

Abstract
Fragment‐based lead generation has proven to be an effective means of identifying high‐quality lead compounds for drug discovery programs. However, the fragment screening sets often used are principally comprised of sp2‐rich aromatic compounds, which limits the structural (and hence biological) diversity of the library. Herein, we describe strategies for the synthesis of a series of partially saturated bicyclic heteroaromatic scaffolds with enhanced sp3 character. Subsequent derivatization led to a fragment collection featuring regio‐ and stereo‐controlled introduction of substituents on the saturated ring system, often with formation of new stereocenters.
Keywords: drug design, drug discovery, fused-ring systems, nitrogen heterocycles, synthetic methods
Fragment‐based drug discovery (FBDD) is a well‐established method for generating high‐quality hits and leads.1 The approval of the B‐Raf kinase inhibitor vemurafenib (Zelboraf) in 20112 and the Bcl‐2 inhibitor venetoclax (Venclexta) in 2016,3 coupled with the ongoing evaluation of over 20 candidates in clinical trials,4 validates this approach as a complementary strategy to other hit‐discovery techniques such as high‐throughput screening.5 While the growing prevalence of fragment‐based approaches is encouraging, evaluation of many existing fragment libraries shows a predominance of (hetero)aromatic, “flat” compounds, with a deficiency of chiral, sp3‐rich examples.6, 7
Studies by Ritchie et al.8 and Lovering et al.9 demonstrate improvements in project progression by, for example, increasing the fraction of sp3 centers within molecules or restricting the number of aromatic rings. Furthermore, computational analysis demonstrates that greater 3D conformational character is observed in compounds that have been clinically evaluated in humans, compared to those found in commercial libraries.10 This indicates the importance of sp3‐richness in both the design of screening collections and the subsequent development of hits to leads.
Examples of previous studies aiming to synthesize collections of sp3‐enriched fragments have been limited. Diversity‐oriented synthesis7, 11 and natural product derivatives12 have been used to generate 3D fragment collections, but there remains an unmet need to access new scaffold types. Recently there have been calls13 for new approaches and methodologies for designing fragments with multiple synthetically accessible growth vectors in three dimensions, to allow rapid and efficient elaboration of hits to leads after initial screening, with some early success.14
With these points in mind, the study described herein was aimed at developing efficient synthetic routes to a series of partially saturated bicyclic heteroaromatic (PSBH) fragments with enhanced sp3 content relative to existing fragment libraries. Compounds featuring PSBHs have been shown to display bioactivity against a range of targets (Figure 1),15 and so a series of related fragments might be expected to serve effectively as a screening collection for FBDD applications.
Figure 1.
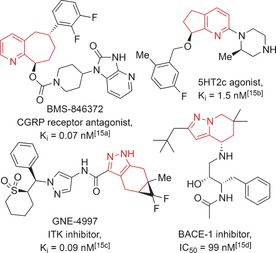
Selected examples of bioactive compounds containing functionalized partially saturated bicyclic heteroaromatics (highlighted in red).
The targets of this study featured a variable aromatic heterocycle fused to a partially saturated carbocycle. The heterocycle could bear either a polar (e.g., amino) group, which should greatly enhance aqueous solubility, a property necessary for fragment screening at higher concentrations,7, 14 or alternatively a hydrophobic (e.g., chloro) group, which can forge key interactions with protein targets.16 The synthetic route (Scheme 1) employed a modular and divergent approach, using simple cross‐coupling and alkylation reactions to install a pair of terminal olefins that could be reliably cyclized through ring‐closing metathesis (RCM).17 This allowed excellent control of the carbocycle ring size and the position and orientation of the resultant endocyclic olefin growth vector, which could undergo subsequent functionalization to produce a range of fragments suitable for screening and/or further elaboration.
Scheme 1.
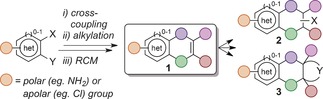
General synthetic strategy toward PSBH scaffolds 1 and subsequent incorporation of new functionalities (2) or rings (3).
