

Abstract
Sphingosine 1-phosphate receptor 1 (S1P1) plays a pivotal signaling role in inflammatory response; because S1P1 modulation has been identified as a therapeutic target for various diseases, a PET tracer for S1P1 would be a useful tool. Fourteen fluorine-containing analogues of S1P ligands were synthesized and their in vitro binding potency measured; four had high potency and selectivity for S1P1 (S1P1 IC50 < 10 nM, >100-fold selectivity for S1P1 over S1P2 and S1P3). The most potent ligand, 28c (IC50 = 2.63 nM for S1P1) was 18F-labeled and evaluated in a mouse model of LPS-induced acute liver injury to determine its S1P1-binding specificity. The results from biodistribution, autoradiography, and microPET imaging showed higher [18F]28c accumulation in the liver of LPS-treated mice than controls. Increased expression of S1P1 in the LPS model was confirmed by immunohistochemical analysis (IHC). These data suggest that [18F]28c is a S1P1 PET tracer with high potential for imaging S1P1 in vivo.
Graphical abstract
1. INTRODUCTION
Sphingosine 1-phosphate (S1P, Figure 1) receptors are a class of G-protein-coupled receptors (GPCRs) with five distinct subtypes, denoted as S1P1–5.1,2 These receptors are regulated by S1P and have important regulatory functions in both normal biological processes and in disease, particularly in pathological processes involving the immune system, the central nervous system (CNS), and the cardiovascular system. Because the S1P/S1P-receptor pathway is especially important in cancer and autoimmune disorders including multiple sclerosis (MS), tremendous development efforts have focused on therapeutically targeted S1P receptor ligands. These efforts discovered promising compounds, 2-amino-2-[2-(4-octyl-phenyl)-ethyl]-propane-1,3-diol 1 (FTY720, Fingolimod, Figure 1), which was approved by the US Food and Drug Administration (FDA) for treatment of relapse remitting multiple sclerosis in 2010. Compound 1 is phosphorylated in vivo by sphingosine kinase 2; this stereospecific process generates the biologically active metabolite (S)-FTY720-Phosphate ((S)-1-P, Figure 1), a potent S1P receptor agonist.3–5 (S)-1-P is a nonselective S1P receptor ligand, which binds to each of the S1P receptor subtypes except S1P2. Since its primary mechanism of action in MS is believed to be through S1P1,4 recent development has focused on ligands having high potency and selectivity for S1P1 with suitable pharmacological properties for therapeutic applications. Although rodent studies using S1P receptor ligands suggested that adverse cardiovascular effects were the result of S1P3 agonism,6,7 the Phase I study of a S1P1 selective therapeutic reported bradycardia associated with the first dose administered to human subjects.8 Because activity at S1P3 has clear effects on the vascular system and does not appear to contribute to therapeutic efficacy of these ligands in the immune system,9 our efforts have focused on identifying subtype-selective ligands. S1P1 ligands are also being evaluated in cancer,10,11 rheumatoid arthritis,12 ulcerative colitis,13,14 as well as liver and lung injury among other therapeutic uses.15–18
Figure 1.
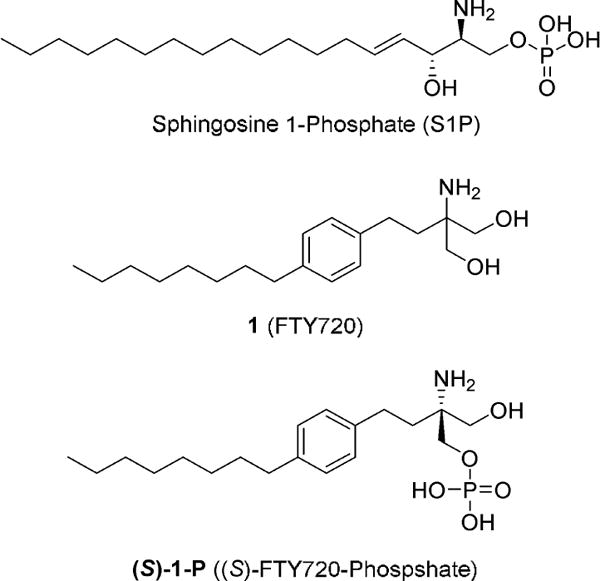
Structures of S1P, FTY720, and (S)-FTY720-phosphate.
