

The maize silkless 1 gene encodes a UDP-glycosyltransferase that protects pistils from elimination by jasmonic acid signaling.
Keywords: Floral development, silkless 1 gene, sex determination, UDP-glycosyltransferase, jasmonic acid, plant hybrids
Abstract
Sex determination in maize involves the production of staminate and pistillate florets from an initially bisexual floral meristem. Pistil elimination in staminate florets requires jasmonic acid signaling, and functional pistils are protected by the action of the silkless 1 (sk1) gene. The sk1 gene was identified and found to encode a previously uncharacterized family 1 uridine diphosphate glycosyltransferase that localized to the plant peroxisomes. Constitutive expression of an sk1 transgene protected all pistils in the plant, causing complete feminization, a gain-of-function phenotype that operates by blocking the accumulation of jasmonates. The segregation of an sk1 transgene was used to effectively control the production of pistillate and staminate inflorescences in maize plants.
INTRODUCTION
Most flowering plants produce hermaphroditic flowers that contain both male (stamen) and female (pistil) reproductive organs. Certain flowering plants produce unisexual flowers, with staminate and pistillate flowers that arise on the same plant (monoecy) or on separate plants (dioecy). The staple crop Zea mays (maize) is monoecious, producing a terminal staminate inflorescence called the tassel and axillary pistillate inflorescences called ears (1). Maize flowers (called florets in grasses) are characteristically arranged in paired spikelets, each having two florets. The florets become staminate in the tassel through the selective elimination of a preformed pistil initial and sexual maturation of stamen. The ear spikelets become pistillate through the maturation of the pistil in the primary floret and the arrest of all stamen initials in both florets (2, 3). Unisexual flowers are highly advantageous for maize and other crop species, enabling hybrid production through outcrossing, with progeny exhibiting heterosis while escaping inbreeding depression.
To generate staminate florets, the elimination of pistils requires a genetic pathway that includes the tasselseed 1 (ts1) and ts2 genes. Mutant plants present a pistillate rather than staminate tassel and double pistils in the ear spikelets (4, 5). The pistil elimination process involves the production of the phytohormone jasmonic acid (JA), which is dependent on a TS1-encoded lipoxygenase localized to plant plastids and the activity of TS2, a short-chain alcohol dehydrogenase whose specific role in the signaling pathway remains elusive (6–9). The ts1 and ts2 genes are proposed to act in association with microRNAs miR156 and miR172 (ts4) to negatively regulate pistillate primary and secondary sex characteristics and promote staminate fate at the tassel inflorescence (10, 11).
The only functional pistils in most lines of maize are found in the primary ear florets. The presence of these functional pistils requires the action of the silkless 1 (sk1) gene. In sk1 mutant plants, all pistils are eliminated (Fig. 1A), a phenotype dependent on the action of the ts1 and ts2 genes (12–14). The epistasis between the ts genes and sk1 suggests that sk1 functions to protect the pistils from the JA-mediated elimination signal encoded by ts1 and ts2 genes.
Fig. 1. sk1 encodes a family 1 UGT.
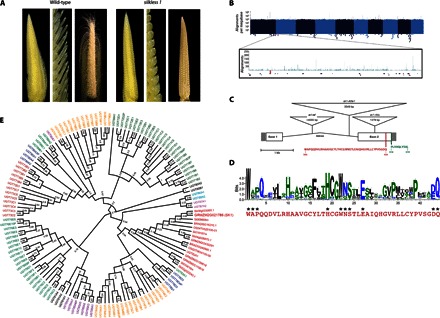
(A) Comparison of wild-type ears (Sk1/sk1-ref; left panels) and sk1 mutant ears (sk1-ref/sk1-ref; right panels). Left panels show respective ears at 2.5 cm in length, middle panels provide higher magnification to resolve individual spikelets, and right panels show ears at 8 cm. (B) Manhattan plot showing the depth of TaqαI read coverage (blue vertical lines) by chromosome position. Each x-axis pixel represents a bin of 1 Mb, and the logarithmic y axis denotes the number of reads mapping to each bin. The sk1 genetic region (see fig. S1) is shown enlarged with the TaqαI read coverage (blue), Mu junction fragments (red triangles), and location of predicted and known genes. (C) Structure of the sk1 gene, mutant alleles, and protein motifs. Filled boxes at the left and right sides indicate the 5′ and 3′ untranslated regions, respectively. Open boxes indicate coding regions, and angled lines indicate the single-intron position. Insertions found in three sk1 mutant alleles are represented by inverted triangles positioned at the corresponding insertion site (see table S1). The UGT signature/PSPG box is shown in red. The C-terminal 10 amino acids contain a PTS1-like domain shown in green. (D) WebLogo displaying the weighted alignment of the PSPG box of 107 identified Arabidopsis UGTs using ClustalW alignment. Conserved residues implicated in UDP-sugar binding are indicated with asterisks. The PSPG box of SK1 is shown in red below the WebLogo. (E) Maximum likelihood tree of SK1 homologs and the Arabidopsis UGT family. SK1 and its homologs cluster with the UGT Group N protein UGT82A1. Group N UGTs are indicated in red, and bootstrapping confidence values are shown at nodes.
RESULTS AND DISCUSSION
To investigate the model for sk1 activity, we identified the maize sk1 gene using a positional interval mapping and next-generation sequencing approach. A genetic (0.2-cM) and physical (700-kb) interval containing the sk1 gene was defined using recombination mapping in an F2 population segregating for the sk1 reference allele (sk1-ref) (fig. S1A) (12). A candidate sk1 gene was identified within this interval by the characterization of a second sk1 allele (sk1-rMu1) derived from active Mutator (Mu) maize lines (15). We used Mu-Taq, a genome sequencing approach that enriches for Mu-chromosomal junction fragments (16). Of the 179 total independent Mu junction fragments identified, 2 mapped within the coding sequence of GRMZM2G021786, a predicted gene located within the sk1 genetic interval, making it a candidate for the sk1 gene (Fig. 1B). The sk1-mu1 allele contained a 1379–base pair (bp) insertion that is 98% identical to the canonical Mu1 element in the second predicted exon of GRMZM2G021786 (Fig. 1C and table S1).