We selected pyrazole and pyridine as representative aromatic heterocycles. Whilst previous studies have shown the synthesis of related structures,18 they have incorporated less control over the position of the olefin and do not feature the amino group found in many of our compounds. Furthermore, there are only very few examples where the olefin is used as a branch point and further functionalized beyond simple reduction.15a, 19
Starting from readily available 3‐nitro‐1H‐pyrazole (4), 2‐(trimethylsilyl)ethoxymethyl (SEM) protection, selective iodination, and subsequent Suzuki coupling with potassium vinyltrifluoroborate gave vinyl derivative 7 a (Table 1). Deprotection followed by N‐alkylation with an alkyl bromide of varying C‐chain length provided metathesis precursors 9 a–c, which upon treatment with either Grubbs’ or Hoveyda‐Grubbs’ 2nd generation catalysts yielded the desired scaffolds 10 a–c. Inclusion of further heteroatoms in the formation of medium‐sized partially saturated rings was achieved through treatment of vinyl intermediate 8 a with either tosylate 11 (leading, after RCM, to O‐containing fragment 10 d), or 3‐Boc‐1,2,3‐oxathiazolidine 2,2‐dioxide 12, which gave access to the N‐containing scaffold 10 e after allylation and metathesis. Use of a different Suzuki coupling partner gave methyl‐substituted product 7 f, which could be elaborated to PSBH fragment 10 f. Alternatively, direct allylation at the C‐5 position of SEM‐protected intermediate 5 could be achieved upon treatment with lithium diisopropylamide (LDA) and allyl bromide. This led, in an analogous way, to scaffolds 10 g–h with non‐conjugated olefins.
Table 1.
Synthesis of pyrazole‐based PSBH scaffolds.
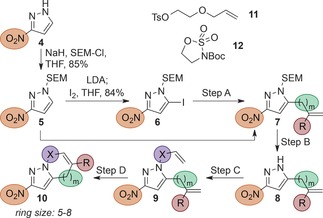
| Step | |||||||
|---|---|---|---|---|---|---|---|
| m | R | X | A (7) | B (8)[c] | C (9)[d] | D (10)[g] | |
| a | 0 | H |

|
85 %[a] | 80 % | 55 % | 45 %[h] |
| b | 0 | H |

|
– | – | 86 % | 84 % |
| c | 0 | H |

|
– | – | 55 % | 89 % |
| d | 0 | H |
|
– | – | 59 %[e] | 65 % |
| e | 0 | H |
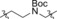
|
– | – | 29 %[f] | 83 % |
| f | 0 | Me |
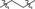
|
76 %[a] | 69 % | 44 % | 90 % |
| g | 1 | H |

|
61 %[b] | 82 % | 70 % | 93 % |
| h | 1 | H |
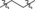
|
– | – | 36 % | 59 %[h] |
Reaction conditions: [a] R′‐BF3K (1.5 equiv), Pd(dppf)Cl2⋅CH2Cl2 (5 mol %), K2CO3 (3.0 equiv), THF/H2O, 70 °C. [b] 5 (1.0 equiv), LDA (1.2 equiv), CuBr (20 mol %), allyl bromide (1.2 equiv), THF, −78 °C to RT. [c] TFA, CH2Cl2, RT. [d] NaH (1.5 equiv), alkyl bromide (1.5 equiv), THF or DMF, 70 °C. [e] NaH (1.5 equiv), 11 (1.5 equiv), THF, 70 °C. [f] 12 (2.0 equiv), K2CO3 (3.0 equiv), DMF, RT, 43 %; then NaH (1.5 equiv), allyl iodide (1.5 equiv), DMF, RT, 68 %. [g] Grubbs II (10 mol %), CH2Cl2, 40 °C. [h] Hoveyda‐Grubbs II (10 mol %), toluene, 110 °C or CH2Cl2, RT. THF=tetrahydrofuran, TFA=trifluoroacetic acid, DMF=N,N‐dimethylformamide.
A similar approach was used to generate PSBH scaffolds from pyridine 13 (Table 2). Attempts to mask the 2‐amino group as a nitro group proved ineffective since, despite successful cross‐coupling reactions, the 2‐nitropyridines were unstable to strong base and did not undergo the desired alkylations at the 4‐methyl position. Mono‐Boc protection was also unsuitable due to poor yields in the cross‐coupling step, possibly due to catalyst chelation. The 2‐amino group could be rendered synthetically tractable, however, either through bis‐Boc protection or through substitution with a 2‐chloro group, which itself can serve as a synthetic handle.20
Table 2.