Positron emission tomography (PET) is a widely utilized imaging modality which can be used to noninvasively quantify molecular targets in vivo. PET is used in drug discovery to assess receptor occupancy, downstream functional changes, and impact on disease pathophysiology.19 A S1P1 specific PET tracer could greatly assist the evaluation of new therapeutics by measuring the relationship between drug exposure (dose administered or plasma concentration) and receptor occupancy, thus enabling accelerated dose selection during early clinical trials.20–22 A successful PET ligand should have a binding potency <10 nM and must be specific to the target versus neighboring nontarget tissues to allow for quantification of the regions of interest during the time frame of clinical studies (0–2 h).23,24
To date, no S1P receptor imaging agent suitable for clinical studies has been reported, although since 2011, several groups have reported their efforts to develop a S1P1 receptor tracer.25–27 The iodinated analogue, 2-amino-2-[2-(2-iodo-4-octylphenyl)ethyl]-1,3-propandiol 2 (BZM055, Figure 2), was 123I-labeled for single photon emission computed tomography (SPECT) studies and could be 124I-labeled for PET imaging.25 Although compound 2 showed pharmacokinetic trends similar to those of compound 1, its lower rate of phosphorylation (required for receptor binding) compared to that of compound 1 limits its effectiveness as an imaging agent. The long half-lives of either iodine-123 (t1/2 = 13.2 h) or iodine-124 (t1/2 = 4.2 days) would enable the study of both the distribution kinetics of the parent ligand and its elimination kinetics. Haufe and coworkers separately reported 18F-labeled PET imaging agents based on the only known S1P1 antagonist, [(3R)-amino-4-[(3-hexylphenyl)amino]-4-oxobutyl]-phosphonic acid, mono-(trifluoroacetate) (W146),26 and based on compound 1.27 The 18F-labeled W146 analogue (R)-1-[[3-(6-fluorohexyl)-phenyl]amino-4-oxobutyl]phosphonic acid, ([18F]3, Figure 2) was shown to bind with S1P1 and unlike 1 does not require a preliminary phosphorylation step for binding potency. However, despite its in vitro stability in mouse serum, PET imaging of [18F]3 in wild-type mice showed rapid total body clearance as well as bone uptake, which is a measure of in vivo defluorination. Their recent report describes the more favorable evaluation of 18F-labeled 2 analogues, with either an 8-carbon (2-amino-2-[4-(8-fluorooctyl)phenethyl]propane-1,3-diol, [18F]4a) or a 6-carbon (2-amino-2-[4-(6-fluorohexyl)-phenethyl]propane-1,3-diol, [18F]4b) aliphatic tail (Figure 2). The ligand with the shorter tail, [18F]4b, showed reduced in vitro activity, but both tracers demonstrated uptake in S1P target organs of wild-type mice with no evidence of in vivo defluorination. Unlike [18F]3, the 1 analogues require phosphorylation before binding to S1P receptors. While potentially useful tools, at this time it is unclear how rapidly and to what extent the ligands undergo phosphorylation during the typical time frame for PET imaging (1–2 h postinjection).
Figure 2.
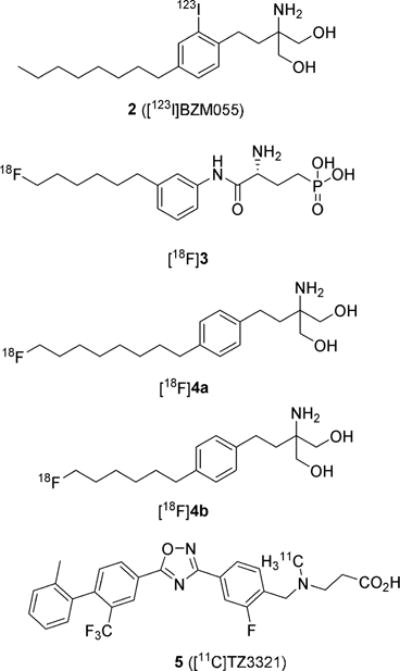
Structures of S1P receptor imaging agents.
