

Serine catabolism results in formate efflux that exceeds anabolic demands for purine synthesis.
Keywords: Serine metabolism, one-carbon metabolism, folate metabolism, overflow metabolism, mitochondria metabolism, metformin
Abstract
Serine catabolism to glycine and a one-carbon unit has been linked to the anabolic requirements of proliferating mammalian cells. However, genome-scale modeling predicts a catabolic role with one-carbon release as formate. We experimentally prove that in cultured cancer cells and nontransformed fibroblasts, most of the serine-derived one-carbon units are released from cells as formate, and that formate release is dependent on mitochondrial reverse 10-CHO-THF synthetase activity. We also show that in cancer cells, formate release is coupled to mitochondrial complex I activity, whereas in nontransformed fibroblasts, it is partially insensitive to inhibition of complex I activity. We demonstrate that in mice, about 50% of plasma formate is derived from serine and that serine starvation or complex I inhibition reduces formate synthesis in vivo. These observations transform our understanding of one-carbon metabolism and have implications for the treatment of diabetes and cancer with complex I inhibitors.
INTRODUCTION
One-carbon units are an essential requirement for the synthesis of nucleotides and methylation (1). Defects in one-carbon metabolism have been implicated in several disease states, including neural tube defects during embryonic development (1) and cancer (2). The nonessential amino acid serine is the major source of one-carbon units in proliferating lymphocytes (3, 4) and cancer cells (5, 6). Serine is broken down into glycine and a one-carbon unit, which can then be incorporated into purines, thymidylate, and methionine. From this point of view, serine one-carbon metabolism has an anabolic function (Fig. 1A).
Fig. 1. Serine catabolism is induced upon energy stress.
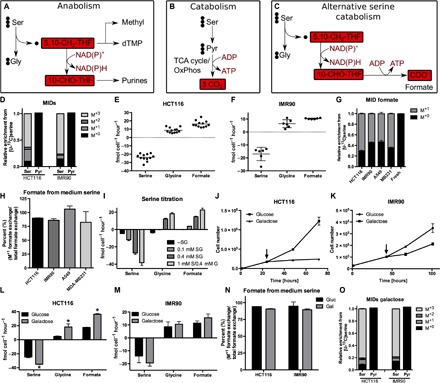
(A) Current model of serine metabolism to support purine synthesis. dTMP, deoxythymidine monophosphate. (B) Current model of serine catabolism. (C) Postulated model of serine one-carbon catabolism with formate overflow. OxPhos, oxidative phosphorylation. (D) MIDs of pyruvate and serine using [U-13C]serine as a tracer in HCT116 colorectal cancer cells and IMR90 human fibroblasts. (E and F) Absolute exchange rates of serine, glycine, and formate in (E) HCT116 and (F) IMR90 cells. Positive values indicate metabolite release and negative value uptake from cells. Each dot indicates an independent experiment (performed with three cultures per condition). (G) MID of extracellular formate using [U-13C]serine as a tracer in HCT116, IMR90, A549, and MDA-MB231 cells. (H) Percentage of formate from medium serine. (I) Serine, glycine, and formate exchange rates at different medium serine concentrations in HCT116 cells (as indicated). S, serine; G, glycine. (J and K) Proliferation of (J) HCT116 and (K) IMR90 cells upon galactose. The arrow indicates start of galactose treatment. (L and M) Absolute exchange rates of serine, glycine, and formate in (L) HCT116 and (M) IMR90 cells upon galactose. (N) Percentage of formate from medium serine in HCT116 and IMR90 cells upon glucose and galactose. (O) MIDs of pyruvate and serine using [U-13C]serine as a tracer in HCT116 and IMR90 cells in galactose medium. Data are presented as means ± SD (n = 3 cultures representative of at least two independent experiments), except for (E) and (F) (see above). *P < 0.05 by Welch’s t test.
Serine could also fulfill a catabolic role for energy generation. The only experimentally verified pathway of serine catabolism for energy production is the conversion of serine to pyruvate followed by pyruvate oxidation (Fig. 1B) (7). However, genome-scale modeling of human cell metabolism predicts an alternative route for serine catabolism via the folate cycle, contributing to energy generation and coupled to one-carbon release as formate (serine one-carbon catabolism with formate overflow; Fig. 1C) (8).
Experiments with isolated mitochondria have shown that the carbon 3 of serine can be oxidized to formate or CO2 (9). However, it is currently assumed that the mitochondria-derived formate supports the synthesis of purines in the cytosol (1), which is an anabolic role. Here, we demonstrate that most formate is released from mammalian cells rather than being incorporated into purines.
RESULTS
Serine catabolism is induced upon energy stress
First, we focused on the canonical serine catabolism pathway via pyruvate (Fig. 1B). We cultured cancer cells and nontransformed lung fibroblasts (IMR90) in medium containing [U-13C]serine and determined the intracellular mass isotopomer distribution (MID) of pyruvate. All the pyruvate was found with three 12C atoms (M+0 fraction; Fig. 1D and fig. S1, A and B), indicating no significant conversion of serine to pyruvate. Therefore, in cancer cells and fibroblasts, there is no significant activity of the canonical serine catabolism pathway for energy generation. Next, we evaluated the putative serine one-carbon catabolism with formate overflow (Fig. 1C). To determine whether and to what extent cells release formate, we developed a gas chromatography–mass spectrometry (GC-MS)–based method for the absolute quantification of formate (fig. S1C) (see Materials and Methods). Using this method, we determined the exchange rates of formate in addition to those of serine and glycine, providing a readout for the rate of serine one-carbon catabolism. We quantified the exchange fluxes of these metabolites in different cancer cell lines and nontransformed fibroblasts (Fig. 1, E and F, and fig. S1, D and E). In all cells tested, formate was released at rates lower but comparable to the rate of serine uptake. Most of the tested cell lines also showed net glycine release under the conditions tested, albeit at different ratios relative to serine uptake. To verify that the formate released is derived from serine, we cultured cells in [U-13C]serine and determined the formate MID in the cell culture medium. After [U-13C]serine cultivation, more than half of the medium formate was labeled (M+1 fraction; Fig. 1G), indicating a significant production of serine-derived formate. On the basis of the total and the M+1 formate exchange rate, we estimated that nearly 100% of the formate released by cells is derived from medium serine (Fig. 1H).
To further demonstrate that serine is the major source of the formate released from cells, we titrated the serine concentration in the cell culture medium. When the medium serine concentration was reduced from 0.4 to 0.1 mM, there was a significant decrease in the rate of formate release, going down to zero when the culture medium contained no serine (Fig. 1I). We also noticed that glycine switched from being released to being taken up when the medium serine concentration was reduced (Fig. 1I). Therefore, the direction of glycine exchange is highly dependent on the extracellular concentration of serine, potentially resulting in both glycine uptake and release as previously reported (6, 10). On the other hand, formate exhibits a net release that rises with increasing serine concentration in the extracellular medium.
