

Abstract
Arsenic and polycyclic aromatic hydrocarbon (PAH) exposures affect many people worldwide leading to cancer and other diseases. Arsenite (As+3) and certain PAHs are known to cause genotoxicity. However, there is limited information on the interactions between As+3 and PAHs at environmentally relevant concentrations. The thymus is the primary immune organ for T cell development in mammals. Our previous studies showed that environmentally relevant concentrations of As+3 induce genotoxicity in mouse thymus cells through Poly(ADP-ribose) polymerase (PARP) inhibition. Certain PAHs, such as the metabolites of benzo(a)pyrene (BaP), are known to cause DNA damage by forming DNA adducts. In the present study, primary mouse thymus cells were examined for DNA damage following 18 hr in vitro treatments with 5 or 50 nM As+3 and 100 nM BaP, benzo[a]pyrene-7,8-dihydrodiol (BP-Diol), or benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide (BPDE). An interactive increase in genotoxicity and apoptosis were observed following treatments with 5 nM As + 3 + 100 nM BP-diol and 50 nM As + 3 + 100 nM BPDE. We attribute the increase in DNA damage to inhibition of PARP inhibition leading to decreased DNA repair. To further support this hypothesis, we found that a PARP inhibitor, 3,4-dihydro-5[4-(1-piperindinyl) butoxyl]-1(2H)-isoquinoline (DPQ), also interacted with BP-diol to produce an increase in DNA damage. Interestingly, we also found that As+3 and BP-diol increased CYP1A1 and CYP1B1 expression, suggesting that increased PAH metabolism may also contribute to genotoxicity. In summary, these results show that the suppression of PARP activity and induction of CYP1A1/CYP1B1 may act together to increase DNA damage produced by As+3 and PAHs.
Keywords: arsenite, benzo(a)pyrene metabolites, primary mouse thymus cells, interactive genotoxicity, CYP1A1, CYP1B1.
INTRODUCTION
Polycyclic aromatic hydrocarbons (PAHs) are widespread organic pollutants, which naturally occur in soil, air, and following the burning of fossil fuels. PAHs are generated from combustion of wood, coal, oil and tobacco, and they are also abundant in overcooked and processed foods. The toxicity of PAHs is dependent on their structures. Benzo(a)pyrene (BaP), a Group One carcinogen listed by International Agency for Research on Cancer, has been associated with increased levels of colon cancer (Le Marchand et al., 2002), as well as genotoxicity in the lung of smokers (Denissenko et al., 1996). BaP is considered a pro-carcinogen, as metabolism and activation by CYP1A1, CYP1B1 and epoxide hydrolase are required to cause cancer (Jones et al., 1995; Shimada and Fujii-Kuriyama, 2004). BaP is first metabolized to benzo[a]pyrene-7,8-dihydrodiol (BP-Diol), which is then converted into benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide (BPDE). BPDE binds covalently to DNA forming adducts resulting in DNA damage and mutation (Kim et al., 1998; Schwarz et al., 2001). The expression of CYP1A1 and CYP1B1 is regulated by aryl hydrocarbon receptor (AhR), which has been shown to be induced by BaP (Hockley et al., 2007).
Arsenic exposure from food and drinking water sources is a world-wide public health concern. The U.S. Environmental Protection Agency maximal level for arsenic in drinking water is 10 ppb (≈ 130 nM). However, many populations in USA and elsewhere are exposed to unregulated drinking water sources that are in excess of 100 ppb (Rahman et al., 2006; Sherwood et al., 2013). The trivalent from of inorganic arsenic, arsenite (As+3), has been associated with many diseases such as diabetes, skin lesions, and cancers (Argos et al., 2010; Schuhmacher-Wolz et al., 2009; Vahter, 2008). One of the primary genotoxic mechanisms of As+3 is the inhibition of DNA repair (Faita et al., 2013). As+3 has been shown to compete with Zn + 2 on C3H1 and C4 zinc fingers, decreasing the activity of zinc finger proteins involved in DNA repair such as Poly(ADP-ribose) polymerase (PARP) and Xeroderma Pigmentosum, Complementation Group A (XPA) (Qin et al., 2012; Zhou et al., 2011 , 2014). Our previous studies demonstrated a dose-dependent increase in DNA damage and PARP inhibition in mouse thymocytes (Xu et al., 2016). At environmentally relevant concentrations, DNA damage induced by As+3 in thymic cells appears to result from PARP inhibition at low exposure levels (e.g. 50 nM As+3). Higher in vitro exposure levels (e.g. 500 nM As+3) result in oxidative stress that is associated with more DNA damage and double strand breaks. The findings are in agreement with those obtained by other groups (Litwin et al., 2013; Qin et al., 2012). There is also evidence showing that PARP contributes to XPA repair of double strand breaks (King et al., 2012).
As+3 has been documented to interact with other environmental agents, such as UVR (Cooper et al., 2009 , 2013; Evans et al., 2004; Zhou et al., 2011). There is also evidence showing that co-exposure with As+3 increases BaP DNA adduct formation and mutations in mouse hepatoma Hepa-1 cells in vitro (Maier et al., 2002). An in vivo study revealed that arsenic co-exposure can increase BaP adducts formation in both lung and skin (Evans et al., 2004). However, studies have not addressed the effects of As+3 and PAH co-exposure on immune cells in the thymus.
Our previous studies revealed that As+3 interacted with PAHs to increase the suppression of progenitor pre-B cell formation in murine bone marrow at very low concentrations both in vitro and in vivo (Ezeh et al., 2014 , 2015). Synergistic immunosuppression of T-dependent antibody responses by different PAHs and As+3 spleen cells were also observed (Li et al., 2010). We also found that human T cells from certain healthy individuals were suppressed by extreme low concentrations (1–100 nM) of As+3 in vitro (Burchiel et al., 2014). Thymocytes are primarily early T cells which have been shown to be very sensitive to As+3 induced genotoxicity, which can occur at low exposure levels not associated with oxidative stress (Xu et al., 2016). Therefore, our hypothesis is that co-exposure of low concentrations of As+3 and PAHs will induce genotoxicity in mouse thymus cells.