We have also used the wealth of knowledge in this field to investigate PET radiotracers for S1P receptors using rodent models of human disease.28–30 Our group previously reported the radiosynthesis of the 11C-labeled S1P1 selective ligand 3-((2-fluoro-4-(5-(2′-methyl-2-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)-1,2,4-oxadiazol-3-yl)benzyl)-(methyl)amino)-propanoic acid 5 ([11C]TZ3321, Figure 2) and demonstrated the feasibility of PET imaging S1P1 as a measure of inflammatory response in both the femoral artery wire-injury mouse model of restenosis29 and in the experimental autoimmune encephalomyelitis (EAE) rat model of multiple sclerosis (MS).30 Here, we present the synthesis and screening of fluorine-containing ligands based on reported compounds with either a benzoxazole core31 or an oxadiazole core.32,33 Compound 28c was identified as a potent and selective ligand for S1P1 over S1P2 or S1P3 in our competitive binding assay,34 so was 18F-labeled for biological evaluation in the mouse model of LPS-induced liver injury.
2. RESULTS AND DISCUSSION
The synthetic strategy is discussed in detail below, but the two lead compounds used for development of an 18F-labeled tracer were selected due to published data indicating that they are not prodrugs which require in vivo phosphorylation and due to their favorable biological behavior.31,32 Our screening focused on S1P1 selective ligands because the efficacy of 2 in MS is attributed to its high binding potency toward to S1P1.6,35 As shown in Table 2, 28c has a high potency (IC50 = 2.63 ± 0.27 nM) and selectivity for S1P1 over S1P2 or S1P3 (IC50 > 1000 nM). The synthesis of [18F]28c was accomplished. The mouse model of LPS-induced acute liver injury was used for the in vivo evaluation of [18F]28c. Biodistribution studies were carried out with both [18F]28c and the liver imaging agent [99mTc]-N-(3-bromo-2,4,6-trimethyacetanilide) iminodiacetic acid ([99mTc]-mebrofenin) in LPS and sham mice.36 The absence of a significant difference in the liver uptake of [99mTc]mebrofenin in LPS-treated mice vs controls at 60 min postinjection suggested that the higher liver uptake of [18F]28c in LPS-treated mice compared to that of control mice was not caused by reduced hepatobiliary clearance in the liver injury model. MicroPET imaging in LPS and sham-treated mice also showed increased retention of [18F]28c in the injured liver which correlated with increased S1P1 expression observed in IHC studies, while no difference in liver uptake of [15O]water was seen between LPS-treated and sham mice,37,38 suggesting that the higher injured liver accumulation of [18F]28c is not due to a change in blood flow in the injured liver. These data indicate that a liver uptake of [18F]28c correlates well with the expression of S1P1 in mouse livers.
Table 2.
Structure–Activity Relationships in the Oxadiazole Core Series

| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | Head Group (R3) | IC50 (nM) | cLogD7.4 | ||
| S1P1 | S1P2 | S1P3 | |||||
| 28a | Et | Η |
|
8.53 ± 3.14 | >1000 | >1000 | 1.10 |
| 28b | FCH2CH2 | Η |
|
9.94 ± 1.03 | >1000 | >1000 | 0.80 |
| 28c | FCH2CH2 | CF3 |
|
2.63 ± 0.27 | >1000 | >1000 | 2.32 |
| 28d | FCH2CH2 | CF3 |
|
45.4 ± 2.7 | >1000 | >1000 | 3.02 |
| 28e | FCH2CH2 | CF3 |
|
509 ± 167 | – | – | 2.08 |
| 28f | Me | CF3 |
|
103 ± 21 | – | – | 2.09 |
| 26c | FCH2CH2 | CF3 |
|
6.67 ± 0.70 | >1000 | >1000 | 4.88 |
2.1. Chemistry
Ligands with a benzoxazole core were prepared as shown in Schemes 1 and 2. The starting halogenated trifluoromethylbenzoic acids were functionalized by either palladium catalyzed Suzuki coupling with the desired aryl boronic acid to provide the biaryl moieties or by nucleophilic aromatic substitution with the desired alcohol to give the ethereal products. The appendant carboxylic acid was transformed to the corresponding acid chloride with oxalyl chloride and catalytic DMF, which was then reacted with a functionalized 2-hydroxyaniline under Schotten–Baumann conditions to give amides 10. These amides were subsequently heated under acidic conditions to cyclize and provide the benzoxazole cores. The installation of the polar head groups was accomplished by a variety of methods. β-Amino acids 15a and 15b, were prepared by reduction of a methyl ester to an alcohol and functional group transformation to a chloride, and subsequently to the azetidine ester, which was unmasked to provide 15. Acid 16 was prepared by a Suzuki coupling with a thiophene boronic acid. Palladium-catalyzed amination with methylpiperidine-4-carboxylate gave the protected products, which were subsequently saponified to give acids 18.