Glycine or formate release could be due to the one-to-one ratio of glycine and one-carbon unit production from serine but different biosynthetic demands of glycine and one-carbon units (11). To reduce the biosynthetic demands and their potential imbalances, we switched to a culture medium where glucose was replaced by galactose. Cells grown in galactose strongly decrease proliferation (Fig. 1, J and K, and fig. S1, F and G), effectively decreasing the biosynthetic demands of glycine and one-carbon units. Although the exchange rates of essential amino acids did not significantly change (fig. S1, H to L), the exchange rates of serine, glycine, and formate significantly increased when cells were switched to galactose (Fig. 1, L and M, and fig. S1, M to O). The switch from glucose to galactose is known to cause energy stress and activate adenosine monophosphate (AMP) kinase (AMPK), a master regulator for switching from anabolism to catabolism (12). We confirmed that AMPK was activated when cells were switched from glucose to galactose, which is indicated by increased phosphorylation of AMPK and the AMPK target acetyl–coenzyme A carboxylase (ACC) (fig. S1P). We note that in one cell line (A549), AMPK phosphorylation did not change, although phosphorylation of the downstream target ACC was increased.
We observed that, under these energy stress conditions, the serine catabolism to glycine and formate exhibits the highest rate among all conditions tested. To confirm that the contribution of medium-derived serine to formate does not decrease under these conditions, we also traced medium [U-13C]serine into medium formate under the galactose condition. As observed for the glucose condition, nearly 100% of the formate released by cells was derived from serine (Fig. 1N). We could not detect any labeling of pyruvate under the galactose condition, further suggesting that serine catabolism takes place via one-carbon metabolism in the cells tested (Fig. 1, D and O).
Serine catabolism is linked to mitochondrial metabolism
Cells grown in galactose have higher oxygen consumption rates (OCRs) and mitochondrial activity than those grown in glucose (fig. S2, A and B) (13). This observation prompted us to investigate a potential link with mitochondrial oxidative phosphorylation. The one-carbon catabolism of serine proceeds in three metabolic steps (Fig. 1C), catalyzed by serine hydroxymethyltransferase, 5,10-methylene-tetrahydrofolate (5,10-CH2-THF) dehydrogenase, and reverse 10-formyl-tetrahydrofolate (10-CHO-THF) synthetase. On the basis of current evidence, these reactions take place predominantly in the mitochondria (1, 14), where they are catalyzed by SHMT2, MTHFD2 or MTHFD2L, and MTHFD1L, respectively. The dehydrogenase activity of MTHFD2 and MTHFD2L can use either NAD+ (nicotinamide adenine dinucleotide, oxidized) or NADP+ (nicotinamide adenine dinucleotidephosphate, oxidized) as a cofactor. MTHFD2 is the predominant enzyme in proliferating cells (15), and its in vitro activity is mostly NAD+-dependent (16). Because formate release increased in the presence of galactose, we hypothesized that the rate of serine one-carbon catabolism with formate overflow depends on the availability of NAD+ and, thus, complex I–dependent NADH (reduced form of NAD+) oxidation.
To test this hypothesis, we used rotenone to inhibit complex I of oxidative phosphorylation (Fig. 2A). We observed a drastic reduction of serine consumption, glycine release, and formate release in all cancer cell lines tested (Fig. 2B and fig. S2, C to E). We note that, although the galactose and rotenone treatment conditions are both characterized by growth inhibition [Figs. 1 (J and K) and 2D, and figs. S1 (F and G) and S2 (J and K)], formate release exhibits opposite changes. Under the galactose condition, where mitochondrial NADH oxidation increases, formate release increases. Upon rotenone treatment, where mitochondrial NADH oxidation is inhibited, formate release decreases. Together, this evidence supports a link between NAD+-dependent MTHFD2 activity, NADH oxidation in the mitochondria, and formate production. These observations are further supported by a recent work by Bao et al. (17), reporting that respiratory chain dysfunction upon mitochondrial DNA depletion causes a reduction of formate release from cells.
Fig. 2. Serine catabolism is linked to mitochondria.
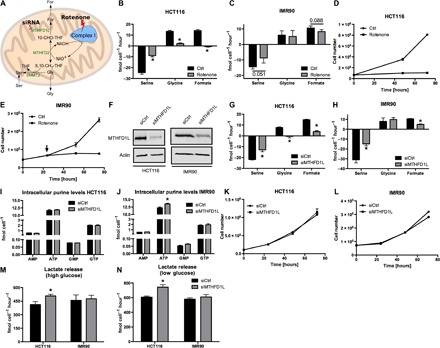
(A) Model illustrating mitochondrial one-carbon metabolism and its potential dependency on complex I. (B and C) Absolute exchange rates of serine, glycine, and formate in (B) HCT116 and (C) IMR90 cells upon rotenone. (D and E) Proliferation of (D) HCT116 and (E) IMR90 cells upon rotenone. (F) Western blot confirming knockdown of MTHFD1L in HCT116 and IMR90 cells. (G and H) Absolute exchange rates of serine, glycine, and formate in (G) HCT116 and (H) IMR90 cells upon MTHFD1L knockdown. (I and J) Absolute intracellular purine levels in (I) HCT116 and (J) IMR90 cells upon MTHFD1L knockdown. GMP, guanosine monophosphate; GTP, guanosine triphosphate. (K and L) Proliferation of (K) HCT116 and (L) IMR90 cells upon MTHFD1L knockdown. (M and N) Lactate release rates of HCT116 and IMR90 cells upon MTHFD1L knockdown under (M) high-glucose (17 mM) and (N) low-glucose (5.5 mM) conditions. Data are presented as means ± SD (n = 3 cultures representative of at least two independent experiments), except for (F) (one culture). *P < 0.05 by Welch’s t test.
In the nontransformed IMR90 fibroblasts, serine consumption and formate release were also reduced upon rotenone treatment, albeit less markedly (Fig. 2C and fig. S2F). Increasing the rotenone concentration from the 250 nM that is used in Fig. 2C to 500 and 1000 nM had no further effect on the rate of formate release by IMR90 cells (fig. S2F). These data indicate that IMR90 cells have a rotenone-insensitive component of formate release. We note that rotenone inhibits the growth (Fig. 2E) and OCR (fig. S2G) of IMR90 cells already at the 250 nM concentration, ruling out the possibility that rotenone is not having an effect on oxidative phosphorylation in these cells. Because the cytosolic dehydrogenase MTHFD1 uses NADP+ and the mitochondrial dehydrogenase MTHFD2L can use NAD+ or NADP+ as a cofactor, we suspected that one or both enzymes could contribute to the 5,10-CH2-THF dehydrogenase activity in IMR90 cells independently of oxidative phosphorylation. However, quantification of gene expression by quantitative polymerase chain reaction revealed a similar pattern of MTHFD2, MTHFD2L, and MTHFD1 expression in HCT116 and IMR90 cells (fig. S2H), with about the same MTHFD2-to-MTHFD2L ratio (fig. S2I). Although this evidence cannot rule out differences in enzymatic activity, it suggests that differences in the dehydrogenase expression are not responsible for the rotenone-insensitive component of formate release in IMR90 cells. We can only speculate that, as a difference to cancer cells (in IMR90 fibroblasts), serine one-carbon catabolism is partially uncoupled from oxidative phosphorylation.