Synthesis of pyridine‐based PSBH scaffolds.
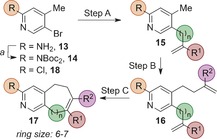
| Step | |||||||
|---|---|---|---|---|---|---|---|
| R | n | R1 | R2 | A (15)[b] | B (16)[e] | C (17)[f] | |
| a | Boc2N | 0 | H | H | 85 % | 60 % | 95 % |
| b | Boc2N | 0 | H | Me | – | 69 % | 94 % |
| c | Boc2N | 0 | H | CF3 | – | 77 % | 54 % |
| d | Boc2N | 0 | Me | H | 87 % | 60 % | 94 % |
| e | Boc2N | 0 | Ph | H | 92 % | 68 % | 83 % |
| f | Boc2N | 1 | H | H | 78 %[c] | 41 % | 65 % |
| g | Cl | 1 | H | H | 73 %[d] | 79 % | 91 % |
Reaction conditions: [a] Boc2O (2.5 equiv), DMAP (0.1 equiv), THF, 70 °C. [b] R′‐BF3K or R′‐B(MIDA) (1.5 equiv), Pd(dppf)Cl2⋅CH2Cl2 (10 mol %), K2CO3 (3.0 equiv), THF/H2O, 70 °C. [c] Allyltributyltin (1.1 equiv), Pd(PPh3)4 (10 mol %), KF (2 equiv), toluene, 110 °C. [d] i‐PrMgCl.LiCl (1.5 equiv), allyl bromide (1.2 equiv), THF, −15 °C to RT. [e] LDA (1.2 equiv), alkyl bromide (1.5 equiv), THF, −78 °C to RT. [f] Grubbs II (5 mol %), CH2Cl2, 40 °C. DMAP=4‐dimethylaminopyridine.
Bis‐Boc substrate 14 (prepared in one step from 13) was functionalized at the 5‐position using either Suzuki coupling (for vinyl substituents) or Stille coupling (for allyl substituents) to produce intermediates 15 a,d–f. Treatment with LDA and trapping of the resultant anion with a variable alkyl bromide electrophile gave a range of metathesis substrates (16 a–f), which under standard ring‐closing metathesis conditions yielded PSBH scaffolds 17 a–f. The 2‐chloro substrate 18 could be allylated in the 5‐position by using an excess of i‐PrMgCl⋅LiCl and trapping the resultant organometallic intermediate with allyl bromide. Allylation at the 4‐methyl position and RCM gave scaffold 17 g in superior yields.
Following PSBH synthesis, a series of simple one‐, two‐, or three‐step functionalizations were performed on selected pyrazole and pyridine scaffolds to demonstrate the synthetic utility of the olefin π‐bond as a growth vector and to generate a variety of new stereocenters (Scheme 2).
Scheme 2.
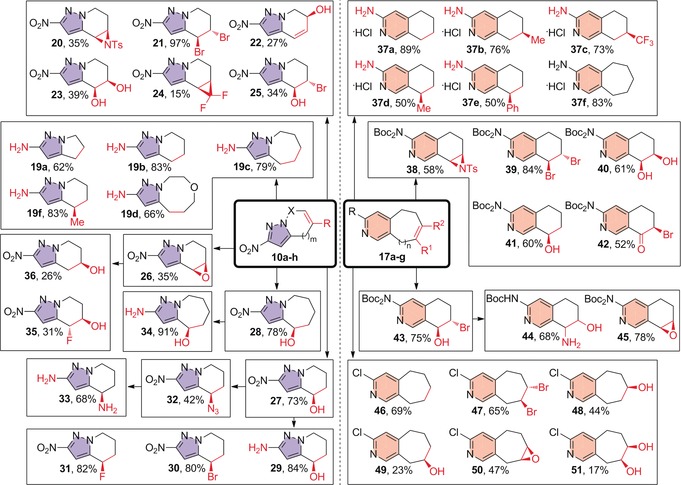
Functionalization of PSBH scaffolds. For reaction conditions, see the Supporting Information.