Scheme 1. Preparation of Benzoxazole Coresa.
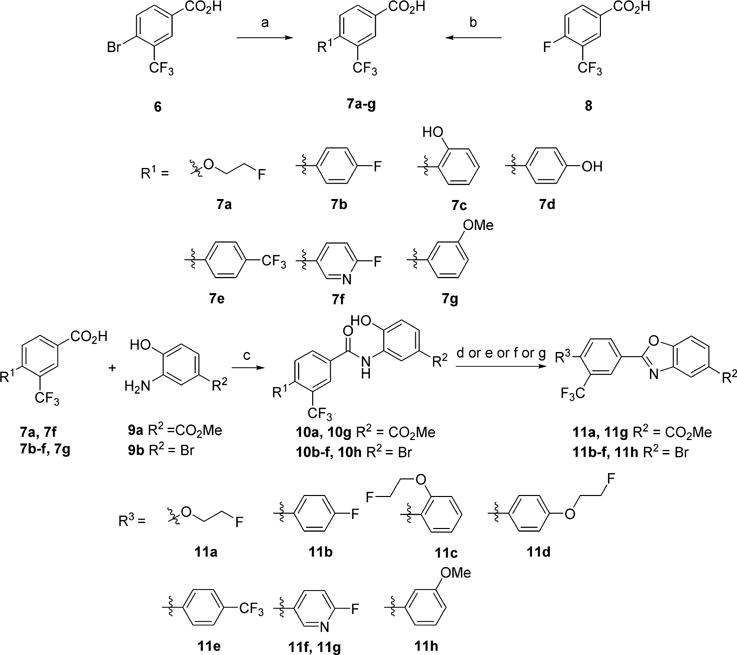
aReagents and conditions: (a) Pd(OAc)2, SPhos, CsF, R1B(OH)2, 1,4-dioxane/H2O for 7b–g; (b) NaH, R1OH, DMSO for 7a; (c) (i) (COCl)2, cat. DMF, CH2Cl2; (ii) Et3N, DMAP, CH2Cl2; (d) p-TsOH, toluene for 11a, 11b, 11e, 11h; (e) (i) p-TsOH, toluene; (ii) Cs2CO3, fluoroethyltosylate, DMF for 11c, 11d; (f) Ph3P, DIAD, THF for 11f; (g) PPTS, toluene for 11g.
Scheme 2. Synthesis of Benzoxazole Targetsa.
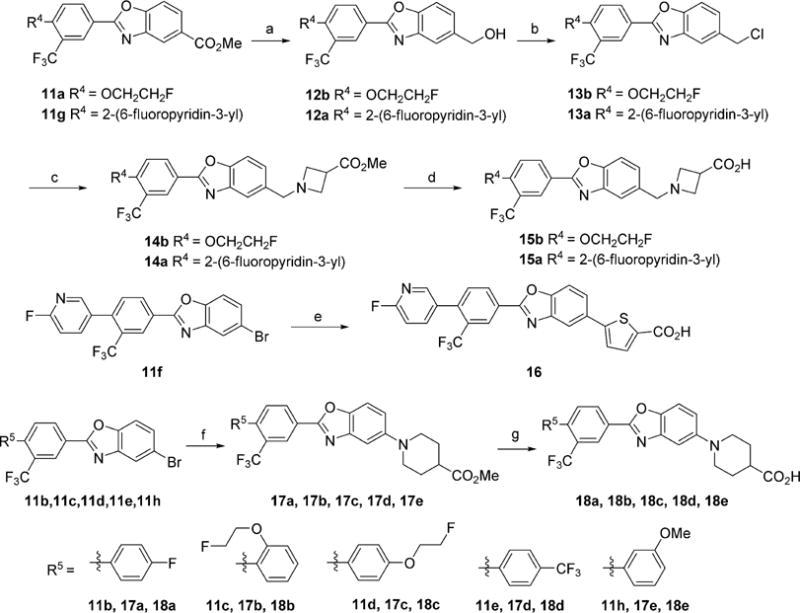
aReagents and conditions: (a) DIBAI-H, THF; (b) cyanuric chloride, DMF, CH2Cl2; (c) methyl azetidine-3-carboxylate hydrochloride, i-Pr2NEt, MeCN; (d) LiOH, THF/H2O; (e) Pd(OAc)2, SPhos, KF, 2-carboxythiophene-5-boronic acid, 1,4-Dioxane/H2O; (f) Pd2(dba)3·CHCl3, XPhos, NaOtBu, methylpiperidine-4-carboxylate; (g) LiOH, THF/H2O.