To verify GRMZM2G021786 as sk1, we examined independent sk1 mutant alleles. In the sk1-ref allele, a Helitron-like transposable element was identified in the intron of GRMZM2G021786 (Fig. 1C). The insertion in sk1 lacked terminal inverted repeats, did not cause a target site duplication, and was inserted between the dinucleotide motif AT, characteristics of other Helitron-induced mutations in maize (17). A third independent allele, sk1-Allie1, contained a novel 3549-bp insertion in the intron of GRMZM2G021786 (Fig. 1C). Together, these results provide independent confirmation of the identity of GRMZM2G021786 as the sk1 gene.
The sk1 gene encodes a 512–amino acid protein with high similarity to family 1 uridine diphosphate (UDP)–glycosyltransferases (UGTs) (Fig. 1, C and D, and fig. S2). Alignment of the SK1 protein to 107 identified Arabidopsis UGTs confirmed the presence of a plant secondary product glycosyltransferase (PSPG) box at amino acids 384 to 434, a conserved motif that is a defining feature of plant UGTs (Fig. 1, C and D) (18). The SK1 PSPG box contains seven conserved amino acids at positions shown to form hydrogen bonds to invariant parts of the UDP-sugar donor in structural studies of other plant UGTs (Fig. 1D) (19). Three other positions—W22, D43, and Q44—are also conserved and have been shown to interact with the variable UDP-sugar moieties of both UDP-galactose and UDP-glucose donor molecules (19–22). A putative peroxisomal targeting sequence (PTS) was identified at the C terminus of SK1 (“-SVL”) that shows some similarity to the canonical -SKL PTS1 motif (Fig. 1C) (23). When compared to all known and predicted Arabidopsis UGTs, SK1 exhibited the greatest similarity to UGT82A1 encoded by At3g22250 (E = 1 × 10−131, with 43% identity), the sole member of the biochemically uncharacterized Arabidopsis UGT Group N (Fig. 1E) (24). All plant UGTs catalyze the transfer of donor UDP-activated sugars (for example, UDP-glucose) to diverse small-molecule acceptor substrates [reviewed by Osmani et al. (19)].
Phytohormones and secondary metabolites have been identified as targets of plant UGT activity (19). In vivo studies have shown that auxin (25), brassinosteroids (26), salicylic acid (27), flavonoids (28), and glucosinolates (29) can all serve as endogenous UGT acceptors. The inhibition of phytohormone signaling by glycosylation has been commonly described, with the glycosylated substrates undergoing sequestration or catabolism to prevent further activity (30). Because sk1 inhibits JA-dependent pistil abortion, its glycosyltransferase activity might inactivate JA or one of its precursors known to be synthesized in peroxisomes (31) to disrupt JA signaling and tasselseed-mediated pistil elimination.
To further investigate the function of sk1, we conducted expression and localization studies. A meta-analysis of RNA sequencing (RNA-seq) data for GRMZM2G021786 revealed extremely low expression across all tissues probed, with no individual sample exceeding a read pseudocount of 10 (Fig. 2A). Consistent with its role in protecting ear pistils, the highest sk1 expression was observed in the immature ear [mean read count of 7.66 ± 1.50 (SE)], a time at which pistil protection takes place. Perhaps because of its extremely low expression, the SK1 RNA was undetectable by in situ hybridization. Next, we examined the localization of the SK1 protein and the role of the putative PTS located at the C terminus of SK1 (-SVL). A fusion of the last 10 amino acids of the SK1 protein, which included the -SVL tripeptide, to the C terminus of the Citrine fluorescent protein (Citrine:SVL) was sufficient to localize Citrine to plant peroxisomes during transient overexpression in Nicotiana benthamiana tissue (Fig. 2B and fig. S3A). However, a fusion of Citrine to the C terminus of the full-length SK1 protein (SK1:Citrine) did not show peroxisomal localization, presumably because the -SVL localization signal was blocked (Fig. 2C and fig. S3B). When the putative PTS domain was relocated to the C terminus of the SK1-Citrine protein fusion constructs (SK1ΔSVL:Citrine:SVL or Citrine:SK1), localization to plant peroxisomes was restored (Fig. 2, D and E). The localization pattern of SK1ΔSVL:Citrine:SVL was confirmed in the leaf tissue of stable transgenic Arabidopsis (fig. S3C), N. benthamiana (fig. S3D), and maize (fig. S3E). Together, these results confirm that the SK1 protein localizes to plant peroxisomes by a requisite C-terminal PTS1-like motif.
Fig. 2. Expression and localization of the SK1 protein.
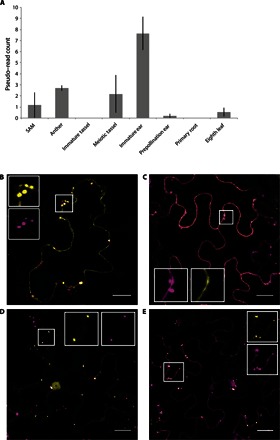
(A) Mean sk1-B73 expression in maize tissues as determined by meta-analysis of RNA-seq data sets. Expression of sk1 in the shoot apical meristem (SAM), anthers, immature tassel, meiotic tassel, immature cob, prepollination cob, primary root, and eighth leaf is shown. Error bars denote SE. Normalized pseudo–read count was determined as described in Materials and Methods. (B) Citrine:SVL (yellow signal) colocalizes with a peroxisomal marker (magenta) when transiently coexpressed in N. benthamiana. Scale bar, 20 μm. Insets show higher magnification. (C) SK1:Citrine (yellow) localizes to the cytoplasm and does not colocalize with a peroxisomal marker (magenta) when transiently coexpressed in N. benthamiana. Scale bar, 20 μm. Insets show higher magnification. (D) Citrine:SK1 (yellow) colocalizes with a peroxisome marker (magenta) when transiently expressed in N. benthamiana. Scale bar, 20 μm. Insets show higher magnification. (E) SK1ΔSVL:Citrine:SVL (yellow) colocalizes with a transiently expressed peroxisomal marker (magenta) in stable transgenic SK1ΔSVL:Citrine:SVL N. benthamiana. Scale bar, 20 μm. Insets show higher magnification.