To further validate whether the serine one-carbon catabolism with formate overflow takes place in the mitochondria, we targeted MTHFD1L, the enzyme catalyzing the step of formate release and adenosine triphosphate (ATP) production (Figs. 1C and 2A). Knocking down MTHFD1L by small interfering RNA (siRNA) (Fig. 2F and fig. S2L), we verified that the formate release from cells is dependent on mitochondrial MTHFD1L abundance in all cell lines tested, including the IMR90 fibroblasts (Fig. 2, G and H, and fig. S2, M and N). The formate generated by MTHFD1L can also be recaptured by cytosolic 10-CHO-THF synthetase (MTHFD1) to satisfy the one-carbon demand for de novo purine synthesis (1). Therefore, reduction of MTHFD1L levels could inhibit purine synthesis. However, knockdown of MTHFD1L did not result in decreased purine levels (Fig. 2, I and J, and fig. S2, O and P) nor in decreased growth (Fig. 2, K and L, and fig. S2, Q and R). The residual activity of the remaining MTHFD1L protein or the cytosolic enzyme (MTHFD1) appears to be sufficient to satisfy the one-carbon unit demand for purine synthesis and thus to sustain growth rates similar to nonsilenced cells. On the basis of recent data from another group, the cytosolic pathway can compensate for the requirement of one-carbon units in cells, where a mitochondrial pathway enzyme (including SHMT2, MTHFD2, and MTHFD1L) has been knocked out (18). Together, these results indicate that the mitochondrial serine catabolism to glycine and formate is dispensable for purine synthesis and growth.
Another potential function of the serine catabolism to glycine and formate could be energy generation. For every formate released from cells, one adenosine diphosphate (ADP) molecule is phosphorylated to form ATP via reverse mitochondrial 10-CHO-THF synthetase (Fig. 1C). The coupling of the mitochondrial NAD+-dependent 5,10-CH2-THF dehydrogenase to mitochondrial oxidative phosphorylation can contribute with an additional 2.5 molecules of ATP per formate molecule released, giving an overall yield of 3.5 ATP per formate. As reported above, MTHFD1L knockdown did not result in decreased purine levels, including ATP (Fig. 2, I and J, and fig. S2, O and P). Therefore, we hypothesized that another pathway for energy generation should compensate for the reduced energy yield by the serine catabolism to formate and glycine. Because glycolysis is a potential candidate to fulfill this role, we quantified the rate of lactate release. MTHFD1L knockdown resulted in increased lactate release in HCT116 cells (Fig. 2, M and N). In these cells, MTHFD1L knockdown decreases formate efflux by ~11 fmol cell−1 hour−1 (Fig. 2G). The expected energy deficit is given by the drop in formate release times 3.5 ATP per formate, that is, about 38.5 fmol cell−1 hour−1 for HCT116 cells upon MTHFD1L knockdown. We measured an increase in lactate release of ~100 fmol cell−1 hour−1, relative to control (Fig. 2, M and N). Because glycolysis has a yield of 1 ATP per lactate, the increase in glycolysis is overcompensating for the reduction in ATP generation by the serine catabolism to formate. In IMR90 cells, formate release is, up to a great extent, rotenone-independent and thus uncoupled from NADH oxidation. In this case, we expect a yield of 1 ATP per formate. In IMR90 cells, formate release goes down by 6 fmol cell−1 hour−1 when MTHFD1L is silenced, and lactate release goes up by 18 fmol cell−1 hour−1 (albeit within the experimental error and not significant). We obtained the same results at physiological glucose conditions (5.5 mM): a significant increase in lactate release in HCT116 cells and no significant increase in IMR90 cells (Fig. 2N). Work from another group has shown that knockdown of MTHFD2 also results in increased lactate release in the MCF7 breast cancer cells (19). Together, the lack of any phenotype regarding purine levels and growth in MTHFD1L knockdown cells, but an increase in glycolysis, suggests a catabolic rather than an anabolic role of the serine one-carbon catabolism to glycine and formate.
Formate efflux exceeds anabolic one-carbon demands
To further demonstrate that formate release is the dominant fate of one-carbon production, we quantified the rate of purine synthesis and compared the result with the rate of formate release (Fig. 3). In the HCT116 colon cancer cell line, we determined a purine synthesis flux of 1.4 fmol cell−1 hour−1 (Fig. 3B), 10-fold lower than the measured average formate release flux (16.18 fmol cell−1 hour−1; Fig. 1E). Similarly, the IMR90 nontransformed lung fibroblasts produce purines at a rate between 0.3 and 0.4 fmol cell−1 hour−1 (Fig. 3C), 25-fold lower than the rate of formate release (10.31 fmol cell−1 hour−1; Fig. 1F). The purine synthesis rates were also 10-fold lower in the two other cell lines tested (fig. S3, G and H). In addition, we examined the effect of rotenone and MTHFD1L knockdown on serine one-carbon metabolism fluxes and revealed that only rotenone decreased the purine synthesis flux, whereas MTHFD1L knockdown did not result in such a strong decrease in this biosynthetic flux (Fig. 3, B and C, and fig. S3, G and H). These results are in line with the observed effect on growth repression upon rotenone, but not MTHFD1L knockdown [Fig. 2D (E, K, and L) and fig. S2 (J, K, Q, and R)]. Because rotenone simultaneously reduces purine synthesis flux and growth, the measured purine levels remained constant in the case of HCT116 and were only moderately reduced in the case of IMR90 (fig. S3, E and F).
Fig. 3. Formate efflux exceeds anabolic one-carbon demands.
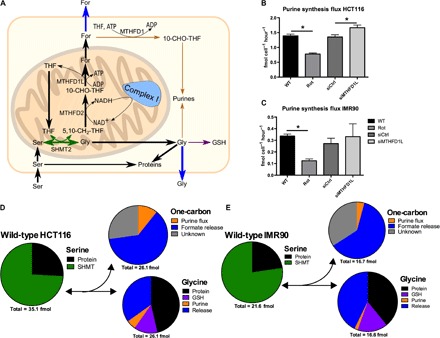
(A) Model summarizing major cellular serine–derived fluxes in mammalian cells. (B and C) Absolute purine synthesis flux in (B) HCT116 and (C) IMR90 cells. (D and E) Pie charts representing fluxes of serine, glycine, and one-carbon moieties in (D) HCT116 and (E) IMR90 cells under normal conditions. The numbers underneath the pie chart represent the summed contribution of all sinks reported in the pie chart above (per cell per hour). We note that these pie charts just reflect the average measurements [see (B) and (C) and fig. S3 (A to D) for the measurement errors of each specific contribution]. SHMT, serine hydroxymethyltransferase. Data are presented as means ± SD (n = 3 cultures representative of at least two independent experiments). *P < 0.05 by Welch’s t test.