MATERIALS AND METHODS
Chemicals and Reagents
Sodium arsenite (As+3, CAS 774-46-5), benzo(a)pyrene (BaP, CAS 50-32-8), benzo(e)pyrene (BeP, CAS 192-97-2), anthracene (ANTH, CAS 120-12-7), dibenz(a,c)anthracene (DAC, CAS 215-58-7), dibenz(a,h)anthracene (DAH, CAS 53-70-3), 3-methylcholanthrene (3-MC, CAS 56-49-5), benz(a)anthracene (BA, CAS 56-55-3), 7,12-dimethylbenz(a)anthracene (DMBA, CAS 57-97-6), 9,10-dimethylanthracene (DMA, CAS 781-43-1) were purchased from Sigma-Aldrich (St. Louis, MO). Dibenzo(def,p)chrysene (DBC, CAS 191-30-0) was purchased from AccuStandard (New Haven, CT). BaP-trans-7,8-dihydrodiol (BP-diol), BaP-trans-7,8-dihydrodiol-9,10-epoxide (BPDE) and DMBA-trans-3,4-dihydrodiol (DMBA-diol), 11,12-dihydroxy-11,12-dihydrodibenzo(def,p)chrysene (DBC-diol) were obtained from National Cancer Institute Chemical Repository (Midwest Research Institute, Kansas City, MO). Hanks Balanced Salt Solution (HBSS) was purchased from Lonza (Walkersville, MD). Penicillin/Streptomycin 10 000 (mg/ml)/10 000 (U/ml) and 200 mM l-glutamine were purchased from Life Technologies (Grand Island, NY). Dulbecco’s phosphate buffered saline w/o Ca + 2 or Mg + 2 (DPBS-) and RPMI 1640 HEPES modified medium base, sodium azide and dimethylsulfoxide (DMSO) were purchased from Sigma-Aldrich. Fetal Bovine Serum (FBS) was purchased from HyClone Laboratories (Logan, UT). 3,4-Dihydro-5[4-(1-piperindinyl) butoxyl]-1(2H)-isoquinoline (DPQ, CAS 129075-73-6) was purchased from Santa Cruz Biotechnology (Dallas, TX). HT chemiluminescent PARP/apoptosis assay kit (Cat. No. 4685-096-K) and Comet Assay 20 well ES starter kit (Cat. No. 4252-040-ESK) were purchased from Trevigen (Gaithersburg, MD). Pierce BCA protein assay kit (Cat. No. 23225) was purchased from Thermo Fisher Scientific (Waltham, MA). FITC Annexin V Apoptosis Detection Kit II (Cat. No. 556570) was purchased from BD Biosciences (San Jose, CA). RNeasy Mini Kit and QIAshredder were purchased from Qiagen (Valencia, CA). High Capacity cDNA reverse transcription kit (Cat. No. 4368814), CYP1A1 (Mm00487218_m1)/CYP1B1 (Mm00487229_m1) TaqMan gene expression assays and TaqMan universal PCR master mix (Cat. No. 4304437) and Dihydroethidium (DHE, Cat. No. D11347) were purchased from Life Technologies. CYP1A1/1B1 P450-Glo assay (Cat. No. V8751) was purchased from Promega (Madison, WI).
Isolation of Primary Mouse Thymus Cells
About 8- to 10-week-old C57BL/6J male mice were purchased from Jackson Laboratory (Bar Harbor, ME). All animal experiments were performed following protocols approved by the Institutional Animal Use and Care Committee at the University of New Mexico Health Sciences Center. Thymuses were harvested and transferred to the laboratory in HBSS. Cell suspensions from three mice were pooled, centrifuged at 200×g for 10 min and resuspended in the mouse medium (RPMI 1640 plus 10% FBS, 2 mM l-glutamine, 100 mg/ml penicillin, 100 units/ml Streptomycin). Cell viability was determined by acridine orange/propidium iodide (AO/PI) staining and counting using the Nexcelom Cellometer 2000.
The Single Cell Gel Electrophoresis Assay (Comet Assay)
Cells were treated in vitro and washed with DPBS-, immobilized in a 1.5-ml microcentrifuge tube containing 250 μl low melting point agarose and then applied to a Trevigen CometSlide. Cells were lysed with Lysis Solution with 10% DMSO, and electrophoresed in NaOH buffer (pH > 13) with 21 V for 30 min. Slides were air dried, stained with Sybr Green and imaged with an Olympus IX70 inverted fluorescence microscope. About 50 cells from each well of the slides were scored using CometScore (TriTek Corp., Sumerduck, VA).
PARP Activity ELISA Assay
After harvest, in vitro treated cells were lysed with Cell Extraction Buffer from the kit and the protein concentration of each lysate was quantified by BCA protein assay. About 200 ng of total protein was combined with activated DNA and nicotinamide adenine dinucleotide (NAD) to form PAR, which became fixed on the bottom of histone-coated strip wells. Anti-PAR monoclonal antibody was added to the wells to target PARP and HRP conjugated secondary antibody was added to bind the primary antibody. TACS-Sapphire was used to generate the chemiluminescence signal. The reaction was stopped by adding 0.2 M HCl. Absorbance was read at 450 nm using SpectraMax 340PC microplate reader (Molecular Devices).
