

Abstract
Objective
To examine the association between PAI-1, an acute phase protein strongly associated with cardiovascular disease risk, and adiposity, insulin resistance, and inflammation among overweight and obese children with a wide range of hepatic steatosis.
Methods
Plasma PAI-1 levels were measured in a prospectively recruited cohort of 39 overweight or obese children who underwent comprehensive anthropometric assessment and metabolic measurements. Hepatic steatosis was quantified using magnetic resonance spectroscopy and participants were divided into 3 groups based on whether they had normal hepatic steatosis (<5%), low hepatic steatosis (≥5–10%) and high hepatic steatosis (>10%).
Results
Plasma PAI-1 levels significantly increased across the severity of hepatic steatosis in overweight and obese children, and this association was independent of BMI z-score, visceral fat, insulin resistance, and inflammatory markers (p < 0.05).
Conclusion
Hepatic steatosis in children is positively associated with circulating levels of PAI-1 independent of BMI, insulin resistance, and inflammatory markers. Further studies are needed to clarify the potential role of PAI-1 as a therapeutic target in pediatric NAFLD.
Keywords: plasminogen activator inhibitor-1, non-alcoholic fatty liver disease, cardiovascular disease risk, children, insulin resistance
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver disease worldwide in adults and children (1, 2) and ranges from simple steatosis to steatohepatitis (NASH), fibrosis and liver cirrhosis (3). NAFLD is closely intertwined with features of metabolic syndrome, most notably obesity, insulin resistance and dyslipidemia (4, 5), as well as an increased risk of cardiovascular disease (CVD) (2, 6–8).
The exact metabolic pathways that underlie the development of hepatic steatosis remain poorly understood, as does the link between NAFLD and CVD (9, 10). Among proposed mechanisms, the fibrinolytic system raises particular interest because of demonstrated association with NAFLD (11), insulin resistance (8), and atherosclerosis (12). Plasminogen activator inhibitor-1 (PAI-1) is an acute-phase protein in the fibrinolytic pathway produced by both hepatocytes and adipocytes, and appears to be elevated in obesity, dyslipidemia, and insulin resistance (8, 13). Recent studies have shown PAI-1 to be associated with hepatic steatosis (14–17) and NAFLD severity (18). Animal models suggest a causal role for PAI-1 and NAFLD onset through suppression of lipid export from the liver (19). However, other studies suggest that the association between elevated PAI-1 and hepatic steatosis may be mediated by the presence of obesity and insulin resistance (20, 21). Therefore, understanding the role of PAI-1 in the development of NAFLD, particularly at this early life stage, is crucial as it may serve as a potential therapeutic target for children with NAFLD.
In the current study, we measured plasma PAI-1 levels in a group of overweight or obese children and assessed its relationship with hepatic steatosis precisely measured by MRS technique, while considering factors such as visceral adiposity, insulin resistance and inflammation. We hypothesized that PAI-1 would be more closely associated with steatosis than with BMI, insulin resistance, or systemic inflammation.
METHODS
Study Design
This study was conducted at the Atlanta Clinical and Translational Science Institute Clinical Research Units at Children’s Healthcare of Atlanta (CHOA) and Emory University Hospital. Subjects were prospectively recruited from pediatric clinics at Emory University Children’s Center and from nearby community centers through flyers and presentations at community events. Written informed consent was obtained from a parent or guardian and written assent was obtained from all children 11 years and older before participation. The protocols were approved by the Institutional Review Boards for Human Subject Research for Emory University and CHOA.