To cover all major sources/sinks of serine and glycine, we also determined the rate of serine synthesis from glucose, serine, and glycine flux to proteins and from glycine to glutathione (GSH) (fig. S3, A to D) to ultimately present an overall picture of major cellular serine/glycine/one-carbon metabolism fluxes (Fig. 3A). We note that there is also a demand of one-carbon units for thymidylate synthesis. However, this demand is significantly lower than that for purine synthesis, and its value is within the error of our measurements. The serine synthesis rates were estimated from the metabolic flux analysis (MFA) model for serine (20, 21). The MFA model takes into account the import of serine, the synthesis of serine from glucose, and the reverse flux of serine hydroxymethyltransferase. In the context of a [U-13C]serine tracer, the latter is reflected in the formation of M+1 serine from the M+1 fraction of 5,10-CH2-THF and M+0 glycine. Under all conditions tested, serine synthesis from glucose represents less than 10% of total serine production (from extracellular glucose and serine). Rotenone treatment decreased the rate of serine synthesis from glucose (fig. S3A). This observation is consistent with the requirement of an NAD+-dependent dehydrogenase step (catalyzed by PHGDH) in the serine biosynthesis and the inhibition of NADH oxidation by rotenone treatment.
We summarized the MFA in a flux pie chart that shows the excessive serine-derived formate efflux, which is about 10-fold above the purine synthesis flux (Fig. 3, D and E). For glycine, we observe two different scenarios. In the A549 lung and MDA-MB231 breast cancer cell lines grown in glucose, the production of glycine from serine matches approximately its demand for biosynthesis, and there is a small rate of glycine release into the extracellular medium (fig. S1, D and E). In these cell lines, excess production of one-carbon units and thus formate release can be explained by the cellular demand of glycine. There is an imbalance between equal rates of glycine and one-carbon unit production from serine and a higher biosynthetic demand of glycine than one-carbon units. This scenario has been hypothesized previously (11), predicting that the excess one-carbon units would be released as CO2 by 10-CHO-THF dehydrogenase. However, as shown here, formate is a major route of one-carbon unit release in A549 and MDA-MB231 cells. In the remaining cell lines, there is an excess production of glycine from serine that is released from cells. In this context, the flux of serine catabolism to glycine and one-carbon unit cannot be explained by biosynthetic imbalances. Our calculations also indicate 25 to 50% of unaccounted one-carbon unit consumption, not explained by the demands for purine synthesis and formate efflux (Fig. 3, D and E). Most likely, this unknown sink represents the overall activity of cytosolic and mitochondrial 10-CHO-THF dehydrogenases.
Serum formate depends on serine catabolism in vivo
Investigations of whole-body formate metabolism indicate that there is a significant amount of formate release into the blood (22). However, the origin of the formate remains largely unconfirmed. The in vitro evidence reported above suggests serine as a potential source. To determine whether serine one-carbon catabolism with formate overflow takes place in vivo, we performed a serine-tracing experiment in wild-type (WT) C57BL/6 mice. This approach was combined with phenformin to test the dependency of serine catabolism to formate on complex I activity. The antidiabetic drugs phenformin and metformin are biguanides with reported inhibitory activity against complex I (23). To exclude carbon contribution from glycine cleavage, which is active in certain organs such as liver and kidneys, we used a [3-13C1]serine tracer. Serine labeled with 13C at carbon 3 transfers 13C to 5,10-CH2-THF only via the serine hydroxymethyltransferase activity, whereas glycine cleavage would donate carbon 2 (12C) 12C of serine. Mice were fasted overnight before the start of the experiment. We administered a bolus of [3-13C1]serine via intraperitoneal injection (±phenformin) and collected blood samples at 15, 30, and 60 min after injection (Fig. 4A), with parallel sampling of four mice per time point. We also conducted an independent pilot experiment with three mice per time point, showing the same overall trend.
Fig. 4. Serum formate depends on serine catabolism in vivo.
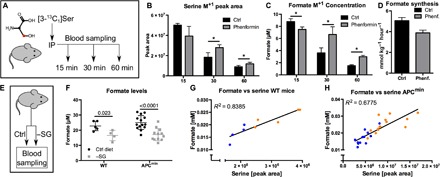
(A) Model illustrating the setup to trace serine-derived formate formation in vivo. (B) Abundance of serine M+1 isotopologues in plasma, 15, 30, and 60 min after injection with or without phenformin. (C) Concentration of formate M+1 isotopologue in plasma, 15, 30, and 60 min after injection with or without phenformin. (D) Estimated serine-derived formate synthesis rate in phenformin-treated or phenformin-untreated mice (see text for reported CIs). (E) Model illustrating the experimental setup for serine/glycine (−SG) starvation, to measure its effect on serum formate level. (F) Formate concentration in the serum of serine- and glycine-starved mice [3 weeks starvation in WT mice (n ≥ 5) and in APCmin mice at clinical end point (n ≥ 12)]. We note the exclusion of one outlier in the 3-week setup (see fig. S4, D and E). (G and H) Correlation between serine and formate levels in (G) WT and in (E) APCmin mice in respect to serine/glycine starvation. Data are presented as means ± SD [(B to D) n = 4 mice per condition and time point]. *P < 0.05 by Welch’s t test.
The plasma levels of serine, glycine, and formate were just slightly above the values in mice injected with a phosphate-buffered saline (PBS) bolus (fig. S4, A to C). We confirmed the appearance of [3-13C1]serine in the plasma following its intraperitoneal injection (Fig. 4B). [3-13C1]serine levels were highest at 15 min after injection and gradually decreased over time. However, clearance rates were different in the control and the phenformin group, resulting in significantly higher M+1 serine levels in the phenformin group at the 30- and 60-min time points (Fig. 4B). We observed the appearance of [13C]formate in plasma (Fig. 4C), supporting the generation of formate from serine in vivo. Fifteen minutes after injection, formate M+1 concentration was lower in the phenformin group, suggesting a decrease in formate generation from serine (Fig. 4C). In contrast, at the 30- and 60-min time points, M+1 formate concentrations were higher in the phenformin group, which could be explained by a concomitant decrease in formate turnover (Fig. 4C). Given the labeling pattern of serine and the observed concentrations of [13C]formate in WT mice, we estimated a rate of serine-derived formate production of 5.1 ± 0.3 mmol kg−1 hour−1, representing about 50% of total formate production (42% in the independent pilot experiment). The remaining 50% could originate from glycine cleavage, cholesterol synthesis, and demethylation. Phenformin treatment resulted in a 1.3-fold [95% confidence interval (CI), 1.1 to 1.5] reduction of the estimated rate of formate generation from serine (Fig. 4D). This observation confirmed the trend observed in the independent pilot experiment, where phenformin treatment resulted in a 1.7-fold (95% CI, 0.7 to 3.3) reduction of the estimated rate of formate generation from serine.