Annexin V/Propidium Iodide Staining
About 1×105 cells were washed twice with DPBS− and resuspended in 100 μl 1× Annexin V Binding Buffer. About 5 μl of FITC Annexin V and 5 µl of Propidium Iodide staining solution were added to each sample. After 15-min incubation at room temperature in dark, 400 µl of 1× Annexin V Binding Buffer was added to each sample. BD Accuri C6 flow cytometer was used to analyze the samples. Thymus cells treated with 10 μM Etoposide for 4 h were used as positive control. For negative control, 5 μg of purified recombinant Annexin V was added to thymus cells to block the FITC conjugated Annexin V binding.
DHE Staining
About 1 mg DHE was suspended with 158 µl DMSO. About 5 µl of the suspended DHE was diluted with 20 ml DPBS− to a final concentration of 5 µM. About 1×106 treated thymus cells were washed twice with cold wash buffer (DPBS- +1% FBS +0.09% sodium azide). Each sample was stained using 100 µl 5 µM DHE in DPBS− with 37°C incubation for 30 min. Cells were then washed with cold wash buffer, resuspended in 500 µl DPBS− and analyzed on BD Accuri C6 flow cytometer. Thymus cells treated with 100 µM H2O2 for 10 min at 37°C were used as positive controls.
RNA Isolation and CYP1A1/1B1 RT-qPCR
RNA isolation was conducted following the instruction manual for the RNeasy Mini Kit and QIAshredder supplied by the manufacturer. The amount of extracted RNA was then quantified on an Agilent Nanodrop spectrophotometer. For each reverse transcriptase (RT) reaction, 60 µl containing a minimum of 1080 ng RNA was run on SimpiAmps thermal cycler (Life Technologies) using the high capacity cDNA reverse transcription kit. Samples were then diluted to approximately 9 ng/µl with RNase, DNase free water and stored at −20°C. Real time PCR (qPCR) reactions were performed with TaqMan gene expression assays of CYP1A1 and CYP1B1 on Applied Biosystem’s 7900HT system. Comparative CT (first amplification cycle exceeding threshold) was applied for relative quantification, and GADPH gene was used as the endogenous control. The method to calculate the comparative CT is described in detail in the manual from Life Technologies.
CYP1A1/1B1 Activity Assay
The CYP1A1/1B1 substrate, Luciferin-CEE was supplied in the P450-Glo assay. Experiment was performed followed the lytic assay protocol provided by Promega. Isolated primary thymus cells were treated with As+3, BP-diol/BPDE and their combinations for 18 h. Luciferin-CEE was then added to the culture, and the cells were incubated for another 3 h at 37°C. Then 50 µl of cells were combined with equal amount of Luciferin substrate resuspended in the reconstitution buffer, transferred to a 96-well, white polystyrene plate, and incubate at RT for 20 min. Luminescence was recorded on a Tecan Infinite 200 (Männedorf, Switzerland).
Statistics
Excel 2010 and Sigma Plot 12.5 software were used for data analysis. One-way analysis of variance (ANOVA) and Dunnett’s t-test were used to determine differences between groups. Three replicates were performed and analyzed for each single dose. CDI (coefficient of drug interaction) was calculated as CDI = AB/(A × B), A, B and AB are the ratios of the treatments to control. Synergistic effect was indicated with CDI > 1 (Xu et al., 2007).
RESULTS
Inhibition of PARP Activity by Different PAHs in Primary Mouse Thymus Cells
As a control for PARP inhibition studies in murine thymocytes, we examined the direct effects of PAH on PARP activity following in vitro exposures. Thymus cells harvested from male C57BL/6J mice were treated with 100 nM BaP, BeP, 3-MC, DMBA, DAC, DAH, DMA, ANTH, BA, DBC, BP-diol, BPDE, DMBA-diol or DBC-diol for 18 h in vitro. As shown in Figure 1, PARP activity was inhibited by 3-MC, DMBA, DAC, DAH, DBC, BPDE, DMBA-diol and DBC-diol, whereas BaP, BeP, DMA, ANTH, BA and BP-diol did not cause PARP inhibition. The mechanism of differential inhibition of PARP activity by PAHs was not determined in this study. Cell recoveries and viabilities after the exposure to PAHs are listed in Table 1. Based on this PARP screening, it appeared that BaP, BP-diol, and BPDE were good candidates for examining interactions with As+3 based on their relative lack of direct PARP inhibition and their environmental relevance.
FIG. 1.
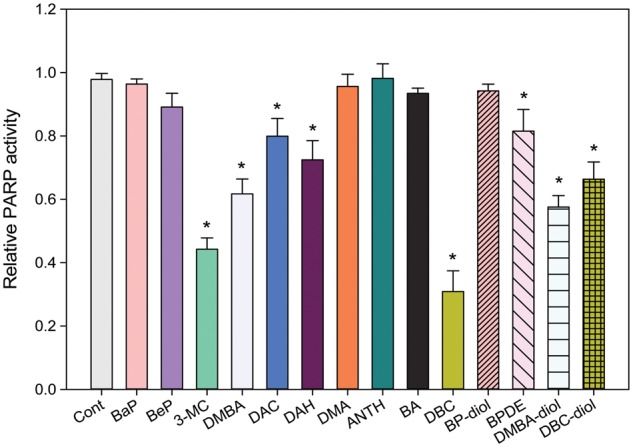
PARP activity in primary mouse thymus cells treated with PAHs in vitro. Primary thymus cells isolated from C57BL/6J male mice were exposed to different PAHs at 100 nM for 18 h in vitro. PARP activity was measured using Trevigen ELISA kits. *Significantly different compared with control (P < 0.05). Results are means ± SD.
TABLE 1.
