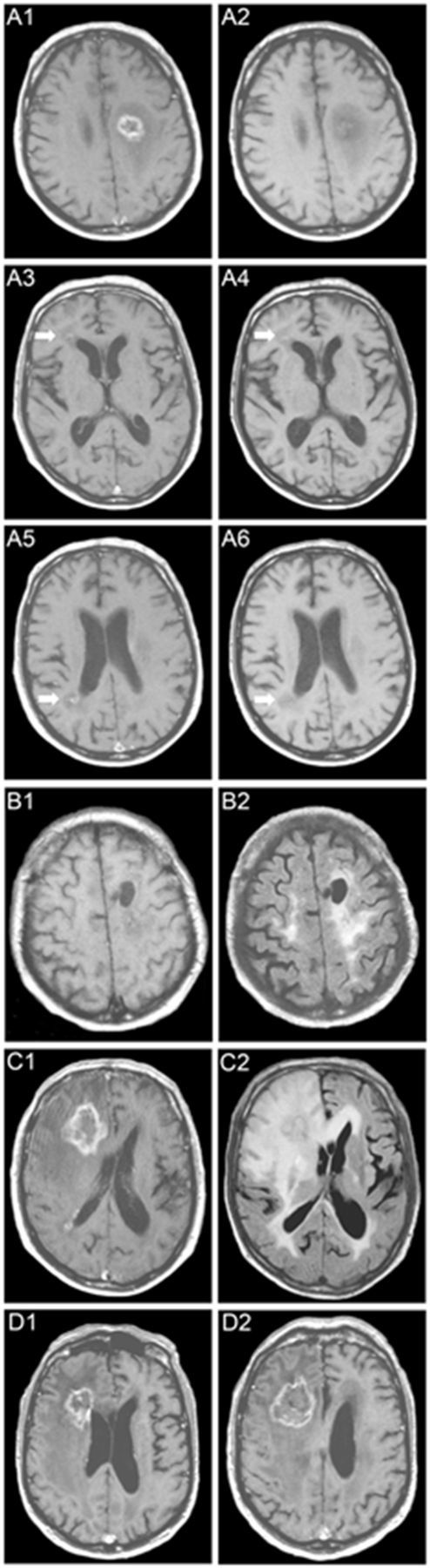
Typical case and clinical course of RVCL-S. At age 52, this male (Family 2, V235fs mutation) noted progressive bilateral loss of vision. Ophthalmologic examination revealed vascular retinopathy. At age 58 he developed slowly progressive right-sided hemiparesis. He became intermittently irritable and passive, and complained of headaches. His medical history revealed Raynaud’s phenomenon and paroxysmal atrial fibrillation. MRI revealed a rim-enhancing lesion with mass-effect and surrounding oedema in the left frontal white matter (A1: gadolinium-enhanced T1-weighted; A2: non-enhanced T1-weighted) with focal calcifications on CT (not shown). Two smaller rim-enhancing lesions were noted periventricularly in the right frontal (arrows, A3: gadolinium-enhanced T1-weighted; A4: non-enhanced T1-weighted) and parietal lobes (arrows, A5: gadolinium enhanced T1-weighted; A6: non-enhanced T1-weighted). Biopsy of the left fronto-parietal lesion revealed tissue necrosis. Dexamethasone (60 mg for 10 days) slightly improved the hemiparesis. Four months later, his headaches became worse and he developed word-finding difficulties and wide-based gait. Routine laboratory investigation revealed mild anaemia and mildly impaired renal and liver function. CSF protein was mildly increased but cell count was normal; there were no oligoclonal bands. Antinuclear antibodies, extractable nuclear antigens, anticardiolipin IgG and IgM, and anti-neutrophilic cytoplasmic antibodies were all negative. The left frontal lesion had diminished in size on MRI and enhancement was only minimal; the surrounding oedema and gliosis, however, had remained as a large zone of confluent T2 hyperintensities with small nodular foci of faint enhancement (B1: gadolinium enhanced T1-weighted; B2: FLAIR). Half a year later his condition worsened and he became easily agitated with emotional lability, disorientation, apathy, and urinary incontinence. Additionally, he developed left-sided hemiparesis with facial weakness and could walk only with assistance. MRI (C1: gadolinium enhanced T1-weighted; C2: FLAIR) revealed, exactly where previous MRI’s had shown a pre-existing punctate enhancing white matter lesion, a large irregularly rim-enhancing lesion with central necrosis and a large zone of surrounding oedema, which extended into the corpus callosum, basal ganglia, and parietal and temporal lobes, and which exerted some mass-effect on the right lateral ventricle. The pre-existing lesion adjacent to the parietal horn of the right lateral ventricle had not changed significantly. A second biopsy showed mainly necrotic tissue. Corticosteroids temporarily improved his gait. Repeat MRI (D1 and D2: gadolinium enhanced T1-weighted) 2 months later showed a persistent right frontal lesion. Open biopsy and partial debulking of the right frontal lesion were performed. Pathology showed largely necrotic tissue with scattered inflammatory cells, mainly around the vessel walls, which were thickened with adventitial fibrosis. In the following year, his condition deteriorated and he died of aspiration pneumonia at age 60. Autopsy was performed and the data are included in Fig. 3.
Retinopathy
Retinopathy was characterized in the early stages by telangiectasias, micro-aneurysms, and cotton wool spots (Fig. 2A and B), and later by perifoveal capillary obliteration and neovascularization (Fig. 2C and D). Histopathological examination of the retina at autopsy (n = 8) was consistent with scattered micro-infarcts. The retinal arteries had thickened hyalinized walls (Fig. 3A) and there were focal areas of disruption of the ganglion cells and inner nuclear layer of the retina, usually accompanied by vascular changes. In some, the pathological process had progressed to retinal haemorrhage and neovascularization.