Patient Characteristics
A total of 39 children participated in our study. Inclusion criteria were ages 11–18 years; self-identified as Hispanic; body mass index (BMI) ≥ 85th percentile for age and gender; and an average self-reported consumption of at least 3 sugar-containing drinks per day. High consumption of sugar-containing beverages was an inclusion criterion in order to increase the likelihood of finding adolescents with significant hepatic steatosis (22). Exclusion criteria included known liver diseases; diabetes or fasting glucose ≥ 126 mg/dL; renal insufficiency (creatinine > 2 mg/dL); any chronic diseases requiring daily medication; acute illness within 2 weeks prior to enrollment (defined by fever > 100.4°F); and supplement or anti-oxidant therapy within 4 weeks prior to enrollment. Body weight, height, and blood pressure were recorded. The age- and gender-specific BMI z-score were calculated (23). To calculate insulin resistance, the homeostasis model assessment of insulin resistance (HOMA-IR) (24), and adipose tissue insulin resistance index (Adipo-IR) (25) were used.
Laboratory Analysis
Blood samples were collected in the morning following a 12-hour overnight fast in EDTA-coated tubes and plasma was separated immediately upon collection. Plasma samples were protected from light and transported in ice packs to the laboratory for further processing (within 4 hours). Plasma concentrations of aspartate aminotransferase (AST), alanine aminotransferase (ALT) and γ-glutamyl transpeptidase (GGT) were measured by routine enzymatic methods at CHOA Clinical Laboratory. Plasma concentrations of PAI-1 and tumor necrosis factor -alpha (TNF-α) were measured by multi-analyte chemiluminescent detection assay using Luminex® xMap technology (Millipore Corporation, St. Louis, MO).
All lipid measurements were performed by the Biomarker Core Laboratory at the Atlanta Veterans Affairs Medical Center using AU480 chemistry analyzer (Beckman Coulter, LaBrea, CA). Total cholesterol and triglycerides (TG) were measured by enzymatic methods using reagents from Beckman (Beckman Diagnostics, Fullerton, CA). Low-density lipoproteins (LDL) and high-density lipoproteins (HDL) were measured by homogeneous enzymatic assays (Sekisui Diagnostics, Exton, PA). Free fatty acids (FFA) and glucose concentrations were determined by colorimetric method (Sekisui Diagnostics, Exton, PA). Plasma fasting insulin level (μU/ml) was assessed using immunoturbidometric methods (Sekisui Diagnostics, Exton, PA).
Measurements of hepatic steatosis and visceral adiposity
Hepatic steatosis was assessed by magnetic resonance spectroscopy (MRS) using our previously described methods (26). Briefly, we used a rapid 15-sec acquisition technique obtained during a single breath hold. Water and lipid magnitude spectra were analyzed by determining the AUC corresponding to a user-defined frequency range surrounding the corresponding water/lipid peaks (water peak: 4.6ppm; lipid peak: 1.3, 2.0ppm). The integrated magnitude signals at each TE were fit to exponential T2 decay curves, whereby the equilibrium signal (M0) and the relaxation rate (R2=1/T2) were determined by least-squares approximation. Using M0 for water and lipid, the T2-corrected hepatic lipid fraction was calculated from: % Hepatic Lipid = M0[lipid]/(M0[lipid] + M0[water]). The calculated percentage of steatosis was then grouped depending on whether the MRS indicated normal hepatic fat content (NHF, < 5%), low hepatic fat content (LHF, ≥ 5–10%), or high hepatic fat content (HHF, > 10%).
Visceral adiposity was calculated using ImageJ software (NIH, Bethesda, MD). From a 3D gradient-echo acquisition with a 3-point Dixon reconstruction (27), a single, “fat-only” image was isolated at the L4 vertebral body. A signal threshold was set manually for each subject such that the subcutaneous fat was completely identified (>90% of maximum signal). This threshold was automatically extended to the visceral region, producing a binary mask of fat and non-fat regions. Manual segmentation was performed to separate subcutaneous and visceral regions by using the intra-abdominal muscle and perineum as boundary landmarks. The vertebra was not included in the segmented region. Visceral adiposity was calculated from the threshold volume of the segmented intra-abdominal region.