Catalytic hydrogenation of nitropyrazoles 10 a–d,f served to reduce both the olefin π‐bond and the nitro group in moderate to good yields, revealing the latent amino functionality and, in the case of 19 f, creating a new stereocenter. Other one‐step reactions include aziridination, dibromination, allylic oxidation dihydroxylation, difluorocyclopropanation, hydroxybromination, epoxidation, and hydroboration (20–28) to introduce functionalities at the 4‐, 5‐ and 6‐positions of the fused pyrazole systems. Demonstrating that these initial products can serve as intermediates to other fragments, the products of hydroboration can react further to incorporate Br, F, and N substituents (30–33), whilst epoxide 26 can be opened by nucleophiles such as fluoride and hydride to form fluorohydrin 35 and alcohol 36. Whilst the yields of some reactions were modest, sufficient material was obtained for full characterization and future screening campaigns.
The pyridine‐based scaffolds 17 a–g can undergo a similar range of transformations. Catalytic hydrogenation and subsequent acid‐mediated deprotection of bis‐Boc compounds 17 a–f generated novel fragments, many of which include new stereocenters (37 a–f). Aziridination, dibromination, dihydroxylation, hydroboration, α‐bromoketone formation, and hydroxybromination were also carried out (38–43). Further reactions included amino‐alcohol (44) and epoxide (45) formation, both from bromohydrin 43. 3‐Chloro fragments 46–51 could also be readily accessed using similar conditions.
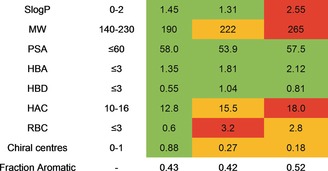
Calculation of a range of physicochemical properties was carried out on all of the PSBH products. Almost all fragments were shown to conform to the so‐called “Rule of Three”, a set of criteria commonly associated with greater hit rates in fragment screening collections.21 Of particular note are the low mean values for molecular weight (190), SlogP (1.45), and “fraction aromatic” (0.43) and the high mean number of chiral centres (0.88), especially when compared to existing commercial libraries (Table 3).
Table 3.
Mean physicochemical properties of fragment collections.
| Property[a] | Ideal Range[b] | This work | Chembridge | Maybridge |
|---|---|---|---|---|
| ||||
[a] MW=molecular weight, PSA=polar surface area, HBA=number of hydrogen‐bond acceptors, HBD=number of hydrogen‐bond donors, HAC=heavy atom count, RBC=rotatable bond count. [b] Based on the guidelines used by Astex Pharmaceuticals.14, 21 Green: within ideal range, orange: at extreme of ideal range, red: outside ideal range. See the Supporting Information for further details.
In conclusion, we have developed simple, scalable routes to a series of partially saturated pyrazole‐ and pyridine‐based scaffolds that can readily undergo a range of synthetic transformations to generate a collection of sp3‐rich fragments, which are suitable for use either as screening members in a library or as intermediates to “higher‐content fragments”. The compounds adhere to recognized guidelines for fragment physicochemical properties, whilst displaying enhanced sp3 character and greater chirality, and providing a range of three‐dimensional growth vectors for synthetic development. It is envisioned that the strategy could be applied to a vast range of analogous scaffolds with varied heterocycles and substituents and that several of the functionalization reactions detailed in Scheme 2 could be rendered asymmetric based on related precedents.19, 22
Dedicated to Professor Stuart L. Schreiber on the occasion of his 60th birthday
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Our research is supported by the EPSRC, BBSRC, MRC, Wellcome Trust, and ERC (FP7/2007‐2013; 279337/DOS). D.G.T. thanks AstraZeneca for funding. S.L.M. thanks BASF for funding. D.R.S. acknowledges support from a Royal Society Wolfson Research Merit award.
D. G. Twigg, N. Kondo, S. L. Mitchell, W. R. J. D. Galloway, H. F. Sore, A. Madin, D. R. Spring, Angew. Chem. Int. Ed. 2016, 55, 12479.
References
- 1.
- 1a. Murray C. W., Rees D. C., Nat. Chem. 2009, 1, 187–192; [DOI] [PubMed] [Google Scholar]
- 1b. Hubbard R. E., Murray J. B. in Methods in Enzymology, Vol. 493 (Ed.: L. C. Kuo), Academic Press, New York, 2011, pp. 509–531; [DOI] [PubMed] [Google Scholar]
- 1c. Erlanson D. in Fragment-Based Drug Discovery and X-Ray Crystallography (Eds.: T. G. Davies, M. Hyvönen), Springer, Berlin, 2012, pp. 1–32; [Google Scholar]
- 1d. Zartler E. R., ACS Med. Chem. Lett. 2014, 5, 952–953; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Congreve M., Chessari G., Tisi D., Woodhead A. J., J. Med. Chem. 2008, 51, 3661–3680; [DOI] [PubMed] [Google Scholar]
- 1f.G. M. Keserű, D. A. Erlanson, G. G. Ferenczy, M. M. Hann, C. W. Murray, S. D. Pickett, J. Med. Chem 2016, DOI: 10.1021/acs.jmedchem.6b00197. [DOI] [PubMed]
- 2.
