

Extracellular ligand-binding inhibitors of receptor tyrosine kinases (eTKIs) in angiogenesis.
Abstract
Receptor tyrosine kinases (RTKs) are key molecules in numerous cellular processes, the inhibitors of which play an important role in the clinic. Among them are the vascular endothelial growth factor (VEGF) family members and their receptors (VEGFR), which are essential in the formation of new blood vessels by angiogenesis. Anti-VEGF therapy has already shown promising results in oncology and ophthalmology, but one of the challenges in the field is the design of specific small-molecule inhibitors for these receptors. We show the identification and characterization of small 6-mer peptides that target the extracellular ligand-binding domain of all three VEGF receptors. These peptides specifically prevent the binding of VEGF family members to all three receptors and downstream signaling but do not affect other angiogenic RTKs and their ligands. One of the selected peptides was also very effective at preventing pathological angiogenesis in a mouse model of retinopathy, normalizing the vasculature to levels similar to those of a normal developing retina. Collectively, our results suggest that these peptides are pan-VEGF inhibitors directed at a common binding pocket shared by all three VEGFRs. These peptides and the druggable binding site they target might be important for the development of novel and selective small-molecule, extracellular ligand-binding inhibitors of RTKs (eTKIs) for angiogenic-dependent diseases.
INTRODUCTION
Angiogenesis is the formation of new blood vessels from preexisting ones, an important process in physiological and pathological conditions (1). For example, tumors cannot grow beyond a few cubic millimeters without proper supply of nutrients and oxygen, and abnormal growth of blood vessels in the retina may result in legal blindness in children and adults (2, 3). Cancer and retinopathy are examples of diseases in which patients already benefit from currently available angiogenesis inhibitors (4, 5), and a growing number of diseases, from tumor formation to other nonneoplastic dysfunctions, share pathological angiogenesis as an underlying cause or as an important component for disease progression (3). Hence, the search for novel therapeutic options, such as antiangiogenic agents, is a valuable approach to improving treatment for these diseases.
Several molecules are important for angiogenesis, but it is well accepted that the vascular endothelial growth factor (VEGF, also known as VEGF-A) and family members [VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PlGF)], along with their corresponding receptors (VEGFR-1, VEGFR-2, and VEGFR-3), are key factors in this process (6–8). Currently available antiangiogenic therapies for cancer and retinopathy are focused on neutralizing VEGF, their receptors, or pathways activated by these growth factors (5, 7, 9).
The three VEGF receptors have been implicated in angiogenesis, and they all belong to the receptor tyrosine kinase (RTK) family. There are more than 50 different RTKs encoded in the human genome, which are organized in 20 different families (10), and because they participate in key cellular processes, RTKs have been of great interest for drug development, including angiogenesis (10–12). Currently, two kinds of RTK inhibitors (TKIs) are in use in the clinic: small molecules directed to the adenosine triphosphatase (ATPase) intracellular kinase domain and antibodies that target the extracellular ligand-binding domain (13, 14). Although small-molecule TKIs offer significant advantages in terms of bioavailability (that is, oral administration), they often lack specificity because of the similarity of the ATPase tyrosine kinase domain shared by all family members (10, 15, 16). Although the broad-spectrum effect of TKI might be desirable in specific settings (for example, oncologic therapy, given the heterogeneous nature of cancer cells), it is also more likely to induce unwanted effects or develop drug resistance due to mutations. Thus, researchers have used different approaches to identifying new druggable sites in the kinase domain to improve selectivity and to overcome drug resistance (16). Although there have been important advances in the field, specificity is still an important challenge for the development of new TKI molecules.
Conversely, the extracellular ligand-binding portion of these receptors is the most diverse region of RTKs in terms of protein structure and, therefore, more attractive for the development of selective drugs (10, 17). In effect, several monoclonal antibodies that target the extracellular domain of an individual RTK have been approved for clinical use, including angiogenesis (5, 7). The trouble with monoclonal antibodies, albeit very specific down to a single-family member, is that they are expensive to produce, lack cell permeability, and have to be administered to patients in hospitals. Orally available molecules would be preferred, so there is a need to find novel alternatives for the development of small selective TKI molecules; targeting the ligand-binding domain of RTKs with small molecules may be an important option (17–19). These extracellular ligand-binding TKIs could offer significant advantages over current drugs by combining the bioavailability and cell permeability of small molecules with the specificity of targeting the ligand-binding domain with monoclonal antibodies (17, 18).
As an example of this strategy, we showed in previous studies that a small tripeptide targeting the extracellular ligand-binding domain of VEGFR-1 and neuropilin-1 (NRP-1) (20, 21) inhibited retinal neovascularization (17). This tripeptide, which can be applied topically to the eye, combines the selectivity of a monoclonal antibody with the advantages of a small-molecule compound, possibly as an unrecognized candidate class of TKI (17). These results have been further validated in a preclinical study that uses a nonhuman primate model of retinal disease, emphasizing the potential for targeting the extracellular domain of transmembrane receptors involved in angiogenesis (19).
Here, we expanded on these previous studies to target another receptor from the VEGF family, the VEGFR-3. We chose this receptor because it is highly expressed by the endothelial tip cells. These are specialized endothelial cells found at the tip of sprouting vessels whose function is believed to be the coordination of vessel formation and migration toward the VEGF gradient (22, 23). Blockage of endothelial tip cells with anti–VEGFR-3 monoclonal antibody prevents retinal angiogenesis (24). Although we were successful in identifying a peptide that targeted the extracellular domain of VEGFR-3 from a phage display library, we noticed that, once the peptides were synthesized and used outside the phage context, they lost their selectivity toward VEGFR-3. The synthetic peptides also interact with VEGFR-1 and VEGFR-2, neutralize binding of VEGF family members to all three receptors, and inhibit neovascularization in vivo in the retina. Hence, it is the first pan-VEGF inhibitor directed at the extracellular domain of an RTK that we have knowledge of. In summary, our study suggests that members of the VEGF receptor family share a common original binding site, which might be important for drug development.
RESULTS
Identification of peptides that target the extracellular domain of VEGFR-3
We used phage display in vitro to isolate peptides that target the extracellular domain of VEGFR-3. Recombinant mouse VEGFR-3 extracellular portion (Tyr25 through Asp770) was immobilized on microtiter plates and incubated with an X6 (X = any amino acids) peptide phage display library. This particular library was chosen because it encodes peptides with relatively small molecular weights (average molecular weight of 6-mer peptides is 660 g/mol) while maintaining diversity greater than 107 possible peptides. The X6 phage display library that we built has an estimated ~109 individual peptides, and it is likely to encode several copies of all conceivable combinations possible for 6-mer peptides (6.4 × 107 unique peptide combinations), excluding peptides toxic to the bacteria or the bacteriophage. After three rounds of selection, we observed an approximately sixfold enrichment in the number of recovered phage compared to the previous round of selection (Fig. 1A), indicating that we had successfully enriched for peptides targeting our ligand. Randomly selected phage clones were analyzed by sequencing to identify their DNA inserts and coding peptides; only two peptide sequences were found encoded in all phage genomes analyzed: PCAIWF (58%) and WVCSGG (42%) (Fig. 1B).
Fig. 1. Isolation of VEGFR-3 binding peptides.
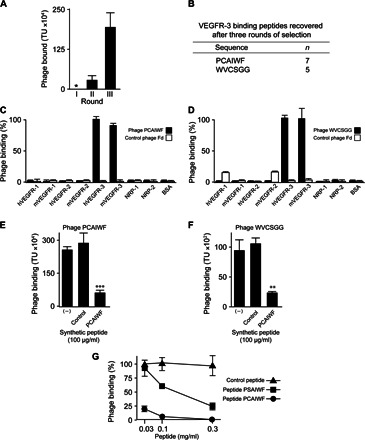
(A) The extracellular domain of mouse VEGFR-3 was immobilized on microtiter wells and incubated with the X6 phage display library. Bar graph shows enrichment in the number of phage recovered [in transducing units (TU)] after consecutive rounds of selection (I, II, and III). (*) Round I was not quantified to prevent the loss of phage displaying unique peptides. (B) Peptide identified by sequencing phage bound to VEGFR-3 (round III) (n, number of phages sequenced). (C and D) Binding of control phage Fd (white bars) and phage PCAIWF (B, black bars) and WVCSGG (C, black bars) to VEGF receptors and co-receptors immobilized on microtiter wells. (E and F) Inhibition of phage PCAIWF (E) or WVCSGG (F) binding to immobilized VEGFR-3 by synthetic peptide PCAIWF or control peptide (CARAC). The minus sign indicates that no synthetic peptide was added to the assay. (G) Dose-response assay. Phage PCAIWF was incubated with immobilized VEGFR-3 in the presence of synthetic peptides PCAIWF, PSAIWF, or CARAC (control). Percentage relative to phage binding in the absence of competing peptide. In all cases, bars represent means ± SEM from triplicate plating. Statistics, Student’s t test (**P ≤ 0.01 and ***P ≤ 0.001).