Figure 2.

Fundoscopic (A and C) and fluorescein angiogram (B and D) images of vascular retinopathy. (A and B) Right eye of a 33-year-old male with cotton-wool spots (arrows, A), extensive areas of capillary obliteration with non-perfusion (arrows, B), and intraretinal microvascular abnormalities (arrow heads, B). (C and D) Right eye of a 48-year-old female with a neovascular membrane (arrowheads, C) and preretinal haemorrhage (arrow, C). Temporal to the macula, vascular sheathing and occlusion is present (asterisk, C). The same eye shows profuse leakage from the membrane on the disc (arrow, D) and a large avascular region involving the fovea (arrow heads, D).
Figure 3.
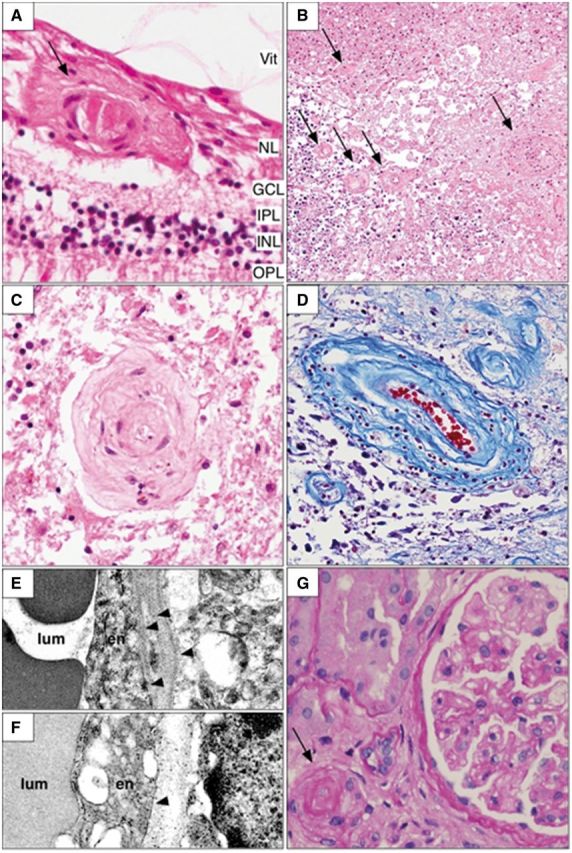
Representative histopathologic findings in the retina, brain and kidney. Microscopic examination of various organs shows a characteristic vasculopathy. The vessels of the inner layers of the retina often demonstrate damage to the walls with occasional deposition of amorphous material [arrow (A)] or thickened collagenous walls. H&E = haematoxylin and eosin stain; Vit = vitreous; NL = nerve fibre layer; GCL = ganglion cell layer; IPL = inner plexiform layer; INL = inner nuclear layer; OPL = outer plexiform layer. The brain also shows a prominent vasculopathy in the white matter, most often adjacent to and in sites of coagulation necrosis. Small to medium sized vessels demonstrate vascular wall thickening with varying degrees of luminal narrowing [arrows; (B) haematoxylin and eosin; brain). In some cases, this progresses to a frank fibrinoid necrosis. In areas with extensive white matter ischaemic damage, granular calcifications are particularly evident (dark blue staining in lower left of B). The vasculopathy may result in luminal obliteration leading to parenchymal necrosis (C; haematoxylin and eosin; brain). There is concentric collagenous thickening of the vessel walls, mostly involving the medial layer of the vessels (D; Trichrome stain; brain). Ultrastructural examination of affected vessel walls in the brain demonstrates multilaminated basement membranes with duplication of the lamina densa [arrowheads; (E) electron microscopy] in contrast to that found in unaffected regions [arrowhead; (F) electron microscopy]. lum = lumen; EN = endothelial cell. In the kidney, the vasculopathy is manifested by arteriosclerosis [arrow; (G) haematoxylin and eosin] and glomerulosclerosis.
Neuroimaging
Neuroimaging was available for 48/65 (74%) of MC+: both CT and MRI in 14, only MRI in 21, and only CT in 13. In all but one, T2 hyperintense white matter lesions (T2-H-WMLs) sparing the grey matter were identified. These could be classified as either: (i) punctate lesions without enhancement; (ii) punctate lesions with nodular enhancement; or (iii) larger mass lesions with rim enhancement, mass effect, and surrounding oedema.
Punctate non-enhancing T2-H-WMLs were detected on MRI in 34/35 (97%) of MC+; the single MC+ without such lesions had been diagnosed with retinopathy for less than a year. These lesions were small- to medium-sized, variable in number and, except for a solitary infratentorial lesion, were always located in the supratentorial periventricular and subcortical white matter. The subcortical U-fibres and corpus callosum were consistently spared. Some lesions were associated with diffusion restriction on MRI or calcifications on CT. Although non-specific as a solitary finding, these white matter changes were in most MC+ remarkably frequent for age.
In a quarter of MC+, punctate T2-H-WMLs showed enhancement sometimes associated with diffusion restriction. Enhancement of punctate T2-H-WMLs was always regarded as incompatible with ‘normal’ age-related lesions and required follow-up imaging. In several MC+ serial MRI demonstrated progression of non-enhancing to enhancing lesions and sometimes even to rim-enhancing mass lesions (Fig. 4A).
Figure 4.