Ligands with an oxadiazole core were synthesized according to Scheme 3. The required carboxylic acids were prepared as described above or from methyl 4-hydroxybenzoate (22) by alkylation and saponification. The desired carboxylic acid was reacted with N′-hydroxy-4-(hydroxymethyl)benzimidamide (25) under standard peptide coupling conditions, and subsequently thermally cyclized to give the oxadiazole cores. O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate (TBTU) was also used as a coupling agent, followed by thermal cyclization.39 The appendant benzyl alcohol was oxidized under Swern conditions to give aldehydes 27. The β-amino acid headgroup was affixed by reductive amination with the desired amine to provide the targeted ligands 28a–f. The lipophilicity of a radiotracer, expressed as logP, impacts nonspecific binding and its ability to cross the BBB, which is a key physical constraint for targets in the CNS and intracellular targets including enzymes.40 The calculated lipophilicity at a pH of 7.4 of the new compounds is reported as cLogD values in Tables 1 and 2. The oxadiazole 28c had a cLogD of 2.32 which is within the desired range for CNS tracers and, as discussed below, a favorable in vitro binding profile; thus, we elected to pursue its development as a radiotracer and synthesized precursors for radiolabeling. Precursor 32 for radiolabeling using [18F]2-fluoroethyltosylate was prepared from phenol 29, as shown in Scheme 4, using a synthetic route similar to that used for the oxadiazole analogues. Formation of the oxadiazole proceeded as expected; however, the oxidation step required different conditions due to the presence of the unprotected phenol. Oxidation with manganese(IV) oxide provided smooth conversion to the desired aldehyde 31. Reductive amination under buffered conditions afforded 32. Precursor 34 for direct labeling was also prepared as shown in Scheme 5. Starting with aldehyde 31, the free phenol was alkylated with ethylene ditosylate to provide compound 33, which was subjected to reductive amination to yield 34. The reductive amination conditions were modified to utilize sodium triacetoxyborohydride because harsher reducing agents tended to reduce the alkyl tosylate.
Scheme 3. Synthesis of Benzoxazole Targetsa.
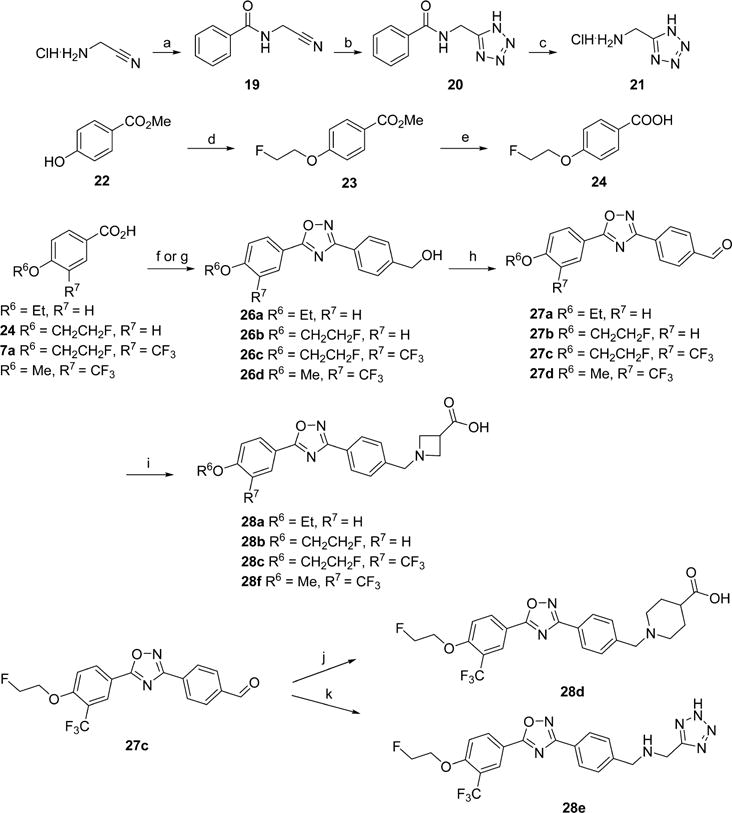
aReagents and conditions: (a) DIBAI-H, THF; (b) cyanuric chloride, DMF, CH2Cl2; (c) methyl azetidine-3-carboxylate hydrochloride, i-Pr2NEt, MeCN; (d) LiOH, THF/H2O; (e) Pd(OAc)2, SPhos, KF, 2-carboxythiophene-5-boronic acid, 1,4-dioxane/H2O; (f) Pd2(dba)3·CHCl3, XPhos, NaOtBu, methylpiperidine-4-carboxylate; (g) LiOH, THF/H2O.