Cell Recovery and Viability of 2×106 (Viability at Plating: 88.7%) of Primary Thymus Cells Following Exposure to Different PAHs at 100 nM In Vitro for 18 h; VIABILITY Was Measured by AO/PI Staining and Cellometera
| Treatments (100 nM) | Cell Counts (106 cells) | Viability (%) |
|---|---|---|
| Control (no treatment) | 1.84±0.07 | 84.2±1.1 |
| BaP | 1.89±0.11 | 83.5±0.8 |
| BeP | 1.81±0.12 | 82.9±1.2 |
| 3-MC | 1.77±0.09 | 81.4±1.1* |
| DMBA | 1.78±0.15 | 80.9±1.2* |
| DAC | 1.76±0.09 | 83.2±1.3 |
| DAH | 1.75±0.14 | 82.7±1.1 |
| DMA | 1.76±0.11 | 84.1±0.9 |
| ANTH | 1.81±0.03 | 84.5±0.8 |
| BA | 1.83±0.09 | 82.9±1.1 |
| DBC | 1.55±0.08* | 79.9±1.7* |
| BP-diol | 1.75±0.13 | 83.2±0.9 |
| BPDE | 1.60±0.05* | 78.9±1.2* |
| DMBA-diol | 1.69±0.08 | 80.1±0.8* |
| DBC-diol | 1.43±0.12* | 72.6±1.5* |
aTriplicate samples were analyzed for each PAH treatment. Results are means ± SD.
*Significantly different from Cont (P < 0.5).
Interactive Genotoxicity Induced by Low Concentrations of As+3 and BP-diol/BPDE
In order to examine the interactions between BaP/BP-diol/BPDE and As+3, primary thymus cells were treated in vitro with 100 nM BaP, BP-diol or BPDE, 5 or 50 nM As+3 and in combinations for 18 h. A Comet assay was used to detect the DNA damage induced by different treatments. A synergistic increase in DNA damage was observed with the combined treatments of 5 nM As + 3 + 100 nM BP-diol and 50 nM As + 3 + 100 nM BPDE (Figure 2A). A significant decrease in PARP activity was also seen with the same treatments (Figure 2B). Thus, As+3 at very low concentrations interacts with BP-diol and BPDE leading to inhibition of PARP and to a synergistic increase in DNA damage. However, since BaP did not cause a positive interactive effect with As+3, the two metabolites, BP-diol and BPDE, we focused on these PAHs in the subsequent studies.
FIG. 2.

DNA damage and PARP activity in primary thymus cells treated with As+3, BaP/BP-diol/BPDE and the combinations in vitro. Primary thymus cells isolated from C57BL/6J male mice were exposed to 5 or 50 nM As+3, 100 nM BaP, BP-diol or BPDE and the combinations of As+3 and BaP/BP-diol/BPDE for 18 h in vitro. A, Comet assay images were scored by CometScore. B, PARP activity measured with Trevigen ELISA kits represented by absorbance at 450 nm. *Significantly different compared to control (p < 0.05). # Synergistic effect compared to 5 nM As+3 and 100 nM BP-diol (CDI > 1). $ Synergistic effect compared to 50 nM As+3 and 100 nM BPDE (CDI > 1). Results are Means ± SD.
Co-exposure to As+3 and BP-diol/BPDE Induced Apoptosis in Thymus Cells
Annexin V staining and flow cytometry were used to see if the interactive genotoxicity induced by the combined treatments caused an increase in apoptosis. Primary thymus cells were treated with 5 or 50 nM As+3, 100 nM BP-diol or BPDE, and their combinations in vitro for 18 h. A decrease in Annexin V-positive and PI-negative cells (e.g. viable cells), and an increase in Annexin V-positive cells (e.g. early and late apoptotic cells) was seen with treatments of 5 nM As + 3 + 100 nM BP-diol and 50 nM As + 3 + 100 nM BPDE (Figs. 3A and B). Again, an apparent effect was observed with combination treatments, as neither As+3 or BP-diol cause any decrease in viability when present as single agents. The increase in the percentage of apoptotic cells was correlated with the decrease of cell viability (Figs. 3A and C). Therefore, the co-exposure of low concentrations of As+3 and BaP metabolites not only induced significant genotoxicity, but also caused cell apoptosis in primary thymus cells.
FIG. 3.

Annexin V and Propidium Iodide staining in primary thymus cells treated with As+3, BP-diol/BPDE and the combinations in vitro. Primary thymus cells isolated from C57BL/6J male mice were exposed to 5 or 50 nM As+3, 100 nM BP-diol or BPDE and the combinations of As+3 and BP-diol/BPDE for 18 h in vitro. A. flow cytometry results showing cells which are Annexin V-PI- (LL), Annexin V+PI- (LR), Annexin V-PI+ (UL), or Annexin V+PI+ (UR). B, viability (% of Annexin V-PI- cells). C. % of early and late apoptotic cells (% of Annexin V+ cells). *Significantly different compared to control (p < 0.05). # Synergistic effect compared to 5 nM As+3 and 100 nM BP-diol (CDI > 1). $ Synergistic effect compared to 50 nM As+3 and 100 nM BPDE (CDI > 1). Results are Means ± SD.
Co-exposure to As+3 and BP-diol/BPDE Did Not Induce Superoxide Production in Thymus Cells
In order to see if the interactive effects observed were caused by an induction of reactive oxygen species (ROS), primary thymus cells were treated with 5 or 50 nM As+3, 100 nM BP-diol or BPDE, and their combinations in vitro for 18 h. DHE staining by flow cytometry was performed to see the superoxide levels in these treatments. As there was no significant change in superoxide production in these treatments (Figure 4), the interactive effects observed in As+3 and BP-diol/BPDE combined treatments were not caused by superoxide production.
FIG. 4.

DHE staining in primary thymus cells treated with As+3, BP-diol/BPDE and the combinations in vitro. Primary thymus cells isolated from C57BL/6J male mice were exposed to 5 or 50 nM As+3, 100 nM BP-diol or BPDE and the combinations for 18 h in vitro. A, flow cytometry results showing unstained cells and positive control (100 μM H2O2 for 10 min). B, DHE mean channel fluorescence.