Neuroimaging in RVCL-S. (A) Characteristic changes over time of contrast-enhancing cerebral mass lesions and white matter hyperintensities indicative of small vessel disease in a patient with RVCL-S. Small to middle-large deep and periventricular non-specific T2 hyperintense white matter lesions, as shown in the first MRI (0 months, axial FLAIR), are seen in the early phases of RVCL-S. There seem to be a predilection for the periventricular white matter next to anterior horn and, to a lesser extent, the posterior horn. The asymmetric pattern of affected white matter might be a hallmark of RVCL-S. Some small lesions showed diffusion restriction and minimal contrast-enhancement (e.g. the periventricular lesion next to the left anterior horn at 19 months). Nineteen months later, a right frontal rim-enhancing lesion with associated calcification (unenhanced CT) and perifocal oedema (gadolinium enhanced T1-weighted MRI) has developed on the location of a pre-existing punctate white matter hyperintensity. MRIs at 2, 3 and 11 months after corticosteroid treatment for several weeks with clinical improvement show progressive reduction of the cerebral oedema and contrast enhancement (gadolinium enhanced T1-weighted MRIs). (B) Cerebral MRI scans of five different patients, each with a different TREX1 frameshift mutation, showing that all known RVCL-S TREX1 mutations are associated with the same type of white matter lesions. V235fs: Axial T1-weighted (left) and axial gadolinium enhanced T1-weighted (right) MRI images of a 59-year-old male (Family 2) with a right frontal rim-enhancing lesion with mass-effect and surrounding oedema. T236fs: Axial T1-weighted (left) and axial gadolinium enhanced T1-weighted (right) images of a 40-year-old male (Family 7) showing a left frontal rim-enhancing lesion, a smaller rim-enhancing lesion left parietal, and an enhancing punctate right frontal periventricular white matter hyperintensity. T249fs: Axial (left) and coronal (right) gadolinium enhanced T1-weighted images (Family 8) show a left frontal rim-enhancing lesion with mass-effect and some enhancing punctate T2 hyperintensities in the right frontal lobes. R284fs: Axial T1-weighted (left) and FLAIR (right) images of a 32-year-old female (Family 10) showing a large right frontal rim-enhancing lesion and non-enhancing periventricular white matter hyperintensities frontal left. L287fs: Sagittal T1-weighted images of a 58-year-old male (Family 11) reveal small and medium-sized non-enhancing periventricular and subcortical white matter hyperintensities.
Rim-enhancing mass lesions were observed in 36/43 (84%) of MC+, sometimes at diagnosis but more frequently with advanced disease. These lesions showed varying patterns of rim enhancement and generally were associated with oedema and mass effect; in five MC+ there were associated areas of diffusion restriction that could persist for months (Figs 1 and 4). The size of the lesions (up to 6 cm diameter) and the amount of adjacent oedema could vary over time depending on treatment with corticosteroids (Mateen et al., 2010).
Focal calcifications were seen on CT in the white matter of 14/27 (52%) MC+, but not in the basal ganglia as is seen in Aicardi-Goutières syndrome (AGS), a rare disease caused by different TREX1 mutations (Crow et al., 2006).
Neuropathology
Neuropathological examination was performed on 13 autopsy and seven biopsy specimens from 20 MC+. Gross pathology at autopsy demonstrated minimal to marked involvement of the periventricular white matter, particularly in the fronto-parietal regions. The brainstem and cerebellum were affected in a few cases. Microscopy showed multiple often confluent foci of ischaemic necrosis of white matter with vessel wall thickening and luminal stenosis. This pattern, with sparing of the grey matter, is also seen in other autosomal dominant small vessel angiopathies such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Larger affected areas had extensive necrosis with focal calcifications (Fig. 3B).
Figure 3B shows vasculopathy of medium and small calibre arteries in these necrotic foci and adjacent white matter. Fibrinoid necrosis, adventitial fibrosis, luminal narrowing and mural hyalinization with collagenous material were hallmarks of the vasculopathy (Fig. 3B–D). Occasional vascular telangiectasias were observed. In some cases, a modest chronic inflammatory cell infiltrate, both perivascular and parenchymal in distribution and composed of lymphocytes and plasma cells, was found near ischaemic lesions. The cellular infiltrate was best interpreted as a reaction to ischaemic brain tissue and there was no evidence of destruction or invasion of vascular walls.
Focal calcifications and reactive astrocytosis were frequent findings. Myelin loss was substantial at autopsy. Neurofilament immunolocalization showed concomitant axon loss, often with large numbers of swollen axonal spheroids, consistent with an ischaemic process. On electron microscopy, irregular thickening and splitting of basement membranes in vessel walls were seen (Fig. 3E), with signs of smooth muscle cell and pericyte degeneration in the media.
Other commonly found features
A number of other disorders occurred remarkably more frequently in MC+ than in the general population (Table 1), suggesting that these disorders might also be part of the clinical RVCL-S spectrum.
Evidence of liver disease was found in 31/40 (78%) MC+. Of these, 28 had (mildly) elevated levels of alkaline phosphatase and gamma-glutamyltransferase, which in 10 was associated with histopathologic changes in biopsy and autopsy specimens. Nodular regenerative hyperplasia was the predominant feature. Micro- and macro-vesicular steatosis, periportal inflammation, and portal and bridging fibrosis were also seen (Supplementary Fig. 2). In three MC+, there was pathologic evidence of liver disease without abnormal laboratory parameters.
Renal disease was present in 27/44 (61%) MC+, which in 22 was associated with proteinuria, mild-to-moderate elevation of serum creatinine, or both. Pathologic examination in 13 of them revealed renal arteriolosclerosis, arteriolonephrosclerosis, and focal or diffuse glomerulosclerosis (Fig. 3G). Similar pathological changes were observed in five MC+ who had normal laboratory values. In one MC+, renal disease was detected prior to retinopathy and cerebral lesions.