Table 1.
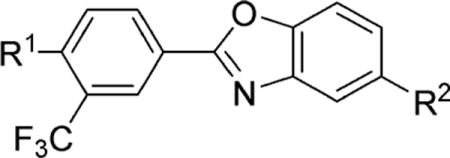
Structure–Activity Relationships in the Benzoxazole Core Series
| |||||
|---|---|---|---|---|---|
| Compound | R1 | R2 | S1P1 IC50 (nM) | S1P1 EC50 (nM) | cLogD7.4 |
| 15a |
|
|
92.7 ± 35.3 | – | 2.08 |
| 15b |
|
|
76.4 ± 21.6 | – | 2.21 |
| 16 |
|
|
334 ± 59 | 2.99 | 4.10 |
| 18a |
|
|
486 ± 69 | 0.76 | 3.72 |
| 18b |
|
|
894 ± 367 | 3.04 | 3.69 |
| 18c |
|
|
114 ± 73 | 11.1 | 3.84 |
| 18d |
|
|
498 ± 335 | 4.47 | 4.76 |
| 18e |
|
|
95.7 ± 25.1 | 4.90 | 3.56 |
Scheme 4. Synthesis of Radiolabeling Precursor 32a.
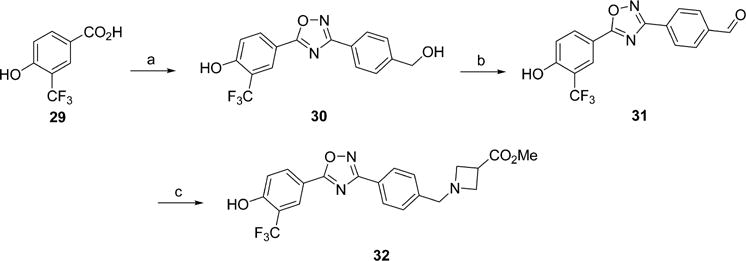
aReagents and conditions: (a) TBTU, HOBt, 25, DMF; (b) MnO2, 1,4-dioxane (c) methyl azetidine-3-carboxylate hydrochloride, NaBH3CN, AcOH, DIPEA, MeOH.
Scheme 5. Preparation of Direct Radiolabeling Precursor 34a.
aReagents and conditions: (a) (CH2OTs)2, K2CO3, MeCN; (b) (i) DIPEA, 1,2-DCE/MeOH; (ii) NaBH(OAc)3, AcOH, 1,2-DCE.
2.2. In Vitro Binding Assays
Competitive inhibition of the binding of radiolabeled [32P]S1P to the new analogues was measured to determine the affinity of the ligands for S1P1, S1P2, and S1P3.34 The results are reported in Tables 1 and 2. The benzoxazole series of ligands displayed nanomolar activity in the functional assay (EC50); however, the ligands displayed lower IC50 value for S1P1 in the competitive binding assay (IC50). Although the reason for this discrepancy in the assay results is not clear, similar results have been observed with other S1P receptor ligands.41 Additionally, while functional assays can be extremely useful in the preliminary stages of imaging agent development, the binding potency of a ligand cannot be determined without a competitive binding assay.42 While none of the ligands in this series had sufficient binding affinity (IC50 < 10 nM) to be pursued as a PET tracer, structure–activity relationships (SAR) became apparent. The residues and shape of the S1P1 binding pocket have been well documented with a published crystal structure.43 The binding pocket consists of two major components, a charged head portion and a lipophilic tail area. The strong recognition for ligand binding is thought to be derived from interactions between the positively charged Arg120 and the phosphate in S1P, and interactions between the negatively charged Glu121 and the ammonium in S1P. The lipophilic part of the binding pocket appears to be quite tolerant of a variety of functional groups, with the noted exception of the ortho-substituted fluoroethyl ether 18b (IC50 = 894 ± 367 nM).