Genetic analysis has shown that sk1 is required to protect functional pistils in ear spikelets from tasselseed-mediated elimination (13, 14). The elimination of all pistils in the tassel and of the secondary ear pistils requires functional ts1 and ts2 genes, and in ts1 and ts2 mutant plants, all pistils in the plant fail to abort. We tested whether ectopic sk1 expression could protect pistils destined to be eliminated by tasselseed action. Maize plants were transformed and regenerated with an sk1 transgene (SK1ΔSVL:Citrine:SVL) driven by a constitutive cauliflower mosaic virus (CaMV) 35S promoter (fig. S4). In transgenic 35S:SK1ΔSVL:Citrine:SVL maize, the SK1-Citrine fusion protein localized to punctate bodies (fig. S3E), mirroring the localization described during heterologous expression. A total of 72 primary transgenic plants (T0) representing 18 independent transformation events were characterized after scoring positively for transgene expression (fig. S4A). All 72 T0 plants displayed complete feminization of the tassel inflorescence (pistillate tassels) and double ear pistils, indicating that all pistils were protected from elimination (Fig. 3A and figs. S4B and S5F). One T0 plant from a nonproductive transgenic event (negative for Citrine fluorescence) produced a wild-type staminate tassel. T0 plants representing 13 independent events were crossed by wild-type males. As expected, most T1 families segregated for the presence of the transgene, as determined by polymerase chain reaction (PCR) and sensitivity to the herbicide phosphinothricin encoded by the selectable marker bar used in the transformation vector (fig. S4, C and D). Two hundred twenty-six of 228 bar-positive plants displayed pistillate tassels, whereas 182 of 182 bar-negative plants were wild type with staminate tassels (Fig. 3B). The T0 and T1 pistillate tassel phenotype was highly penetrant and expressive in transgenic 35S:SK1ΔSVL:Citrine:SVL maize, with all tassel spikelets displaying complete feminization (fig. S5, A to C). Partial expressivity was only rarely observed among hundreds of plants in a few most apical spikelets as the presence of rudimentary anthers (fig. S5, D and E). Together, these results indicate that sk1 expression is sufficient to block the tasselseed-mediated elimination of pistils in both ear and tassel spikelets, resulting in a completely feminized plant.
Fig. 3. sk1 gain-of-function pistillate phenotype in transgenic maize.

(A) Wild-type maize terminal inflorescence (left) and 35S::SK1ΔSVL:Citrine:SVL maize terminal inflorescence (right). The 35S::SK1ΔSVL:Citrine:SVL transgenic maize T0 plants display a pistillate phenotype, where the tassel inflorescence is completely feminized. (B) Representative T1 plants segregating for the presence and absence of the 35S::SK1ΔSVL:Citrine:SVL transgene. All plants displaying a pistillate phenotype tested positive for the presence of the transgene (see fig. S4). (C) Box plot summarizing the distribution of OPDA and JA in T1 plants segregating for the 35S::SK1ΔSVL:Citrine:SVL transgene. Jasmonates were measured in the staminate terminal inflorescence of plants without the transgene (+/+) and in the pistillate terminal inflorescence of plants containing the 35S::SK1ΔSVL:Citrine:SVL transgene (SK1-CIT/+). Open circles represent individual measurements. Whiskers extend to minimum and maximum values. FW, fresh weight.
The tasselseed genes eliminate pistils by stimulating the production of jasmonates (8). Therefore, we investigated whether the protection mediated by sk1 was accompanied by altered JA levels. Jasmonate levels were examined in both wild-type and pistillate tassels of T1 plants segregating 1:1 for the SK1ΔSVL:Citrine:SVL transgene. As expected, JA and its precursor molecule 12-oxo-phytodienoic acid (OPDA) were readily detected in developing staminate tassels that did not express the sk1 transgene (+/+; Fig. 3C). However, OPDA was strongly reduced (~50-fold), and JA was undetectable in sibling transgenics with pistillate tassels (SK1-CIT/+). These results indicate that sk1 expression strongly attenuates JA levels and its immediate precursor OPDA, implying a mechanism of sk1 protection by preventing JA-mediated pistil elimination.
Recent reports have demonstrated the attenuation of JA signaling by catabolism of biologically active JA–l-isoleucine via cytochrome P450 hydroxylases or indole-3-acetic acid amidohydrolases localized in the endoplasmic reticulum (32–34). Such attenuation may prevent the persistence of costly stress-activated JA responses. The homology of SK1 to UGTs, another type of small molecule–modifying enzymes, raises the possibility of another mechanism for JA signaling control in the developmental process of floral sexuality, in this case through the modification of JA synthesis intermediaries localized in the peroxisomes.
We performed untargeted metabolite profiling using high-resolution mass spectrometry to attempt to identify modified JA intermediates specific to SK1-Citrine activity in SK1ΔSVL:Citrine:SVL tassels, but we were unable to identify a putative SK1 target in these experiments (35). The low native expression level of sk1 at the developing ear (Fig. 2A) suggests that only a small amount of SK1 may be required for inhibition of JA signaling, and a SK1-dependent intermediate may exist below our detection limits. However, we cannot exclude the possibility that SK1-Citrine acts upstream of JA biosynthesis and that the changes in OPDA and JA levels in SK1ΔSVL:Citrine:SVL tassels are an indirect consequence of SK1-mediated sex determination.
Maize is one of several grasses with a sex determination system that results in imperfect florets. Yet, many related grasses, such as sorghum, bear perfect rather than imperfect florets. Four of these related grasses with complete genome sequences available—Brachypodium distachyon, Oryza sativa, Setaria italica, and Sorghum bicolor—were examined for potential sk1 orthologs. Single-copy orthologs of sk1 were identified in each of these four grasses, although they have perfect florets (fig. S2). The orthologs of sk1 were analyzed for the presence of a PTS1 domain similar to the -SVL domain required for maize sk1 to localize to peroxisomes. The C-terminal -SVL tripeptide was also found in the S. bicolor sk1, whereas another previously reported PTS1 sequence, -STL, was identified in B. distachyon sk1. No PTS1 or PTS1-like sequence could be identified in the S. italica or O. sativa sk1 orthologs. Only one other UGT, the sterol glucosyltransferase UGT51 (ATG26), has previously been shown to localize to peroxisomes, where it has been shown to promote peroxisomal degradation by autophagy (36). UGT51 does not have a PTS motif, instead associating with the peroxisome membrane via protein-protein interactions with the micropexophagic apparatus. No homologs of UGT51 have been identified outside of yeast, and UGT51 does not show significant homology to SK1. We hypothesize that the acquisition of a PTS1 motif at the SK1 C terminus may have played a significant role in the evolution of the sex determination pathway in maize because without the ability to selectively block JA-mediated pistil elimination the staminate sex determination process would be irreversible.