To further demonstrate that serine is a significant source of formate in vivo, we investigated the impact of a serine/glycine-deficient diet (−SG diet) on plasma formate levels. We set C57BL/6 WT mice on a serine/glycine-deficient diet for 3 weeks to monitor serine and formate levels in the blood (Fig. 4E). After 3 weeks of starvation, we observed a significant reduction of serum formate concentration (Fig. 4F). We also observed a linear correlation between serine and formate levels, independently of the diet, with the −SG diet mice shifted to lower serine/formate levels (Fig. 4G). In an alternative attempt, we took advantage of a parallel experiment testing the impact of serine deprivation on cancer-related survival in the APCmin mouse model for colorectal cancer. APCmin mice die of tumor burden at 130 days of age on average. Eighty-day-old APCmin mice were set on a −SG diet until clinical end point (tumor-related survival, approximately 50 days). At the time of death, blood samples were collected. Using these samples, we retrospectively analyzed serum formate levels. Serine-starved APCmin mice had a 32% lower plasma formate concentration on average compared to the control group, fed a diet containing serine and glycine (Fig. 4F). As in WT mice, formate levels also linearly correlated with plasma serine levels (Fig. 4H). These data indicate that serine availability determines formate levels in blood, providing further support for serine as a significant source of formate in vivo.
DISCUSSION
Here, we demonstrate that cells run serine one-carbon catabolism in excess of the biosynthetic demand of one-carbon units. Most of the excess one-carbon units are released as formate under the conditions tested. For every formate molecule released from cells, one ADP molecule is phosphorylated to form ATP via reverse mitochondrial 10-CHO-THF synthetase (Fig. 1C). The coupling of the mitochondrial NAD+-dependent 5,10-CH2-THF dehydrogenase to mitochondrial oxidative phosphorylation can contribute with an additional 2.5 molecules of ATP per formate molecule released. The only cell-autonomous phenotype we have observed in MTHFD1L knockdown cells is an increase in glycolysis, a canonical pathway for energy generation, in the HCT116 colorectal cancer cells but not in the IMR90 nontransformed lung fibroblasts. Although this evidence is not conclusive, it supports the idea that serine one-carbon catabolism with formate overflow can contribute to energy generation. Further work is required to investigate whether this contribution is enhanced in certain cancers relative to normal tissues.
Some cancer cells exhibit a high rate of formate release but almost no glycine release when growing in standard culture medium. Therefore, we cannot exclude the possibility that excess one-carbon unit production is a side effect of glycine generation from serine and a higher metabolic demand of glycine than one-carbon units (11). However, if such hypothesis is correct, then we would need to resort to additional hypotheses to explain why mammalian cells evolved to generate glycine from serine when glycine is available in the extracellular medium and why the excess one-carbon units are mostly released as formate instead of as less toxic CO2.
The demonstration of serine catabolism with formate overflow has several implications to our understanding of mammalian metabolism in normal physiology and disease states. Of greatest importance is the use of metformin, a less potent complex I inhibitor than phenformin, for the treatment of type 2 diabetes. Although metformin has different organ-specific pharmacokinetics than phenformin, both drugs target complex I. Our data suggest a reduction in plasma formate levels as a consequence of treatment with complex I inhibitors. It will be interesting to investigate whether formate levels are reduced in type 2 diabetes patients under long-term metformin treatment and whether lower formate levels are simply a consequence of the treatment or whether they might even have a direct impact on the phenotype.
Metformin is also currently being investigated for the treatment of cancer. A study in mice has shown that metformin treatment synergizes with serine deprivation in the growth inhibition of mouse xenograft tumors (24). The evidence reported here indicates that both treatment strategies inhibit the serine one-carbon catabolism to formate, potentially contributing to the observed synergy.
The phenomenon uncovered here, serine one-carbon catabolism with formate overflow, resembles the well-known phenotype of glucose catabolism with lactate overflow [the Warburg effect (25)]. Both phenotypes are characterized by the apparent “waste” of carbon atoms and energy production. They are different with respect to localization. The Warburg effect contributes to the cytosolic, and the serine one-carbon catabolism contributes to the mitochondrial energy generation. They also differ in the magnitude of the pathway rate, with glucose catabolism being significantly higher than serine catabolism. However, this rate difference is not unexpected, considering that mitochondria represent a low percentage of the biomass content of normal proliferating cells and cancer cells [table S1 in Vazquez et al. (26)]. Finally, similar to the resynthesis of glucose from lactate in the liver via the Cori cycle, serine could be resynthesized in the liver, contributing to the organism balance of energy and one-carbon units.
MATERIALS AND METHODS
Cell culture
All cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) 5030 (Thermo Fisher Scientific) with addition of 2 mM glutamine, 17 mM glucose, and 10% fetal bovine serum (FBS) at 37°C and 5% CO2. IMR90 cells were cultured identically with the exception of 20% FBS. In the case of galactose experiments, glucose was replaced by 17 mM galactose at the start of the experiment. HCT116 and A549 cells were obtained from the group of K. Vousden (Cancer Research U.K. Beatson Institute), and MDA-MB231 and K562 cells were obtained from the group of E. Gottlieb (Cancer Research U.K. Beatson Institute). IMR90 fibroblasts were obtained from the group of P. Adams (University of Glasgow). All cell lines were checked for mycoplasma contaminations. All experiments were performed in triplicate wells per condition, unless otherwise stated, and were repeated at least twice to monitor biological variance in addition to phenotype verification across different genetic backgrounds (different cell lines). Rotenone (Sigma-Aldrich) was used at a final concentration of 250 nM. For low-glucose conditions, cells were cultured in minimum essential medium (MEM) (Thermo Fisher Scientific, 11090-081; 5.5 mM glucose) with the addition of 2 mM glutamine, 400 μM serine, and 400 μM glycine.
siRNA-mediated gene silencing
To silence MTHFD1L, 10 pmol of SMARTpool siRNA (GE Dharmacon, L-009949-01-0005), 2.5 μl of Lipofectamine 2000 (Invitrogen), and 200 μl of Opti-MEM (Thermo Fisher Scientific) were mixed and incubated at room temperature for 20 min. As a control, a nontargeting siRNA control was used instead (GE Dharmacon, D-001810-10-05). During 20 min of incubation, cells were trypsinized, counted, and diluted to the desired concentration (150 to 250 K cells per well). After 20 min of siRNA incubation, 200 μl of the siRNA-Lipofectamine mix was aliquoted in 1 well of a 12-well plate. Next, 800 μl of cell suspension was added to each well. The next day, medium was replaced by complete medium. Knockdown was verified 48 to 72 hours after transfection by Western blot. In the case of growth assays, time point 0 corresponds to the time of medium replacement after transfection. Cells were counted with a CASY cell counter (OLS).