The oxadiazole ligand series showed much more promise than the benzoxazole series, as seen in Table 2. We explored the effects of modifying the lipophilic side chain of 28a (IC50 = 8.53 ± 3.14 nM). Addition of a fluorine (28b, IC50 = 9.94 ± 1.03 nM) had no significant change on the binding potency, while an ortho-trifluoromethyl group noticeably improved the affinity to 2.63 ± 0.27 nM (28c). Next, we turned to the polar headgroup. While some S1P1 ligands without a polar headgroup have a high degree of potency, we elected to maintain this property in our explorations. Changing the azetidine ring of 28c to a piperidine (28d) gave a 20-fold decrease in binding affinity to 45.4 ± 2.7 nM. Bioisosteric substitution of the acid to a tetrazole resulted in significant loss of activity of over 100-fold to 509 ± 167 nM (28e), while shortening the two-carbon tail to a methyl group adversely impacted the binding potency giving an IC50 > 100 nM (28f). Finally, the benzyl alcohol 26c was synthesized to determine how critical the amino acid headgroup was; surprisingly, this headgroup was not required as 26c was quite potent with an IC50 of 6.67 ± 0.70 nM. All compounds with a IC50 value under 50 nM were screened for selectivity versus S1P2 and S1P3; no tested compounds showed detectable binding with S1P2 or S1P3. 28c had a high potency for S1P1 (IC50 = 2.63 ± 0.27 nM) with no measurable potency for S1P2 (IC50 > 1000 nM) or S1P3 (IC50 > 1000 nM) with a cLogD of 2.32; thus, we elected to pursue 28c as our lead compound for 18F-labeling.
2.3. Radiochemistry
The radiosynthesis of 28c was initially attempted using the versatile indirect approach of labeling with [18F]2-fluoroethyltosylate. The precursor 32 was prepared as shown in Scheme 4. Unfortunately, attempts to label 32 indirectly resulted only in extremely low yields under a variety of conditions (<5% radiochemical yield for the alkylation step), likely due to the low nucleophilicity of the electron-deficient phenol. Because of this concern, as well as a desire to avoid a three-step, two-pot radiosynthesis, we subsequently pursued a direct labeling approach starting with the tosylate precursor (34) as shown in Scheme 6.
Scheme 6.
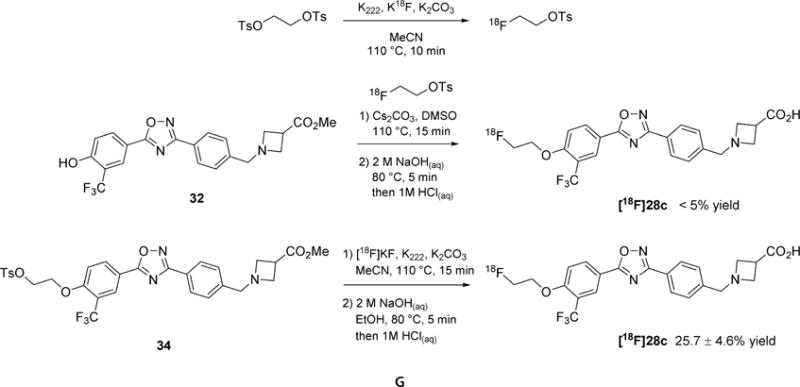
Radiosynthesis of [18F]28c
The tosylate precursor (34) was reacted with Kryptofix 222 (K222) and [18F]KF in acetonitrile for 15 min at 110 °C to install the fluoroethyl tail. The carboxylic acid headgroup was unmasked by saponification with sodium hydroxide. The resulting crude product was purified by high performance liquid chromatography (HPLC) to give [18F]28c in 25.7 ± 4.6% (n = 10, decay corrected to start of synthesis), with >98% chemical and radiochemical purity, and a high specific activity (1.43 ± 0.12 Ci/μmol).
2.4. Biological Studies
2.4.1. Biodistribution in Normal Rats
To investigate the tissue distribution of [18F]28c in living subjects, a biodistribution study was performed in adult male Sprague–Dawley (SD) rats. As shown in Table 3, the results showed significant uptake and retention in the liver, without evidence of in vivo defluorination (no increased accumulation in bone was observed from 5 to 120 min postinjection). Relatively high uptake was observed for the heart, lungs, pancreas, and spleen, all organs known to have high S1P1 expression. The surprisingly low brain uptake of [18F]28c as shown in Table 3 may potential limit its utility for CNS applications.