PCAIWF and WVCSGG peptides are selective and bind to the same site in VEGFR-3
To validate the interaction of phage PCAIWF and WVCSGG and determine its specificity for VEGFR-3, we used a phage binding assay. All three VEGF receptors were individually immobilized on a plate and incubated with phage PCAIWF or WVCSGG or with a control insertless phage (Fd). We observed that phages PCAIWF and WVCSGG bind to VEGFR-3 but not to the other receptors, VEGFR-1 and VEGFR-2 (Fig. 1, C and D), including the two other non-RTKs, NRP-1 and NRP-2, which have been described as co-receptors for VEGF. Moreover, binding was independent of the receptor’s species of origin because both phages bound to mouse and human VEGFR-3. No binding was observed when control phage Fd was used in the assays. Because all three receptors used in our assays are produced as recombinant proteins fused to the Fc domain of human immunoglobulin G1 (IgG1), these results also rule out the possibility that PCAIWF and WVCSGG phages bind to the Fc fusion portion present in these receptors. Finally, to exclude the possibility that random mutations in phage are responsible for receptor interaction, we found that binding of phage PCAIWF to VEGFR-3 is mediated specifically by the peptide because phage binding was inhibited by the cognate synthetic PCAIWF peptide (Fig. 1E). A control peptide had no effect on phage binding to this receptor.
Peptides PCAIWF and WVCSGG have no obvious motif in common or sequence similarity, but they both share two residues, a tryptophan and a cysteine. To assess whether they actually bind to the same site in VEGFR-3, we performed a competition assay. When synthetic peptide PCAIWF in solution was added to our binding assay, it prevented the binding of phage WVCSGG to VEGFR-3 (Fig. 1F). These results indicate that both peptides target the same site in VEGFR-3. Inhibition by peptide PCAIWF was dose-dependent with a median inhibitory concentration (IC50) below 30 μg/ml (Fig. 1G, black circles). Because both peptides have an unpaired cysteine residue with a free sulfhydryl group, we pondered whether a disulfide bridge formed between the peptide and VEGFR-3 was actually responsible for the interaction between these two molecules. To test this, we synthesized a new version of the peptide by replacing serine with cysteine. Although the peptide PSAIWF can no longer form a disulfide bridge, it was also effective at preventing the binding of phage PCAIWF to VEGFR-3, albeit with a lower efficiency (IC50 of ~200 μg/ml) (Fig. 1G, black squares). These results suggest that the cysteine residue is important for binding but does not form a covalent bond with the receptor or the ligand. In summary, we have identified two peptides that target the same binding site within the extracellular portion of VEGFR-3.
PCAIWF interacts with the ligand-binding domain of VEGFR-3
To map the binding site of peptide PCAIWF within VEGFR-3, we performed a competition assay. Phage PCAIWF was incubated with VEGFR-3 in the presence or absence of its natural ligand, VEGF-C. As control, we use VEGF-A, which does not bind to this receptor. Only VEGF-C prevents the binding of phage PCAIWF to the receptor (Fig. 2A) in a dose-dependent manner (Fig. 2B). To corroborate these findings, we observed that binding of phage WVCSGG to VEGFR-3 is also inhibited by VEGF-C but not by VEGF-A. These data suggest that phage PCAIWF binds to the ligand-binding domain of VEGFR-3. To further validate these results, we produced the ligand-binding domain of VEGFR-3 as a recombinant protein to test for PCAIWF binding. The extracellular portion of VEGFR-3 is composed of seven immunoglobulin-like (Ig) domains, and the Ig domain 2 (IgD2) has been identified as the main binding site for VEGF-C and VEGF-D, with IgD3 also contributing to the interaction (Fig. 2C) (25). To confirm that peptide PCAIWF interacts with this domain, we produced IgD2 and IgD2-3 domains from human VEGFR-3 (Fig. 2D) and used the recombinant proteins in our phage assay. Phage PCAIWF binds to VEGFR-3 IgD2 and IgD2-3 domains with similar levels compared to the full-length extracellular VEGFR-3 protein, whereas the control phage Fd did not bind to any of the protein tested. Binding to the recombinant Ig domains could be prevented by the synthetic peptide PCAIWF but not by the control peptide (Fig. 2E). To confirm that our recombinant IgD2 and IgD2-3 domains were functional, we tested for the capacity to bind VEGF-C. VEGF-C binds to both recombinant ligand-binding domains IgD2 (Fig. 2F) and IgD2-3 (Fig. 2G). We observed that, when synthetic peptides PCAIWF and WVCSGG were added to the assay, both prevented binding of VEGF-C to IgD2 and IgD2-3 (Fig. 2, F and G). Together, these results indicate that peptide PCAIWF interacts with the ligand-binding domain of VEGFR-3 and prevents VEGF-C interaction with the receptor.
Fig. 2. Peptides PCAIWF and WVCSGG interact with the ligand-binding domain of VEGFR-3.
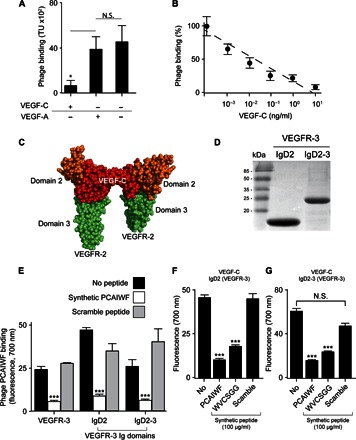
(A) Binding of phage PCAIWF to immobilized VEGFR-3 in the presence or absence of VEGF-A or VEGF-C (10 ng/ml). (B) Binding of phage PCAIWF to immobilized VEGFR-3 in the presence of increasing concentrations of VEGF-C. Percentage relative to phage binding in the absence of VEGF-C. (C) Cartoon showing the three-dimensional structure of the complex VEGF-C (red) bound to VEGFR-2 IgD2-3 (shown in orange and green, respectively) (Protein Data Bank #2X1W). (D) Analysis by SDS–polyacrylamide gel electrophoresis of purified recombinant IgD2 and IgD2-3 proteins containing the ligand-binding domain of VEGFR-3. (E) Binding of phage PCAIWF to VEGFR-3 and its recombinant Ig domains immobilized on microtiter wells in the presence or absence of the synthetic peptide PCAIWF or its scramble version, IFCAPW (100 μg/ml). Phage binding was quantified by FLISA using an anti-bacteriophage sera. (F) Binding of VEGF-C to microtiter wells coated with immobilized recombinant ligand binding domains IgD2 and IgD2-3 of VEGFR-3 in the presence or absence of synthetic peptides PCAIWF and WVCSGG or the scramble control peptide (IFCAPW). For phage experiments (A and B), bars represent mean ± SEM from triplicate plating; for FLISA assays (E to G), bars represent means ± SEM from duplicate wells. Statistics, Student’s t test [not significant (N.S.), P > 0.05; *P ≤ 0.05 and ***P ≤ 0.001].