Stable isotope labeling
Stable isotope experiments were performed in MEM (Thermo Fisher Scientific, 11090-081), with the addition of 400 μM glycine, 2 mM glutamine, 17 mM glucose, 400 μM serine, and 10% FBS. To allow for adaptation to the different cell culture media, we cultured cells for at least one passage in complete MEM before the start of the experiment. At the start of the experiment, 150,000 cells were seeded in 12-well plates. In the case of MTHFD1L knockdown, transfection was performed as described in the previous section. Extra triplicate wells were seeded accordingly to count cells at the start and end of the experiment. The next day, medium was replaced, and cells were cultured for an additional 24 hours. At the start of the experiment, three wells per condition were counted to assess cell number. For tracing, cells were washed once with PBS, and medium was replaced with 400 μM [U-13C]serine-containing medium. Cells were incubated for the indicated times (8 hours for HCT116 and A549 and 16 hours for MDA-MD231 and IMR90). At the extraction time point, triplicate wells were counted to assess cell number at the end of the experiment, and one set of triplicate wells was used for metabolite extraction. Medium of the extracted cells was collected and analyzed for exchange rates. To determine consumption rates, the same medium had been incubated in empty wells for the same time in the incubator.
Metabolite extraction
For the extraction of intracellular metabolites and the quantification of exchange rates, medium was collected and, for the extraction of intracellular metabolites, cells were washed with PBS, and 400 μl of ice-cold extraction fluid [acetonitrile/H2OMQ/methanol (ratio, 3:2:5); liquid chromatography–MS (LC-MS) grade solvents)] was added immediately to each well. Plates were incubated at 4°C for 5 min on a rocking shaker. After 5 min, supernatant was collected in 1.5-ml Eppendorf tubes, vortexed, and subsequently centrifuged for 5 min at 13,000g at 4°C. Two hundred microliters of supernatant was transferred into LC-MS glass vials with inserts and stored at −80°C until measurement.
Collected medium was centrifuged at 300g for 5 min at 4°C to pellet cellular debris. Medium supernatant was collected in Eppendorf tubes and stored at −80°C until extraction of metabolites. For the extraction of metabolites, 10 μl of medium was diluted in 990 μl of ice-cold extraction fluid, vortexed, and centrifuged at 13,000g for 5 min at 4°C. For the specific quantification of lactate, samples were extracted with extraction solvent containing 100 μM [U-13C]lactate as internal standard. Supernatant was transferred into LC-MS glass vials and stored at −80°C until measurement. For the measurement of serum- or plasma-derived metabolites, 5 μl of sample was mixed with 250 μl of extraction fluid and vortexed for 10 min at 4°C. Further procedures were equivalent to medium-derived metabolite extraction.
Formate extraction, derivatization, and quantification
Forty microliters of sample was added to a 1.5-ml microfuge tube, followed by 20 μl of 1 mM internal standard sodium 13C,2H-formate (M+2) (Sigma-Aldrich, CAS 1215684-17-5), 10 μl of benzyl alcohol (Sigma-Aldrich, CAS 100-51-6), 10 μl of 1 M sodium hydroxide, and 50 μl of pyridine. The tube was then placed on ice for 5 min. Derivatization was started by adding 20 μl of methyl chloroformate with vigorous vortexing for 20 s. Note that derivatization should be performed in a fume hood as gases were produced during the reaction. After vortexing, 200 μl of water and 100 μl of methyl tertiary butyl ether were added, and the sample was vortexed for another 20 s and centrifuged at 10,000g for 5 min. Forty-five microliters of the resulting upper layer containing formate derivative (benzyl formate) was transferred to a GC glass vial and stored at −80°C until analysis. For the quantification of formate in mouse serum samples, we used the same procedure with the exception of the concentration of the internal standard at 50 μM.
Determination of intracellular purine and GSH levels
For the determination of absolute intracellular purine and GSH levels, the standard addition method was used. Briefly, respective standards were prepared in extraction solvent and spiked in different concentrations into extracts of WT cells. Instead of 400 μl of extraction volume, cells were extracted with 380 μl of extraction volume and 20 μl of a standard mix containing a defined concentration of ATP, and guanosine triphosphate and GSH were added. The following standards were used: 0, 40, 80, and 200 μM.
Protein quantification and determination of serine and glycine flux into total protein
For quantification of intracellular protein content, a Bradford assay (Sigma-Aldrich) was used according to the manufacturer’s instructions. Signal was recorded in 96-well plates at 595 nm. Protein content was determined in microgram per cell.
Oxygen consumption rates
To measure OCRs, we used the Extracellular Flux Analyzer (Seahorse Biosciences). Briefly, 30,000 cells per well of a 96-well plate (Seahorse Bioscience) were seeded the day before the experiment. The culture medium was DMEM 5030 (as described in the “Cell Culture” section). Immediately before the start of the experiment, 8 wells from the same 96-well plate were trypsinized and counted to assess cell number per well. OCR was determined according to the manufacturer’s conditions.
Western blot
Proteins were extracted using radioimmunoprecipitation assay buffer (Thermo Fisher Scientific) with the addition of Halt Protease and Phosphatase Inhibitor Cocktail (100×) (Thermo Fisher Scientific) at a 1× final concentration. Approximately 100 μl of extraction buffer was used per 100,000 cells. Cells were cultured in 12-well plates. Proteins were separated in 10% precast SDS gels (Bio-Rad) at 200 V for 40 min. Proteins were transferred to nitrocellulose membrane (Amersham) and blocked with 7% milk powder in tris-buffered saline–Tween 20 (TBS-T) for at least 1 hour (at room temperature) or overnight (at 4°C) on a rocking shaker. Blots were washed with TBS-T to remove residual milk powder and were incubated with primary antibody (1:1000 in TBS-T) for 1 hour at room temperature on a rocking shaker: anti–β-actin (Cell Signaling, 8457P), anti-MTHFD1L (Proteintech, 16113-1-AP), anti-AMPKα [AMPKα, D5A2; monoclonal antibody (mAb) #5831, Cell Signaling], anti–phospho-AMPKα (Thr172, 40H9; mAb #2535, Cell Signaling), ACC (C83B10; mAb #3676), and anti–phospho-ACC (Ser79, D7D11; mAb #11818). After incubation, blots were washed three times for 10 min with TBS-T and incubated with the secondary antibody [IRDye 800CW donkey anti-rabbit immunoglobulin G (H + L); 1:5000 (LI-COR, 926-32213)] for 1 hour at room temperature on a rocking shaker. After incubation, blots were washed three times for 10 min with TBS-T. Fluorescence signals were recorded using the Odyssey analyzer (LI-COR) with respective software package (Image Studio Lite). LI-COR software was also used for image processing before exporting as TIFF files. Final image assembly was carried out using Inkscape software package (https://inkscape.org). Images were exported as .eps files and imported into Prism 6 for final figure assembly.