- 2a. Bollag G., Tsai J., Zhang J., Zhang C., Ibrahim P., Nolop K., Hirth P., Nat. Rev. Drug Discovery 2012, 11, 873–886. [DOI] [PubMed] [Google Scholar]
- 3. Souers A. J., Leverson J. D., Boghaert E. R., Ackler S. L., Catron N. D., Chen J., Dayton B. D., Ding H., Enschede S. H., Fairbrother W. J., Huang D. C., Hymowitz S. G., Jin S., Khaw S. L., Kovar P. J., Lam L. T., Lee J., Maecker H. L., Marsh K. C., Mason K. D., Mitten M. J., Nimmer P. M., Oleksijew A., Park C. H., Park C. M., Phillips D. C., Roberts A. W., Sampath D., Seymour J. F., Smith M. L., Sullivan G. M., Tahir S. K., Tse C., Wendt M. D., Xiao Y., Xue J. C., Zhang H., Humerickhouse R. A., Rosenberg S. H., Elmore S. W., Nat. Med. 2013, 19, 202–208. [DOI] [PubMed] [Google Scholar]
- 4. Baker M., Nat. Rev. Drug Discovery 2013, 12, 5–7. [DOI] [PubMed] [Google Scholar]
- 5. Barelier S., Krimm I., Curr. Opin. Chem. Biol. 2011, 15, 469–474. [DOI] [PubMed] [Google Scholar]
- 6. Hajduk P. J., Galloway W. R. J. D., Spring D. R., Nature 2011, 470, 42–43. [DOI] [PubMed] [Google Scholar]
- 7. Hung A. W., Ramek A., Wang Y., Kaya T., Wilson J. A., Clemons P. A., Young D. W., Proc. Natl. Acad. Sci. USA 2011, 108, 6799–6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ritchie T. J., MacDonald S. J. F., Drug Discovery Today 2009, 14, 1011–1020. [DOI] [PubMed] [Google Scholar]
- 9. Lovering F., Bikker J., Humblet C., J. Med. Chem. 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- 10. Morley A. D., Pugliese A., Birchall K., Bower J., Brennan P., Brown N., Chapman T., Drysdale M., Gilbert I. H., Hoelder S., Jordan A., Ley S. V., Merritt A., Miller D., Swarbrick M. E., Wyatt P. G., Drug Discovery Today 2013, 18, 1221–1227. [DOI] [PubMed] [Google Scholar]
- 11. Damerla V. S. B., Tulluri C., Gundla R., Naviri L., Adepally U., Iyer P. S., Murthy Y. L. N., Prabhakar N., Sen S., Chem. Asian J. 2012, 7, 2351–2360. [DOI] [PubMed] [Google Scholar]
- 12. Over B., Wetzel S., Grutter C., Nakai Y., Renner S., Rauh D., Waldmann H., Nat. Chem. 2013, 5, 21–28. [DOI] [PubMed] [Google Scholar]
- 13. Murray C. W., Rees D. C., Angew. Chem. Int. Ed. 2016, 55, 488–492; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 498–503. [Google Scholar]
- 14. Palmer N., Peakman T. M., Norton D., Rees D. C., Org. Biomol. Chem. 2016, 14, 1599–1610. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Luo G., Chen L., Conway C. M., Denton R., Keavy D., Gulianello M., Huang Y., Kostich W., Lentz K. A., Mercer S. E., Schartman R., Signor L., Browning M., Macor J. E., Dubowchik G. M., ACS Med. Chem. Lett. 2012, 3, 337–341; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Liu K. K.-C., Cornelius P., Patterson T. A., Zeng Y., Santucci S., Tomlinson E., Gibbons C., Maurer T. S., Marala R., Brown J., Kong J. X., Lee E., Werner W., Wenzel Z., Vage C., Bioorg. Med. Chem. Lett. 2010, 20, 266–271; [DOI] [PubMed] [Google Scholar]