This study shows that the simple segregation of a gain-of-function sk1 transgene can be used to effectively control sexuality in maize. When this transgene is expressed in the sk1 mutant background, production of staminate and pistillate maize plants can be stably maintained even in open pollinated field conditions. Moreover, the physical linkage of an herbicide resistance trait to the sk1 transgene can be used to completely feminize a maize population by herbicide application in the field. Future studies will examine whether transgene control of sexuality can be extended to staple cereal crops, such as sorghum and rice, and for hybrid seed development to enhance food security.
MATERIALS AND METHODS
Genetic stocks
The sk1-ref and sk1-mu1 alleles were obtained from the Maize Genetics Cooperation Stock Center (http://maizecoop.cropsci.uiuc.edu/). Several sk1 alleles were originally found as segregating silkless plants arising from the active Mutator lines from the RescueMu project (15). Because these plants were derived from a population of Mutator plants with common parents, it was likely that they represented a single recessive mutation, which we refer to as the sk1-mu1 allele. To determine allelism with the sk1 reference allele, we crossed sk1-mu1/sk1-mu1 pollen to female Sk1-W22/sk1-ref plants. The progeny of this cross segregated 1:1 for silkless, confirming that sk1-mu1 is allelic to sk1-ref. The sk1-Allie1 allele was recovered by testcrossing sk1-ref/sk1-ref plants to females homozygous for b-Peru:dSpm with an active Spms. Kernels showing a high degree of instability were selected and planted. In a population of approximately 6000, three silkless mutant plants that failed to complement the sk1-ref mutation, including the sk1-Allie1 mutant, were found.
Selection and design of molecular markers for sk1-ref mapping
Molecular markers were initially selected from the IBM2 2004 Neighbors genetic map of maize chromosome 2 available at www.maizegdb.org. This tool was developed by the Maize Mapping Project, and at the time that this study began, in 2009, it was the best resolved genetic map of maize with ~2000 loci. Table S2 presents a list of the markers used. In most cases, these were simple sequence repeats (SSRs) that had previously been developed into PCR-based assays. In other cases, as noted in table S2, the genomic sequence of the markers was obtained from W22 and sk1-ref lines and used to design cleaved amplified polymorphic sequence (CAPS) assays (37). Additional CAPS markers were designed from predicted gene sequences previously filtered for repetitive DNA in the TIGR Maize Database.
Physical mapping of sk1-ref genetic interval
A physical map position of sk1 was initially defined using a population of 198 testcross individuals segregating 1:1 for wild-type (sk1-ref/Sk1-W22) and mutant (sk1-ref/sk1-ref) plants. The molecular marker umc34 was identified as located proximal and the closest to sk1, at ~1.5 cM (fig. S1A). The search for distal markers to umc34 led to the creation of a CAPS marker from AY107034, an expressed sequence tag anchored in the maize physical map. This CAPS marker was tested in the initial recombinant population, and 3 of 20 distal recombination breakpoints were shown to map proximal to AY107034, which indicated that this was the closest distal marker to sk1, at ~1.5 cM, identified so far (fig. S1A). The flanking markers AY107034 and phi109642 (an SSR marker used as an alternative to umc34 because it showed complete linkage to umc34 in our mapping population and was simpler to score in genotyping experiments) were used in high-throughput screenings in a mapping population expanded to 634 individuals. This analysis resulted in the identification of 44 individuals showing a recombination breakpoint between these flanking markers. These recombinant individuals were later scored for the silkless phenotype at maturity. Markers umc1769, umc1555, and bnlg1064 were subsequently evaluated in all 44 recombinant individuals. The refined genetic map (fig. S1B) was derived on the basis of data from all mapping populations analyzed up to this point (n = 832). The distal end of the sk1 genetic interval was delimited by bnlg1064 at 0.1 cM (one crossover), whereas the proximal end marked by phi109642 remained at ~1.9 cM from sk1 (fig. S1A), refining the sk1 physical interval to ~1 Mb. A closer sk1 proximal marker was sought among the predicted genes of the bacterial artificial chromosome clone Z377J20 available at that time. A CAPS marker from the predicted gene FG06631 was designed and tested in the 16 proximal sk1 recombinants. Seven of these recombinants mapped distal to FG06631, establishing this marker as the new proximal boundary in the sk1 genetic interval at 0.8 cM (fig. S1B). This interval was found to contain 13 putative genes based on the 2008 maize filtered gene set, a number that expanded to ~30 upon the release of the B73 reference genome.
Mu-Taq library construction and identification of sk1-mu1
All genomic DNA was isolated as described (38). Genomic sk1-rMu1 DNA was digested with TaqαI, end-repaired, adenylated, and ligated to custom Illumina paired-end adapters, as described by Howard et al. (16), with the modifications described below. TaqαI libraries were created with genomic DNA extracted from four independent plants homozygous for the sk1-rMu1 allele. The custom adapters used for sk1-rMu1 cloning were of an earlier iteration than those described by Howard et al. (16). One adapter incorporated a 4-bp barcode index, whereas the other was a common adapter (table S2). These adapters were essentially identical to those described previously (39). Each genomic sample was associated with a unique barcoded adapter. End-repaired, adenylated TaqαI fragments (18 μl) were ligated to adapters by 5 μl of Quick T4 DNA Ligase (New England BioLabs) in 50-μl reactions containing 28 nM each of adapter in 1× Quick Ligase Buffer (New England BioLabs). Reactions were incubated at 20°C for 20 min. Excess adapters were removed using Microcon YM-50 columns (Millipore), as described by Howard et al. (16). Duplicate 50-μl PCRs were performed to enrich each sample for sequencing. Reactions contained 1× Phusion High-Fidelity PCR Master Mix with HF Buffer (New England BioLabs), 500 nM of each primer (see table S2), and ~100 ng of adapted DNA. Cycling instructions were as follows: 98°C (2 min); 15 cycles of 98°C (10 s), 65°C (30 s), 72°C (30 s); and 72°C (5 min). All barcoded, amplified samples were multiplexed (pooled), and the buffer was exchanged to 1× tris-EDTA using Microcon YM-30, as described by Howard et al. (16). No gel extraction step or quantitative PCR step was performed to normalize the concentrations of each sample before pooling. Sequencing was performed using Illumina Genome Analyzer IIx at the Yale Center for Genome Analysis.
Genome walking and PCR-based fine mapping of sk1 alleles
Identification and fine mapping of the sk1-mu1, sk1-ref, and sk1-Allie1 alleles were performed by PCR using insertion-specific primer pairs with Phusion DNA polymerase. Identification of the Helitron-like insertion in sk1-ref plants was mediated by NaeI, SfoI, and StuI GenomeWalker (Clontech) libraries using nested PCRs. PCR primers were designed on the basis of the B73 reference genome. Primers and PCR conditions used for the mapping of individual sk1 alleles are available upon request.
Phylogenetic analysis of sk1
Phylogeny was determined using a two-step analysis. First, the top five most related proteins to SK1 (GRMZM2G021768) were determined by Blastp score under default Gramene settings (allowing some local misalignments) for B. distachyon, O. sativa, S. italica, S. bicolor, and Z. mays. Analysis of two top hits from rice, Os04T0525100 and Os04T0525200, suggested that they were two exons of the same gene, and thus, these two sequences were combined into one for the final phylogeny (Os04T0525100-200). Amino acid sequences were aligned using the ClustalW module [BLOSUM (blocks substitution matrix), gap open penalty = 3.0, gap extension penalty = 1.8] in MEGA6 (Molecular Evolutionary Genetics Analysis version 6.0) (40). Regions with missing sequence were trimmed visually. A maximum likelihood method in MEGA was used to determine the optimal amino acid substitution model. An initial tree was built with MrBayes (41), using a Whelan and Goldman substitution model, four chains, heat 0.5, and 1,000,000 iterations. The final SD of split frequencies was 0.002696. Genes separated into two general clusters in this tree (fig. S2A). All genes in the SK1 cluster were retained for further analysis.
A second phylogenetic tree was generated to better quantify the relationships between sk1 and its homologs (fig. S2B). This tree was generated with the coding sequences of selected monocot genes plus the Arabidopsis gene AT3G22250, which was identified as the closest homolog to maize SK1 via Blastp and was set as an outgroup. Alignment was performed in MEGA6 via ClustalW (codon alignment, gap open penalty = 3.0, gap extension penalty = 1.8). Aligned sequence was then trimmed visually to remove regions with excessive missing sequence. A nucleotide substitution model was selected using a maximum likelihood method in MEGA6. The final tree was built in MrBayes using a general time-reversible model with gamma distribution (1,000,000 iterations, four chains, temp = 0.1, sumt burnin = 1000). The final SD of split frequencies was 0.003863.
A third phylogenetic tree was developed to identify the relationship of SK1 with 107 UGT proteins identified in Arabidopsis (Fig. 1E). The amino acid sequences of all proteins used in the second tree were aligned using ClustalW (codon alignment, gap open penalty = 3.0, gap extension penalty = 1.8). Gaps in the sequence were visually identified and trimmed from the alignment. To build the final tree, the maximum likelihood algorithm in MEGA6 with 100 bootstraps was used with a Le and Gascuel amino acid substitution model with gamma distribution. ClustalW amino acid sequence alignment of SK1 (GRMZM2G021768) to the nearest Arabidopsis homolog UGT82A1 (AT3G22250) is shown in fig. S2C.
Fluorescent protein fusion constructs
Citrine:SVL (pYU2969) was created by fusing the coding sequence (CDS) of the last 10 amino acids of the SK1 protein (“-SVL domain”) to the 3′ end of the Citrine CDS. SK1ΔSVL:Citrine:SVL (pYU2996) was created by fusing the full-length SK1 CDS, excluding the -SVL domain, to the 5′ end of pYU2969. SK1:Citrine (pYU3103) and Citrine:SK1 (pYU3119) contained 3′- or 5′-end fusions of Citrine to the full-length SK1 CDS. For all four constructs, these coding sequences were placed under control of the single CaMV 35S promoter with a tobacco etch viral 5′ leader and the 35S terminator. These expression cassettes were then cloned into the plant expression vector pPZP200 (42). Plasmid construction details are available upon request. The peroxisomal marker used in this paper (peroxisome-mCherry) was obtained from the Arabidopsis Biological Resource Center stock CD3-983 (43).
Transient expression by agroinfiltration
Agrobacterium strain GV2260 containing the appropriate plasmid expression vectors was grown, as previously described (44). Briefly, Agrobacterium was grown overnight, pelleted, and resuspended in infiltration medium containing 10 mM MgCl2, 10 mM MES, and 200 mM acetosyringone. Strains were induced at room temperature for 4 hours, followed by vacuum infiltration into 4- to 5-week-old N. benthamiana leaves at OD600 (optical density at 600 nm) of 1.2 to 1.4. For co-infiltration, equal volumes of Agrobacterium were mixed at OD600 of 1.6 to 1.8. A further 1:10 or 1:100 dilution of Agrobacterium in infiltration medium before infiltration was sometimes used to produce optimum expression levels for confocal microscopy. All fusion proteins expressed transiently in N. benthamiana tissue were confirmed by Western blotting (fig. S3F).
Fluorescence microscopy
Live tissue microscopy was performed on a Zeiss LSM 510 META confocal microscope (Carl Zeiss) using a 40× C-Apochromat water immersion objective lens. For transient expression experiments, tissue samples were cut from N. benthamiana leaves at approximately 42 hours after infiltration. Transgenic Arabidopsis, N. benthamiana, or maize leaves were sampled from 3- to 6-week-old plants. The 488-nm laser line of a 25-mW argon laser (Coherent) with a band-pass (BP) 500- to 550-nm infrared (IR) emission filter was used to image Citrine, and the same laser line with META detector (651 to 683 nm) was used to image chloroplasts. The 561-nm laser line of a diode-pumped solid-state laser with a BP 575- to 630-nm IR emission filter was used to image mCherry.