Hypertension was found in 30/50 (60%) of all MC+ and in 18/20 (90%) of MC+ with renal dysfunction. Mild-to-moderate normocytic and normochromic anaemia was present in 25/34 (74%) MC+ and was associated with microscopic gastrointestinal bleeding or telangiectasias in six. Positive autoimmune markers were detected in 3/18 (17%) MC+: anti-nuclear antibodies by immunofluorescence on Hep2 substrate in two and anti-cardiolipin IgG by ELISA in one.
Migraine was diagnosed (Headache Classification Subcommittee of the International Headache Society, 2004) in 24/41 (59%) MC+. In 14/17 (82%) migraineurs with a recorded age of migraine onset, the attacks had begun about two decades before characteristic RVCL-S manifestations. Raynaud’s phenomenon, typically mild, without ischaemic injury, and not requiring treatment, was present in 24/60 (40%) of MC+.
Mutation carriers without retinopathy or brain lesions
The 13 mutation carriers without retinopathy or brain lesions were aged 35.1 ± 10.6 years (range 18–58), on average 8 years younger than MC+. Except for mild Raynaud’s phenomenon (7/13; 54%), migraine (5/12; 42%), and depression or anxiety (3/13; 23%), no other features were present.
Review of five additional cases from the literature
We identified five additional genetically confirmed cases from the literature, which were not included in the present study (Winkler et al., 2008; Schuh et al., 2014; Dhamija et al., 2015; DiFrancesco et al., 2015; Vodopivec et al., 2016). The clinical features reported for these five cases (Table 3) are consistent with those observed in our own population with the exception of two additional features: avascular necrosis of the femur head (DiFrancesco et al., 2015) and dilated (hypertensive) cardiomyopathy (Winkler et al., 2008; DiFrancesco et al., 2015; Vodopivec et al., 2016).
Table 3.
Clinical features of five RVCL-S TREX1 mutation carriers reported in literature but not included in the present study
| Reference | DiFranscecso et al. | DiFrancesco et al. | Vodopivec et al. | Schuh et al. | Dhamija et al. |
|---|---|---|---|---|---|
| Winkler et al. | |||||
| Mutation (protein) | V235fs | T270fs | D278fs | P275fs | E285fs |
| Mutation carriers (n) | + | + | + | + | + |
| Age at diagnosis of retinopathy | ? | 26 | 38 | ? | ? |
| Age at last follow-up | ? | 36 | 45 | 39 | ? |
| Age at death | NA | NA | 45 | NA | ? |
| Survival time from onset (years) | NA | NA | 7 | NA | ? |
| Major features | |||||
| Retinopathy | + | + | + | + | + |
| Cerebral features | |||||
| Focal brain features | + | + | + | + | + |
| Cognitive impairment | + | + | + | ? | ? |
| Psychiatric disease | + | + | ? | ? | ? |
| Seizures | + | + | ? | ? | ? |
| Neuroimaging evidence of white matter disease | |||||
| Rim-enhancing mass lesions | + | + | + | + | + |
| Punctate non-enhancing and/ornodular contrast-enhancing lesions | ? | ? | + | + | + |
| Calcifications on CT | + | + | + | + | ? |
| Other commonly found features | |||||
| Liver disease | + | + | ? | ? | ? |
| Kidney disease | + | + | + | + | ? |
| Hypertension | ? | + | + | + | ? |
| Anaemia | ? | ? | ? | ? | ? |
| Gastrointestinal bleeding | + | ? | ? | ? | ? |
| Migraine with or without aura | + | ? | H | H | H |
| Raynaud’s phenomenon (mild) | ? | ? | ? | ? | ? |
+ The clinical feature is present.
? Unknown whether the clinical feature was present.
H = headache, which was not further classified; NA = ‘not applicable’ as the patient was still alive at the time of the report.
Cases reported by Gruver et al. (2011) and Mateen et al. (2010) are included in our own dataset (Families 9 and 1, respectively).
Discussion
We present here the genetic and clinicopathological spectrum of RVCL-S, a frequently misdiagnosed autosomal dominant systemic small vessel disease caused by C-terminal frameshift mutations in TREX1. We identified five different mutations in 78 subjects from 11 unrelated families and, unlike previous case reports (Grand et al., 1988; Storimans et al., 1991; Jen et al., 1997; Terwindt et al., 1998; Richards et al., 2007; Mateen et al., 2010), found a consistent disease profile across patients, families, mutations, and countries.
Based on our findings and long-term clinical experience with many patients with RVCL-S, we propose the following tentative clinical course for this disease, which needs to be confirmed in prospective studies. Between age 35 and 50, visual impairment due to progressive vascular retinopathy will become apparent, soon followed by clinical manifestations of progressive focal and global brain disease. Typically, brain imaging will reveal rim-enhancing mass lesions and/or punctuate hyperintense white matter lesions with nodular enhancement, often in combination with focal calcifications in the cerebral white matter. Most if not all patients will also reveal, for age remarkably many, punctate hyperintense non-enhancing white matter lesions. Although non-specific as a solitary finding, their presence should raise suspicion of RVCL-S if in combination with retinopathy or positive family history. In addition to visual and cerebral manifestations, many patients will also have or develop one or more of the following features: liver disease, nephropathy, anaemia with or without gastrointestinal bleeding, hypertension, migraine, and mild Raynaud’s phenomenon. Ultimately patients will die between age 50 and 60, usually due to progressive brain failure. To aid the clinical recognition of RVCL-S we propose operational diagnostic criteria (Table 2). Demonstration of C-terminal frameshift mutations in TREX1 will confirm the diagnosis.
Table 2.