LC-MS measurements
The LC-MS method has been previously described in the studies by Tedeschi et al. (21, 27) and Patella et al. (21, 27). A Q Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific) was used, together with a Thermo UltiMate 3000 high-performance liquid chromatography (HPLC) system. The HPLC setup consisted of a ZIC-pHILIC column (SeQuant, 150 mm × 2.1 mm; Merck KGaA, 5 μm), with a ZIC-pHILIC guard column (SeQuant, 20 mm × 2.1 mm) and an initial mobile phase of 20% of 20 mM ammonium carbonate (pH 9.4) and 80% acetonitrile. Cell and medium extracts (5 μl) were injected, and metabolites were separated over a 15-min mobile phase gradient, decreasing the acetonitrile content to 20%, at a flow rate of 200 μl/min and a column temperature of 45°C. The total analysis time was 23 min [adapted from the study by Tedeschi et al. (21)]. All metabolites were detected across a mass range of 75 to 1000 mass/charge ratio (m/z) using the Exactive mass spectrometer at a resolution of 25,000 (at 200 m/z), with electrospray ionization and polarity switching to enable both positive and negative ions to be determined in the same run. Lock masses were used, and the mass accuracy obtained for all metabolites was below 5 parts per million. Data were acquired using Thermo Xcalibur software.
The peak areas of different metabolites were determined using Thermo TraceFinder software, where metabolites were identified by the exact mass of the singly charged ion and by known retention time on the HPLC column. Commercial standards of all metabolites detected had been previously analyzed on this LC-MS system with the ZIC-pHILIC column. The 13C-labeling patterns were determined by measuring the peak areas for the accurate mass of each isotopologue of a given metabolite [adapted from the study by Patella et al. (27)]. Integrated peak areas had been extracted and used for further quantifications. To determine the relative intracellular metabolite abundance, we normalized integrated peak areas to the cell count at the end of the experiment.
GC-MS measurements
Derivatized formate samples were analyzed with an Agilent 7890B GC system coupled to a 7000 triple quadrupole MS system. The column was a Phenomenex ZB-1701 column (30 m × 0.25 mm × 0.25 μm). Samples (2 μl) were injected into the GC-MS using split mode [0.5 bars; split flow (25 ml/min)]. Gas flow through the column was held constant at 1.0 ml of He per minute. The temperature of the inlet was 280°C, the interface temperature was 230°C, and the quadrupole temperature was 200°C. Reduced electron energy (60 eV) was used for analyte ionization. The column was equilibrated for 3 min before each analysis. The mass spectrometer was operated in selected ion monitoring (SIM) mode between 3.0 and 4.3 min with SIM masses of 136, 137, and 138 for M0, M+1, and M+2 (internal standard) formate, respectively. The utilized oven program is described in table S1.
The total run time of a sample was 5.97 min. Natural isotope abundance was taken into account and subtracted, as described in the Supplementary Materials.
Recorded data were processed using the MassHunter Software (Agilent). Integrated peak areas were extracted and used for further quantifications using in-house software packages.
In vivo work
All animal work was carried out under PPL 70/8468 in line with the Animals (Scientific Procedures) Act 1986 and the European Union Directive 2010 and was sanctioned by the local ethical review process (University of Glasgow). Male C57BL/6J mice (stock number, 000664; at least 20 g in weight) were purchased from Charles River. Thirty milligrams of [3-13C1]serine was dissolved in 6 ml of PBS. The 6 ml was split into 2 ml × 3 ml, and 30 mg of phenformin was added to one tube. Both tubes were sterile-filtered and used for intraperitoneal injection. As a control, only one group was injected with PBS, and 200 μl of solution was injected per mouse. Mice were randomly assigned to seven groups (four mice per group: three groups for [3-13C1]serine, three groups for [3-13C1]serine + phenformin, and one group for PBS) and were injected intraperitoneally. Mice were sacrificed 15, 30, or 60 min after injection (15 min only for PBS group) by Schedule 1 method. Whole blood was collected and centrifuged at 13,000 rpm for 5 min, and sera were transferred to clean tubes and snap-frozen on dry ice.
For serine starvation, APCmin (28) or C57BL/6 mice were housed under same conditions. APCmin mice were housed until day 80. At that time point, the cohort was split into a control group (full diet) and a serine/glycine-deficient diet (−SG) group. The control group consisted of 14 mice, and the −SG group consisted of 12 mice. Mice were sacrificed individually, depending on the progression of tumor burden. WT mice were split into two groups, consisting of six mice each, and were kept on specific diets for 3 weeks. Whole blood was collected and processed to serum, as described in the previous section. Formate analysis in sera from the serine starvation experiment was performed in a blinded manner. Mouse IDs were assigned to the results, retrospectively.
Supplementary Material
Acknowledgments
We acknowledge the Cancer Research U.K. Glasgow Centre (C596/A18076) and the Biological Services Unit (BSU) facilities at the Cancer Research U.K. Beatson Institute (C596/A17196). Funding: This work was supported by the Cancer Research U.K. grant no. C596/A21140. J.M. was supported by a Deutsche Forschungsgemeinschaft Fellowship grant no. ME 4636/2-1. Author contributions: J.M. performed all in vitro experiments and analyzed data. S.T. and J.J.K. developed the GC-MS method for formate. O.M., C.F.L., and K.V. conceived mice serine starvation experiments. D.A. and K.B. performed in vivo mice serine starvation experiments and in vivo 13C serine tracing. N.V.D.B. and G.M.M. measured LC-MS samples and developed analysis methods. E.G. provided guidance. A.V. developed software algorithms for data analysis and analyzed data. J.M. and A.V. designed experiments and wrote the manuscript. All authors read the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/10/e1601273/DC1
Supplementary Text
fig. S1 Serine catabolism is induced upon energy stress.
fig. S2. Serine catabolism is linked to mitochondria.
fig. S3. Formate efflux exceeds anabolic one-carbon demands.
fig. S4. Serum formate depends on serine catabolism in vivo.
table. S1. GC temperature program for formate analysis.