Interactive Effects on DNA Damage were Induced by As+3 Direct PARP Inhibition
To further test our hypothesis that As+3 induced interactive genotoxicity with BP-diol/BPDE by PARP inhibition, we utilized a potent specific PARP inhibitor, DPQ (Suto et al., 1991), to treat primary thymus cells together with BP-diol/BPDE in vitro for 18 h. A significant increase in DNA damage was observed in cells treated with 1 μM DPQ + 100 nM BP-diol and 1 μM DPQ + 100 nM BPDE (Figure 5), indicating that PARP inhibition is associated with a synergistic increase in DNA damage produced by BP-diol/BPDE.
FIG. 5.
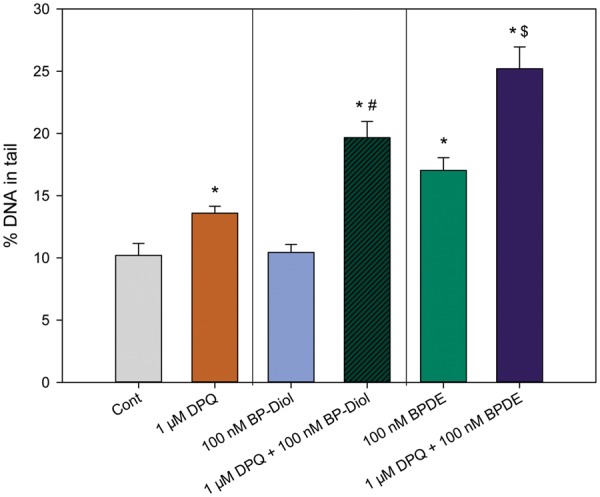
DNA damage in primary thymus cells treated with DPQ (a known PARP inhibitor), BP-diol/BPDE and the combinations in vitro for 18 h. Primary thymus cells isolated from C57BL/6J male mice were exposed to 1 μM DPQ, 100 nM BP-diol or BPDE and the combinations for 18 h in vitro. DNA damage was measured by percentage of DNA in tail using alkaline Comet assay *Significantly different compared to control (p < 0.05). # Synergistic effect compared to 1 μM DPQ and 100 nM BP-diol (CDI > 1). $ Synergistic effect compared to 1 μM DPQ and 100 nM BPDE (CDI > 1). Results are Means ± SD.
CYP1A1 and CYP1B1 RNA Expressions and Activities Were Altered by As+3 and BP-diol/BPDE Co-exposures
The metabolism of BaP and BP-diol is dependent on CYP1A1 and CYP1B1. In order to see if the co-exposures might affect the metabolism of BaP, primary thymus cells were treated with As+3, BP-diol/BPDE and the combinations for 18 h in vitro. Expression of CYP1A1 and CYP1B1 was analyzed by qPCR. We found that As+3 increased the expression of CYP1A1 and CYP1B1 only when combined with BP-diol (Figs. 6A and B). Interestingly, BPDE (100 nM) decreased the expression of CYP1A1 and CYP1B1 when combined with 50 nM As+3 (Figs. 6A and B). The induction of CYP1A1 and CYP1B1 suggests that As+3 may also increase BPDE adduct formation by BP-diol through enzyme induction, which is also supported by a trend of increase of BPDE adduct formation measured by a chemiluminescence immunoassay in As+3 and BP-diol 18 h co-treatment (Supplementary Figure 1). In order to confirm the activities of CYP1A1 and CYP1B1 were altered by these combined treatments, a luminescent CYP1A1/1B1 activity assay was performed using a prolyciferin CYP1A1/1B1 common substrate-Luciferin-CEE. Again, significant increase of CYP1A1/1B1 activity was observed in 5 nM As+3 and 100 nM BP-diol combined treatments, and 50 nM As+3 + 100 nM BPDE decreased the activity (Figure 7), indicating the alteration of CYP1A1 and CYP1B1 mRNA expression did affect their activities.
FIG. 6.

mRNA expression of CYP1A1 and CYP1B1 in primary thymus cells treated with As+3, BP-diol/BPDE and the combinations in vitro. Primary thymus cells isolated from C57BL/6J male mice were exposed to 5 or 50 nM As+3, 100 nM BP-diol or BPDE and the combinations of As+3 and BP-diol/BPDE for 18 h in vitro. Expression of CYP1A1 and CYP1B1 were examined by RT-qPCR. A, CYP1A1 expression. B, CYP1B1 expression. *Significantly different compared to control (p < 0.05). # Synergistic effect compared to 5 nM As+3 and 100 nM BP-diol (CDI > 1). $ Synergistic effect compared to 50 nM As+3 and 100 nM BPDE (CDI > 1). Results are Means ± SD.
FIG. 7.
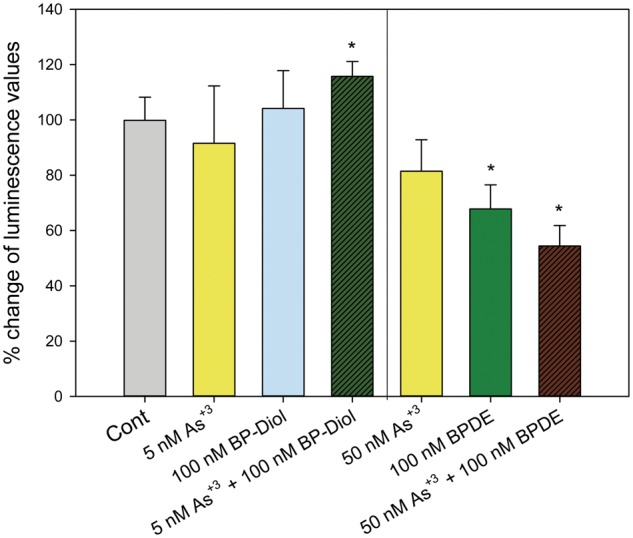
CYP1A1/1B1 activities in primary thymus cells treated with As+3, BP-diol/BPDE and the combinations in vitro. Primary thymus cells isolated from C57BL/6J male mice were exposed to 5 or 50 nM As+3, 100 nM BP-diol or BPDE and the combinations of As+3 and BP-diol/BPDE for 18 h in vitro. CYP1A1/1B1 activities were examined using a Luciferin substrate. After 3 h incubation, the Luciferin substrate was added and luminescence was measured. Data was presented as % of changes in luminescence compared to Cont. *Significantly different compared to control (p < 0.05).