Proposed diagnostic criteria to recognize and select patients with possible RVCL-S for diagnostic TREX1
| Major diagnostic criteria |
| (1) Vascular retinopathy (which in the early phases is associated with retinal haemorrhages, intraretinal microvascular abnormalities, and/or cotton wool spots) |
| (2) Features of focal and/or global brain dysfunction associated on MRI with (i) punctate T2 hyperintense white matter lesions with nodular enhancement; and/or (ii) larger T2 hyperintense white matter mass lesions with rim-enhancement, mass effect, and surrounding oedema |
| (3) Family history of autosomal dominant inheritance with middle-age onset of disease manifestationsa |
| (4) Demonstration of a C-terminal frame-shift mutation in TREX1 to confirm the diagnosis |
| Supportive features |
| (1) On CT focal white matter calcifications and/or on MRI non-enhancing punctate T2 hyperintense white matter lesions at an age that non-specific age-related white matter hyperintensities are infrequent |
| (2) Microvascular liver disease (nodular regenerative hyperplasia) |
| (3) Microvascular kidney disease (arterio- or arteriolonephrosclerosis, glomerulosclerosis) |
| Possibly associated features |
| (1) Anaemia consistent with blood loss and/or chronic disease |
| (2) Microscopic gastrointestinal bleeding |
| (3) Hypertension |
| (4) Migraine with or without aura |
| (5) Raynaud’s phenomenon (typically mild) |
aDe novo mutations may be possible although none have been reported to date.
Migraine and Raynaud’s phenomenon were remarkably common among both MC+ and MC−, and typically preceded the onset of retinopathy and cerebral lesions by two decades. Compared with the general population (Launer et al., 1999; Jensen and Stovner, 2008), the prevalence of migraine was in females three times and in males five times higher, using the same validated diagnostic criteria (Headache Classification Subcommittee of the International Headache Society, 2004), while the prevalence of mild Raynaud’s phenomenon was twice as high (Palmer et al., 2000). These data suggest a causal relationship between TREX1 RVCL-S mutations and both migraine and Raynaud’s phenomenon, as was previously suggested in a genetic study of Family 2 (Hottenga et al., 2005). Whether mutations that cause other TREX1-related diseases (see below) have similar effects is unknown (Mitsikostas et al., 2004; Hanly et al., 2013; Lee-Kirsch et al., 2014). The association of migraine with RVCL-S is reminiscent of the association of migraine with another neurovascular disorder, CADASIL. Here also, migraine is highly prevalent and precedes the typical disease features by many years. Unravelling the mechanisms for migraine in both angiopathies might help to resolve the debate about the role of blood vessels in the pathogenesis of migraine (Schoonman et al., 2008; Amin et al., 2013).
The differential diagnosis of RVCL-S is challenging. The retinopathy can be confused with diabetic, hypertensive or radiation retinopathy, branch retinal vein occlusion, infectious retinitis, sickle cell disease, and collagen vascular disease (Grand et al., 1988; Gass et al., 1993). Early white matter lesions were often confused with multiple sclerosis, multi-infarct dementia, vasculitis, and other hereditary small vessel diseases with prominent white matter lesions such as CADASIL (Joutel et al., 1996; Plaisier et al., 2007). Rim-enhancing mass lesions were mistaken for neoplasms or tumefactive multiple sclerosis, with brain biopsies and surgical resection sometimes undertaken. Patients with acute focal neurological deficits could be diagnosed with ischaemic stroke. Interestingly, diffusion restriction signifying cytotoxic oedema was observed in the centre of some mass lesions.
The clinical and histopathologic findings are consistent with the hypothesis that RVCL-S is a systemic small vessel vasculopathy caused by an endotheliopathy that disrupts the vascular basal membrane and leads to progressive loss of microvascular blood flow. The histopathological findings in the brain are reminiscent of delayed radiation necrosis, which is believed to result from endothelial cell dysfunction (Nordal et al., 2005). Electron microscopy in RVCL-S shows multi-laminated capillary basement membranes in the brain, kidney, stomach, appendix, omentum, and skin (Jen et al. 1997). The presence of retinal and cerebral vessel wall thickening and lumen obliteration, nodular regenerative liver hyperplasia suggestive of diminished hepatic blood flow (Reshamwala et al., 2006), renal arteriolosclerosis and glomerulosclerosis, and microscopic gastrointestinal bleeding favours a small vessel disease. Although largely non-specific (perhaps with the exception of the basement membrane changes), the pathological changes should point to RVCL-S when present in combination with characteristic clinical and neuroimaging features of RVCL-S.
How TREX1 mutations lead to RVCL-S is unknown. TREX1 has been implicated in immunity. In Trex1 knock-out mice, there was cellular accumulation of single-stranded DNA and multi-organ inflammation (Morita et al., 2004; Yang et al., 2007; Stetson et al., 2008). Mutations abolishing TREX1 exonuclease activity have been associated with three autoimmune diseases in humans: AGS, autosomal dominant familial chilblain lupus (FCL), and systemic lupus erythematosus (SLE). AGS, in most cases, is caused by homozygous missense or compound heterozygous mutations leading to complete absence of exonuclease function. The disease mimics an in utero viral encephalopathy, possibly secondary to activation of the immune system by host DNA (Morita et al., 2004). Heterozygous TREX1 missense mutations are found in FCL, an autoimmune disease that primarily affects the skin (Lee-Kirsch et al. 2007a; Rice et al., 2007). Rare heterozygous (missense, frameshift, and 3’-UTR) TREX1 variants have also been associated with SLE (Lee-Kirsch et al., 2007b; Namjou et al., 2011). Further support for a role in innate immunity comes from studies demonstrating that TREX1 degrades HIV-1 DNA generated during infection, thereby preventing interferon-mediated immune responses (Yan et al., 2010).