Synthetic PCAIWF is a pan-VEGF inhibitor
Having shown that phage PCAIWF binds to the ligand-binding domain of VEGFR-3 but not to other VEGF receptors and modulates the binding of VEGF-C to this receptor, we asked whether peptides PCAIWF and WVCSGG were selective for the binding of VEGF-C to VEGFR-3. To answer this question, we set up a fluorophore-linked immunosorbent assay (FLISA) to evaluate the effect of PCAIWF on the binding of the three main angiogenic ligands (VEGF-A, PlGF, and VEGF-C) to all three RTK VEGF receptors (Fig. 3A). Because peptides PCAIWF and WVCSGG target the same site in the receptor, we decide to concentrate our studies on the PCAIWF peptide. The VEGF receptors were individually immobilized on plates and incubated with their respective ligands in the presence or absence of the synthetic peptide PCAIWF or PSAIWF. As control, we used a synthetic peptide containing the same amino acid residues of PCAIWF but in a distinct sequence (scramble). To our surprise, we observed that the synthetic peptide PCAIWF inhibits the binding of all VEGF members that we tested to their respective receptors (Fig. 3, B to F). Briefly, peptide PCAIWF inhibited the binding of VEGF-C to VEGFR-3 (Fig. 3B) and VEGFR-2 (Fig. 3C), prevented the binding of PlGF to VEGFR-1 (Fig. 3D), and also blocked the binding of VEGF-A to VEGFR-2 (Fig. 3E) and VEGFR-1 (Fig. 3F). These inhibitions were specific and were not affected by the control scramble peptide. Moreover, the synthetic peptide PSAIWF containing a serine residue instead of cysteine also inhibited the binding to all VEGF factors tested (albeit with lower affinity), again indicating that the sulfhydryl residue is important, but not essential, for binding (Fig. 3, B to F). Notably, we could block 80 to 90% binding of all three VEGF family members using peptide PCAIWF (100 μg/ml) and 30 to 50% using peptide PSAIWF (100 μg/ml), except for binding of VEGF-A to VEGFR-1, which required higher concentrations of both peptides (400 μg/ml) for similar levels of inhibition. This agrees with the fact that VEGFR-1 is the high-affinity receptor for VEGF-A (26).
Fig. 3. PCAIWF is a pan-VEGF inhibitor.
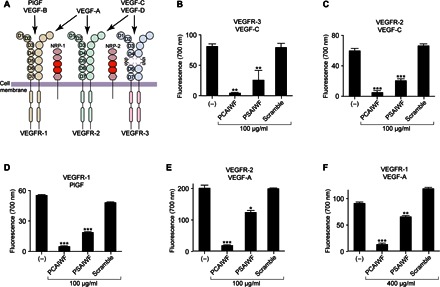
(A) Representation of the VEGF family, their receptors, and pattern of interaction. (B to F) Recombinant proteins for the human VEGFR-3 (B), VEGFR-2 (C and E), and VEGFR-1 (D and F) extracellular domains were immobilized on microtiter wells and incubated with the human ligands VEGF-C (B and C), PlGF (D), and VEGF-A (E and F) in the presence or absence of synthetic peptides PCAIWF and PSAIWF or the scramble control peptide (IFCAPW). Growth factors bound to the wells were quantified by FLISA using immunospecific antibodies and fluorescent detection. Bars represent means ± SEM from duplicate wells. Statistics, analysis of variance (ANOVA) (Tukey’s multiple comparison test) (*P ≤ 0.05; **P ≤ 0.01 and ***P ≤ 0.001).
To assess whether the inhibitory effect of peptides PCAIWF and WVCSGG was specific for receptors of the VEGF family, we performed a similar FLISA-based binding assay using other angiogenic RTK receptors with their cognate ligands. We observed no effect of peptide PCAIWF or WVCSGG on the binding of basic human fibroblast growth factor (FGF) to its receptor (FGFR-1α) (Fig. 4A) or on the binding of platelet-derived growth factor B (PDGF-BB) to PDGFR-β (Fig. 4B). Like the VEGFR, these receptors are also important in angiogenesis and have similar structural architecture compared to the VEGFR: ligand-binding moieties constituted of immunoglobulin folds. Neuropilins have also been described as ligands for the VEGF receptors (27, 28). It has been described that NRP-1 binds with high affinity to VEGFR-1 (Kd ~ 1.8 nM) (27). In agreement with these studies, we observed a strong interaction between NRP-1 and NRP-2 with VEGFR-1 using the FLISA binding assay. These interactions were not affected by peptide PCAIWF or WVCSGG (Fig. 4, C and D).
Fig. 4. PCAIWF does not affect other angiogenic RTK or neuropilin binding.
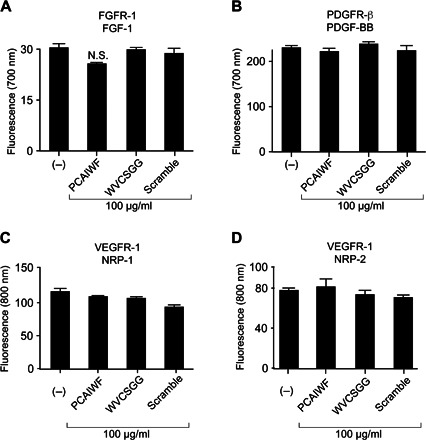
(A to D) Recombinant proteins for the human FGFR-1 (A), PDGFR-β (B), and VEGFR-1 (C and D) extracellular domains were immobilized on microtiter wells and incubated with the human ligands FGF-1 (A), PDGF-BB (B), human NRP-1 (C), and rat NRP-2 (D) in the presence or absence of synthetic peptides PCAIWF and WVCSGG or the scramble control peptide (IFCAPW). Growth factor and neuropilin bound to the wells were quantified by FLISA using immunospecific antibodies and fluorescent detection. Bars represent means ± SEM from duplicate wells. Statistics, ANOVA (Tukey’s multiple comparison test) (N.S., P > 0.05).
Binding of growth factors to RTKs triggers the activation of signaling events that are important for cell growth and survival. To assess whether peptide PCAIWF could prevent VEGFR-mediated activation of downstream signaling, lymphatic endothelial cells (LECs) were stimulated with VEGF-A, VEGF-C, or FGF-1. LECs were selected because they express high levels of VEGFR-3, VEGFR-2, and FGFR-1 and respond to these three growth factors by activating the extracellular signal–regulated protein kinases 1 and 2 (ERK1/2) pathway (29, 30). We observed that phosphorylation of ERK1/2 increases upon stimulation of LECs with VEGF-A, VEGF-C, or FGF-1; as expected, peptide PCAIWF inhibited the phosphorylation of ERK1/2 induced by VEGF-A and VEGF-C but had no effect on the phosphorylation of ERK1/2 induced by FGF-1 (Fig. 5, A and B). In all cases, the scramble synthetic control peptides had no effect on this downstream signaling event. Together, these results indicate that the effect of peptide PCAIWF is specific and selective for RTKs of the VEGF family.
Fig. 5. Effect of PCAIWF on VEGF induced ERK1/2 pathway activation.
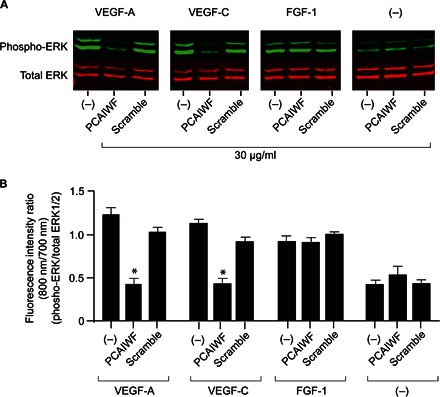
(A) Immunoblot analysis of phosphorylated and total forms of ERK1/2 in LECs incubated with VEGF-A, VEGF-C, or FGF (100 ng/ml) in the presence or absence of peptide PCAIWF or scramble (IFCAPW) (30 μg/ml). (B) Ratio of fluorescent intensity for phosphorylated and total ERK1/2. Bars represent means ± SEM from three independent measurements of the immunoblot membrane. Two independent experiments were performed with similar results. Bars represent means ± SEM from triplicate readings. Statistics, ANOVA (Tukey’s multiple comparison test) (*P ≤ 0.05).
Pan-VEGF inhibitor PCAIWF prevents retinal neovascularization in a mouse model
VEGF inhibitors have shown promising results for the treatment of ocular diseases with an angiogenic component. To assess whether synthetic peptide PCAIWF could inhibit neovascularization, we performed two angiogenesis assays: endothelial tube formation in Matrigel and the oxygen-induced retinopathy (OIR) model (in vivo). For the endothelial cell tubulogenesis assay, peptides PCAIWF and scramble were embedded in the Matrigel layer, and human umbilical vein endothelial cells (HUVECs) were stimulated with VEGF-A or VEGF-C (31). In both cases, we observed inhibition of tube formation by peptide PCAIWF but not by the control scramble peptide or vehicle alone (Fig. 6A). Tube formation was reduced by ~50% in both cases (VEGF-A and VEGF-C), suggesting that other growth factors present in the Matrigel, which are not affected by peptide PCAIWF, might be stimulating the endothelial cells (Fig. 6B) and preventing the full inhibition of tubulogenesis. Next, we assessed the effect of PCAIWF in an in vivo mouse model.
Fig. 6. Effect of PCAIWF on endothelial tube formation.
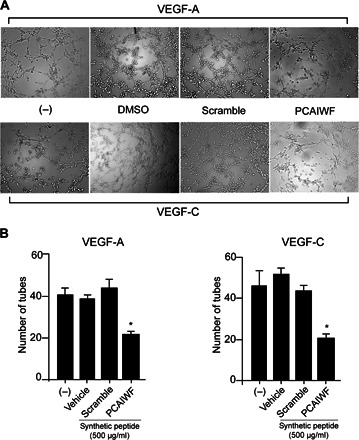
(A) Tube formation by HUVECs in Matrigel induced by VEGF or VEGF-C in the presence or absence of peptide PCAIWF or scramble (500 μg/ml, embedded in the Matrigel layer). (B) Number of tubes formed between endothelial cells. Bars represent means ± SEM from triplicate wells. Statistics, Student’s t test (*P ≤ 0.05). Two independent experiments were performed with similar results.
The OIR model is a well-accepted animal model for the study of human diseases, such as retinopathy of prematurity and, to a certain extent, diabetic retinopathy (32, 33). The retinal vasculature in mice develops and matures after birth, a process that is controlled by oxygen tension and, therefore, VEGF levels (34). By exposing mice at postnatal day 7 (P7) to 75% oxygen, VEGF expression is down-regulated, and its concentration in the eye is substantially reduced. This inhibits neovascularization that would otherwise be active in physiological retinas, with the formation of a central zone of vaso-obliteration. Once the mice at P12 are returned to room air (normal oxygen levels, 21%), vascularization has stopped and the retina experiences severe hypoxia; VEGF expression rises above normal, resuming vascularization, but it is now in a pathological state that exacerbates the angiogenic process, resulting in a retinopathic condition, which peaks at P17 (32–34). In mice, it eventually resolves within a few days, but in premature babies exposed to high oxygen concentrations in neonatal care units, if severe enough, the ongoing retinopathy may result in retinal detachment and blindness (retinopathy of prematurity) (35).
To assess whether our pan-VEGF peptide inhibitor PCAIWF could prevent pathological angiogenesis in the OIR model, mice at P15 (3 days after exposure to 75% oxygen) were treated with a single intravitreal injection of peptide PCAIWF. Peptide treatment could not be performed as early as P12 because young mice have their eyelids shut and they are not fully open until P14 to P15 (36). Two days after treatment, the retinas were analyzed, and neovascularization was quantified (37). We observed that the retinas of animals treated with peptide PCAIWF showed a significant reduction in the amount of blood vessels (total vascular area) (Fig. 7, A and B) and vascular sprouting and ramifications (Fig. 7, C and D). No alteration in the number of vessels and their ramifications was observed when the animals were treated with the scramble control peptide or vehicle only.
Fig. 7. PCAIWF inhibits neovascularization in vivo.
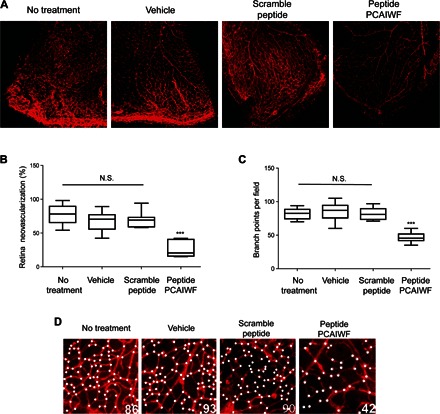
Neonatal C57BL/6 mice with OIR at P15 were treated or not treated (n = 9 retinas) with a single intravitreal injection (1 μl) of vehicle only [dimethyl sulfoxide (DMSO)] (n = 11 retinas), peptide PCAIWF (30 μg) (n = 6 retinas), or scramble peptide IFCAPW (30 μg) (n = 7 retinas). Whole-mount retinas were stained with isolectin-B4 conjugated to Alexa Fluor 594 red-fluorescent dye. (A) Representative confocal microscopy images of retinas of OIR neonatal C57Bl/6 mice at P17. (B) Quantification of neovascularization in the retinas of OIR neonatal C57BL/6 mice treated or not treated with peptides. (C) High-magnification images (×200) of the retinas at P17 of OIR neonatal C57BL/6 mice treated or not treated with peptides. White dots indicate vessel sprouts or bifurcations. The numbers of sprouts/bifurcations determined for each image are indicated at the bottom right corner. (D) Quantification of vessel sprout or bifurcations. Statistics, ANOVA (Tukey’s multiple comparison test) (N.S., P > 0.05; ***P < 0.005). Box plots in which the boxes define the 25th and 75th percentiles, with a line at a median and error bars defining the 10th and 90th percentiles.
One of the hallmarks of the OIR model is the formation of tufts due to the outgrowth of blood vessels protuberating into the vitreous cavity. We noticed that treatment with peptide PCAIWF significantly reduced tuft formation in number and size (Fig. 8A). To further evaluate the effect of the treatment with the peptide on tuft formation, we performed confocal laser scanning microscopy to gain information on the vascular layer deep in the retina (Fig. 8B). Confocal images were obtained at 2.4-μm intervals and assembled with the aid of computer software (Fig. 8C), which allowed us to determine the thickness of the vascular layer and to visualize blood vessels migrating toward the vitreous chamber (tufts). As expected, normal C57BL/6 mice that were not subjected to the OIR model have a homogeneous vascular layer of approximately 39 ± 1.3 μm (Fig. 8D) that is positioned between the ganglion and the outer plexiform layers of the normal retina (19). When mice are treated by OIR, the vascular layer changes shape and becomes thicker (75 ± 2.7 μm) because of a series of tufts of vessels extending toward the vitreous humor (Fig. 8, D and E). These projections are almost absent in OIR mice treated with peptide PCAIWF in which the thickness of the vascular layer reduces to 44 ± 3.9 μm, similar to normally developing retinas in C57BL/6 mice (Fig. 8, D and E). Animals treated with vehicle only showed no significant reduction (67 ± 3.7 μm) in vascular layer thickness (Fig. 8D). In summary, the small-molecule pan-VEGF inhibitory peptide PCAIWF prevents pathological angiogenesis in vivo in one of the most widely used animal models for retinopathy of prematurity, suggesting that this peptide might have important uses in the development of novel antiangiogenic inhibitors for retinopathy and other human diseases.
Fig. 8. PCAIWF prevents neovascularization in the vitreal cavity.
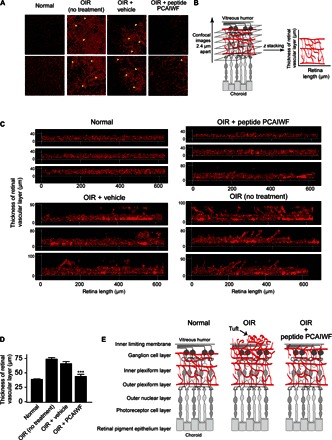
(A) Assembly of confocal microscopy images of P17 retinas of normal neonatal C57BL/6 mice or with OIR treated with PCAIWF (n = 4 retinas) or not (n = 10 retinas) by a single intravitreal injection (1 μl) of vehicle only (DMSO) or peptide PCAIWF (30 μg). Yellow arrowheads indicate vascular complexity (tufts). (B) Schematic representation of a mouse retina showing the location of the retinal vascular layer and the images obtained by confocal laser scanning microscopy to gain information on vascular depth. (C) Computer-assisted assembly of the laser scanning confocal images of P17 retinas (obtained at 2.4-μm intervals) of neonatal C57BL/6 mice with OIR treated or not treated with vehicle or peptide PCAIWF. (D) Quantification of the retinal vascular thickness. (E) Drawing outlining the retinal, its layers, and blood vessels protuberating or not into the humor vitreous. Statistics, ANOVA (Tukey’s multiple comparison test) (***P < 0.005). Error bars represent means ± SEM.
DISCUSSION
Antiangiogenic therapy has been firmly established by numerous clinical trials using drugs aimed at the central factors involved in this process: VEGF and its receptors. Unfortunately, despite the significant benefits of these drugs, there are important questions in antiangiogenic therapy that have not been solved. For example, not all patients respond to anti-VEGF therapy, and others eventually develop resistance (38). Why do some tumors respond to antiangiogenic therapy (colorectal) and others do not (pancreatic)? The ingenious hypothesis [proposed by Folkman (39)] that tumors would starve to death if not properly nourished by blood vessels is an impeccable and compelling concept, but researchers are still puzzled as to why antiangiogenic compounds have not achieved the expected efficacy (5, 7). Although this conundrum is likely to be multifaceted, targeting molecules other than VEGF-A and VEGFR-2 might help improve efficacy. In effect, in ophthalmology, clinical studies with aflibercept have shown better efficacy compared to bevacizumab and ranibizumab (40). The latter two drugs exclusively target VEGF-A, whereas aflibercept is the only one that, in addition to VEGF-A, neutralizes two other members of the family, VEGF-B and PlGF. Drugs with a broader effect may do better in angiogenesis therapy.
All VEGF family members and the three receptors are expressed in the retina and play distinct and important roles in angiogenesis. For example, studies with animal models indicate that VEGF-B is dispensable for neovascularization but is essential for retinal endothelial cell survival, and antibodies targeting VEGF-B inhibit choroid and retina neovascularization (41). PlGF has also been implicated in pathological angiogenesis (42, 43) and is said to have contributed to the development of diabetic retinopathy (44). VEGF-C and VEGF-D are expressed in the ischemic retina (45) and the subretinal vascular membrane of an age-related macular degeneration patient (46), and blockage of VEGFR-3 with monoclonal antibodies prevents neovascularization in the retina (24), suggesting that they also contribute to neovascularization in the retina. Furthermore, these growth factors have different effects in the type of newly formed blood vessels and in their stability and permeability (47).
However, targeting all members of the VEGF family is challenging. Monoclonal antibodies are selective to a single receptor. Similarly, molecules like VEGF-trap (aflibercept), which is composed of the ligand-binding domain of VEGFR-1 fused to an Ig constant region, will target multiple members of the VEGF family (VEGF-A, VEGF-B, and PlGF) but will miss others (VEGF-C and VEGF-D) (48). An alternative is to use small TKIs, such as sunitinib, which is a pan-VEGF inhibitor. Unfortunately, these molecules also affect other angiogenic RTKs, resulting in important side effects, such as cardiotoxicity, such as in the case of sunitinib and PDGFR-β (49). Therefore, drugs with similar activity to peptides PCAIWF and WVCSGG (neutralization of all VEGFRs through the extracellular domain) could have important applications in the clinic for the development of true pan-VEGF inhibitors.
In a previous study (19), we have successfully developed a small tripeptide (now denominated Vasotide) into a prodrug candidate. Nevertheless, it is not very often that peptides are suitable for drug development. They are prone to proteolysis and suffer the same limitations in terms of biodistribution as monoclonal antibodies. For instance, peptides PCAIWF and WVCSGG are not found in the vitreous humor if applied topically to the retina, which seems to preclude their use in eye drop formulations. Because they are small molecules, they probably will have short half-lives in the eye and will not be good drug candidates for repeated intravitreal injections (granting that we have not performed pharmacokinetic studies yet). The hydrophobicity and relative small molecular weight of peptides PCAIWF and WVCSGG (736 and 608 Da, respectively) are not far from the rule of five (Lipinski’s rule), which suggests that their binding site is likely a druggable region in these receptors. With better knowledge of the structural requirements for VEGFR binding, these peptides might be important leads for drug development. Small molecules, such as peptides PCAIWF and WVCSGG, might comprise a new class of drugs, the extracellular ligand-binding inhibitors of receptor tyrosine kinases (eTKIs). Despite all the difficulties associated with peptide drug development, given the strong antiangiogenic effect of PCAIWF in vivo following a single dose and without any optimization, it is possible that, if properly formulated (that is, slow-release formulations), peptide PCAIWF might be a drug candidate for the treatment of human retinopathy. Notably, peptides PCAIWF and WVCSGG are fairly soluble in Matrigel compared to aqueous media. Matrigel has a composition similar to that of the vitreous humor, rich in collagen, collagen-like proteins (vitrosin), and glycosaminoglycans, which may help solubilize the peptides in the eye. Collectively, drugs based on PCAIWF and WVCSGG might be interesting alternatives for antiangiogenic therapy because the neutralization of all VEGFRs could be achieved with a single compound.
The change of specificity of peptides PCAIWF and WVCSGG depending on the context in which they are presented to the receptors is noteworthy. These peptides are fairly specific for VEGFR-3 if fused to the bacteriophage coat protein but become more permissive in solution as synthetic-free peptide molecules. These data suggest that peptides that are identified by combinatorial approaches (such as phage display) should be carefully validated, and one should not assume the peptide’s specificity without considering the contexts in which the peptide is presented (bacteriophage or in solution). It also highlights another important aspect: Distinct families of RTK might retain an original binding site shared by family members. Peptides PCAIWF and WVCSGG prevent ligand binding and inhibit angiogenesis in vivo (PCAIWF). These results suggest that, if these binding sites are present in other RTKs, original sites might be important druggable domains. Studies using the FGF receptor family corroborate this idea. A small experimental TKI molecule (SSR128129E) binds to and prevents the activation of all four FGF receptors (17). Different from peptides PCAIWF and WVCSGG, this FGF receptor inhibitor does not interact with the ligand-binding domain of the receptors and is likely to exert its effect through a putative allosteric domain. The exact binding site and mechanism by which PCAIWF and WVCSGG exert their effect and prevent ligand interaction are still unknown; whether they bind directly to the ligand domains of VEGFRs or to an allosteric adjacent binding site remains to be determined. An important difference is that peptides identified by phage display often mimic natural ligands for their target (20, 50–53). Peptide PCAIWF does not share significant primary sequence similarity to any of the VEGF family members. Thus, if peptide PCAIWF were to bind to an allosteric binding site in the VEGF receptor family, it would be interesting to identify the natural ligand for this putative domain. Further studies will be necessary to answer these questions.
In summary, and in a larger context, our work suggests that RTKs might have family-specific common binding sites that modulate ligand interaction with the receptors. Notwithstanding the great advances in drug design for better RTK inhibitors, selectivity is still an important challenge to the field (16). Identification of these original binding sites shared by members of RTK-distinct families may contribute well to this milestone and to the design of novel and better molecules with the exquisite selectivity necessary to target this important superfamily of receptors that are highly relevant in many human diseases.
MATERIALS AND METHODS
Materials
All recombinant growth factors and their respective receptors were obtained commercially from R&D Systems. Antibodies and other reagents were obtained commercially: anti-human VEGF (AF-293-NA), anti-human VEGF-C (AF752), anti-human PlGF (AF-264-PB), anti-human PDGF-BB (AF-220-NA), anti-human FGF-basic (AF-233-NA), anti-mouse/rat NRP-1 (AF566), and anti-mouse/rat NRP-2 (AF567) were from R&D Systems; anti-fd Bacteriophage-Biotin Conjugate (B2661) was from Sigma-Aldrich; secondary antibodies IRDye 680LT Donkey anti-goat IgG and IRDye 680LT Streptavidin were from LI-COR. The gas admixture (75% oxygen and 25% nitrogen) used for the OIR mouse model was obtained from Air Products.
Phage library construction
The linear hexapeptide phage display (X6) library was built as previously described (54) but with modifications. Briefly, the fUSE55 vector (provided by G. Smith, University of Missouri, Columbia, MO) was prepared in large scale using the Maxiprep kit (Qiagen) followed by two consecutive CsCl equilibrium gradient purifications. Equimolar amounts of oligonucleotides 5′-CACTCGGCCGACGGGGCTNNKNNKNNKNNKNNKNNKGGGGCCGCTGGGGCCGAA-3′ and 5′-TTCGGCCCCAGCGGC-3′ (where N = any nucleotide and K = T or G) were converted to double-stranded DNA with Klenow enzyme (as recommended by the manufacturer) (New England Biolabs) and purified using a P500 Maxiprep column (Qiagen). The vector (500 μg) and oligonucleotide insert (20 μg) digested with the restriction enzyme Bg lI were ligated using T4 DNA ligase (New England Biolabs). The ligation product was purified using a P500 Maxiprep column and transformed into electrocompetent Escherichia coli MC1061 cells, resulting in 1.4 × 109 transformants, of which 1.2 × 109 contained inserts coding for peptides. Bacteria were cultured for ~20 hours, and phages were purified from culture supernatants by the polyethylene glycol/NaCl method (20).
Synthetic peptides
Peptides were synthesized and purified by high-performance liquid chromatography to a purity greater than 95% by Bachem or Chinese Peptide Company. Two control peptides were used in this study: peptide CARAC (referred to as control) (20) and the scramble version of the peptide PCAIWF, sequence IFCAPW (referred to as scramble).
Biopanning on VEGFR-3 and phage binding assays
Phage assays were performed as previously described (20, 50). To isolate peptides targeting VEGFR-3, microtiter wells were coated with recombinant mouse VEGFR-3/Fc [1 μg in 50 μl of phosphate-buffered saline (PBS)] [10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, and 2.7 mM KCl (pH 7.4)], blocked with PBS supplemented with 3% bovine serum albumin (BSA), and incubated with the X6 phage peptide library (109 TU) in 50 μl of PBS supplemented with 1% BSA (PBS/BSA) for 2 hours at room temperature. The wells were then washed, and bound phages were recovered by bacterial infection and amplified for the next cycle of selection. After the third round of selection, random bacterial colonies were selected for DNA sequencing to identify the phage coding peptides. Briefly, each bacterial colony was dispersed in 50 μl of PBS, and 2 μl was used to subject the pII gene encoding the random DNA insert [5′-GCAAGCTGATAAACCGATACAATT-3′ (forward) and 5′-CCCTCATAGTTAGCGTAACGATCT-3′ (reverse)] to polymerase chain reaction (PCR) using Taq DNA polymerase (Thermo Fisher Scientific) in a thermal cycler (Bio-Rad) (94°C for 2 min, followed by 35 cycles at 94°C for 15 s, 60°C for 20 s, and 72°C for 45 s). DNA sequencing was performed by the Sanger method in the DNA sequencing facility at the Chemistry Institute, University of São Paulo. For the phage binding assays, the procedure was similar. Wells of microtiter plate were coated with the specified ligand (overnight at 4°C), incubated with the specified phage (108 TU in 50 μl of PBS) for 2 hours at room temperature, and washed, and bound phage was quantified by either colony count or phage-FLISA. The latter was performed using an anti-Fd bacteriophage conjugated to biotin, followed by IRDye 680LT Streptavidin, and plates were scanned in the Odyssey Infrared Imaging Scanner. For the competition assays, phage incubation was performed in the presence or absence of different concentrations of the specified synthetic peptide or growth factor. In all experiments, the insertless phage Fd was used as control.
Recombinant VEGFR-3 ligand-binding domain
The complementary DNA encoding the full-length human VEGFR-3 (pDON233-FLT4) was obtained from Addgene and used to produce by PCR amplicons encoding the ligand-binding domains of human VEGFR-3 [IgD2: 5′-GTGAGACATATGGAGCAGCCATTCATC-3′ (forward) and 5′-GATCTCGAGGAGCTCGTTGCCTGTGAT-3′ (reverse); IgD2-3: same forward primer used for IgD2 and 5′-GACCTCGAGGAAGGGATTTTCATGCAC-3′ (reverse)]. The amplicons were cloned into pET21a and used to produce the corresponding recombinant proteins (IgD2, residues 136 to 229; IgD2-3, residues 136 to 332) using Rosetta(DE3)pLysS cells (EMD Millipore). The recombinant proteins were purified from inclusion bodies. Briefly, cells were lysed by sonication and freeze/thaw and were centrifuged (10,000g for 10 min at 4°C), and the insoluble pellets were washed twice with wash buffer [50 mM tris, 0.5% (v/v) Triton X-100, 100 mM NaCl, and 1 mM EDTA (pH 8.0)]. They were resuspended in denaturing buffer [20 mM NaH2PO4, 8 M urea, 100 mM NaCl, and 5 mM β-mercaptoethanol (pH 8.0)], and the IgD2 and IgD2-3 proteins were purified using a Ni-NTA agarose column (Qiagen). To refold, the recombinant proteins were slowly diluted to a final concentration of 50 μg/ml in refolding buffer [20 mM NaH2PO4 and 50 mM NaCl (pH 7.3)] (with agitation), followed by purification in a Ni-NTA agarose column under native conditions. The recombinant proteins were finally dialyzed against refolding buffer.
Growth factor binding assay
Binding of select growth factors to their corresponding receptors was performed using a FLISA-based assay. Human VEGFR-1, VEGFR-2, VEGFR-3, FGFR-1, or PDGFR-β was individually immobilized on microtiter wells (200 ng in 50 μl of PBS, overnight at 4°C), blocked with Odyssey Blocking Buffer (LI-COR), and incubated with the cognate human ligand [VEGF-A (60 ng/ml), PlGF (20 ng/ml), VEGF-C (20 ng/ml), FGF-1 (20 ng/ml), PDGF-BB (20 ng/ml), and NRP-1 or NRP-2, all in PBS supplemented with 10% (v/v) DMSO and 10% (v/v) Odyssey Blocking Buffer] in the presence or absence of the synthetic peptide PCAIWF, PSAIWF, or IFCAPW (100 μg/ml) (and WVCSGG when indicated). Ligand binding to each receptor was assessed using specific anti-sera against the ligand [goat anti–hVEGF-A, anti–hVEGF-C, anti-PlGF, anti–NRP-1, and anti–NRP-2 (1:100 dilution); anti–PDGF-BB and anti–FGF-basic (1:200 dilution); all from R&D Systems] followed by incubation with donkey anti-goat Ig conjugated to IRDye 680LT (LI-COR). Plates were quantified using the Odyssey Infrared Imaging system (LI-COR). DMSO had no effect on the binding of any of the ligands analyzed in this study.
VEGF signaling
Human lymphatic microvascular dermal endothelial cells (Lonza, CC-2810) were seeded into six-well plates (106 cells per well), cultured overnight in endothelial basal medium-2 (EBM2) complete medium supplemented with 2% fetal bovine serum plus bullet kits (Lonza, CC-3156), and then cultured for 18 hours in EBM2 medium supplemented only with 0.2% BSA (Sigma Merck). Cells were then treated with VEGF-A, VEGF-C, or FGF-1 (100 ng/ml) in EBM2 medium containing 1% DMSO and heparin (10 U/ml; Sigma Merck, H3393) with or without peptide PCAIWF or scramble (30 μg/ml) for 10 min at 37°C. The cells were washed with ice-cold PBS and lysed with 50 mM tris-HCl at pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate, 1 mM EGTA, 1 mM EDTA, 2.5 mM sodium pyrophosphate, 1 mM NaF, 1 mM glycerophosphate, 1 mM Na3VO4, and the protease and phosphatase inhibitor cocktail (Sigma). The detection of phosphorylated and total forms of ERK1/2 was performed by Western blotting using specific antibodies (mouse IgG anti-p44/42 MAPK ERK1/2, 3A7, #9107; rabbit IgG anti–phospho-p44/42 MAPK ERK1/2 Thr202/Tyr204, 197G2, #4377; Cell Signaling) according to the manufacturer’s recommendations. Total ERK was detected using goat anti-mouse Ig conjugated to IRDye 680LT (LI-COR), and phospho-ERK1/2 was detected using donkey anti-rabbit Ig conjugated to IRDye 800CW (LI-COR). The immunoblots were quantified using the Odyssey Infrared Imaging system (LI-COR).
Endothelial cell tube formation assay
HUVECs (HUVEC-C, American Type Culture Collection CRL-1730) were seeded onto 96-well plates coated with 80 μl of Matrigel (BD Biosciences, 354234) containing 1% DMSO and supplemented or not supplemented with peptide PCAIWF or scramble (500 μg/ml). Cells (1.5 × 104 cells per well) were incubated in RPMI 1640 (Thermo Fisher Scientific) containing VEGF-A (30 ng/ml) or VEGF-C (100 ng/ml). Quantification of endothelial network formation was performed by counting the number of tubes formed per field (objective, 4×) using an inverted (bright-field) microscope (Nikon) (31).
Animals
The Institutional Animal Care and Use Committees at the Chemistry Institute of the University of São Paulo approved all animal experimentation (protocol number 10/2010). This study followed the Association for Research in Vision and Ophthalmology statement for the use of animals in ophthalmic and vision research. C57BL/6 mice (Taconic) maintained at the animal facility of the Chemistry Institute and Pharmacy School of the University of São Paulo were used for all experiments.
OIR neovascularization and peptide treatment
The OIR mouse model was performed as described previously (17, 32, 37). Briefly, the animals were exposed to 75% oxygen from P7 to P12, along with their nursing mothers. Four experiments were performed with similar results. The initial three experiments were performed with a homemade oxygen chamber, and the last experiment was performed on a Biospherix Hyperoxia Chamber equipped with a ProOx 110 oxygen controller (Biospherix). At P12, the animals were returned to room air (20.8% oxygen) and organized in groups, and 3 days later (P15), they were treated with vehicle (DMSO) only or with 30 μg of each individual peptide by intravitreal injection (peptides were solubilized in DMSO solution at 30 mg/ml; injection of 1 μl per eyeball). Animals weighing less than 6 g were not used for the study.
Whole-mount retina preparation and neovascularization quantification
At P17 (2 days after treatment), the animals were enucleated, their eyeballs were fixed in PBS containing 4% paraformaldehyde, and the retina was dissected under a stereomicroscope. The blood vessels in the retinas were then stained with isolectin B4 conjugated with Alexa Fluor 594 (Life Technologies), mounted with VECTASHIELD (Vector Laboratories), and examined in an epifluorescent microscope (Nikon). The neovascularization was quantified as previously described (32). Briefly, images from whole-mount retinas were obtained at ×100 magnification using the same exposition time for all samples. Images for the same retina were merged into a single picture using Adobe Photoshop CS3. Quantification of total neovascularization (NV) was performed by selecting the fluorescent areas containing the neovessels and counting the total number of pixels. A similar procedure was used to determine the total number of pixels in the whole retina (RA). The percentage of retinal vascularization was then calculated with the formula (NV/RA) × 100. Representative confocal images of retinas were performed on a Zeiss LSM510 META microscope and used to determine vessel sprouts and bifurcations by counting the number of branch points per identical 167-μm × 167-μm fields (two fields per quadrant and a total of eight fields per retina) for each animal retina. For the laser scanning microscopy, confocal images spanning the whole retina were obtained at 2.4-μm intervals using the 10× objective (final magnification, ×70). The three-dimensional retina was reconstructed with the Zeiss Zen software.
Statistical analysis
Statistics were performed using GraphPad Prism software. Error bars are presented as means ± SD. Statistical significance was determined by Student’s t test or the two-way ANOVA test set at P < 0.05.
Acknowledgments
We thank M.L. Baldini for assistance with the experiments and phage library construction and C.A.L. Braga for technical assistance. Funding: This work was supported by research grants from São Paulo Research Foundation (FAPESP) (grant 2009/54.806-8 to R.J.G. and grant 2009/54844-0 to J.S.M.) and the National Council for Scientific and Technological Development (CNPq) (to R.J.G.). Author contributions: J.S.M. and R.J.G. designed the research. J.S.M. performed most of the experiments, with the assistance of A.R.R., L.S.M., and C.C.C. J.S.M. and R.J.G. analyzed the data and wrote the manuscript. All authors approved the final manuscript. Competing interests: The authors declare that they have no competing interests. University of São Paulo has filed a patent application in Brazil. R.J.G., J.S.M., and C.C.C. are entitled to standard royalties if licensing or commercialization occurs. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from R.J.G. (giordano@iq.usp.br).
REFERENCES AND NOTES
- 1.Chung A. S., Ferrara N., Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 27, 563–584 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D., Folkman J., Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86, 353–364 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Folkman J., Angiogenesis: An organizing principle for drug discovery?. Nat. Rev. Drug Discov. 6, 273–286 (2007). [DOI] [PubMed] [Google Scholar]
- 4.Miller J. W., Le Couter J., Strauss E. C., Ferrara N., Vascular endothelial growth factor A in intraocular vascular disease. Ophthalmology 120, 106–114 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Ferrara N., Adamis A. P., Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 15, 385–403 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Ferrara N., Vascular endothelial growth factor. Arterioscler. Thromb. Vasc. Biol. 29, 789–791 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Jain R. K., Antiangiogenesis strategies revisited: From starving tumors to alleviating hypoxia. Cancer Cell 26, 605–622 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dvorak H. F., Discovery of vascular permeability factor (VPF). Exp. Cell Res. 312, 522–526 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Tah V., Orlans H. O., Hyer J., Casswell E., Din N., Sri Shanmuganathan V., Ramskold L., Pasu S., Anti-VEGF therapy and the retina: An update. J. Ophthalmol. 2015, 627674 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lemmon M. A., Schlessinger J., Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunter T., Tyrosine phosphorylation: Thirty years and counting. Curr. Opin. Cell Biol. 21, 140–146 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeltsch M., Leppänen V.-M., Saharinen P., Alitalo K., Receptor tyrosine kinase-mediated angiogenesis. Cold Spring Harb. Perspect. Biol. 5, a009183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Traxler P., Tyrosine kinases as targets in cancer therapy—Successes and failures. Expert Opin. Ther. Targets 7, 215–234 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Krause D. S., Van Etten R. A., Tyrosine kinases as targets for cancer therapy. N. Engl. J. Med. 353, 172–187 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Davis M. I., Hunt J. P., Herrgard S., Ciceri P., Wodicka L. M., Pallares G., Hocker M., Treiber D. K., Zarrinkar P. P., Comprehensive analysis of kinase inhibitor selectivity. Nat. Biotechnol. 29, 1046–1051 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Norman R. A., Toader D., Ferguson A. D., Structural approaches to obtain kinase selectivity. Trends Pharmacol. Sci. 33, 273–278 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Giordano R. J., Cardó-Vila M., Salameh A., Anobom C. D., Zeitlin B. D., Hawke D. H., Valente A. P., Almeida F. C., Nör J. E., Sidman R. L., Pasqualini R., Arap W., From combinatorial peptide selection to drug prototype (I): Targeting the vascular endothelial growth factor receptor pathway. Proc. Natl. Acad. Sci. U.S.A. 107, 5112–5117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bono F., De Smet F., Herbert C., De Bock K., Georgiadou M., Fons P., Tjwa M., Alcouffe C., Ny A., Bianciotto M., Jonckx B., Murakami M., Lanahan A. A., Michielsen C., Sibrac D., Dol-Gleizes F., Mazzone M., Zacchigna S., Herault J.-P., Fischer C., Rigon P., de Almodovar C. R., Claes F., Blanc I., Poesen K., Zhang J., Segura I., Gueguen G., Bordes M.-F., Lambrechts D., Broussy R., van de Wouwer M., Michaux C., Shimada T., Jean I., Blacher S., Noel A., Motte P., Rom E., Rakic J.-M., Katsuma S., Schaeffer P., Yayon A., Van Schepdael A., Schwalbe H., Gervasio F. L., Carmeliet G., Rozensky J., Dewerchin M., Simons M., Christopoulos A., Herbert J.-M., Carmeliet P., Inhibition of tumor angiogenesis and growth by a small-molecule multi-FGF receptor blocker with allosteric properties. Cancer Cell 23, 477–488 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Sidman R. L., Li J., Lawrence M., Hu W., Musso G. F., Giordano R. J., Cardó-Vila M., Pasqualini R., Arap W., The peptidomimetic Vasotide targets two retinal VEGF receptors and reduces pathological angiogenesis in murine and nonhuman primate models of retinal disease. Sci. Transl. Med. 7, 309ra165 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giordano R. J., Cardó-Vila M., Lahdenranta J., Pasqualini R., Arap W., Biopanning and rapid analysis of selective interactive ligands. Nat. Med. 7, 1249–1253 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Giordano R. J., Anobom C. D., Cardó-Vila M., Kalil J., Valente A. P., Pasqualini R., Almeida F. C. L., Arap W., Structural basis for the interaction of a vascular endothelial growth factor mimic peptide motif and its corresponding receptors. Chem. Biol. 12, 1075–1083 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Gerhardt H., Golding M., Fruttiger M., Ruhrberg C., Lundkvist A., Abramsson A., Jeltsch M., Mitchell C., Alitalo K., Shima D., Betsholtz C., VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 161, 1163–1177 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herbert S. P., Stainier D. Y. R., Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell Biol. 12, 551–564 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tammela T., Zarkada G., Wallgard E., Murtomäki A., Suchting S., Wirzenius M., Waltari M., Hellström M., Schomber T., Peltonen R., Freitas C., Duarte A., Isoniemi H., Laakkonen P., Christofori G., Ylä-Herttuala S., Shibuya M., Pytowski B., Eichmann A., Betsholtz C., Alitalo K., Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature 454, 656–660 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Leppänen V.-M., Tvorogov D., Kisko K., Prota A. E., Jeltsch M., Anisimov A., Markovic-Mueller S., Stuttfeld E., Goldie K. N., Ballmer-Hofer K., Alitalo K., Structural and mechanistic insights into VEGF receptor 3 ligand binding and activation. Proc. Natl. Acad. Sci. U.S.A. 110, 12960–12965 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shibuya M., Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2, 1097–1105 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuh G., Garcia K. C., de Vos A. M., The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor Flt-1. J. Biol. Chem. 275, 26690–26695 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Favier B., Alam A., Barron P., Bonnin J., Laboudie P., Fons P., Mandron M., Herault J.-P., Neufeld G., Savi P., Herbert J.-M., Bono F., Neuropilin-2 interacts with VEGFR-2 and VEGFR-3 and promotes human endothelial cell survival and migration. Blood 108, 1243–1250 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Kodera Y., Katanasaka Y., Kitamura Y., Tsuda H., Nishio K., Tamura T., Koizumi F., Sunitinib inhibits lymphatic endothelial cell functions and lymph node metastasis in a breast cancer model through inhibition of vascular endothelial growth factor receptor 3. Breast Cancer Res. 13, R66 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao R., Ji H., Feng N., Zhang Y., Yang X., Andersson P., Sun Y., Tritsaris K., Hansen A. J., Dissing S., Cao Y., Collaborative interplay between FGF-2 and VEGF-C promotes lymphangiogenesis and metastasis. Proc. Natl. Acad. Sci. U.S.A. 109, 15894–15899 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arnaoutova I., Kleinman H. K., In vitro angiogenesis: Endothelial cell tube formation on gelled basement membrane extract. Nat. Protoc. 5, 628–635 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Smith L. E., Wesolowski E., McLellan A., Kostyk S. K., D’Amato R., Sullivan R., D’Amore P. A., Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis. Sci. 35, 101–111 (1994). [PubMed] [Google Scholar]
- 33.Stahl A., Connor K. M., Sapieha P., Chen J., Dennison R. J., Krah N. M., Seaward M. R., Willett K. L., Aderman C. M., Guerin K. I., Hua J., Löfqvist C., Hellström A., Smith L. E. H., The mouse retina as an angiogenesis model. Invest. Ophthalmol. Vis. Sci. 51, 2813–2826 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pierce E. A., Foley E. D., Smith L. E. H., Regulation of vascular endothelial growth factor by oxygen in a model of retinopathy of prematurity. Arch. Ophthalmol. 114, 1219–1228 (1996). [DOI] [PubMed] [Google Scholar]
- 35.Hellström A., Smith L. E. H., Dammann O., Retinopathy of prematurity. Lancet 382, 1445–1457 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Findlater G. S., McDougall R. D., Kaufman M. H., Eyelid development, fusion and subsequent reopening in the mouse. J. Anat. 183, 121–129 (1993). [PMC free article] [PubMed] [Google Scholar]
- 37.Connor K. M., Krah N. M., Dennison R. J., Aderman C. M., Chen J., Guerin K. I., Sapieha P., Stahl A., Willett K. L., Smith L. E. H., Quantification of oxygen-induced retinopathy in the mouse: A model of vessel loss, vessel regrowth and pathological angiogenesis. Nat. Protoc. 4, 1565–1573 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jayson G. C., Hicklin D. J., Ellis L. M., Antiangiogenic therapy—Evolving view based on clinical trial results. Nat. Rev. Clin. Oncol. 9, 297–303 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Folkman J., Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 285, 1182–1186 (1971). [DOI] [PubMed] [Google Scholar]
- 40.Diabetic Retinopathy Clinical Research Network, Wells J. A., Glassman A. R., Ayala A. R., Jampol L. M., Aiello L. P., Antoszyk A. N., Arnold-Bush B., Baker C. W., Bressler N. M., Browning D. J., Elman M. J., Ferris F. L., Friedman S. M., Melia M., Pieramici D. J., Sun J. K., Beck R. W., Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema. N. Engl. J. Med. 372, 1193–1203 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang F., Tang Z., Hou X., Lennartsson J., Li Y., Koch A. W., Scotney P., Lee C., Arjunan P., Dong L., Kumar A., Rissanen T. T., Wang B., Nagai N., Fons P., Fariss R., Zhang Y., Wawrousek E., Tansey G., Raber J., Fong G. H., Ding H., Greenberg D. A., Becker K. G., Herbert J.-M., Nash A., Yla-Herttuala S., Cao Y., Watts R. J., Li X., VEGF-B is dispensable for blood vessel growth but critical for their survival, and VEGF-B targeting inhibits pathological angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 6152–6157 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luttun A., Tjwa M., Carmeliet P., Placental growth factor (PlGF) and its receptor Flt-1 (VEGFR-1): Novel therapeutic targets for angiogenic disorders. Ann. N. Y. Acad. Sci. 979, 80–93 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Fischer C., Mazzone M., Jonckx B., Carmeliet P., FLT1 and its ligands VEGFB and PlGF: Drug targets for anti-angiogenic therapy?. Nat. Rev. Cancer 8, 942–956 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Huang H., He J., Johnson D., Wei Y., Liu Y., Wang S., Lutty G. A., Duh E. J., Carmeliet P., Semba R. D., Deletion of placental growth factor prevents diabetic retinopathy and is associated with Akt activation and HIF1α-VEGF pathway inhibition. Diabetes 64, 200–212 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simpson D. A. C., Murphy G. M., Bhaduri T., Gardiner T. A., Archer D. B., Stitt A. W., Expression of the VEGF gene family during retinal vaso-obliteration and hypoxia. Biochem. Biophys. Res. Commun. 262, 333–340 (1999). [DOI] [PubMed] [Google Scholar]
- 46.Ikeda Y., Yonemitsu Y., Onimaru M., Nakano T., Miyazaki M., Kohno R.-i., Nakagawa K., Ueno A., Sueishi K., Ishibashi T., The regulation of vascular endothelial growth factors (VEGF-A, -C, and -D) expression in the retinal pigment epithelium. Exp. Eye Res. 83, 1031–1040 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Nagy J. A., Dvorak A. M., Dvorak H. F., VEGF-A164/165 and PlGF: Roles in angiogenesis and arteriogenesis. Trends. Cardiovasc. Med. 13, 169–175 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Holash J., Davis S., Papadopoulos N., Croll S. D., Ho L., Russell M., Boland P., Leidich R., Hylton D., Burova E., Ioffe E., Huang T., Radziejewski C., Bailey K., Fandl J. P., Daly T., Wiegand S. J., Yancopoulos G. D., Rudge J. S., VEGF-Trap: A VEGF blocker with potent antitumor effects. Proc. Natl. Acad. Sci. U.S.A. 99, 11393–11398 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chintalgattu V., Rees M. L., Culver J. C., Goel A., Jiffar T., Zhang J., Dunner K. Jr, Pati S., Bankson J. A., Pasqualini R., Arap W., Bryan N. S., Taegtmeyer H., Langley R. R., Yao H., Kupferman M. E., Entman M. L., Dickinson M. E., Khakoo A. Y., Coronary microvascular pericytes are the cellular target of sunitinib malate–induced cardiotoxicity. Sci. Transl. Med. 5, 187ra69 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arap W., Kolonin M. G., Trepel M., Lahdenranta J., Cardó-Vila M., Giordano R. J., Mintz P. J., Ardelt P. U., Yao V. J., Vidal C. L., Chen L., Flamm A., Valtanen H., Weavind L. M., Hicks M. E., Pollock R. E., Botz G. H., Bucana C. D., Koivunen E., Cahill D., Troncoso P., Baggerly K. A., Pentz R. D., Do K. A., Logothetis C. J., Pasqualini R., Steps toward mapping the human vasculature by phage display. Nat. Med. 8, 121–127 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Cardó-Vila M., Zurita A. J., Giordano R. J., Sun J., Rangel R., Guzman-Rojas L., Anobom C. D., Valente A. P., Almeida F. C. L., Lahdenranta J., Kolonin M. G., Arap W., Pasqualini R., A ligand peptide motif selected from a cancer patient is a receptor-interacting site within human interleukin-11. PLOS ONE 3, e3452 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sergeeva A., Kolonin M. G., Molldrem J. J., Pasqualini R., Arap W., Display technologies: Application for the discovery of drug and gene delivery agents. Adv. Drug Deliv. Rev. 58, 1622–1654 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Staquicini F. I., Ozawa M. G., Moya C. A., Driessen W. H. P., Barbu E. M., Nishimori H., Soghomonyan S., Flores L. G. II, Liang X., Paolillo V., Alauddin M. M., Basilion J. P., Furnari F. B., Bogler O., Lang F. F., Aldape K. D., Fuller G. N., Höök M., Gelovani J. G., Sidman R. L., Cavenee W. K., Pasqualini R., Arap W., Systemic combinatorial peptide selection yields a non-canonical iron-mimicry mechanism for targeting tumors in a mouse model of human glioblastoma. J. Clin. Invest. 121, 161–173 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith G. P., Scott J. K., Libraries of peptides and proteins displayed on filamentous phage. Methods Enzymol. 217, 228–257 (1993). [DOI] [PubMed] [Google Scholar]