Statistical Analysis
Statistical analyses were performed using SAS 9.3 (SAS Institute, Cary, NC). To compare variables across three categories of hepatic steatosis (NHF, LHF, and HHF), ANOVA tests or Kruskal-Wallis tests (for variables without normal distribution) were used. Data are expressed as mean (± standard error), with statistical significance considered as a p-value ≤ 0.05. Linear regression models were performed to determine the independent association between plasma PAI-1 levels and hepatic steatosis, after adjusting for covariates including BMI z-score, visceral adiposity, insulin resistance, and inflammatory markers.
RESULTS
Baseline Characteristics of Participants
Demographic characteristics and baseline variables are presented in Table 1. Study participants were categorized into three groups based upon quantification of hepatic steatosis. Of the 39 Hispanic, overweight or obese children, 12 (30.8%) had normal hepatic fat (NHF, < 5%), 14 (35.9%) had low hepatic fat (LHF, ≥ 5–10%), and 13 (33.3%) had high hepatic fat (HHF, > 10%). Significant differences were observed in BMI z-score, visceral adiposity, and percent of hepatic steatosis between the groups. Additionally, indices of insulin resistance, plasma triglyceride and free fatty acid content were significantly different across the three categories of hepatic steatosis (Table 1). No statistically significant differences were observed for glucose, cholesterol, LDL, HDL measurements, or for inflammatory markers (Table 1).
Table 1.
Demographic and laboratory characteristics of the study population
| Parameters | NHF (N=12) | LHF (N=14) | HHF (N=13) | p-value |
|---|---|---|---|---|
| Age (yrs) | 14.6 (0.54) | 13.9 (0.62) | 13.7 (0.78) | 0.621 |
| Male (n, %) | 5 (41.7) | 2 (14.3) | 9 (69.2) | 0.015 |
| BMI z-score | 1.95 (0.08) | 2.01 (0.08) | 2.30 (0.12) | 0.030 |
| Visceral adiposity (cm2)ⱡ | 89.7 (11.4) | 72.8 (7.08) | 129 (13.8) | 0.010 |
| Hepatic steatosis (%) | 3.96 (0.19) | 7.26 (1.65) | 15.9 (4.48) | <0.001 |
| ALT (U/L) | 17.3 (2.15) | 17.1 (1.05) | 94.3 (36.2) | <0.001 |
| Glucose (mg/dl) | 72.8 (7.31) | 122 (14.4) | 201 (32.7) | 0.769 |
| Insulin (mU/L)* | 17.1 (2.15) | 27.7 (22.1) | 46.3 (12.9) | 0.012 |
| HOMA-IR* | 3.92 (0.44) | 6.89 (1.66) | 11.3 (4.01) | 0.028 |
| Adipo-IR* | 13.1 (1.60) | 23.8 (5.99) | 85.3 (46.7) | 0.001 |
| Cholesterol (mg/dl) | 157 (22.1) | 161 (8.78) | 176 (12.0) | 0.354 |
| TG (mg/dl) | 72.8 (7.31) | 122 (14.4) | 201 (32.7) | 0.001 |
| LDL (mg/dl) | 99.3 (8.28) | 105 (6.56) | 109 (9.69) | 0.720 |
| HDL (mg/dl) | 46.9 (2.51) | 42.9 (7.98) | 44.1 (10.8) | 0.542 |
| FFA (mEq/L) | 0.81 (0.08) | 0.85 (0.07) | 1.31 (0.80) | 0.032 |
| hs-CRP (mg/L) | 3.46 (1.03) | 3.12 (0.64) | 6.52 (2.18) | 0.315 |
| TNF-α (pg/mL)* | 4.54 (0.61) | 4.92 (0.46) | 6.34 (0.62) | 0.081 |
Data are expressed as mean (SE). p-values were generated by ANOVA or Kruskal-Wallis tests (for variables not normally distributed). Significant p-values (p < 0.05) marked in bold.
NHF, n=10; LHF, N=13; HHF, N=8;
HHF, n=11.
Abbreviations: NHF, normal hepatic fat (≤5%); LHF, low hepatic fat (5–10%); HHF, high hepatic fat (>10%); ALT, alanine aminotransferase; HOMA-IR, homeostatic model assessment for insulin resistance index; Adipo-IR, adipose insulin resistance index; TG, triglycerides; LDL, low-density lipoprotein; HDL, high-density lipoprotein; FFA, free fatty acids; hs-CRP, high sensitivity C-reactive protein; TNF-α, tumor necrosis factor-α.
Association between plasma PAI-1 and Hepatic Steatosis
Figure 1 illustrates the difference in PAI-1 plasma concentration across the groups with variable hepatic steatosis. PAI-1 levels were significantly lower in individuals with normal hepatic fat (8.78 ± 1.17 ng/mL) when compared to those in individuals with low hepatic fat (11.6 ± 0.99 ng/mL, p = 0.04) and high hepatic fat (20.8 ± 2.26 ng/mL, p < 0.001). Additionally, significant differences in PAI-1 plasma levels were observed between low and high hepatic fat groups (p = 0.002).
Figure 1. Plasma concentration of PAI-1 significantly differed across the severity of hepatic steatosis.
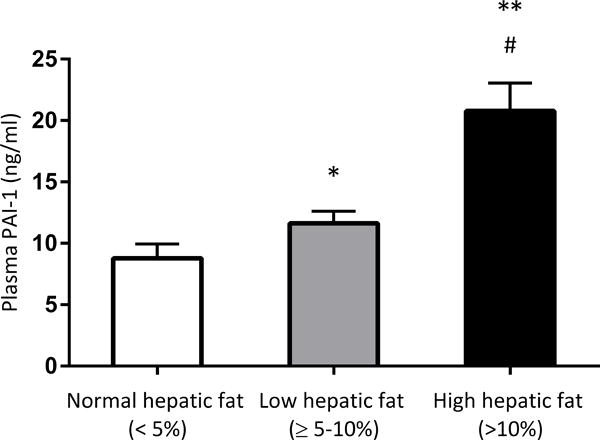
*p = 0.04 vs. normal hepatic fat group. ** p < 0.001 vs. normal hepatic fat group. # p = 0.002 vs. low hepatic fat group.
To further assess the relationship between PAI-1 and hepatic steatosis, linear regression models were performed. Plasma PAI-1 levels were independently associated with hepatic steatosis after adjusting for BMI z-score, visceral adiposity, insulin resistance indices, ALT, and TNF-α (Table 2). When further adjustment was made for plasma TG, the association between plasma PAI-1 levels and hepatic steatosis was attenuated and lost statistical significance.
Table 2.
β coefficients for the association between plasma PAI-1 concentrations and hepatic steatosis.
| Linear regression covariates | β (SEE) | p-value |
|---|---|---|
| Model 1: BMI z-score | 0.614 (0.165) | <0.001 |
| Model 2: BMI z-score, visceral adiposity | 0.686 (0.253) | <0.001 |
| Model 3: BMI z-score, visceral adiposity, insulin resistance indices | 0.599 (0.279) | 0.004 |
| Model 4: BMI z-score, viceral adiposity, insulin resistance indices, ALT, TNF-α | 0.575 (0.320) | 0.013 |
| Model 5: BMI z-score, viceral adiposity, insulin resistance indices, ALT, TNF-α, plasma TG | 0.292 (0.334) | 0.204 |
Insulin resistance indices include plasma insulin, HOMA-IR, and Adipo-IR index.
DISCUSSION
By using a well characterized cohort of overweight or obese children with a wide range of hepatic steatosis, precisely quantified by MRS, we demonstrated that plasma PAI-1 levels were related to the amount of hepatic steatosis independent of BMI, visceral fat, insulin resistance indices, and inflammatory markers. This finding is in contrast to previous conclusions that PAI-1 is more closely related to body weight and insulin resistance. Our study supports an independent role for PAI-1 in the mechanisms of NAFLD and its consideration as a possible therapeutic target for both NAFLD and the NAFLD-associated CVD risk.
Until the development of MRI-based hepatic steatosis quantification methods, it was difficult to characterize hepatic steatosis precisely, which could in part explain previously reported conflicting results in regards to PAI-1 levels and hepatic steatosis. A study by de Larrañaga et al. in which adult patients at risk for metabolic syndrome underwent liver ultrasound scans to measure hepatic steatosis, concluded that plasma PAI-1 levels were better determined by whole-body fat content than liver steatosis content, after controlling for features of metabolic syndrome (28). Targher et al. showed that in healthy adult men the association between ultrasound-diagnosed NAFLD and PAI-1 was no longer apparent after adjusting for CT-measured visceral adiposity (29). Alessi et al. countered by showing that PAI-1 levels were more closely related to liver steatosis than visceral adiposity in obese versus healthy adults who underwent liver biopsy, however it is unclear how many cases of fatty liver were alcohol-induced (17). Previous studies in children found an association between PAI-1 and hepatic steatosis similar to ours, however they did not control for BMI in their analyses (30, 31).
Previous studies using a murine cell culture model of hepatocytes suggest that the relationship of PAI-1 and hepatic steatosis is via suppression of microsomal triglyceride transfer protein (MTTP) activity by PAI-1 and resultant accumulation of triglycerides in cells (32). Similarly, PAI-1 knockout mice were protected from fructose-induced hepatic steatosis and liver damage through effects on hepatic lipid export (33). Pharmaceutical treatment with losartan, a medication that reduces PAI-1 levels, has been shown to strongly reduce hepatic PAI-1 expression and reverse hepatic steatosis and lipid accumulation in rats (34). Thus, while PAI-1 is best known for its other biologic roles in the fibrinolytic pathway, it appears to be active in modification of lipid regulation in the liver as well. This makes PAI-1 a promising therapeutic target in adults and children with NAFLD.
The major strengths of this study include the use of MRS to precisely quantify hepatic and visceral steatosis in children with NAFLD and overweight or obese controls. MRS methodology provides a precise and non-invasive measure of hepatic fat and has been shown to have close correlation with the gold standard procedure of liver biopsy (r = 0.9) (35). All data collection and evaluation were performed in a dedicated pediatric research center and the children were extensively characterized allowing comparisons between the liver, whole body adiposity, and insulin sensitivity. Our study is subjected to limitations given its cross-sectional nature. By adjusting for adiposity, insulin resistance, and inflammation, we found an independent association between the elevated PAI-1 levels and hepatic steatosis but we are not able to identify the causality. In addition, the sample was made up of overweight and obese Hispanic children, thereby potentially limiting the generalizability of our results to other ethnic groups.
In conclusion, our results show that hepatic steatosis in children is positively associated with plasma levels of PAI-1 independent of body weight, insulin resistance indices, and inflammatory markers. Our findings support further investigation of PAI-1 in NAFLD, particularly since it could serve as a therapeutic target benefiting both hepatic steatosis and the associated cardiovascular disease risk.
What is Known.
Nonalcoholic fatty liver disease is associated with increased risk of cardiovascular disease.
Plasminogen activator inhibitor-1 (PAI-1) is an acute phase protein that is elevated in obesity and has been suggested to mediate the development of cardiovascular disease in NAFLD.
What is New.
PAI-1 is closely correlated with the quantity of hepatic steatosis in children independent of adiposity, insulin resistance indices, and inflammatory markers, supporting a possible role of PAI-1 in mediating NAFLD development and contributing to the increased cardiovascular risk as commonly seen in NAFLD.
Acknowledgments
Support for these studies included grants from NIH K23 DK080953 (Vos), and the North American Society of Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN) Foundation Nestle Young Investigator Award (Vos). Also supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under Award Number UL1TR000454. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Financial Support: The study was funded, in part, from grants from the National Institutes of Health K23 DK080953, R01DK096157-01 (MBV), K24 DK096574 (TRZ), 1R01AA018869-01, 1U01AA021901-01 (CJM) and DoD W81XWH-11-1-0595 (CJM) and ACTSI grant UL1 TR000454. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Trial identification number and URL: NCT01188083 https://clinicaltrials.gov/ct2/show/NCT01188083?term=miriam+vos&rank=3
CONFLICT OF INTEREST/FINANCIAL DISCLOSURE STATEMENT
All authors have no personal or financial conflicts of interest to declare.
Author contributions: Study design (MV, JH, RJ, CM, GA), data analysis (MV, JH, RJ) interpretation of data (MV, JH, CM, GA, NL, RJ, EB), drafting of manuscript (JH), manuscript revision and final approval (MV, JK, RJ). All authors were involved in writing the manuscript and had final approval of the submitted and published versions.
Contributor Information
Jeffrey R. Holzberg, Email: jholzberg@gmail.com.
Ran Jin, Email: rjin@emory.edu.
Ngoc-Anh Le, Email: anh.le@va.gov.
Thomas R. Ziegler, Email: tzieg01@emory.edu.
Elizabeth M. Brunt, Email: EBrunt@path.wustl.edu.
Craig J. McClain, Email: craig.mcclain@louisville.edu.
Gavin E. Arteel, Email: gavin.arteel@louisville.edu.
Miriam B. Vos, Email: mvos@emory.edu.
References
- 1.Schwimmer JB, Deutsch R, Kahen T, et al. Prevalence of fatty liver in children and adolescents. Pediatrics. 2006;118:1388–93. doi: 10.1542/peds.2006-1212. [DOI] [PubMed] [Google Scholar]
- 2.Smith BW, Adams LA. Non-alcoholic fatty liver disease. Crit Rev Clin Lab Sci. 2011;48:97–113. doi: 10.3109/10408363.2011.596521. [DOI] [PubMed] [Google Scholar]
- 3.Ratziu V, Bellentani S, Cortez-Pinto H, et al. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J Hepatol. 2010;53:372–84. doi: 10.1016/j.jhep.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 4.Fishbein MH, Miner M, Mogren C, et al. The spectrum of fatty liver in obese children and the relationship of serum aminotransferases to severity of steatosis. J Pediatr Gastroenterol Nutr. 2003;36:54–61. doi: 10.1097/00005176-200301000-00012. [DOI] [PubMed] [Google Scholar]
- 5.Schwimmer JB, Deutsch R, Rauch JB, et al. Obesity, insulin resistance, and other clinicopathological correlates of pediatric nonalcoholic fatty liver disease. J Pediatr. 2003;143:500–5. doi: 10.1067/S0022-3476(03)00325-1. [DOI] [PubMed] [Google Scholar]
- 6.Pacifico L, Di Martino M, De Merulis A, et al. Left ventricular dysfunction in obese children and adolescents with nonalcoholic fatty liver disease. Hepatology. 2014;59:461–70. doi: 10.1002/hep.26610. [DOI] [PubMed] [Google Scholar]
- 7.Targher G, Bertolini L, Padovani R, et al. Relations between carotid artery wall thickness and liver histology in subjects with nonalcoholic fatty liver disease. Diabetes care. 2006;29:1325–30. doi: 10.2337/dc06-0135. [DOI] [PubMed] [Google Scholar]
- 8.Asplund-Carlson A, Hamsten A, Wiman B, et al. Relationship between plasma plasminogen activator inhibitor-1 activity and VLDL triglyceride concentration, insulin levels and insulin sensitivity: studies in randomly selected normo- and hypertriglyceridaemic men. Diab tologia. 1993;36:817–25. doi: 10.1007/BF00400356. [DOI] [PubMed] [Google Scholar]
- 9.Santoro N, Caprio S. Nonalcoholic fatty liver disease/nonalcoholic steatohepatitis in obese adolescents: A looming marker of cardiac dysfunction. Hepatology. 2014;59:372–4. doi: 10.1002/hep.26663. [DOI] [PubMed] [Google Scholar]
- 10.Starley BQ, Calcagno CJ, Harrison SA. Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology. 2010;51:1820–32. doi: 10.1002/hep.23594. [DOI] [PubMed] [Google Scholar]
- 11.Verrijken A, Francque S, Mertens I, et al. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2014;59:121–9. doi: 10.1002/hep.26510. [DOI] [PubMed] [Google Scholar]
- 12.Ha H, Oh EY, Lee HB. The role of plasminogen activator inhibitor 1 in renal and cardiovascular diseases. Nature reviews. Nephrology. 2009;5:203–11. doi: 10.1038/nrneph.2009.15. [DOI] [PubMed] [Google Scholar]
- 13.Schneiderman J, Sawdey MS, Keeton MR, et al. Increased type 1 plasminogen activator inhibitor gene expression in atherosclerotic human arteries. Proc Natl Acad Sci U S A. 1992;89:6998–7002. doi: 10.1073/pnas.89.15.6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thuy S, Ladurner R, Volynets V, et al. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J Nutr. 2008;138:1452–5. doi: 10.1093/jn/138.8.1452. [DOI] [PubMed] [Google Scholar]
- 15.Ardigo D, Franzini L, Valtuena S, et al. The increase in plasma PAI-1 associated with insulin resistance may be mediated by the presence of hepatic steatosis. Atherosclerosis. 2010;208:240–5. doi: 10.1016/j.atherosclerosis.2009.06.026. [DOI] [PubMed] [Google Scholar]
- 16.Targher G, Bertolini L, Rodella S, et al. NASH predicts plasma inflammatory biomarkers independently of visceral fat in men. Obesity (Silver Spring) 2008;16:1394–9. doi: 10.1038/oby.2008.64. [DOI] [PubMed] [Google Scholar]
- 17.Alessi MC, Bastelica D, Mavri A, et al. Plasma PAI-1 levels are more strongly related to liver steatosis than to adipose tissue accumulation. Arterioscler Thromb Vasc Biol. 2003;23:1262–8. doi: 10.1161/01.ATV.0000077401.36885.BB. [DOI] [PubMed] [Google Scholar]
- 18.Verrijken A, Francque S, Mertens I, et al. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2014;59:121–9. doi: 10.1002/hep.26510. [DOI] [PubMed] [Google Scholar]
- 19.Kanuri G, Spruss A, Wagnerberger S, et al. Fructose-induced steatosis in mice: role of plasminogen activator inhibitor-1, microsomal triglyceride transfer protein and NKT cells. Lab Invest. 2011;91:885–95. doi: 10.1038/labinvest.2011.44. [DOI] [PubMed] [Google Scholar]
- 20.Janand-Delenne B, Chagnaud C, Raccah D, et al. Visceral fat as a main determinant of plasminogen activator inhibitor 1 level in women. Int J Obes Relat Metab Disord. 1998;22:312–7. doi: 10.1038/sj.ijo.0800585. [DOI] [PubMed] [Google Scholar]
- 21.Alessi MC, Peiretti F, Morange P, et al. Production of plasminogen activator inhibitor 1 by human adipose tissue: possible link between visceral fat accumulation and vascular disease. Diabetes. 1997;46:860–7. doi: 10.2337/diab.46.5.860. [DOI] [PubMed] [Google Scholar]
- 22.Maersk M, Belza A, Stodkilde-Jorgensen H, et al. Sucrose-sweetened beverages increase fat storage in the liver, muscle, and visceral fat depot: a 6-mo randomized intervention study. Am J Clin Nutri. 2012;95:283–9. doi: 10.3945/ajcn.111.022533. [DOI] [PubMed] [Google Scholar]