Unlike TREX1 mutations causing AGS, FCL or SLE, the TREX1 mutations that cause RVCL-S preserve the enzymatic function of the TREX1 protein but alter its intracellular localization because the C-terminus that anchors the protein to the endoplasmic reticulum is absent (Richards et al., 2007). Nonsense-mediated RNA decay is unlikely because TREX1 is a single-exon gene and truncating RVCL-S TREX1 mutations consequently escape such decay. Haploinsufficiency is also an unlikely explanation as parents of children with AGS, presumably with half-normal levels of functional exonuclease, may have an increased incidence of autoimmune disease but do not develop RVCL-S (Schmidt et al., 2012). Instead the mutations in RVCL-S are hypothesized to lead to a toxic gain-of-function that primarily affects the microvasculature through a mechanism that remains to be determined. A recent report points at a novel second function of TREX1, especially its ‘tail’ that is truncated in RVCL-S, in regulatory glycosylation in the endoplasmic reticulum (Hasan et al., 2015). Unlike AGS, FCL, and SLE, RVCL-S displays no features of vasculitis and does not seem to be primarily an autoimmune disorder. However, autoimmunity may play a role at certain stages of the disease, for instance during development of rim-enhancing cerebral mass lesions.
There is no specific treatment for RVCL-S. Immunosuppressive agents such as cyclophosphamide were given to a few individuals, but without benefit (J.P.A., unpublished data). In two patients, intra-vitreal bevacizumab improved vision and reduced the retinal neovascularization, vascular leakage, and exudation (Kernt et al., 2010; G.D., personal observation). Corticosteroids temporarily reduced cerebral vasogenic oedema but had no effect on the underlying lesions.
Although not all data were available for all subjects, the emerging clinical picture is consistent across patients and families, and well in line with our own clinical experience and the clinical reports of five additional genetically confirmed patients from the literature (Table 3) (Winkler et al., 2008; Schuh et al., 2014; Dhamija et al., 2015; DiFrancesco et al., 2015; Vodopivec et al., 2016). In these reports, two features were noted that were not observed in our cases: avascular necrosis of the femur head in one (DiFrancesco et al., 2015) and dilated (hypertensive) cardiomyopathy in three (Winkler et al., 2008; DiFrancesco et al., 2015; Vodopivec et al., 2016). It is possible that gaps in the retrospectively acquired data on RVCL-S have restricted our understanding of: (i) the early course of the disease; (ii) timing and degree of involvement of the various features; (iii) mortality; and (iv) penetrance. As MC− were on average 8 years younger than MC+ and, like in MC+, migraine and Raynaud’s phenomenon are far more prevalent in MC− than in the general population, we suspect that all MC− will ultimately transform to full-blown RVCL-S. To date, no known mutation carrier has lived a normal lifespan without developing RVCL-S, suggesting high penetrance and mortality. More detailed information will come from prospective, long-term follow-up research for which the present study has provided an indispensable first step.
Compared to previous (Grand et al., 1988; Storimans et al., 1991; Jen et al., 1997; Terwindt et al., 1998; Weil et al., 1999; Ophoff et al., 2001; Cohn et al., 2005; Gruver et al., 2011) and more recent (Winkler et al., 2008; Schuh et al., 2014; Dhamija et al., 2015; DiFrancesco et al., 2015; Vodopivec et al., 2016) case reports, the present study has important advantages. These include: (i) large study sample of genetically verified and thus reliably diagnosed mutation carriers across a wide range of ages and disease stages; (ii) 11 unrelated families carrying five different mutations enabling clinical comparison between families and mutations; (iii) detailed follow-up over several decades of still living and already deceased patients from several extended multigenerational families; (iv) unbiased clinical assessment by using a standard checklist and generally accepted diagnostic criteria, enabling [in combination with (i) and (iii)] comprehensive description of both the typical clinical picture and full clinical spectrum across different disease stages; (v) extensive neuroimaging data of 48 affected members from 11 families enabling: (a) detailed characterization of the brain changes; (b) documentation of full development of typical enhancing brain mass lesions; and (c) contrary to what the previous term RVCL suggested, exclusion of leukodystrophy; (vi) extensive pathological data from brain, retina, kidney, and liver from many different patients enabling detailed pathological characterization of involvement of retina, brain and other organs; and (vii) multidisciplinary input from a wealth of international experts, many of whom have long-term involvement in the care of patients with RVCL-S.
Initially described as distinct entities, we show here that cerebroretinal vasculopathy (CRV; Grand et al., 1988), hereditary vascular retinopathy (HVR; Storimans et al., 1991; Terwindt et al., 1998), and hereditary endotheliopathy, retinopathy, nephropathy and stroke (HERNS; Jen et al., 1997) represent the same systemic angiopathy caused by C-terminal frameshift mutations in TREX1 (Ophoff et al., 2001; Richards et al., 2007). Although OMIM originally and autonomously had coined the eponym RVCL (www.ncbi.nlm.nih.gov/omim)—without consulting any of the authors of the gene discovery papers (Ophoff et al., 2001; Richards et al., 2007)—we now propose, after careful analysis of all data, to rename the disease to RVCL-S to better recognize the (i) main clinical characteristics; (ii) underlying neurovascular mechanism; (iii) cerebral and systemic involvement; (iv) genetic cause; and (v) absence of leukodystrophy.
The results of our study and proposed diagnostic criteria will: (i) guide physicians to recognize and select patients with possible RVCL-S for diagnostic TREX1 mutation screening; (ii) help physicians to diagnose RVCL-S while avoiding potentially hazardous diagnostic procedures such as brain biopsies; (iii) enable genetic counselling; and (iv) facilitate future clinical, pathophysiological, and therapeutical research into this devastating disorder.
Funding
The Netherlands Organization for Scientific Research (NWO) (907-00-217, and Vidi 917-11-31 (G.M.T.), 920-03-473 (A.H.S.), Vici 918-56-602, Spinoza 2009 and European Community (EC) FP7-EUROHEADPAIN - no. 602633 (M.D.F.), the Center of Medical System Biology (CMSB) 050-060-409, the European Union Seventh Framework Programme NIMBL 241779, the Deutsche Forschungsgemeinschaft (German Research Foundation) within the framework of the Munich Cluster for Systems Neurology (EXC 1010 SyNergy), the National Institutes of Health R01 NS062069 (A.S., J.C.J., J.P.A.) Private donations from cure CRV Research, Energy 4A Cure foundation, the Robert G Clark family and Clayco corporation (P.H.K., A.S., J.P.A.) and NIH/NHLB1 HL083822 (PHK). None of the funders had a role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; and preparation, review, or approval of the manuscript.
Conflict of interest
Anine H. Stam has received independent support from NWO (nr 920-03-473). Paulus T.V.M. de Jong received unrestricted grants from Alcon for research not related to this manuscript. Greet Dijkman received travel grants and consultancy fees from Novartis and Bayer. Mark C. Kruit has, in the past 3 years, received research funding from NIH for research not related to this manuscript. Joost Haan has, in the past 3 years, received consultancy fees from Merck. Gisela M. Terwindt received consultancy or industry support from Menarini and independent support from NWO. Michel D. Ferrari has, in the past 3 years, received grants and consultancy or industry support from Medtronic and independent support from NWO, NIH, European Community, and the Dutch Heart and Brain Foundations.
Supplementary material
Supplementary material is available at Brain online.
Supplementary Material
Glossary
Abbreviations
- AGS
Aicardi-Goutières syndrome
- MC
mutation carrier
- RVCL(-S)
retinal vasculopathy with cerebral leukoencephalopathy (and systemic manifestations)
References
- Amin FM, Asghar MS, Hougaard A, Hansen AE, Larsen VA, de Koning PJ, et al. Magnetic resonance angiography of intracranial and extracranial arteries in patients with spontaneous migraine without aura: a cross-sectional study. Lancet Neurol 2013; 12: 454–61. [DOI] [PubMed] [Google Scholar]
- Cohn AC, Kotschet K, Veitch A, Delatycki MB, McCombe MF. Novel ophthalmological features in hereditary endotheliopathy with retinopathy, nephropathy and stroke syndrome. Clin Exp Ophthalmol 2005; 33: 181–83. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet 2006; 38: 917–20. [DOI] [PubMed] [Google Scholar]
- DiFrancesco JC, Novara F, Zuffardi O, Forlino A, Gioia R, Cossu F, et al. TREX1 C-terminal frameshift mutations in the systemic variant of retinal vasculopathy with cerebral leukodystrophy. Neurol Sci 2015; 36: 323–30. [DOI] [PubMed] [Google Scholar]
- Dhamija R, Schiff D, Lopes MB, Jen JC, Lin DD, Worrall BB. Evolution of brain lesions in a patient with TREX1 cerebroretinal vasculopathy. Neurology 2015; 85: 1633–4. [DOI] [PubMed] [Google Scholar]
- Gass JD, Blodi BA. Idiopathic juxtafoveolar retinal telangiectasis. Update of classification and follow-up study. Ophthalmology 1993; 100: 1536–46. [PubMed] [Google Scholar]
- Grand MG, Kaine J, Fulling K, Atkinson J, Dowton SB, Farber M, et al. Cerebroretinal vasculopathy. A new hereditary syndrome. Ophthalmology 1988; 95: 649–59. [DOI] [PubMed] [Google Scholar]
- Gruver AM, Schoenfield L, Coleman JF, Hajj-Ali R, Rodriguez ER, Tan CD. Novel ophthalmic pathology in an autopsy case of autosomal dominant retinal vasculopathy with cerebral leukodystrophy. J Neuroophthalmol 2011; 31: 20–4. [DOI] [PubMed] [Google Scholar]
- Hanly JG, Urowitz MB, O'Keeffe AG, Gordon C, Bae SC, Sanchez-Guerrero J, et al. Headache in systemic lupus erythematosus: results from a prospective, international inception cohort study. Arthritis Rheum 2013; 65: 2887–97. [DOI] [PubMed] [Google Scholar]
- Hasan M, Fermaintt CS, Gao N, Sakai T, Miyazaki T, Jiang S, et al. Cytosolic nuclease TREX1 regulates oligosaccharyltransferase activity independent of nuclease activity to suppress immune activation. Immunity 2015; 43: 463–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headache Classification Subcommittee of the International Headache Society. The international classification of headache disorders: 2nd edition. Cephalalgia 2004; 24 (Suppl 1): 9–160. [DOI] [PubMed] [Google Scholar]
- Hottenga JJ, Vanmolkot KR, Kors EE, Kheradmand Kia S, de Jong PT, Haan J, et al. The 3p21.1-p21.3 hereditary vascular retinopathy locus increases the risk for Raynaud's phenomenon and migraine. Cephalalgia 2005; 25: 1168–72. [DOI] [PubMed] [Google Scholar]
- Jen J, Cohen AH, Yue Q, Stout JT, Vinters HV, Nelson S, et al. Hereditary endotheliopathy with retinopathy, nephropathy, and stroke (HERNS). Neurology 1997; 49: 1322–30. [DOI] [PubMed] [Google Scholar]
- Jensen R, Stovner LJ. Epidemiology and comorbidity of headache. Lancet Neurol 2008; 7: 354–61. [DOI] [PubMed] [Google Scholar]
- Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 1996; 383: 707–10. [DOI] [PubMed] [Google Scholar]
- Kernt M, Gschwendtner A, Neubauer AS, Dichgans M, Haritoglou C. Effects of intravitreal bevacizumab treatment on proliferative retinopathy in a patient with cerebroretinal vasculopathy. J Neurol 2010; 257:1213–4. [DOI] [PubMed] [Google Scholar]
- Launer LJ, Terwindt GM, Ferrari MD. The prevalence and characteristics of migraine in a population-based cohort: the GEM study. Neurology 1999; 53: 537–42. [DOI] [PubMed] [Google Scholar]
- Lee-Kirsch MA, Chowdhury D, Harvey S, Gong M, Senenko L, Engel K, et al. A mutation in TREX1 that impairs susceptibility to granzyme A-mediated cell death underlies familial chilblain lupus. J Mol Med 2007a; 85: 531–7. [DOI] [PubMed] [Google Scholar]
- Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, et al. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet 2007b; 39: 1065–7. [DOI] [PubMed] [Google Scholar]
- Lee-Kirsch MA, Wolf C, Günther C. Aicardi–Goutières syndrome: a model disease for systemic autoimmunity. Clin Exp Immunol 2014; 175: 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateen FJ, Krecke K, Younge BR, Ford AL, Shaikh A, Kothari PH, et al. Evolution of a tumor-like lesion in cerebroretinal vasculopathy and TREX1 mutation. Neurology 2010; 75: 1211–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsikostas DD, Sfikakis PP, Goadsby PJ. A meta-analysis for headache in systemic lupus erythematosus: the evidence and the myth. Brain 2004; 127 (Pt 5): 1200–9. [DOI] [PubMed] [Google Scholar]
- Morita M, Stamp G, Robins P, Dulic A, Rosewell I, Hrivnak G, et al. Gene-targeted mice lacking the Trex1 (DNase III) 3'–>5' DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol 2004; 24: 6719–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, et al. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun 2011; 12: 270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordal RA, Wong CS. Molecular targets in radiation-induced blood-brain barrier disruption. Int J Radiat Oncol Biol Phys 2005; 62: 279–87. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, DeYoung J, Service SK, Joosse M, Caffo NA, Sandkuijl LA, et al. Hereditary vascular retinopathy, cerebroretinal vasculopathy, and hereditary endotheliopathy with retinopathy, nephropathy, and stroke map to a single locus on 6. chromosome 3p21.1-p21.3. Am J Hum Genet 2001; 69: 447–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer KT, Griffin MJ, Syddall H, Pannett B, Cooper C, Coggon D. Prevalence of Raynaud's phenomenon in great Britain and its relation to hand transmitted vibration: a national postal survey. Occup Environ Med 2000; 57: 448–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaisier E, Gribouval O, Alamowitch S, Mougenot B, Prost C, Verpont MC, et al. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N Engl J Med 2007; 357: 2687–95. [DOI] [PubMed] [Google Scholar]
- Reshamwala PA, Kleiner DE, Heller T. Nodular regenerative hyperplasia: not all nodules are created equal. Hepatology 2006; 44: 7–14. [DOI] [PubMed] [Google Scholar]
- Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, et al. Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutieres syndrome. Am J Hum Genet 2007; 80: 811–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards A, van den Maagdenberg AM, Jen JC, Kavanagh D, Bertram P, Spitzer D, et al. C-terminal truncations in human 3'-5' DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat Genet 2007; 39: 1068–70. [DOI] [PubMed] [Google Scholar]
- Schuh E, Ertl-Wagner B, Lohse P, Wolf W, Mann JF, Lee-Kirsch MA, et al. Multiple sclerosis-like lesions and type I interferon signature in a patient with RVCL. Neurol Neuroimmunol Neuroinflamm 2014;2:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JL, Olivieri I, Vento J, Fazzi E, Gordish-Dressman H, Orcesi S, et al. Family history of autoimmune disease in patients with Aicardi-Goutieres syndrome. Clin Dev Immunol 2012; 2012: 206730 doi: 10.1155/2012/206730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonman GG, van der Grond J, Kortmann C, van der Geest RJ, Terwindt GM, Ferrari MD. Migraine headache is not associated with cerebral or meningeal vasodilatation–a 3T magnetic resonance angiography study. Brain 2008; 131 (Pt 8): 2192–200. [DOI] [PubMed] [Google Scholar]
- Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 2008; 134: 587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storimans CW, Van Schooneveld MJ, Oosterhuis JA, Bos PJ. A new autosomal dominant vascular retinopathy syndrome. Eur J Ophthalmol 1991; 1: 73–8. [DOI] [PubMed] [Google Scholar]
- Terwindt GM, Haan J, Ophoff RA, Groenen SM, Storimans CW, Lanser JB, et al. Clinical and genetic analysis of a large Dutch family with autosomal dominant vascular retinopathy, migraine and Raynaud's phenomenon. Brain 1998; 121 (Pt 2): 303–16. [DOI] [PubMed] [Google Scholar]
- Vodopivec I, Oakley DH, Perugino CA, Venna N, Hedley-Whyte ET, Stone JH. A 44-year-old man with eye, kidney, and brain dysfunction. Ann Neurol 2016; 79: 507–19. doi: 10.1002/ana.24583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil S, Reifenberger G, Dudel C, Yousry TA, Schriever S, Noachtar S. Cerebroretinal vasculopathy mimicking a brain tumor: a case of a rare hereditary syndrome. Neurology 1999; 53: 629–31. [DOI] [PubMed] [Google Scholar]
- Winkler DT, Lyrer P, Probst A, Devys D, Haufschild T, Haller S, et al. Hereditary systemic angiopathy (HSA) with cerebral calcifications, retinopathy, progressive nephropathy, and hepatopathy. J Neurol 2008; 255: 77–88. [DOI] [PubMed] [Google Scholar]
- Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat Immunol 2010; 11: 1005–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 2007; 131: 873–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.