REFERENCES AND NOTES
- 1.Tibbetts A. S., Appling D. R., Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 30, 57–81 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Locasale J. W., Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 13, 572–583 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen R. W., Moskowitz M., Arrest of cell growth in G1 phase of cell cycle by serine deprivation. Exp. Cell Res. 116, 127–137 (1978). [DOI] [PubMed] [Google Scholar]
- 4.Rowe P. B., Sauer D., Fahey D., Craig G., Mccairns E., One-carbon metabolism in lectin-activated human-lymphocytes. Arch. Biochem. Biophys. 236, 277–288 (1985). [DOI] [PubMed] [Google Scholar]
- 5.Maddocks O. D. K., Berkers C. R., Mason S. M., Zheng L., Blyth K., Gottlieb E., Vousden K. H., Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542−546 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Labuschagne C. F., van den Broek N. J. F., Mackay G. M., Vousden K. H., Maddocks O. D. K., Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 7, 1248–1258 (2014). [DOI] [PubMed] [Google Scholar]
- 7.D. Voet, J. G. Voet, Biochemistry (John Wiley & Sons Inc., ed. 4, 2011). [Google Scholar]
- 8.Vazquez A., Markert E. K., Oltvai Z. N., Serine biosynthesis with one carbon catabolism and the glycine cleavage system represents a novel pathway for ATP generation. PLOS ONE 6, e25881 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Martinez L. F., Appling D. R., Characterization of the folate-dependent mitochondrial oxidation of carbon 3 of serine. Biochemistry 32, 4671–4676 (1993). [DOI] [PubMed] [Google Scholar]
- 10.Jain M., Nilsson R., Sharma S., Madhusudhan N., Kitami T., Souza A. L., Kafri R., Kirschner M. W., Clish C. B., Mootha V. K., Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336, 1040–1044 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brosnan M. E., MacMillan L., Stevens J. R., Brosnan J. T., Division of labour: How does folate metabolism partition between one-carbon metabolism and amino acid oxidation?. Biochem. J. 472, 135–146 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Hardie D. G., Ross F. A., Hawley S. A., AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson B. H., Petrova-Benedict R., Buncic J. R., Wallace D. C., Nonviability of cells with oxidative defects in galactose medium: A screening test for affected patient fibroblasts. Biochem. Med. Metab. Biol. 48, 122–126 (1992). [DOI] [PubMed] [Google Scholar]
- 14.Lewis C. A., Parker S. J., Fiske B. P., McCloskey D., Gui D. Y., Green C. R., Vokes N. I., Feist A. M., Vander Heiden M. G., Metallo C. M., Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 55, 253–263 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meiser J., Vazquez A., Give it or take it: The flux of one-carbon in cancer cells. Febs J., 10.1111/febs.13731 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Smith G. K., Banks S. D., Monaco T. J., Rigual R., Duch D. S., Mullin R. J., Huber B. E., Activity of an NAD-dependent 5,10-methylenetetrahydrofolate dehydrogenase in normal tissue, neoplastic cells, and oncogene-transformed cells. Arch. Biochem. Biophys. 283, 367–371 (1990). [DOI] [PubMed] [Google Scholar]
- 17.Bao X. R., Ong S. E., Goldberger O., Peng J., Sharma R., Thompson D. A., Vafai S. B., Cox A. G., Marutani E., Ichinose F., Goessling W., Regev A., Carr S. A., Clish C. B., Mootha V. K., Mitochondrial dysfunction remodels one-carbon metabolism in human cells. eLife 5, pii: e10575 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ducker G. S., Chen L., Morscher R. J., Ghergurovich J. M., Esposito M., Teng X., Kang Y., Rabinowitz J. D., Reversal of cytosolic one-carbon flux compensates for loss of the mitochondrial folate pathway. Cell Metab. 23, 1140–1153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koufaris C., Gallage S., Yang T., Lau C. H., Valbuena G. N., Keun H. C., Suppression of MTHFD2 in MCF-7 breast cancer cells increases glycolysis, dependency on exogenous glycine, and sensitivity to folate depletion. J. Proteome Res. 15, 2618–2625 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Fan J., Kamphorst J. J., Mathew R., Chung M. K., White E., Shlomi T., Rabinowitz J. D., Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 9, 712 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tedeschi P. M., Johnson-Farley N., Lin H., Shelton L. M., Ooga T., Mackay G., Van Den Broek N., Bertino J. R., Vazquez A., Quantification of folate metabolism using transient metabolic flux analysis. Cancer Metab. 3, 6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morrow G. P., MacMillan L., Lamarre S. G., Young S. K., MacFarlane A. J., Brosnan M. E., Brosnan J. T., In vivo kinetics of formate metabolism in folate-deficient and folate-replete rats. J. Biol. Chem. 290, 2244–2250 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bridges H. R., Jones A. J. Y., Pollak M. N., Hirst J., Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 462, 475–487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gravel S.-P., Hulea L., Toban N., Birman E., Blouin M.-J., Zakikhani M., Zhao Y., Topisirovic I., St-Pierre J., Pollak M., Serine deprivation enhances antineoplastic activity of biguanides. Cancer Res. 74, 7521–7533 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Warburg O., On the origin of cancer cells. Science 123, 309–314 (1956). [DOI] [PubMed] [Google Scholar]
- 26.Vazquez A., Liu J., Zhou Y., Oltvai Z. N., Catabolic efficiency of aerobic glycolysis: The Warburg effect revisited. BMC Syst. Biol. 4, 58 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patella F., Schug Z. T., Persi E., Neilson L. J., Erami Z., Avanzato D., Maione F., Hernandez-Fernaud J. R., Mackay G., Zheng L., Reid S., Frezza C., Giraudo E., Fiorio Pla A., Anderson K., Ruppin E., Gottlieb E., Zanivan S., Proteomics-based metabolic modeling reveals that fatty acid oxidation (FAO) controls endothelial cell (EC) permeability. Mol. Cell. Proteomics 14, 621–634 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fodde R., Smits R., Clevers H., APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer 1, 55–67 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Dolfi S. C., Chan L. L.-Y., Qiu J., Tedeschi P. M., Bertino J. R., Hirshfield K. M., Oltvai Z. N., Vazquez A., The metabolic demands of cancer cells are coupled to their size and protein synthesis rates. Cancer Metab. 1, 20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan J., Ye J. B., Kamphorst J. J., Shlomi T., Thompson C. B., Rabinowitz J. D., Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510, 298–302 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang J. H., Wada A., Yoshida K., Miyoshi Y., Sayano T., Esaki K., Kinoshita M. O., Tomonaga S., Azuma N., Watanabe M., Hamase K., Zaitsu K., Machida T., Messing A., Itohara S., Hirabayashi Y., Furuya S., Brain-specific Phgdh deletion reveals a pivotal role for l-serine biosynthesis in controlling the level of d-serine, an N-methyl-d-aspartate receptor co-agonist, in adult brain. J. Biol. Chem. 285, 41380–41390 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/10/e1601273/DC1
Supplementary Text
fig. S1 Serine catabolism is induced upon energy stress.
fig. S2. Serine catabolism is linked to mitochondria.
fig. S3. Formate efflux exceeds anabolic one-carbon demands.
fig. S4. Serum formate depends on serine catabolism in vivo.
table. S1. GC temperature program for formate analysis.