- 15c. Burch J. D., Barrett K., Chen Y., DeVoss J., Eigenbrot C., Goldsmith R., Ismaili M. H. A., Lau K., Lin Z., Ortwine D. F., Zarrin A. A., McEwan P. A., Barker J. J., Ellebrandt C., Kordt D., Stein D. B., Wang X., Chen Y., Hu B., Xu X., Yuen P.-W., Zhang Y., Pei Z., J. Med. Chem. 2015, 58, 3806–3816; [DOI] [PubMed] [Google Scholar]
- 15d. Sund C., Belda O., Borkakoti N., Lindberg J., Derbyshire D., Vrang L., Hamelink E., Åhgren C., Woestenenk E., Wikström K., Eneroth A., Lindström E., Kalayanov G., Bioorg. Med. Chem. Lett. 2012, 22, 6721–6727. [DOI] [PubMed] [Google Scholar]
- 16. Tan Y. S., Spring D. R., Abell C., Verma C., J. Chem. Inf. Model. 2014, 54, 1821–1827. [DOI] [PubMed] [Google Scholar]
- 17.RCM has been used in many diversity-generating libraries and applications. Selected examples:
- 17a. Herndon J. W., Coord. Chem. Rev. 2015, 286, 30–150; [Google Scholar]
- 17b. Collins S., Bartlett S., Nie F., Sore H. F., Spring D. R., Synthesis 2016, 1457–1473; [Google Scholar]
- 17c. Grossmann A., Bartlett S., Janecek M., Hodgkinson J. T., Spring D. R., Angew. Chem. Int. Ed. 2014, 53, 13093–13097; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13309–13313; [Google Scholar]
- 17d. Morton D., Leach S., Cordier C., Warriner S., Nelson A., Angew. Chem. Int. Ed. 2009, 48, 104–109; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 110–115. [Google Scholar]
- 18.
- 18a. Fustero S., Román R., Asensio A., Maestro M. A., Aceña J. A., Simón-Fuentes A., Eur. J. Org. Chem. 2013, 7164–7174; [Google Scholar]
- 18b. Eberhart A. J., Cicoira C., Procter D. J., Org. Lett. 2013, 15, 3994–3997; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18c. Moss T. A., Tetrahedron Lett. 2013, 54, 993–997; [Google Scholar]
- 18d. Ahamad S., Gupta A. K., Kant R., Mohanan K., Org. Biomol. Chem. 2015, 13, 1492–1499; [DOI] [PubMed] [Google Scholar]
- 18e. Velcicky J., Pflieger D., Synlett 2010, 1397–1401; [Google Scholar]
- 18f. Luo J., Huo Z., Fu J., Jin F., Yamamoto Y., Org. Biomol. Chem. 2015, 13, 3227–3235; [DOI] [PubMed] [Google Scholar]
- 18g. van den Hoogenband A., den Hartog J. A. J., Faber-Hilhorst N., Lange J. H. M., Terpstra J. W., Tetrahedron Lett. 2009, 50, 5040–5043. [Google Scholar]
- 19.
- 19a. Fackler P., Huber S. M., Bach T., J. Am. Chem. Soc. 2012, 134, 12869–12878; [DOI] [PubMed] [Google Scholar]
- 19b. Mukherjee P., Roy S. J. S., Sarkar T. K., Org. Lett. 2010, 12, 2472–2475. [DOI] [PubMed] [Google Scholar]
- 20. Pelletier G., e-EROS Encycl. Reagents Org. Synth. 2011, 1–5. [Google Scholar]
- 21. Congreve M., Carr R., Murray C., Jhoti H., Drug Discovery Today 2003, 8, 876–877. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Aaseng J. E., Melnes S., Reian G., Gautun O. R., Tetrahedron 2010, 66, 9790–9797; [Google Scholar]
- 22b. Doucet H., Fernandez E., Layzell T. P., Brown J. M., Chem. Eur. J. 1999, 5, 1320–1330; [Google Scholar]; Friedfeld M. R., Shevlin M., Margulieux G. W., Campeau L.-C., Chirik P. J., J. Am. Chem. Soc. 2016, 138, 3314–3324. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary