

Abstract
In this manuscript we expand significantly on our earlier communication by investigating the bilayer self-assembly of eight different types of phospholipids in unbiased molecular dynamics (MD) simulations using three widely used all-atom lipid force fields. Irrespective of the underlying force field, the lipids are shown to spontaneously form stable lamellar bilayer structures within 1 microsecond, the majority of which display properties in satisfactory agreement with the experimental data. The lipids self-assemble via the same general mechanism, though at formation rates that differ both between lipid types, force fields and even repeats on the same lipid/force field combination. In addition to zwitterionic phosphatidylcholine (PC) and phosphatidylethanolamine (PE) lipids, anionic phosphatidylserine (PS) and phosphatidylglycerol (PG) lipids are represented. To our knowledge this is the first time bilayer self-assembly of phospholipids with negatively charged head groups is demonstrated in all-atom MD simulations.
Introduction
Biological membranes are omnipresent in the body and have a wide range of functions. It has been estimated that over 50% of all proteins interact with membranes.1 Membranes are also important in pharmacokinetics and -dynamics. Drug molecules usually have to penetrate membrane barriers to reach their site of action, and transmembrane proteins comprise a significant portion of the targets for marketed drugs.2,3 Detailed structural studies on membranes are therefore of high relevance. However, the fluid nature of biological membranes often complicates high-resolution experimental studies, providing a strong argument for theoretical simulations that can complement and build upon the experimental data.
Because of the duality in their chemical structure and the hydrophobic effect, phospholipids have the inherent ability to self-aggregate into lamellar bilayer structures, the fundamental structural basis of biological membranes. Thermodynamically, this is often the most favourable spatial arrangement for these amphiphilic lipids, allowing them to minimize the highly unfavourable contact between their long, aliphatic hydrocarbon tails and polar molecules by directing their hydrophilic head groups towards the aqueous surroundings.
Applying united-atom4–8 or coarse-grained9–13 models, the self-assembly of phospholipids has previously been demonstrated in molecular dynamics (MD) simulations, both the assembly into bilayers4,6–8,13 and vesicles5,11,13 and into bilayers formed around peptides,6,9 proteins9,12 and DNA.10 In a recently published communication,14 we showed for the first time bilayer self-assembly in unbiased MD simulations where all atoms are explicitly treated. Four types of zwitterionic phospholipids assembled from random configurations into stable bilayers characterized by structural properties in good agreement with experiment. The paper included a comparison between the AMBER Lipid1415 and the Charmm C3616 lipid force fields with regards to the self-assembly process and the properties of the resulting membranes.
In the present work we significantly expand upon the subject introduced in the communication14 through the inclusion of a broader range of lipids, a more comprehensive structural analysis of assembled bilayers (on the level of lipid force field validation papers) and the addition of a third all-atom lipid force field, Slipids.17–19 The selection of lipids has been extended to include four types of negatively charged phospholipids – palmitoyl-oleoyl-phosphatidylserine (POPS), palmitoyl-oleoyl-phosphatidylglycerol (POPG), dioleoyl-phosphatidylserine (DOPS) and dioleoyl-phosphatidylglycerol (DOPG) – which together with the original set (dipalmitoyl-phosphatidylcholine (DPPC), palmitoyl-oleoyl-phosphatidylcholine (POPC), dioleoyl-phosphatidylcholine (DOPC) and palmitoyl-oleoyl-phosphatidylethanolamine (POPE)) ensures that head groups of varying charge (zwitterionic/anionic) and size are represented, as well as hydrophobic tail portions with varying degrees of unsaturation. At the same time, the introduction of the Slipids force field means that three major all-atom lipid force fields are represented. The anionic head groups simulated here have, along with several other residues, been recently parameterized and added to the Lipid14 force field, whose module-based parameterizations will be published elsewhere.
All simulations were performed using version 14 of the AMBER molecular dynamics software suite.20,21 For each of the three force fields, a minimum of three repeats were performed for each lipid (with the exception of POPS which is not included in the Slipids force field) of 1 μs duration each. The total accumulated simulation time is 75 μs. In addition to providing a thorough comparison between the three major all-atom lipid force fields in view of self-assembly, this paper also contributes the first example of self-aggregation of negatively charged all-atom phospholipids into stable and structurally relevant bilayer structures. It also sheds some light on the influence of ion parameters on the self-assembly process.
Methods
Simulation conditions
All simulations were performed using version 14 of the AMBER molecular dynamics software suite,20,21 the GPU-accelerated AMBER PMEMD implementation22,23 and the SPFP precision model.24 Temperature was regulated by a Langevin thermostat25 with a 1.0 ps−1 collision frequency, and a reference pressure of 1.0 bar was maintained using the Berendsen coupling scheme.26 The SHAKE algorithm27 constrained the bond lengths involving hydrogen atoms. Periodic boundary conditions were employed, with the particle mesh Ewald (PME) method28 (4th order B-spline interpolation and a grid spacing of 1.0 Å) evaluating the electrostatic interactions. The direct space sum and the van der Waals interactions were truncated by an applied cut-off of 10 Å.
Self-assembly simulations
Each of the lipid systems in Table 1 was subjected to three simulation repeats with Lipid14 parameters. The same initial random configuration of lipids, ions and water was used in all three repeats, but different random seeds were generated in each case. The following strategy was applied:14 (I) 10000 steps of minimization; (II) 10 ns simulation at production temperature with isotropic pressure scaling (NPT) and a time step of 0.5 fs; (III) 10 ns simulation at production temperature with isotropic pressure scaling (NPT) and a time step of 1.0 fs; (IV) simulation at production temperature with anisotropic pressure scaling (NPT) and 2.0 fs time step. The production temperature (Table 1) was kept above the phase transition temperature of the relevant phospholipid across all three simulation steps. In step (IV) the simulation settings correspond to the ones applied in the production stage of the Lipid14 validation simulations.15
Table 1. Simulation system details.
| System | Number of lipids | TIP3P water/lipid ratio | Number of K+/Cl− | Number of Na+ | Simulation time per repeata (ns) | Simulation temperature (K) |
|---|---|---|---|---|---|---|
| DOPCb | 128 | 32.8 | 12/12 | — | 1000 | 303.0 |
| POPC | 128 | 31.0 | 11/11 | — | 1000 | 303.0 |
| POPE | 128 | 32.0 | 12/12 | — | 1000 | 310.0 |
| DPPCb | 128 | 30.1 | 11/11 | — | 1000 | 323.0 |
| POPS | 128 | 50 | — | 128 | 1000 | 303.0 |
| POPG | 128 | 50 | — | 128 | 1000 | 303.0 |
| DOPS | 128 | 50 | — | 128 | 1000 | 303.0 |
| DOPG | 128 | 50 | — | 128 | 1000 | 303.0 |
For each of the three lipid force fields – Lipid14, Charmm C36 and Slipids – three simulation repeats of 1 μs duration each were run on each system.
Three additional 1 μs repeats were performed for the C36 DPPC and the C36 DOPC systems using a van der Waals force switch function over 8 to 12 Å (all other simulations were run with a strict 10 Å cut-off).
Systems with the same number of lipids, water and ions as listed in Table 1 but described by Charmm C36 parameters16 were generated and converted to AMBER topology and coordinate files by means of the CHAMBER program from AmberTools v14.29 In terms of the anionic C36 systems, modified Lennard-Jones radii for the interaction between sodium ions and lipid oxygen atoms30 were subsequently introduced into the topology files using the ParmEd module of AmberTools v14.20
The C36 lipid force field also functioned as the starting point for the parameterizations that became the Slipids force field.17–19 The two force fields share the same nomenclature and many of the parameters, including all the bond and angle parameters as well as Lennard-Jones and torsional parameters for the glycerol portion and the head groups. Slipids has been made available in the Gromacs format31 and was ported to AMBER by (i) introducing the Slipids-specific bonded and non-bonded parameters (except for 1–4 van der Waals parameters) into the C36 force field parameter files; (ii) generating new Slipids-specific psf files in Charmm32 from all the C36 systems already created (see above) except for POPS, which is not included in Slipids; (iii) converting the psf files to AMBER topology files using CHAMBER;29 (iv) introducing the Slipids 1–4 van der Waals interaction parameters as well as AMBER 1–4 scaling factors for van der Waals and electrostatic interactions (as used in Slipids17–19) using ParmEd.20 The C36 Charmm-to-AMBER and Slipids Gromacs-to-AMBER lipid parameter conversions were verified by comparison of the single point energies calculated in AMBER, Charmm and Gromacs.
In accordance with the Slipids validation simulations of anionic phospholipids,19 AMBER ff99 Na+ parameters33 were used in the anionic Slipids self-assembly systems. No ions were present in the original Slipids simulations of zwitterionic lipids,17,18 but in order to be consistent with the use of KCl in the Lipid14 and C36 simulations and with the fact that ff99 sodium ion parameters are used in Slipids,19 ff99 parameters were applied for the potassium33 and chloride34 ions in the self-assembly simulations of zwitterionic Slipids. In three simulation repeats per lipid, the same procedure used for the Lipid14 systems was followed for the C36 and Slipids systems and the same simulation settings were applied.
Analysis
In most of the simulations the lipids partitioned asymmetrically between the two leaflets of the self-assembled bilayer (Table 2a). Hence the area per lipid (AL) was calculated by doubling the lateral area of the simulation box (Abox) and dividing by the total number of lipids (nlipid):
Table 2. (a) Formation time, number of lipids per leaflet, area and volume per lipid for self-assembled bilayers (b) thicknesses and isothermal compressibility moduli for the self-assembled bilayers.
| (a) | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||||||
| Bilayer formation timea (ns) | No. of lipids per leaflet | Area per lipidd (Å2) | Volume per lipidd (Å3) | ||||||||||||||||
|
|
|
|
|
||||||||||||||||
| C36b | C36 | C36 | C36 | ||||||||||||||||
|
|
|
|
|
||||||||||||||||
| Lipid | Sim. no. |
Lipidl4 | cut | fsw | Slipidsc | Lipidl4 | cut | fsw | Slipids | Lipidl4 | cut | fsw | Slipids | Exp. | Lipidl4 | cut | fsw | Slipids | Exp. |
| DOPC | 1 | 150 | 135 | 152 | 246 | 66/62 | 61/67 | 64/64 | 65/63 | 69.3 ± 1.2 | 67.8 ± 1.2 | 69.1 ± 1.1 | 69.1 ± 1.0 | 67.4,39 72.544 | 1251.5 ± 4.4 | 1238.1 ± 4.2 | 1280.9 ± 4.8 | 1271.7 ± 4.4 | 130344 |
| 2 | 285 | 145 | 385 | 717 | 62/66 | 67/61 | 64/64 | 64/64 | 69.2 ± 1.1 | 67.8 ± 1.1 | 69.0 ± 1.1 | 69.2 ± 1.1 | 1251.5 ± 4.4 | 1238.1 ± 4.2 | 1280.9 ± 4.7 | 1271.6 ± 4.4 | |||
| 3 | 720 | 160 | — | 941 | 63/65 | 65/63 | — | 61/67 | 69.0 ± 1.2 | 67.6 ± 1.1 | — | 69.7 ± 1.0 | 1251.3 ± 4.4 | 1238.1 ± 4.2 | — | 1271.7 ± 4.5 | |||
| POPC | 1 | 375 | 160 | 94 | 64/64 | 66/62 | 63/65 | 65.5 ± 1.2 | 63.8 ± 1.2 | 66.5 ± 1.1 | 64.3,40 68.341 | 1207.3 ± 4.3 | 1191.9 ± 4.2 | 1224.8 ± 4.3 | 125641 | ||||
| 2 | 535 | 325 | 228 | 63/65 | 66/62 | 67/61 | 65.7 ± 1.3 | 63.7 ± 1.2 | 66.6 ± 1.1 | 1207.4 ± 4.3 | 1191.7 ± 4.2 | 1224.7 ± 4.4 | |||||||
| 3 | 755 | 425 | — | 68/60 | 62/66 | — | 65.6 ± 1.3 | 63.8 ± 1.1 | — | 1207.1 ± 4.4 | 1191.9 ± 4.1 | — | |||||||
| POPE | 1 | 70 | 95 | 89 | 62/66 | 61/67 | 69/59 | 56.0 ± 1.1 | 56.9 ± 1.1 | 60.7 ± 1.0 | 56.6,49 58,43 59–6050 | 1141.1 ± 4.4 | 1134.9 ± 4.3 | 1171.5 ± 4.5 | 1175.1,43 118050 | ||||
| 2 | 100 | 115 | 153 | 63/65 | 62/66 | 58/70 | 56.3 ± 1.1 | 56.5 ± 1.1 | 60.8 ± 1.0 | 1141.2 ± 4.5 | 1134.7 ± 4.5 | 1171.3 ± 4.5 | |||||||
| 3 | 125 | 205 | 245 | 71/57 | 67/61 | 63/65 | 57.2 ± 1.3 | 56.9 ± 1.1 | 59.8 ± 1.1 | 1140.4 ± 4.8 | 1135.1 ± 4.3 | 1171.4 ± 4.5 | |||||||
| DPPC | 1 | 230 | 35 | 89 | 75 | 65/63 | 66/62 | 64/64 | 62/66 | 62.2 ± 1.4 | 54.4 ± 0.6 | 62.3 ± 1.2 | 65.0 ± 1.1 | 63.1,40 64.342 | 1178.8 ± 5.1 | 1099.6 ± 4.6 | 1203.2 ± 5.2 | 1196.1 ± 4.6 | 123244 |
| 2 | 350 | 85 | 117 | 157 | 64/64 | 64/64 | 63/65 | 66/62 | 62.3 ± 1.3 | 52.2 ± 0.6 | 62.4 ± 1.3 | 65.0 ± 1.1 | 1179.4 ± 4.9 | 1098.8 ± 4.6 | 1203.3 ± 5.3 | 1196.2 ± 4.6 | |||
| 3 | 440 | 325 | 123 | 302 | 60/68 | 62/66 | 63/65 | 63/65 | 62.3 ± 1.4 | 54.8 ± 0.7 | 62.2 ± 1.2 | 64.7 ± 1.2 | 1178.7 ± 5.0 | 1102.6 ± 4.6 | 1203.1 ± 5.2 | 1196.1 ± 4.6 | |||
| POPS | 1 | 70 | 136 | N/A | 64/64 | 68/60 | N/A | 58.2 ± 1.2 | 57.5 ± 1.2 | N/A | 62.745 | 1147.1 ± 4.8 | 1120.5 ± 4.8 | N/A | 1198.545 | ||||
| 2 | 84 | 156 | N/A | 63/65 | 63/65 | N/A | 58.4 ± 1.4 | 57.7 ± 1.3 | N/A | 1147.2 ± 4.7 | 1121.6 ± 4.7 | N/A | |||||||
| 3 | 160 | — | N/A | 62/66 | — | N/A | 58.1 ± 1.1 | — | N/A | 1146.9 ± 4.8 | — | N/A | |||||||
| POPG | 1 | 47 | 106 | 90 | 64/64 | 61/67 | 66/62 | 66.7 ± 1.3 | 67.2 ± 1.4 | 68.9 ± 1.3 | 64.3,47 66.146 | 1163.9 ± 4.7 | 1151.0 ± 4.6 | 1192.6 ± 4.8 | 1208.746 | ||||
| 2 | 106 | 330 | 133 | 64/64 | 64/64 | 64/64 | 66.7 ± 1.3 | 67.4 ± 1.3 | 68.8 ± 1.3 | 1164.1 ± 4.8 | 1151.2 ± 4.6 | 1192.5 ± 4.8 | |||||||
| 3 | 155 | 350 | 856 | 62/66 | 62/66 | 65/63 | 66.8 ± 1.3 | 67.3 ± 1.6 | 68.7 ± 1.3 | 1163.9 ± 4.7 | 1151.0 ± 4.6 | 1192.5 ± 4.8 | |||||||
| DOPS | 1 | 46 | 79 | 98 | 65/63 | 65/63 | 61/67 | 63.7 ± 1.0 | 63.1 ± 1.3 | 66.3 ± 1.1 | 64.148 | 1191.0 ± 4.7 | 1170.6 ± 4.6 | 1229.1 ± 4.9 | 122848 | ||||
| 2 | 53 | 127 | 99 | 69/59 | 63/65 | 62/66 | 64.3 ± 1.2 | 63.3 ± 1.2 | 65.8 ± 1.2 | 1191.0 ± 4.8 | 1170.8 ± 4.6 | 1229.1 ± 4.9 | |||||||
| 3 | 68 | 242 | 107 | 67/61 | 65/63 | 67/61 | 63.9 ± 1.1 | 62.8 ± 1.2 | 65.9 ± 1.2 | 1191.0 ± 4.7 | 1170.7 ± 4.6 | 1229.2 ± 4.9 | |||||||
| DOPG | 1 | 63 | 251 | 86 | 67/61 | 62/66 | 64/64 | 70.6 ± 1.2 | 71.2 ± 1.3 | 71.8 ± 1.2 | 69.1,47 70.846 | 1206.7 ± 4.8 | 1198.4 ± 4.6 | 1238.6 ± 4.9 | 126546 | ||||
| 2 | 66 | 323 | 90 | 65/63 | 62/66 | 63/65 | 70.5 ± 1.3 | 70.9 ± 1.2 | 71.6 ± 1.2 | 1206.8 ± 4.8 | 1198.3 ± 4.5 | 1238.5 ± 4.9 | |||||||
| 3 | 202 | 448 | 326 | 62/66 | 65/63 | 67/61 | 70.6 ± 1.3 | 71.0 ± 1.4 | 72.0 ± 1.2 | 1206.8 ± 4.8 | 1198.4 ± 4.6 | 1238.6 ± 4.9 | |||||||
|
| |||||||||||||||||||
| (b) | |||||||||||||||||||
|
| |||||||||||||||||||
| Bilayer thickness DHHh (Å) | Luzzati thickness DBh (Å) | Isothermal compressibility modulus KAh (mN m−1) | |||||||||||||||||
|
|
|
|
|||||||||||||||||
| C36f | C36f | C36f | |||||||||||||||||
|
|
|
|
|||||||||||||||||
| Lipid | Sim. no.e |
Lipidl4 | cut | fsw | Slipidsg | Exp. | Lipidl4 | cut | fsw | Slipidsg | Exp. | Lipidl4 | cut | fsw | Slipidsg | Exp. | |||
|
| |||||||||||||||||||
| DOPC | 1 | 35.3,53 36.7,39 | 35.9,44 36.1,52 | 265,57 300,56 | |||||||||||||||
| 2 | 37.3 ± 0.3 | 37.9 ± 0.4 | 38.1 ± 0.2 | 36.0 ± 0.4 | 36.9,44 37.152 | 36.2 ± 0.1 | 36.6 ± 0.1 | 37.1 ± 0.0 | 36.7 ± 0.2 | 38.739 | 320 ± 30 | 350 ± 10 | 370 ± 40 | 430 ± 40 | 31855 | ||||
| 3 | |||||||||||||||||||
| POPC | 1 | 36.8,41 | |||||||||||||||||
| 2 | 37.3 ± 0.0 | 38.3 ± 0.3 | 36.3 ± 0.4 | 3741 | 36.8 ± 0.1 | 37.4 ± 0.0 | 36.8 ± 0.0 | 39.140 | 270 ± 30 | 300 ± 20 | 360 ± 40 | 180-33054 | |||||||
| 3 | |||||||||||||||||||
| POPE | 1 | ||||||||||||||||||
| 2 | 41.9 ± 0.4 | 41.1 ± 0.4 | 37.7 ± 0.5 | 39.550 | 40.4 ± 0.4 | 40.0 ± 0.2 | 38.8 ± 0.4 | 40.543 | 290 ± 40 | 310 ± 0 | 370 ± 10 | 23349 | |||||||
| 3 | |||||||||||||||||||
| DPPC | 1 | ||||||||||||||||||
| 2 | 37.8 ± 0.1 | 43.8 ± 2.4 | 39.1 ± 0.4 | 36.2 ± 0.1 | 38,39 38.344 | 37.9 ± 0.0 | 40.9 ± 1.0 | 38.6 ± 0.1 | 36.9 ± 0.1 | 39.040 | 230 ± 20 | 930 ± 30 | 290 ± 20 | 350 ± 20 | 23144 | ||||
| 3 | |||||||||||||||||||
| POPS | 1 | N/A | N/A | N/A | — | ||||||||||||||
| 2 | 42.3 ± 0.1 | 42.4 ± 0.2 | N/A | 42.245 | 39.4 ± 0.1 | 39.0 ± 0.1 | N/A | 38.245 | 250 ± 50 | 250 ± 20 | N/A | ||||||||
| 3 | N/A | N/A | N/A | ||||||||||||||||
| POPG | 1 | 36.6,46 | — | ||||||||||||||||
| 2 | 36.8 ± 0.0 | 36.3 ± 0.0 | 34.8 ± 0.3 | 37.351 | 34.9 ± 0.0 | 34.2 ± 0.0 | 34.7 ± 0.1 | 37.647 | 270 ± 10 | 220 ± 40 | 260 ± 10 | ||||||||
| 3 | |||||||||||||||||||
| DOPS | 1 | — | |||||||||||||||||
| 2 | 40.6 ± 0.1 | 41.3 ± 0.3 | 38.1 ± 0.1 | 39.048 | 37.3 ± 0.2 | 37.1 ± 0.1 | 37.3 ± 0.1 | 38.348 | 340 ± 40 | 270 ± 20 | 320 ± 10 | ||||||||
| 3 | |||||||||||||||||||
| DOPG | 1 | 35.7,46 | — | ||||||||||||||||
| 2 | 36.3 ± 0.3 | 35.9 ± 0.1 | 34.6 ± 0.1 | 34.2 ± 0.0 | 33.8 ± 0.1 | 34.5 ± 0.1 | 36.647 | 290 ± 10 | 280 ± 40 | 320 ± 20 | |||||||||
| 3 | |||||||||||||||||||
Both the Lipidl4, C36 and Slipids repeats for each lipid type are sorted in ascending order based on bilayer formation time.
cut refers to C36 simulations performed with a strict 10 Å cut-off, and fsw refers to C36 simulations run with a van der Waals force switch function over 8 to 12 Å. The lipids did not fully assemble into bilayers within 1 μs of simulation time in the last C36 DOPC fsw repeat and in the last C36 POPS cut-off repeat.
N/A refers to the fact that POPS is not included in the Slipids force field. The lipids did not fully assemble into bilayers within 1 μs of simulation time in the last Slipids POPC repeat. The last Slipids DOPC and POPG repeats were prolonged to 1100 ns and 1010 ns, respectively, in order to obtain at least 100 ns of simulation time for analysis of bilayer properties (see footnote d).
Areas and volumes per lipid are given as average ± standard deviation and were calculated from the interval from 50 ns after bilayer was fully formed until 1 μs of total simulation time. The exceptions are the three C36 DPPC repeats run with strict cut-off, for which analyses were done on the portion of each simulation where the overly ordered structure (described in the main text and visualized in Fig. S3, ESI) had been adopted.
Repeats listed in the same order as in (a).
cut refers to C36 simulations performed with a strict 10 Å cut-off, and fsw refers to C36 simulations run with a van der Waals force switch function over 8 to 12 Å. The lipids did not fully assemble into bilayers within 1 μs of simulation time in the last C36 DOPC fsw repeat and in the last C36 POPS cut-off repeat; these repeats are therefore not included in the relevant calculated averages.
N/A refers to the fact that POPS is not included in the Slipids force field. The lipids did not fully assemble into a bilayer within 1 μs of simulation time in the last Slipids POPC repeat; this repeat is therefore not included in the relevant calculated averages. The last Slipids DOPC and POPG repeats were prolonged to 1100 ns and 1010 ns, respectively, in order to obtain at least 100 ns of simulation time for the analysis of bilayer properties (see footnote h and (a)).
Properties given as the average across repeats ± standard deviation, where the average value from each individual repeat was calculated from the interval from 50 ns after bilayer was fully formed until 1 μs of total simulation time. The exceptions are the three C36 DPPC repeats run with strict cut-off, for which analyses were done on the portion of each simulation where the overly ordered structure (described in the main text and visualized in Fig. S3, ESI) had been adopted.
| (1) |
The volume per lipid (VL) was obtained using the following equation:15,35
| (2) |
Vbox is the volume of the simulation box, nw corresponds to the number of water molecules and Vw is the temperature-dependent volume of a TIP3P water molecule.
Bilayer thickness (DHH) refers to the distance between the phosphate peaks in the time-averaged electron density profile calculated from the simulation trajectory. Subtracting the integral of the probability distribution of the water density (ρw(bn)) along the bilayer normal dimension (bn) from the time-averaged bilayer normal dimension dbn gave rise to the Luzzati thickness (DB):8,15,17
| (3) |
Deuterium order parameters (SCD) quantify the degree of order in the aliphatic acyl chains comprising the hydrophobic core region of a bilayer, with lower values implying more disorder. Ensemble- and time-averaged order parameters for the assembled bilayers were calculated as a function of θ, the angle between the C–H vector of a carbon atom in the acyl chain and the bilayer normal, using
| (4) |
The order parameters were averaged over the two C–H bonds for each carbon atom along the aliphatic tail and then averaged across all repeats for each lipid/force field combination, producing the profiles presented in Fig. 2 and Fig. S4 and S5 (ESI†).
Fig. 2.
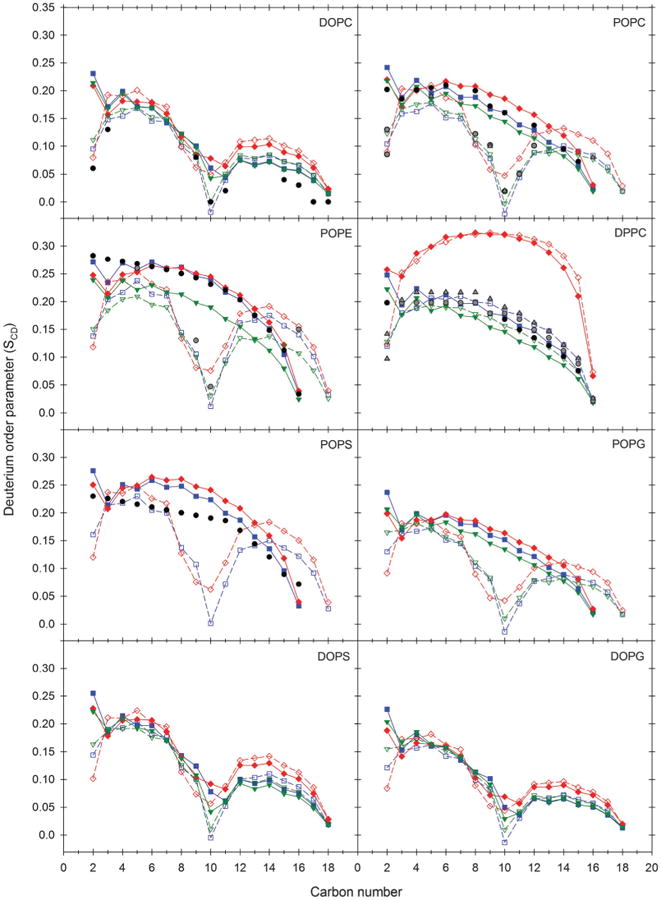
Deuterium order parameters (SCD) for self-assembled bilayers and comparison with experiment. Simulation values for each lipid/force field combination were calculated as averages across all repeats. The Lipid14 profiles are shown as blue squares, Charmm C36 (simulated with strict cut-off and denoted cut in Table 2a and b) as red diamonds and Slipids as downward green triangles. The sn-1 acyl chain is indicated by filled symbols and solid lines, while sn-2 is represented by open symbols and dashed lines. For each repeat, the analysis was done on the interval from 50 ns after the bilayer was fully formed to the end of the simulation. Experimental data,58–64 where available, are given as black spheres for the sn-1 and gray spheres or upward triangles for the sn-2 acyl chain.
Isothermal compressibility moduli (KA) were derived by inserting the Boltzmann constant (kB), the simulation temperature (T), the mean area per lipid (〈AL〉), the variance of the area per lipid (σA2) and the number of lipids (nlipid) into eqn (5):15, 35
| (5) |
X-ray and neutron scattering form factors were calculated from the simulations by Fourier transformation of electron density profiles using the SIMtoEXP software.36 Since electron densities along the bilayer normal form the basis of these calculations, asymmetry will affect the resulting form factor profiles. For each lipid/force field combination, the repeat with the most symmetrical inter-leaflet lipid distribution or, if equal symmetries, the repeat with the shortest bilayer formation time (Table 2a) was used for generating the simulation profiles in Fig. S1, S2 and S4 (ESI†). Asymmetrical distributions up to a ratio of 66/62 were found to influence the form factor profiles only marginally, so the plots representing the three force fields should still be comparable for each lipid.
The bulk of the analyses described above was conducted using PTRAJ/CPPTRAJ.20,37 Snapshots from the simulations were generated in VMD.38
Results/discussion
Self-assembly mechanism
All the phospholipids in the present work (Table 1) showed the ability to self-assemble into bilayers in simulations irrespective of the underlying all-atom lipid force field. POPS, which is not included in the Slipids force field, formed bilayers when described using Lipid14 or Charmm C36. The lipids aggregated into bilayers within 1 μs of simulation time in all but two repeats (one Slipids POPC and one C36 POPS repeat, see Table 2a). In general the self-assembly process followed the same general mechanism as was described in our previous work,14 the characteristic stages of which are presented in Fig. 1. Starting from an initial random “solution” of lipids, ions and water (stage 1), the hydrocarbon tails aggregate to form one big micelle-like lipid assembly within tens of nanoseconds. “Lipid bridges” are present in the interface between the lipid assembly and its periodic images (stage 2). Subsequently the lipid bridge phospholipids are inserted into the lipid assembly, resulting in a water pore-containing lamellar configuration (stage 3). When the lipid head groups lining the pore have retreated from the hydrophobic interior of the lamellar lipid structure and into the water–lipid interface, a bilayer has been fully formed (stage 4). The mechanism is consistent with what has been shown in united-atom self-assembly simulations.4,7,8 However, it should be noted that in some of the fastest self-assembly processes there is significant overlap between the stages, making it difficult to distinguish between them.
Fig. 1.
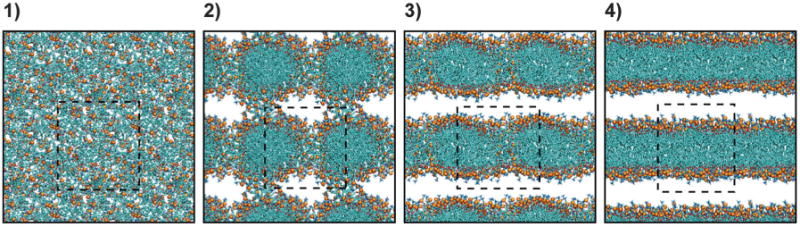
General mechanism of all-atom bilayer self-assembly. Four characteristic stages were observed during the self-assembly process (see main text for details) and are illustrated here by representative snapshots from one of the simulations. Phospholipids are shown as stick models, with the phosphorus atoms in the constituent head groups represented by orange spheres. Water, ions and hydrogen atoms have been omitted for clarity. Please note that the snapshots include portions of neighbouring periodic images in addition to the simulation unit cell, which is indicated by dashed-lined squares.
Visualization of the individual simulations reveals a difference between C36 versus Lipid14 and Slipids. In a significant proportion of the simulations of C36 PC lipids – in two DOPC, two POPC and all three DPPC repeats – pore closure was finalized before all the lipid bridge phospholipids were incorporated into the lipid assembly. However, this scenario occurs in only one of the corresponding Lipid14 repeats and in one of the eight Slipids PC simulations in which a bilayer was formed (Table 2a). The Lipid14/Slipids simulations appear to be more in line with united-atom self-assembly mechanisms where the closure of the water pore is characterized as the last and often time-limiting step in the bilayer formation.4,7,8 The discrepancy may in part be related to cut-off conditions. Consistent with the Lipid14 and Slipids simulations in the present work, the C36 lipids were simulated using a strict van der Waals cut-off (Table 1, denoted cut in Table 2a and b), while force switching schemes were applied in the original validation of the C36 force field.16 As will be discussed in detail later, additional C36 DPPC and DOPC simulations were performed using a similar force switch function as in the original C36 paper (Table 1, denoted as fsw in Table 2a and b). One out of three DPPC force switch repeats showed the possibly premature pore closure compared to all three repeats with the cut-off, suggesting that the treatment of van der Waals forces might influence not only bilayer properties but also the self-assembly pathway in simulations and might have contributed to the observed difference in mechanism. Nonetheless, three out of the six force switch simulations still displayed the early pore closure described above.
There are large variations in bilayer formation times (Table 2a), both between phospholipids, between force fields and between repeats for a specific lipid using the same force field. Substantial differences in bilayer formation times have also been established in self-assembly studies using united-atom models.6–8 Drawing any conclusions is therefore difficult, but certain trends can be identified and perhaps more so for Lipid14 than for the other two force fields. In terms of the Lipid14 zwitterionic lipids in Table 2a the rate of self-assembly appears to be higher for POPE than for POPC. One explanation might be that the phosphatidylcholine head group is bigger and bulkier than phosphatidylethanolamine and therefore faces more significant steric challenges upon retreating from the hydrophobic region of the lipid assembly to the lipid-water interface. Indeed, Marrink et al.7 pointed to steric hindrance as a plausible factor contributing to lengthy pore lifetimes. Secondly the timings for Lipid14 suggest that also the anionic PS and PG lipids self-assemble faster than the PC lipids. Electrostatically and in light of the hydrophobic effect it seems reasonable that charged, more polar anionic head groups escape the hydrophobic environment more easily than the neutral zwitterionic PC head groups. The results indicate that the rate of Lipid14 self-assembly is dependent on both the size and charge of the head group for the phospholipids under investigation. However, it is important to mention that the ion concentration was significantly higher in the simulations of the anionics than was the case for the zwitterionics (Table 1), which might have had an influence.
Structural properties of self-assembled bilayers
Self-assembled bilayers were allowed to relax and equilibrate for 50 ns, and the remaining portion of each of the 1 microsecond simulations was used for calculating average structural bilayer properties. These properties are featured together with experimental counterparts in Table 2a and b and Fig. 2 and include areas and volumes per lipid,39–50 bilayer (DHH) and Luzzati (DB) thicknesses,39–41,43–48,50–53 isothermal compressibility moduli (KA)44,49,54–57 and deuterium order parameter (SCD) profiles.58–64 Of the most robust structural data to validate lipid simulations against are X-ray and neutron scattering form factors directly derived from experiments that can be directly compared to simulation without requiring modelling or fitting of the experimental data.36,40 X-ray and neutron form factor profiles are presented in Fig. S1 and S2 (ESI†), respectively, for the lipids for which experimental data are available.
Overall, all three force fields give good agreement with experimental observables for the assembled bilayers. The notable exception is DPPC modeled with the C36 force field and a non-bonded cut-off of 10 Å, where the bilayers in all three repeats eventually adopt a highly ordered configuration with partial overlap between the tails from opposite leaflets (Fig. S3, ESI†). The resulting static and compressed nature of these bilayers is reflected by low areas and volumes per lipid, very high KA values, overestimated thicknesses, very high order parameters and misplaced form factor profiles. Various reasons were considered for this behaviour, the main ones being the use of a strict van der Waals cut-off and asymmetry in the distribution of lipids between the two leaflets. The latter is unlikely given that one of the three DPPC repeats showed a symmetrical lipid distribution (Table 2a) while the former has been raised as a concern in correspondence with the C36 authors. In respect of the observed anomalous behaviour of DPPC, three additional C36 DPPC self-assembly repeats (1 μs each) were performed but this time using a force switch cut-off scheme recently implemented in AMBER (Table 1, and denoted as fsw in Table 2a and b). A force switch function over 8 to 12 Å for the van der Waals forces, as in the original C36 validation paper,16 replaced the 10 Å cut-off applied for all other systems. Notably, this change in cut-off conditions resulted in a 28% decrease in simulation speed on a GeForce GTX TITAN X card (∼41 versus ∼57 ns per day). The overly ordered configuration described above did not appear during the course of any of the force switch repeats, and the structural properties of the self-assembled bilayers agree well with experimental observables (Table 2a and b and Fig. S4, ESI†). Furthermore, the area per lipid, bilayer thickness and order parameters for both tails are close to the corresponding values reported for 420 ns simulations of DPPC with force switching applied in Charmm.65,66 These observations suggest that C36 DPPC simulation requires force switching of van der Waals interactions and is highly sensitive to changes in cut-off conditions, particularly to the use of a strict cut-off, and more so than its Slipids counterpart (originally validated with a switch function over 14 to 15 Å17). To verify that sensitivity to modifications in cut-off scheme may be less of an issue with the other C36 lipids and that the force switch does not drastically change bilayer properties compared to the 10 Å cut-off simulations, three C36 DOPC self-assembly repeats were run with the same force switch function as for DPPC. As we will detail later on in this work there is only slight perturbation of calculated properties for C36 DOPC with and without the application of the force switch. It is, perhaps, worth reiterating that for seven of the eight C36 lipids and all seven of the Slipids investigated the vast majority of the experimental observables are reproduced to a high degree of fidelity with a 10 Å cut-off applied.
For the other lipid/force field combinations, areas per lipid, often the first port of call in lipid bilayer structural analysis, are generally close to the value or within the range of values determined experimentally for all the lipids, though slightly underestimated for POPS. SCD values for the carbon atoms along the palmitoyl chain of POPS lie higher than the experimental profile (Fig. 2), indicating that both the Lipid14 and C36 POPS bilayers are slightly too ordered (POPS is not included in the Slipids force field).
The level of agreement with experiment for the volumes per lipid in Table 2a follows the order Slipids > Lipid14 > C36 (simulated with strict cut-off), but all three force fields qualitatively capture the differences in volume across the collection of simulated lipids. With the force switch, the volumes per lipid for C36 DPPC and DOPC are closest to the corresponding experimental values relative to Lipid14/Slipids. The isothermal compressibility moduli for the Slipids zwitterionic bilayers are significantly overestimated while Lipid14 and C36 KA values are more in line with the available experimental data. The differences arise from lower variances in area per lipid in the Slipids simulations, which in turn elevate KA (see eqn (5)). While the Luzzati thicknesses for the most part are similar across the three force fields and are in reasonable agreement with experiment, Slipids DHH thicknesses are consistently lower than the Lipid14 and C36 counterparts across all the lipids. In the cases where these differences are most pronounced, i.e. for POPE and the PG lipids, X-ray form factor minima also move towards higher q values compared to the Lipid14, C36 and experimental profiles, which correspond to thinner bilayers (Fig. S1, ESI†).
Fig. 2 shows that Lipid14 provides very good agreement with the experimental SCD profiles for the zwitterionic lipids. While the Slipids order parameters nearly overlap with Lipid14 for DOPC and for the oleoyl chain of POPC, a higher degree of disorder than observed experimentally is spotted for DPPC, POPE and the sn-1 palmitoyl chain of POPC. That the Slipids and Lipid14 profiles appear to be in closer proximity for the unsaturated than for the saturated tails also holds true for the anionic PG and DOPS lipids, for which no experimental data were found in the literature. The C36 lipids (simulated with the strict cut-off) tend to be more ordered than their Lipid14 equivalents, at least along portions of the acyl chains.
All the X-ray form factor profiles calculated for the self-assembled bilayers – when considering the force switch results for C36 DPPC (Fig. S4, ESI†) – capture the characteristics of those derived experimentally to a high degree, both in terms of the placement and magnitude of the different lobes, with Lipid14 arguably the most consistent among the three force fields (Fig. S1, ESI†). The simulation neutron form factors in Fig. S2 (ESI†) also reproduce the experimental data well. In summary, the self-assembled bilayers largely exhibit properties in accordance with those determined experimentally, implying that all three force fields are capable of reproducing the structural features of pure bilayers made up of biologically relevant phospholipids.
Comparisons with results in the original Slipids papers17–19 and previous Charmm C36 validations16,30,65 also indicate that our conversions of the lipid parameters to AMBER format are valid. Areas per lipid calculated for the Slipids PC and anionic lipids are about 1–2 Å2 higher than those in the original papers,17–19 and the other properties are consistent with the self-assembled bilayers being slightly more disordered. These discrepancies can be ascribed to differences in cut-off schemes. A longer van der Waals cut-off of 15 Å (with a force switch function starting at 14 Å) was used in the Slipids validation simulations, and it has been recently demonstrated in bilayer simulations that the area per lipid increases in a systematic fashion as the van der Waals cut-off decreases.67 For Slipids POPE, the area is 3.5–4.5 Å2 higher than in the Slipids validation.18 In addition to the cut-off difference, a 7 K higher temperature in the self-assembly simulations (310 versus 303 K) could have contributed to the increased disorder of the POPE bilayers.
Of the phospholipids represented in the present work, the zwitterionic lipids,16,65 POPG65 and POPS30 have previously been validated by bilayer simulations with the C36 force field. As discussed above, DPPC simulated with the force switch function provides good agreement with earlier simulations of C36 DPPC bilayers in Charmm. The POPG, POPS and DOPC bilayers self-assembled using the 10 Å non-bonded cut-off display areas that are close to and only 0.7–1.4 Å2 lower than the C36 validations. The SCD order parameters for the palmitoyl chain of POPS and for both tails of DOPC (Fig. 2) are also very similar to the ones reported for Charmm simulations.30,65 While the force switch improves the area and volume per lipid of DOPC, the other bilayer properties remain largely the same as those derived from the cut-off simulations (see below). Areas per lipid calculated for POPC simulated with cut-off are just 1 Å below the value in the original C36 force field paper,16 but 2.2 Å lower than the area derived from a more recent Charmm simulation.65 The areas per lipid from the C36 POPE self-assembly simulations are around 2 to 2.4 Å2 below C36 validation results.16,65 The discrepancies can be explained by slight differences in simulation conditions relative to the C36 validation simulations. In the original C36 paper16 Klauda et al. reported two areas per lipid for DPPC of 62.9 Å2 and 59.1 Å2 derived using Charmm and NAMD, respectively, and argue that the 3.8 Å2 difference, which is greater than the area per lipid divergences described above, resulted from minor differences in simulation conditions. To conclude, our C36 Charmm-to-AMBER and Slipids Gromacs-to-AMBER lipid parameter conversions and self-assembly simulation settings appear to be reasonable.
As mentioned above we ran three repeats of C36 DOPC using the same force switch as for DPPC. While the lipids did not form a bilayer within 1 μs of simulation time in one of the repeats, bilayers were formed after 152 and 385 ns in the other two repeats. The average structural properties computed for these two membranes are presented in Table 2a and b (denoted as fsw) and Fig. S5 (ESI†). The area per lipid is roughly 1.3–1.5 Å2 higher than with the 10 Å cut-off (Table 2a), an increase ascribable to the change in cut-off conditions. At the same time the area is in excellent agreement with the values derived previously from DOPC simulations with the C36 force field.16,65 Discrepancies in volume per lipid with the force switch relative to the cut-off simulations (Table 2a) are directly related to the area per lipid increase, while the bilayer and Luzzati thicknesses as well as the isothermal compressibility modulus are close to the values obtained with the strict cut-off (Table 2b). Direct comparison of the SCD order parameters between the two cut-off approaches (Fig. S5, ESI†) reveals only minor differences. The two order parameter profiles overlap very well from carbon number 2 to 11, beyond which the acyl chains in the force switch repeats are marginally more disordered. Compared to a 10 Å cut-off, the application of a force switch over 8 to 12 Å does not significantly change the properties of self-assembled C36 DOPC bilayers beyond what can be expected from the change in cut-off scheme.
Areas per lipid, volumes per lipid and thicknesses computed for the Lipid14 zwitterionic lipids are very close to the averages reported in the original validation of the Lipid14 force field,15 and the SCD order parameter and form factor profiles also match very well. Interestingly the Lipid14 isothermal compressibility moduli in Table 2b generally show better agreement with experiment relative to the Lipid14 validation results.15 Such bilayer characteristics might affect the interplay between the phospholipids and other molecules. Our results suggest that self-assembly may be a more effective strategy than starting simulations from preformed bilayers in some cases, particularly when the aim is to introduce proteins or other interaction partners into the membrane environment. Indeed, self-assembly of united-atom or coarse-grained phospholipids around peptides and proteins has been performed as an unbiased approach to obtain protein/membrane complexes and for predicting the position of proteins or peptides in bilayers,6,9,12 as opposed to inserting them “manually” into premade bilayers before simulation. Nevertheless, full atomic resolution might be required for accurately modelling the interactions between the membrane proteins and the surrounding self-assembled lipid environment. In terms of the lipid component in membranes, mixtures of different types of lipids are potentially important targets for self-assembly simulation strategies. Simulations of all-atom lipid mixtures carefully validated against experimental data represent crucial steps on the path towards the ultimate goal of simulating realistic biological membranes. Yet it is difficult to predict the “real” inter-leaflet distribution of each lipid type when building the model bilayer, and the high-energy barrier to lipid flip-flop prevents “equilibration” of the distribution. Self-assembly would help attenuate any bias caused by the starting configuration.
Influence of ion parameters
Ions and choice of ion parameters can influence lipid bilayer properties in simulations, especially when anionic lipids are among the membrane constituents and high concentrations of positively charged ions are used.30,68 Lipid14, together with the various AMBER ion parameters, is no exception in that regard. Monovalent counterions described by parameters recently developed by Joung and Cheatham69 have been found to condense anionic Lipid14 bilayers to areas per lipid well below experimental values (results not shown) due to strong interactions with the negatively charged lipid head groups. The condensing effect is avoided by using the older Amber ff99 sodium parameters,33 as a greater Lennard-Jones radius for the sodium ions most likely prevents them from engaging in strong interactions within the lipid–water interface region.
Without counterions, each of the anionic lipid systems in Table 1 would give a total charge of −128. The application of counterions ensures that the system is neutral, which is a prerequisite for PME,28 but also results in an unrealistic system setup that is far from experimental conditions. At the same time, experimental data for anionic lipids have generally been obtained “in the absence of salt”. As such, using the ff99 ions provides better agreement with experiment as the ions to a higher degree remain in the water phase and interact less strongly with the head groups than the Joung/Cheatham ions. The latter parameter set might be just as valid, but the unrealistically high concentration of positive ions in the system and the resulting ordering of the membrane make comparison with experiment “in the absence of salt” difficult. Hence, and also for consistency, ff99 parameters are used in all the Lipid14 (and Slipids) simulations.
A similar condensing effect has been observed in Charmm.30 Too compact anionic bilayers arising from strong binding of ions to the lipid head groups prompted the modification of Lennard-Jones radii for pair-specific interactions between sodium ions and lipid oxygen atoms.30 The purpose was to weaken the interactions between the ions and the anionic head groups which in turn gives better agreement with experiment. For the present work several attempts were made at self-assembly of C36 anionic lipids using sodium ion parameters from Noskov and Roux30,70 without the aforementioned sodium–oxygen radii modifications, in which the lipids did not form bilayers but rather remained trapped in non-lamellar, possibly non-physical configurations not seen in any of the other self-assembly simulations (results not shown). The results presented for the C36 anionics in this paper are with the radii revisions30 applied, suggesting that the choice of ion parameters can significantly influence not only the properties of pre-formed bilayers, but also the lipid self-assembly process.
Conclusions
Lipid bilayer self-assembly will be a valuable addition to the area of all-atom MD simulations, in particular as a means to avoid biased starting structures for the simulation of membrane-related systems such as transmembrane proteins and peptides or even bi- or multicomponent lipid mixtures. Self-assembly simulations can also offer additional validation of the underlying lipid force field. In this paper we subjected eight types of phospholipids described by each of the three major all-atom lipid force fields to self-assembly simulations in AMBER, running three repeats per lipid/force field combination. Four of the types of simulated lipids are negatively charged and to our knowledge this is the first time bilayer self-assembly of anionic phospholipids has been shown in MD simulations. In all but three repeats (force switch simulations included) the lipids assembled into stable bilayers within 1 μs of simulation that, with the exception of C36 DPPC when simulated with a 10 Å cutoff, displayed structural properties in good agreement with the available experimental data. We therefore recommend for C36 DPPC simulations using the AMBER GPU code that the force switch implementation be applied; for all other lipids presented in this work using any of the force fields, Lipid14, C36 and Slipids, one can reliably utilize the 10 Å cut-off.
A unique advantage of Lipid14 is that it is the only modular all-atom force field for lipids and thus it facilitates creation of any lipid from the underlying phospholipid head groups and tails which have already been developed. It is also fully compatible with the other AMBER force fields for proteins, carbohydrates, nucleic acids and small molecules. Recently the force field has also been extended to include more lipid types, such as cholesterol, sphingomyelin and the anionic head groups applied in the present work. The self-assembly simulations and the structural analyses in this paper further validate both the original Lipid14 force field and its extension to anionic lipids and lend more evidence in support of the underpinning module-based parameterization strategy.
Each simulation system in the current work contained one type of phospholipid that self-assembled into pure bilayers. Future work could involve the application of the self-assembly strategy to the study of other relevant and more complex systems, such as transmembrane proteins and peptides as well as lipid mixtures containing several types of phospholipids, cholesterol and sphingomyelin, all of which are important constituents of biological membranes.
Supplementary Material
Acknowledgments
We are very grateful to Dr Hannes Loeffler of the Science and Technology Facilities Council, UK, for writing and maintaining the modified PTRAJ/CPPTRAJ routines that were used in this work. ÅAS and K. T. acknowledge the support of Centre for Pharmacy, University of Bergen. IRG and CJD thank the Institute of Chemical Biology, UK Biotechnology and Biological Sciences Research Council (BBSRC) and GlaxoSmithKline for the award of a studentship to CJD. IRG would also like to acknowledge funding from the EU in the form of the project “HeCaToS – Hepatic and Cardiac Toxicity Systems modeling” FP7-HEALTH-2013-INNOVATION-1 (Project number 602156). BDM and CL would like to acknowledge funding for this work provided by the NIH Molecular Biophysics Training Grant (T32 GM008326) and the NVIDIA Graduate Fellowship Program. RCW and ÅAS acknowledge funding through NSF SI2-SSE grant (NSF-1148276) to RCW. RCW also acknowledges funding through fellowships from Intel Corp. and NVIDIA, Inc.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp07379k
Contributor Information
Ross C. Walker, Email: ross@rosswalker.co.uk.
Ian R. Gould, Email: i.gould@imperial.ac.uk.
References
- 1.Lomize AL, Pogozheva ID, Lomize MA, Mosberg HI. BMC Struct Biol. 2007;7:44. doi: 10.1186/1472-6807-7-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garland SL. J Biomol Screening. 2013;18:947–966. doi: 10.1177/1087057113498418. [DOI] [PubMed] [Google Scholar]
- 3.Overington JP, Al-Lazikani B, Hopkins AL. Nat Rev Drug Discovery. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 4.de Vries AH, Mark AE, Marrink SJ. J Phys Chem B. 2004;108:2454–2463. [Google Scholar]
- 5.de Vries AH, Mark AE, Marrink SJ. J Am Chem Soc. 2004;126:4488–4489. doi: 10.1021/ja0398417. [DOI] [PubMed] [Google Scholar]
- 6.Esteban-Martín S, Salgado J. Biophys J. 2007;92:903–912. doi: 10.1529/biophysj.106.093013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marrink SJ, Lindahl E, Edholm O, Mark AE. J Am Chem Soc. 2001;123:8638–8639. doi: 10.1021/ja0159618. [DOI] [PubMed] [Google Scholar]
- 8.Poger D, van Gunsteren WF, Mark AE. J Comput Chem. 2010;31:1117–1125. doi: 10.1002/jcc.21396. [DOI] [PubMed] [Google Scholar]
- 9.Bond PJ, Holyoake J, Ivetac A, Khalid S, Sansom MSP. J Struct Biol. 2007;157:593–605. doi: 10.1016/j.jsb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Khalid S, Bond PJ, Holyoake J, Hawtin RW, Sansom MSP. J R Soc, Interface. 2008;5:S241–S250. doi: 10.1098/rsif.2008.0239.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marrink SJ, Mark AE. J Am Chem Soc. 2003;125:15233–15242. doi: 10.1021/ja0352092. [DOI] [PubMed] [Google Scholar]
- 12.Scott KA, Bond PJ, Ivetac A, Chetwynd AP, Khalid S, Sansom MSP. Structure. 2008;16:621–630. doi: 10.1016/j.str.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 13.Shinoda W, DeVane R, Klein ML. J Phys Chem B. 2010;114:6836–6849. doi: 10.1021/jp9107206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skjevik ÅA, Madej BD, Dickson CJ, Teigen K, Walker RC, Gould IR. Chem Commun. 2015;51:4402–4405. doi: 10.1039/c4cc09584g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dickson CJ, Madej BD, Skjevik ÅA, Betz RM, Teigen K, Gould IR, Walker RC. J Chem Theory Comput. 2014;10:865–879. doi: 10.1021/ct4010307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klauda JB, Venable RM, Freites JA, O'Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Jr, Pastor RW. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jämbeck JPM, Lyubartsev AP. J Phys Chem B. 2012;116:3164–3179. doi: 10.1021/jp212503e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jämbeck JPM, Lyubartsev AP. J Chem Theory Comput. 2012;8:2938–2948. doi: 10.1021/ct300342n. [DOI] [PubMed] [Google Scholar]
- 19.Jämbeck JPM, Lyubartsev AP. J Chem Theory Comput. 2013;9:774–784. doi: 10.1021/ct300777p. [DOI] [PubMed] [Google Scholar]
- 20.Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, Cheatham TE, III, Darden TA, Duke RE, Gohlke H, Götz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, Le Grand S, Luchko T, Luo R, Madej BD, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling C, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Kollman PA. AMBER 14. University of California; San Francisco: 2014. [Google Scholar]
- 21.Salomon-Ferrer R, Case DA, Walker RC. Wiley Interdiscip Rev: Comput Mol Sci. 2013;3:198–210. [Google Scholar]
- 22.Götz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. J Chem Theory Comput. 2012;8:1542–1555. doi: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. J Chem Theory Comput. 2013;9:3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 24.Le Grand S, Götz AW, Walker RC. Comput Phys Commun. 2013;184:374–380. [Google Scholar]
- 25.Loncharich RJ, Brooks BR, Pastor RW. Biopolymers. 1992;32:523–535. doi: 10.1002/bip.360320508. [DOI] [PubMed] [Google Scholar]
- 26.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 27.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 28.Darden T, York D, Pedersen L. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 29.Crowley MF, Williamson MJ, Walker RC. Int J Quantum Chem. 2009;109:3767–3772. [Google Scholar]
- 30.Venable RM, Luo Y, Gawrisch K, Roux B, Pastor RW. J Phys Chem B. 2013;117:10183–10192. doi: 10.1021/jp401512z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.http://mmkluster.fos.su.se/slipids/Downloads.html.
- 32.Brooks BR, Brooks CL, III, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Åqvist J. J Phys Chem. 1990;94:8021–8024. [Google Scholar]
- 34.Smith DE, Dang LX. J Chem Phys. 1994;100:3757–3766. [Google Scholar]
- 35.Dickson CJ, Rosso L, Betz RM, Walker RC, Gould IR. Soft Matter. 2012;8:9617–9627. [Google Scholar]
- 36.Kučerka N, Katsaras J, Nagle JF. J Membr Biol. 2010;235:43–50. doi: 10.1007/s00232-010-9254-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roe DR, Cheatham TE., III J Chem Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- 38.Humphrey W, Dalke A, Schulten K. J Mol Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 39.Kučerka N, Nagle JF, Sachs JN, Feller SE, Pencer J, Jackson A, Katsaras J. Biophys J. 2008;95:2356–2367. doi: 10.1529/biophysj.108.132662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kučerka N, Nieh MP, Katsaras J. Biochim Biophys Acta. 2011;1808:2761–2771. doi: 10.1016/j.bbamem.2011.07.022. [DOI] [PubMed] [Google Scholar]
- 41.Kučerka N, Tristram-Nagle S, Nagle JF. J Membr Biol. 2005;208:193–202. doi: 10.1007/s00232-005-7006-8. [DOI] [PubMed] [Google Scholar]
- 42.Kučerka N, Tristram-Nagle S, Nagle JF. Biophys J. 2006;90:L83–L85. doi: 10.1529/biophysj.106.086017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kučerka N, van Oosten B, Pan J, Heberle FA, Harroun TA, Katsaras J. J Phys Chem B. 2015;119:1947–1956. doi: 10.1021/jp511159q. [DOI] [PubMed] [Google Scholar]
- 44.Nagle JF, Tristram-Nagle S. Biochim Biophys Acta. 2000;1469:159–195. doi: 10.1016/s0304-4157(00)00016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pan J, Cheng X, Monticelli L, Heberle FA, Kučerka N, Tieleman DP, Katsaras J. Soft Matter. 2014;10:3716–3725. doi: 10.1039/c4sm00066h. [DOI] [PubMed] [Google Scholar]
- 46.Pan J, Heberle FA, Tristram-Nagle S, Szymanski M, Koepfinger M, Katsaras J, Kučerka N. Biochim Biophys Acta. 2012;1818:2135–2148. doi: 10.1016/j.bbamem.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pan J, Marquardt D, Heberle FA, Kučerka N, Katsaras J. Biochim Biophys Acta. 2014;1838:2966–2969. doi: 10.1016/j.bbamem.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 48.Petrache HI, Tristram-Nagle S, Gawrisch K, Harries D, Parsegian VA, Nagle JF. Biophys J. 2004;86:1574–1586. doi: 10.1016/S0006-3495(04)74225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rand RP, Parsegian VA. Biochim Biophys Acta. 1989;988:351–376. [Google Scholar]
- 50.Rappolt M, Hickel A, Bringezu F, Lohner K. Biophys J. 2003;84:3111–3122. doi: 10.1016/S0006-3495(03)70036-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kučerka N, Holland BW, Gray CG, Tomberli B, Katsaras J. J Phys Chem B. 2012;116:232–239. doi: 10.1021/jp208920h. [DOI] [PubMed] [Google Scholar]
- 52.Liu Y, Nagle JF. Phys Rev E: Stat, Nonlinear, Soft Matter Phys. 2004;69:040901. doi: 10.1103/PhysRevE.69.040901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tristram-Nagle S, Petrache HI, Nagle JF. Biophys J. 1998;75:917–925. doi: 10.1016/S0006-3495(98)77580-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Binder H, Gawrisch K. J Phys Chem B. 2001;105:12378–12390. [Google Scholar]
- 55.Evans E. personal communication – DOPC isothermal compressibility modulus from X-ray data at 293 K. 2014 [Google Scholar]
- 56.Evans E, Rawicz W, Smith BA. Faraday Discuss. 2013;161:591–611. doi: 10.1039/c2fd20127e. [DOI] [PubMed] [Google Scholar]
- 57.Rawicz W, Olbrich KC, McIntosh T, Needham D, Evans E. Biophys J. 2000;79:328–339. doi: 10.1016/S0006-3495(00)76295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abu-Baker S, Qi X, Lorigan GA. Biophys J. 2007;93:3480–3490. doi: 10.1529/biophysj.107.107789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Douliez JP, Léonard A, Dufourc EJ. Biophys J. 1995;68:1727–1739. doi: 10.1016/S0006-3495(95)80350-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lafleur M, Cullis PR, Fine B, Bloom M. Biochemistry. 1990;29:8325–8333. doi: 10.1021/bi00488a018. [DOI] [PubMed] [Google Scholar]
- 61.Perly B, Smith ICP, Jarrell HC. Biochemistry. 1985;24:1055–1063. doi: 10.1021/bi00325a038. [DOI] [PubMed] [Google Scholar]
- 62.Petrache HI, Dodd SW, Brown MF. Biophys J. 2000;79:3172–3192. doi: 10.1016/S0006-3495(00)76551-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Seelig J, Waespe-Šarčević N. Biochemistry. 1978;17:3310–3315. doi: 10.1021/bi00609a021. [DOI] [PubMed] [Google Scholar]
- 64.Warschawski DE, Devaux PF. Eur Biophys J. 2005;34:987–996. doi: 10.1007/s00249-005-0482-z. [DOI] [PubMed] [Google Scholar]
- 65.Venable RM, Brown FLH, Pastor RW. Chem Phys Lipids. 2015;192:60–74. doi: 10.1016/j.chemphyslip.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Venable RM, Sodt AJ, Rogaski B, Rui H, Hatcher E, MacKerell AD, Jr, Pastor RW, Klauda JB. Biophys J. 2014;107:134–145. doi: 10.1016/j.bpj.2014.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang K, García AE. J Chem Phys. 2014;141:105101. doi: 10.1063/1.4893965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vila-Viçosa D, Teixeira VH, Santos HAF, Baptista AM, Machuqueiro M. J Chem Theory Comput. 2014;10:5483–5492. doi: 10.1021/ct500680q. [DOI] [PubMed] [Google Scholar]
- 69.Joung IS, Cheatham TE., III J Phys Chem B. 2008;112:9020–9041. doi: 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Noskov SY, Roux B. J Mol Biol. 2008;377:804–818. doi: 10.1016/j.jmb.2008.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.