Analysis of sk1 gene expression in maize tissues
The expression of sk1-B73 was determined by an in silico analysis of 24 RNA-seq samples from eight distinct tissue types—stem shoot apical meristem, anthers, immature tassel, meiotic tassel, immature cob, prepollination cob, primary root, and eighth leaf (table S2). RNA-seq data were acquired from the National Center for Biotechnology Information Short Read Archive study SRP014652. This study was selected because of the availability of three replicates for each tissue. Reads were initially aligned to Z. mays AGP version 3.22 reference genome and then counted against Z. mays version 3.22 transcriptome annotation using the TopHat pipeline default parameters with the -b2-very-sensitive option. Read counts were obtained using HTSeq with default parameters. Read counts were normalized via edgeR, and normalized pseudocounts were used for analysis.
Generation of transgenic plants
To generate stable transgenic SK1ΔSVL:Citrine:SVL Arabidopsis, pYU2996 was first transformed into the Agrobacterium strain GV3101. Transgenic Arabidopsis lines were then generated using the floral dip method (45). Arabidopsis transformants were selected by 0.02% Finale herbicide spray. Stable transgenic SK1ΔSVL:Citrine:SVL N. benthamiana plants were generated, as described by Clemente (46), with modifications to the medium as described below. Agrobacterium cultures were grown in LB medium with appropriate antibiotics. Cocultivation medium contained 1/10 Murashige and Skoog (MS) basal medium and vitamins, 30 mM MES, 3% sucrose, 6-benzylaminopurine (BAP) (1 μg/ml), naphthaleneacetic acid (NAA) (100 ng/ml), and 200 μM acetosyringone. Selection medium contained 1× MS basal medium and vitamins, 3% sucrose, BAP (1 μg/ml), NAA (100 ng/ml), Timentin (500 μg/ml), and glufosinate (3 μg/ml). Rooting medium contained 1/2 MS basal medium and vitamins, 1% sucrose, NAA (100 ng/ml), Timentin (500 μg/ml), and glufosinate (3 μg/ml). To generate SK1ΔSVL:Citrine:SVL maize transgenics, pYU2996 was moved into the Agrobacterium strain EHA101 via electroporation. Sixty-three independent transgenic maize events were produced following the procedure by Vega et al. (47), with modifications to the medium described below. Plant tissue culture grade agar (8 g/liter) was used in place of Gelrite until plant regeneration. To eliminate Agrobacteria after cocultivation, carbenicillin (150 mg/liter) was used in conjunction with vancomycin (100 mg/liter) instead of cefotaxime. During plant regeneration, myo-inositol (100 mg/liter) was added to the medium and bialaphos (3 mg/liter) was maintained until transplantation. SK1ΔSVL:Citrine:SVL expression in stable transgenic Arabidopsis and N. benthamiana and maize was confirmed by Western blotting (fig. S3F).
Screening of transgenic SK1ΔSVL:Citrine:SVL maize
Transgenic maize plants were screened for the presence of the transgene cassettes via swabbing of mature leaves with 3% Finale herbicide (48). Resistance or sensitivity to the herbicide was scored after 4 days. Leaves of T0 plants were screened for SK1ΔSVL:Citrine:SVL transgene expression using the 532-nm laser line of a Typhoon 9400 fluorescence imager with a 526-nm short-pass filter. A subset of T1 plants were further screened for the presence of the SK1ΔSVL:Citrine:SVL transgene by PCR assay with primers targeting either (i) the 3′ end of the SK1ΔSVL:Citrine:SVL coding sequence, including the 35S terminator, or (ii) the bar selectable marker (fig. S4, H and I). PCRs were performed using Phusion DNA polymerase (New England BioLabs).
Monitoring protein levels
Plant tissue expressing the proteins of interest was collected and ground in liquid nitrogen. Protein was extracted with buffer containing 50 mM NaCl, 20 mM tris-HCl (pH 7.5), 1 mM EDTA (pH 8.0), 0.75% Triton X-100, 10% glycerol, 2 mM dithiothreitol, 4 mM NaF, 2 mM phenylmethylsulfonyl fluoride, and cOmplete protease inhibitors (Roche). To facilitate detection of SK1-Citrine in SK1ΔSVL:Citrine:SVL maize leaf tissue, crude immunoprecipitation was performed to concentrate the protein using green fluorescent protein (GFP)–binding magnetic beads (Allele). The appropriate volume of 2× SDS loading buffer was added to each sample, and samples were heated at 90°C for 10 min before loading. Protein was run on polyacrylamide gels and transferred to a polyvinylidene difluoride membrane (Millipore) for Western blot analysis, and Citrine fusions were detected using mouse anti-GFP (Covance) and rabbit anti-mouse horseradish peroxidase (Sigma).
Quantification of jasmonates in terminal inflorescences
The developing terminal inflorescence of SK1ΔSVL:Citrine:SVL T1 maize plants of +/+ and SK1-CIT/+ genotypes was dissected between 2.5 and 13 cm in length and rapidly frozen in liquid nitrogen. Tissue samples were stored at −80°C before metabolite extraction. Jasmonate quantification was performed, as described by Glauser and Wolfender (49), with minor modifications. Briefly, plant tissues were ground under liquid nitrogen, and 200 mg of fresh frozen powder was weighed in microcentrifuge tubes. Acidified isopropanol (1.5 ml), internal standard (10 μl; d5-JA), and glass beads (5 to 10) were added to the tubes. Extraction was performed in a paint shaker for 3 min, followed by centrifugation and evaporation to dryness. The extract was purified by solid-phase extraction, dried again, and reconstituted in 300 μl of methanol/H2O [85:15 (v/v)] before analysis. Jasmonate profiling was achieved by ultrahigh-pressure liquid chromatography coupled to high-resolution mass spectrometry. Concentrations of jasmonates were calculated by normalizing the obtained peaks to that of the internal standard.
Supplementary Material
Acknowledgments
We thank Y. Tong for technical assistance, C. Bolick for greenhouse support, and A. Bentley for field support in isolating sk1 mutations. Funding: This study was supported by the NSF (award nos. 0965420 and 14444787) and the Bill and Melinda Gates Foundation to S.L.D., M.A.M., and A.P.K. Sequencing services were provided by the Keck Center for Biotechnology and Center for Genome Analysis at Yale University. Computational analyses were performed on the Yale University Biomedical High Performance Computing Cluster, which is supported by NIH grants RR19895 and RR029676-01. Author contributions: A.P.H., M.A.M., T.P.H., C.H., and S.R. conducted the molecular experiments to clone and characterize the sk1 gene; J.P.M. and I.F.A. conducted molecular genetic experiments to isolate and characterize sk1 alleles; J.H., K.N., and A.P.K. conducted maize transgenic work; G.G. conducted metabolomics to characterize jasmonates; and A.P.H., A.P.K., G.G., and S.L.D. were involved in data analysis and in writing the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/10/e1600991/DC1
fig. S1. Identification of the sk1 genetic and genomic interval.
fig. S2. sk1 encodes a Group N UGT.
fig. S3. sk1 localizes to peroxisomes.
fig. S4. Pistillate phenotype cosegregates with the SK1-GoF transgene in maize.
fig. S5. Characterization of SK1-GoF transgenic maize plants.
table S1. sk1 alleles used in this study.
table S2. Selected primers used in this study.
REFERENCES AND NOTES
- 1.T. A. Kiesselbach, The Structure and Reproduction of Corn (University of Nebraska, 1949). [Google Scholar]
- 2.Bonnet O. T., Development of the staminate and pistillate inflorescences of sweet corn. J. Agric. Res. 60, 25–37 (1940). [Google Scholar]
- 3.Cheng P. C., Gryson R. I., Walden D. B., Organ initiation and the development of unisexual flowers in the tassel and ear of Zea Mays. Am. J. Bot. 70, 450–462 (1983). [Google Scholar]
- 4.Emerson R. A., Heritable characters of maize. II. Pistillated flowered maize plants. J. Hered. 11, 65–76 (1920). [Google Scholar]
- 5.Nickerson N. H., Dale E. E., Tassel modification in Zea mays. Ann. Missouri Bot. Gard. 42, 195–211 (1955). [Google Scholar]
- 6.DeLong A., Calderon-Urrea A., Dellaporta S. L., Sex determination gene TASSELSEED2 of maize encodes a short-chain alcohol dehydrogenase required for stage-specific floral organ abortion. Cell 74, 757–768 (1993). [DOI] [PubMed] [Google Scholar]
- 7.Calderon-Urrea A., Dellaporta S. L., Cell death and cell protection genes determine the fate of pistils in maize. Development 126, 435–441 (1999). [DOI] [PubMed] [Google Scholar]
- 8.Acosta I. F., Laparra H., Romero S. P., Schmelz E., Hamberg M., Mottinger J. P., Moreno M. A., Dellaporta S. L., tasselseed1 is a lipoxygenase affecting jasmonic acid signaling in sex determination of maize. Science 323, 262–265 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Wu X., Knapp S., Stamp A., Stammers D. K., Jörnvall H., Dellaporta S. L., Oppermann U., Biochemical characterization of TASSELSEED 2, an essential plant short-chain dehydrogenase/reductase with broad spectrum activities. FEBS J. 274, 1172–1182 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Hultquist J. F., Dorweiler J. E., Feminized tassels of maize mop1 and ts1 mutants exhibit altered levels of miR156 and specific SBP-box genes. Planta 229, 99–113 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Chuck G., Meeley R., Irish E., Sakai H., Hake S., The maize tasselseed4 microRNA controls sex determination and meristem cell fate by targeting Tasselseed6/indeterminate spikelet1. Nat. Genet. 39, 1517–1521 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Jones D. F., Heritable characters of maize. XXIII—Silkless. J. Hered. 16, 339–342 (1925). [Google Scholar]
- 13.Jones D. F., The interaction of specific genes determining sex in dioecious maize. Proc. Sixth Int. Cong. Genet. 2, 104–107 (1932). [Google Scholar]
- 14.Irish E. E., Langdale J. A., Nelson T. M., Tassel seed double mutants of maize. Dev. Genet. 15, 175–171 (1994). [Google Scholar]
- 15.Fernandes J., Dong Q., Schneider B., Morrow D. J., Nan G.-L., Brendel V., Walbot V., Genome-wide mutagenesis of Zea mays L. using RescueMu transposons. Genome Biol. 5, R82 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Howard T. P. III, Hayward A. P., Tordillos A., Fragoso C., Moreno M. A., Tohme J., Kausch A. P., Mottinger J. P., Dellaporta S. L., Identification of the maize gravitropism gene lazy plant1 by a transposon-tagging genome resequencing strategy. PLOS ONE 9, e87053 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta S., Gallavotti A., Stryker G. A., Schmidt R. J., Lal S. K., A novel class of Helitron-related transposable elements in maize contain portions of multiple pseudogenes. Plant Mol. Biol. 57, 115–127 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Gachon C. M. M., Langlois-Meurinne M., Saindrenan P., Plant secondary metabolism glycosyltransferases: The emerging functional analysis. Trends Plant Sci. 10, 542–549 (2005). [DOI] [PubMed] [Google Scholar]
- 19.Osmani S. A., Bak S., Møller B. L., Substrate specificity of plant UDP-dependent glycosyltransferases predicted from crystal structures and homology modeling. Phytochemistry 70, 325–347 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Kubo A., Arai Y., Nagashima S., Yoshikawa T., Alteration of sugar donor specificities of plant glycosyltransferases by a single point mutation. Arch. Biochem. Biophys. 429, 198–203 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Offen W., Martinez-Fleites C., Yang M., Kiat-Lim E., Davis B. G., Tarling C. A., Ford C. M., Bowles D. J., Davies G. J., Structure of a flavonoid glucosyltransferase reveals the basis for plant natural product modification. EMBO J. 25, 1396–1405 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shao H., He X., Achnine L., Blount J. W., Dixon R. A., Wang X., Crystal structures of a multifunctional triterpene/flavonoid glycosyltransferase from Medicago truncatula. Plant Cell 17, 3141–3154 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu J., Baker A., Bartel B., Linka N., Mullen R. T., Reumann S., Zolman B. K., Plant peroxisomes: Biogenesis and function. Plant Cell 24, 2279–2303 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross J., Li Y., Lim E.-K., Bowles D. J., Higher plant glycosyltransferases. Genome Biol. 2, reviews3004.1 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jackson R. G., Kowalczyk M., Li Y., Higgins G., Ross J., Sandberg G., Bowles D. J., Over-expression of an Arabidopsis gene encoding a glucosyltransferase of indole-3-acetic acid: Phenotypic characterisation of transgenic lines. Plant J. 32, 573–583 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Poppenberger B., Fujioka S., Soeno K., George G. L., Vaistij F. E., Hiranuma S., Seto H., Takatsuto S., Adam G., Yoshida S., Bowles D., The UGT73C5 of Arabidopsis thaliana glucosylates brassinosteroids. Proc. Natl. Acad. Sci. U.S.A. 102, 15253–15258 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dean J. V., Delaney S. P., Metabolism of salicylic acid in wild-type, ugt74f1 and ugt74f2 glucosyltransferase mutants of Arabidopsis thaliana. Physiol. Plant. 132, 417–425 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Tohge T., Nishiyama Y., Hirai M. Y., Yano M., Nakajima J.-i., Awazuhara M., Inoue E., Takahashi H., Goodenowe D. B., Kitayama M., Noji M., Yamazaki M., Saito K., Functional genomics by integrated analysis of metabolome and transcriptome of Arabidopsis plants over-expressing an MYB transcription factor. Plant J. 42, 218–235 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Grubb C. D., Zipp B. J., Ludwig-Müller J., Masuno M. N., Molinski T. F., Abel S., Arabidopsis glucosyltransferase UGT74B1 functions in glucosinolate biosynthesis and auxin homeostasis. Plant J. 40, 893–908 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Ostrowski M., Jakubowska A., UDP-glycosyltransferases of plant hormones. Adv. Cell Biol. 4, 43–60 (2014). [Google Scholar]
- 31.Wasternack C., Kombrink E., Jasmonates: Structural requirements for lipid-derived signals active in plant stress responses and development. ACS Chem. Biol. 5, 63–77 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Heitz T., Widemann E., Lugan R., Miesch L., Ullmann P., Désaubry L., Holder E., Grausem B., Kandel S., Miesch M., Werck-Reichhart D., Pinot F., Cytochromes P450 CYP94C1 and CYP94B3 catalyze two successive oxidation steps of plant hormone jasmonoyl-isoleucine for catabolic turnover. J. Biol. Chem. 287, 6296–6306 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koo A. J. K., Cooke T. F., Howe G. A., Cytochrome P450 CYP94B3 mediates catabolism and inactivation of the plant hormone jasmonoyl-L-isoleucine. Proc. Natl. Acad. Sci. U.S.A. 108, 9298–9303 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koo A. J., Thireault C., Zemelis S., Poudel A. N., Zhang T., Kitaoka N., Brandizzi F., Matsuura H., Howe G. A., Endoplasmic reticulum-associated inactivation of the hormone jasmonoyl-L-isoleucine by multiple members of the cytochrome P450 94 family in Arabidopsis. J. Biol. Chem. 289, 29728–29738 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glauser G., Veyrat N., Rochat B., Wolfender J.-L., Turlings T. C. J., Ultra-high pressure liquid chromatography–mass spectrometry for plant metabolomics: A systematic comparison of high-resolution quadrupole-time-of-flight and single stage Orbitrap mass spectrometers. J. Chromatogr. A 1292, 151–159 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Oku M., Warnecke D., Noda T., Müller F., Heinz E., Mukaiyama H., Kato N., Sakai Y., Peroxisome degradation requires catalytically active sterol glucosyltransferase with a GRAM domain. EMBO J. 22, 3231–3241 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konieczny A., Ausubel F. M., A procedure for mapping Arabidopsis mutations using co-dominant ecotype-specific PCR-based markers. Plant J. 4, 403–410 (1993). [DOI] [PubMed] [Google Scholar]
- 38.J. Chen, S. Dellaporta, in The Maize Handbook, M. Freeling, V. Walbot, Eds. (Springer-Verlag, 1993), pp. 526–527. [Google Scholar]
- 39.Elshire R. J., Glaubitz J. C., Sun Q., Poland J. A., Kawamoto K., Buckler E. S., Mitchell S. E., A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLOS ONE 6, e19379 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tamura K., Stecher G., Peterson D., Filipski A., Kumar S., MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ronquist F., Huelsenbeck J. P., MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Hajdukiewicz P., Svab Z., Maliga P., The small, versatile pPZP family of Agrobacterium binary vectors for plant transformation. Plant Mol. Biol. 25, 989–994 (1994). [DOI] [PubMed] [Google Scholar]
- 43.Nelson B. K., Cai X., Nebenführ A., A multicolored set of in vivo organelle markers for co-localization studies in Arabidopsis and other plants. Plant J. 51, 1126–1136 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Hayward A., Padmanabhan M., Dinesh-Kumar S. P., Virus-induced gene silencing in Nicotiana benthamiana and other plant species. Methods Mol. Biol. 678, 55–63 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Zhang X., Henriques R., Lin S. S., Niu Q. W., Chua N. H., Agrobacterium-mediated transformation of Arabidopsis thaliana using the floral dip method. Nat. Protoc. 1, 641–646 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Clemente T., Nicotiana (Nicotiana tobaccum, Nicotiana benthamiana). Methods Mol. Biol. 343, 143–154 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Vega J. M., Yu W., Kennon A. R., Chen X., Zhang Z. J., Improvement of Agrobacterium-mediated transformation in Hi-II maize (Zea mays) using standard binary vectors. Plant Cell Rep. 27, 297–305 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Gordon-Kamm W. J., Spencer T. M., Mangano M. L., Adams T. R., Daines R. J., Start W. G., O’Brien J. V., Chambers S. A., Adams W. R. Jr, Willetts N. G., Rice T. B., Mackey C. J., Krueger R. W., Kausch A. P., Lemaux P. G., Transformation of maize cells and regeneration of fertile transgenic plants. Plant Cell 2, 603–618 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glauser G., Wolfender J.-L., A non-targeted approach for extended liquid chromatography-mass spectrometry profiling of free and esterified jasmonates after wounding. Methods Mol. Biol. 1011, 123–134 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/10/e1600991/DC1
fig. S1. Identification of the sk1 genetic and genomic interval.
fig. S2. sk1 encodes a Group N UGT.
fig. S3. sk1 localizes to peroxisomes.
fig. S4. Pistillate phenotype cosegregates with the SK1-GoF transgene in maize.
fig. S5. Characterization of SK1-GoF transgenic maize plants.
table S1. sk1 alleles used in this study.
table S2. Selected primers used in this study.