DISCUSSION
Environmental exposures to As+3 and PAHs such as BaP are common and are known to produce adverse health effects in millions of people throughout the world. The interactions between these chemicals and related family members have not been well studied. BaP and its metabolites are known to cause DNA damage through DNA adduct formation that induce strand breaks (Hockley et al., 2007). Our previous reports indicated that As+3 induces genotoxicity in mouse primary thymus cells at environmentally relevant levels through PARP inhibition associated with DNA repair which may lead to immunotoxicity (Xu et al., 2016). As many people are co-exposed to PAHs and arsenic, it is important to understand their potential mechanism(s) of interaction.
Previous studies on the interactions between BaP and As+3 have shown that As+3 potentiates BaP toxicity (Lewińska et al., 2007; Maier et al., 2002). However, these studies were conducted at high concentrations that exceeded those expected to result from environmental exposures. Our previous studies demonstrated that DBC is a strong immunosuppressant of murine spleen cells (Lauer et al., 2013), and that As+3 interacts with DBC at extremely low concentrations to suppress mouse bone marrow pre-B cells (Ezeh et al., 2015). In the present study, the interactions between As+3 and the metabolites of BaP on genotoxicity in mouse thymus cells were analyzed within the nanomolar range of exposure, which are more representative of environmental exposures.
PARP is the initiator of base excision repair of DNA damage. Inhibition of PARP is known to cause DNA damage during cell replication (Dale Rein et al., 2015). Based on our previous findings on genotoxicity of arsenic and PAHs (Harper et al., 2015; Li et al., 2010; Xu et al., 2016), we proposed that As+3 potentiates the DNA damage induced by PAHs through PARP inhibition. Our preliminary experiments revealed that certain PAHs did not interact with low concentrations of As+3, which might have been due to their ability to inhibit PARP activity on their own (data not shown). Therefore, we screened different PAHs for their ability to inhibit PARP, and the results indicated that some PAHs (such as DBC, 3-MC, DMBA, and DAC) can inhibit PARP activity (Figure 1). We selected the BaP family in subsequent studies based on the lack of PARP inhibition by its two metabolites (BP-diol and BPDE) and their environmental relevance. The inhibition of PARP by DBC, 3-MC, DAC and DMBA confounds the ability to see their interactive effects based on DNA damage alone. We did not investigate the mechanism(s) of inhibition of PARP by these PAHs in the present study. Further mechanistic studies need to be conducted in order to form a complete picture of the genotoxicity induced by certain PAHs and their potential interactions with As+3.
CYP1A1 and CYP1B1 are essential for the metabolism of BaP and BP-diol to BPDE. Previous studies on the effect of arsenic on CYP1A1 and CYP1B1 expression have yield mixed results. Some studies showed that As+3 diminished CYP1A1 and CYP1B1 induction in breast cancer cells in vitro (Spink et al., 2002), whereas others revealed that As+3 could increase their expression by AhR induction in lung cells after in vivo exposure (Wu et al., 2009). In our study, we showed differential effects of As+3 + BP-diol and As+3 + BPDE (Figs. 6A and B). It is very interesting that As+3 potentiated CYP1A1 and CYP1B1 expression following the co-treatment with 100 nM BP-diol at 5 nM As+3. BP-diol is known to be an agonist of the AhR (Chen et al., 2003), but the mechanism by which As+3 increases CYP1A1 and CYP1B1 in thymus cells is unknown and needs to be identified in future studies. Also, we found a trend of increase in BPDE adduct formation in thymus cells treated with low concentrations of As+3 and BP-diol (Supplementary Figure 1), using a highly sensitive chemiluminescence immunoassay (Divi et al., 2002). Therefore, it is likely that low concentrations of As+3 induce interactive genotoxicity with BP-diol not only by inhibiting DNA repair, but also by increasing the formation of DNA adducts resulting from the conversion of BP-diol to BPDE.
The decrease in expression of CYP1A1 and CYP1B1 that occurred following co-treatments with As+3 and BPDE may be the result of apoptosis. We found that low concentrations of As+3 and BP-diol/BPDE produced apoptosis following co-treatments, which correlated with an increase in DNA damage (Figs. 2A and 3A). The concentrations of As+3 and BP-diol/BPDE used in our study were very low and had limited cytotoxicity on their own. However, the fact that As+3 and BP-diol/BPDE co-treatments induced significant apoptosis in primary thymus cells at such low concentrations is significant.
In summary, As+3 potentiates the DNA damage induced by the metabolites of BaP at environmentally relevant concentrations through PARP inhibition. Interactive genotoxicity and apoptosis were observed at low concentrations of As+3 in combination with BP-diol/BPDE. The induction of CYP1A1 and CYP1B1 expression by As+3 and BP-diol may play a partial role in the synergistic genotoxicity found in these studies. These findings show that As+3 and certain PAHs may interact with thymus cells at extremely low concentrations. These interactions should be considered in the assessment of risk associated with human exposures.