- 23.Kuczmarski RJ, Ogden CL, Guo SS, et al. 2000 CDC Growth Charts for the United States: methods and development. Vital Health Stat 11. 2002:1–190. [PubMed] [Google Scholar]
- 24.Matthews DR, Hosker JP, Rudenski AS, et al. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diab tologia. 1985;28:412–9. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 25.Gastaldelli A, Harrison SA, Belfort-Aguilar R, et al. Importance of changes in adipose tissue insulin resistance to histological response during thiazolidinedione treatment of patients with nonalcoholic steatohepatitis. Hepatology. 2009;50:1087–93. doi: 10.1002/hep.23116. [DOI] [PubMed] [Google Scholar]
- 26.Sharma P, Pineda-Alonso N, Vos M, et al. High-Speed (T2)-Corrected Multi-Echo (HISTO) Magnetic Resonance Spectroscopy: A Fast, Reproducible, Non-invasive Technique for Accurate Hepatic Lipid Quantification. Hepatology. 2008:995A. [Google Scholar]
- 27.Glover GH, Schneider E. Three-point Dixon technique for true water/fat decomposition with B0 inhomogeneity correction. Magnetic resonance in medicine. 1991;18:371–83. doi: 10.1002/mrm.1910180211. [DOI] [PubMed] [Google Scholar]
- 28.de Larranaga G, Wingeyer SP, Graffigna M, et al. Plasma plasminogen activator inhibitor-1 levels and nonalcoholic fatty liver in individuals with features of metabolic syndrome. Clin Appl Thromb Hemost. 2008;14:319–24. doi: 10.1177/1076029607304094. [DOI] [PubMed] [Google Scholar]
- 29.Targher G, Bertolini L, Scala L, et al. Non-alcoholic hepatic steatosis and its relation to increased plasma biomarkers of inflammation and endothelial dysfunction in non-diabetic men. Role of visceral adipose tissue. Diabet Med. 2005;22:1354–8. doi: 10.1111/j.1464-5491.2005.01646.x. [DOI] [PubMed] [Google Scholar]
- 30.Alisi A, Manco M, Devito R, et al. Endotoxin and plasminogen activator inhibitor-1 serum levels associated with nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr. 2010;50:645–9. doi: 10.1097/MPG.0b013e3181c7bdf1. [DOI] [PubMed] [Google Scholar]
- 31.Fitzpatrick E, Dew TK, Quaglia A, et al. Analysis of adipokine concentrations in paediatric non-alcoholic fatty liver disease. Pediatr obes. 2012;7:471–9. doi: 10.1111/j.2047-6310.2012.00082.x. [DOI] [PubMed] [Google Scholar]
- 32.Kanuri G, Spruss A, Wagnerberger S, et al. Fructose-induced steatosis in mice: role of plasminogen activator inhibitor-1, microsomal triglyceride transfer protein and NKT cells. Lab Invest. 2011;91:885–95. doi: 10.1038/labinvest.2011.44. [DOI] [PubMed] [Google Scholar]
- 33.Ma LJ, Mao SL, Taylor KL, et al. Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes. 2004;53:336–46. doi: 10.2337/diabetes.53.2.336. [DOI] [PubMed] [Google Scholar]
- 34.Rosselli MS, Burgueno AL, Carabelli J, et al. Losartan reduces liver expression of plasminogen activator inhibitor-1 (PAI-1) in a high fat-induced rat nonalcoholic fatty liver disease model. Atherosclerosis. 2009;206:119–26. doi: 10.1016/j.atherosclerosis.2009.01.026. [DOI] [PubMed] [Google Scholar]
- 35.Thomsen C, Becker U, Winkler K, et al. Quantification of liver fat using magnetic resonance spectroscopy. Magn Reson Imaging. 1994;12:487–95. doi: 10.1016/0730-725x(94)92543-7. [DOI] [PubMed] [Google Scholar]