SUPPLEMENTARY DATA
Supplementary data are available online at http://toxsci.oxfordjournals.org/.
ACKNOWLEDGMENT
We would like to thank Dr Regina M. Santella at Columbia University Mailman School of Public Health for analyzing the BP adducts.
FUNDING
This work was supported by National Institute of Environmental Health Sciences at the National Institutes of Health (R01 ES019968).
REFERENCES
- Argos M., Kalra T., Rathouz P. J., Chen Y., Pierce B., Parvez F., Islam T., Ahmed A., Rakibuz-Zaman M., Hasan R., et al. (2010). Arsenic exposure from drinking water, and all-cause and chronic-disease mortalities in Bangladesh (HEALS): A prospective cohort study. Lancet 376, 252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchiel S. W., Lauer F. T., Beswick E. J., Gandolfi A. J., Parvez F., Liu K. J., Hudson L. G. (2014). Differential susceptibility of human peripheral blood T cells to suppression by environmental levels of sodium arsenite and monomethylarsonous acid. PLoS One 9, e109192.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Nguyen N., Tamura K., Karin M., Tukey R. H. (2003). The role of the Ah receptor and p38 in benzo[a]pyrene-7,8-dihydrodiol and benzo[a]pyrene-7,8-dihydrodiol-9,10-epoxide-induced apoptosis. J. Biol. Chem. 278, 19526–19533. [DOI] [PubMed] [Google Scholar]
- Cooper K. L., King B. S., Sandoval M. M., Liu K. J., Hudson L. G. (2013). Reduction of arsenite-enhanced ultraviolet radiation-induced DNA damage by supplemental zinc. Toxicol. Appl. Pharmacol. 269, 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper K. L., Liu K. J., Hudson L. G. (2009). Enhanced ROS production and redox signaling with combined arsenite and UVA exposures: Contributions of NADPH oxidase. Free Radic. Biol. Med. 47, 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale Rein I., Solberg Landsverk K., Micci F., Patzke S., Stokke T. (2015). Replication-induced DNA damage after PARP inhibition causes G2 delay, and cell line-dependent apoptosis, necrosis and multinucleation. Cell Cycle 14, 3248–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denissenko M. F., Paom A., Tang M., Pfeifer G. P. (1996). Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science 274, 430–432. [DOI] [PubMed] [Google Scholar]
- Divi R. L., Beland F. A., Fu P. P., Von Tungeln L. S., Schoket B., Camara J. E., Ghei M., Rothman N., Sinha R., Poirier M. C. (2002). Highly sensitive chemiluminescence immunoassay for benzo[a]pyrene-DNA adducts: Validation by comparison with other methods, and use in human biomonitoring. Carcinogenesis 23, 2043–2049. [DOI] [PubMed] [Google Scholar]
- Evans C. D., LaDow K., Schumann B. L., Savage R. E., Jr., Caruso J., Vonderheide A., Succop P., Talaska G. (2004). Effect of arsenic on benzo[a]pyrene DNA adduct levels in mouse skin and lung. Carcinogenesis 25, 493–497. [DOI] [PubMed] [Google Scholar]
- Ezeh P. C., Lauer F. T., Liu K. J., Hudson L. G., Burchiel S. W. (2015). Arsenite interacts with dibenzo[def,p]chrysene (DBC) at low levels to suppress bone marrow lymphoid progenitors in mice. Biol. Trace. Elem. Res. 166, 82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezeh P. C., Lauer F. T., MacKenzie D., McClain S., Liu K. J., Hudson L. G., Gandolfi A. J., Burchiel S. W. (2014). Arsenite selectively inhibits mouse bone marrow lymphoid progenitor cell development in vivo and in vitro and suppresses humoral immunity in vivo. PLoS One 9, e93920.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faita F., Cori L., Bianchi F., Andreassi M. G. (2013). Arsenic-induced genotoxicity and genetic susceptibility to arsenic-related pathologies. Int. J. Environ. Res. Publ. Health 10, 1527–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper T. A., Jr., Morré J., Lauer F. T., McQuistan T. J., Hummel J. M., Burchiel S. W., Williams D. E. (2015). Analysis of dibenzo[def,p]chrysene-deoxyadenosine adducts in wild-type and cytochrome P450 1b1 knockout mice using stable-isotope dilution UHPLC-MS/MS. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 782, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockley S. L., Arlt V. M., Brewer D., Te Poele R., Workman P., Giddings I., Phillips D. H. (2007). AHR- and DNA-damage-mediated gene expression responses induced by benzo(a)pyrene in human cell lines. Chem. Res. Toxicol. 20, 1797–1810. [DOI] [PubMed] [Google Scholar]
- Jones J. P., Shou M., Korzekwa K. R. (1995). Stereospecific activation of the procarcinogen benzo[a]pyrene: A probe for the active sites of the cytochrome P450 superfamily. Biochemistry 34, 6956–6961. [DOI] [PubMed] [Google Scholar]
- Kim J. H., Stansbury K. H., Walker N. J., Trush M. A., Strickland P. T., Sutter T. R. (1998). Metabolism of benzo[a]pyrene and benzo[a]pyrene-7,8-diol by human cytochrome P450 1B1. Carcinogenesis 19, 1847–1853. [DOI] [PubMed] [Google Scholar]
- King B. S., Cooper K. L., Liu K. J., Hudson L. G. (2012). Poly(ADP-ribose) contributes to an association between poly(ADP-ribose) polymerase-1 and xeroderma pigmentosum complementation group A in nucleotide excision repair. J. Biol. Chem. 287, 39824–39833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer F. T., Walker M. K., Burchiel S. W. (2013). Dibenzo[def,p]chrysene (DBC) suppresses antibody formation in spleen cells following oral exposures of mice. J. Toxicol. Environ. Health A. 76, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Marchand L., Hankin J. H., Pierce L. M., Sinha R., Nerurkar P. V., Franke A. A., Wilkens L. R., Kolonel L. N., Donlon T., Seifried A., et al. (2002). Well-done red meat, metabolic phenotypes and colorectal cancer in Hawaii. Mutat. Res. 506–507, 205–214. [DOI] [PubMed] [Google Scholar]
- Lewińska D., Arkusz J., Stańczyk M., Palus J., Dziubałtowska E., Stepnik M. (2007). Comparison of the effects of arsenic and cadmium on benzo(a)pyrene-induced micronuclei in mouse bone-marrow. Mutat. Res. 632, 37–43. [DOI] [PubMed] [Google Scholar]
- Li Q., Lauer F. T., Liu K. J., Hudson L. G., Burchiel S. W. (2010). Low-dose synergistic immunosuppression of T-dependent antibody responses by polycyclic aromatic hydrocarbons and arsenic in C57BL/6J murine spleen cells. Toxicol. Appl. Pharmacol. 245, 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litwin I., Bocer T., Dziadkowiec D., Wysocki R. (2013). Oxidative stress and replication-independent DNA breakage induced by arsenic in Saccharomyces cerevisiae. PLoS Genet. 9, e1003640.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier A., Schumann B. L., Chang X., Talaska G., Puga A. (2002). Arsenic co-exposure potentiates benzo[a]pyrene genotoxicity. Mutat. Res. 517, 101–111. [DOI] [PubMed] [Google Scholar]
- Qin X. J., Liu W., Li Y. N., Sun X., Hai C. X., Hudson L. G., Liu K. J. (2012). Poly(ADP-ribose) polymerase-1 inhibition by arsenite promotes the survival of cells with unrepaired DNA lesions induced by UV exposure. Toxicol. Sci. 127, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman M., Vahter M., Wahed M. A., Sohel N., Yunus M., Streatfield P. K., El Arifeen S., Bhuiya A., Zaman K., Chowdhury A. M., et al. (2006). Prevalence of arsenic exposure and skin lesions. A population based survey in Matlab, Bangladesh. J. Epidemiol. Commun. Health 60, 242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmacher-Wolz U., Dieter H. H., Klein D., Schneider K. (2009). Oral exposure to inorganic arsenic: Evaluation of its carcinogenic and non-carcinogenic effects. Crit. Rev. Toxicol. 39, 271–298. [DOI] [PubMed] [Google Scholar]
- Schwarz D., Kisselev P., Cascorbi I., Schunck W. H., Roots I. (2001). Differential metabolism of benzo[a]pyrene and benzo[a]pyrene-7,8-dihydrodiol by human CYP1A1 variants. Carcinogenesis 22, 453–459. [DOI] [PubMed] [Google Scholar]
- Sherwood C. L., Lantz R. C., Boitano S. (2013). Chronic arsenic exposure in nanomolar concentrations compromises wound response and intercellular signaling in airway epithelial cells. Toxicol. Sci. 132, 222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T., Fujii-Kuriyama Y. (2004). Metabolic activation of polycyclic aromatic hydrocarbons to carcinogens by cytochromes P450 1A1 and 1B1. Cancer Sci. 95, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spink D. C., Katz B. H., Hussain M. M., Spink B. C., Wu S. J., Liu N., Pause R., Kaminsky L. S. (2002). Induction of CYP1A1 and CYP1B1 in T-47D human breast cancer cells by benzo[a]pyrene is diminished by arsenite. Drug Metab. Dispos. 30, 262–269. [DOI] [PubMed] [Google Scholar]
- Suto M. J., Turner W. R., Arundel-Suto C. M., Werbel L. M., Sebolt-Leopold J. S. (1991). Dihydroisoquinolinones: The design and synthesis of a new series of potent inhibitors of poly(ADP-ribose) polymerase. Anticancer Drug Des. 6, 107–117. [PubMed] [Google Scholar]
- Vahter M. (2008). Health effects of early life exposure to arsenic. Basic Clin. Pharmacol. Toxicol. 102, 204–211. [DOI] [PubMed] [Google Scholar]
- Wu J. P., Chang L. W., Yao H. T., Chang H., Tsai H. T., Tsai M. H., Yeh T. K., Lin P. (2009). Involvement of oxidative stress and activation of aryl hydrocarbon receptor in elevation of CYP1A1 expression and activity in lung cells and tissues by arsenic: An in vitro and in vivo study. Toxicol. Sci. 107, 385–393. [DOI] [PubMed] [Google Scholar]
- Xu H., Zhou X., Wen X., Lauer F. T., Liu K. J., Hudson L. G., Aleksunes L. M., Burchiel S. W. (2016). Environmentally-relevant concentrations of arsenite induce dose-dependent differential genotoxicity through poly(ADP-ribose) polymerase (PARP) inhibition and oxidative stress in mouse thymus cells. Toxicol. Sci. 149, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S. P., Sun G. P., Shen Y. X., Peng W. R., Wang H., Wei W. (2007). Synergistic effect of combining paeonol and cisplatin on apoptotic induction of human hepatoma cell lines. Acta. Pharmacol. Sin. 28, 869–878. [DOI] [PubMed] [Google Scholar]
- Zhou X., Sun X., Mobarak C., Gandolfi A. J., Burchiel S. W., Hudson L. G., Liu K. J. (2014). Differential binding of monomethylarsonous acid compared to arsenite and arsenic trioxide with zinc finger peptides and proteins. Chem. Res. Toxicol. 27, 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Sun X., Cooper K. L., Wang F., Liu K. J., Hudson L. G. (2011). Arsenite interacts selectively with zinc finger proteins containing C3H1 or C4 motifs. J. Biol. Chem. 286, 22855–22863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.