Table 3.
Biodistribution of [18F]28c in Adult Male SD Rats (% ID/g ± SD)(n = 4)
| organ | 5 min | 30 min | 60 min | 120 min |
|---|---|---|---|---|
| blood | 0.304 ± 0.058 | 0.240 ± 0.024 | 0.223 ± 0.007 | 0.178 ± 0.039 |
| lung | 0.753 ± 0.097 | 0.413 ± 0.005 | 0.341 ± 0.024 | 0.272 ± 0.053 |
| liver | 6.076 ± 1.134 | 5.509 ± 0.567 | 5.081 ± 0.324 | 3.355 ± 0.772 |
| spleen | 0.589 ± 0.067 | 0.282 ± 0.030 | 0.251 ± 0.014 | 0.202 ± 0.033 |
| kidney | 2.026 ± 0.250 | 1.080 ± 0.000 | 1.117 ± 0.052 | 0.845 ± 0.091 |
| muscle | 0.121 ± 0.013 | 0.145 ± 0.013 | 0.156 ± 0.010 | 0.135 ± 0.007 |
| fat | 0.051 ± 0.011 | 0.061 ± 0.003 | 0.051 ± 0.003 | 0.059 ± 0.011 |
| heart | 0.811 ± 0.167 | 0.445 ± 0.030 | 0.379 ± 0.015 | 0.316 ± 0.061 |
| brain | 0.027 ± 0.004 | 0.018 ± 0.002 | 0.017 ± 0.002 | 0.015 ± 0.002 |
| bone | 0.215 ± 0.027 | 0.114 ± 0.002 | 0.105 ± 0.100 | 0.100 ± 0.012 |
| pancreas | 0.755 ± 0.207 | 0.320 ± 0.047 | 0.298 ± 0.089 | 0.265 ± 0.128 |
| thymus | 0.150 ± 0.023 | 0.151 ± 0.010 | 0.161 ± 0.007 | 0.182 ± 0.013 |
2.4.2. In Vitro Autoradiography and IHC in LPS-Treated Mice
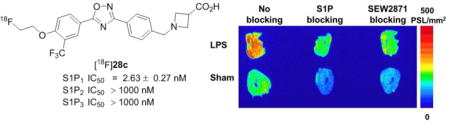
We utilized the mouse model of LPS-induced liver injury and inflammation, in which inflammation caused by LPS is known to cause increased liver expression of S1P1 after ∼24 h to evaluate [18F]28c in tissue from an animal model of inflammatory disease.44,45 In vitro autoradiographic data showed increased uptake of [18F]28c in the liver of LPS-treated mice, compared with the sham liver shown in Figure 3A. The specificity of [18F]28c for S1P1 was demonstrated by incubation of adjacent mouse liver slices with the tracer in the presence of either the native ligand (S1P) or the S1P1 selective compound, SEW2871.46 The upregulation of S1P1 in the liver at the 24 h time point following LPS treatment was further confirmed by IHC staining as shown in Figure 3B. These blocking studies in conjunction with the IHC results suggest that the binding of [18F]28c in the liver of LPS-treated mice is the result of specific binding to the receptor and is increased when S1P1 expression is increased.
Figure 3.
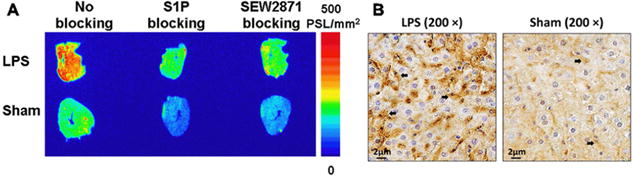
(A) Representative in vitro autoradiographic images of serial liver sections from LPS-treated and sham mice after incubation with [18F]28c under baseline or blocking conditions. (B) IHC staining for S1P1 in mouse liver sections. Positive staining for S1P1 (brown, indicated by black arrows) was observed in the liver of both LPS-treated (left) and sham (right) mice. The number of S1P1-positive cells in the liver of LPS-treated mice was much greater than that in sham mice.
2.4.3. MicroPET Imaging of LPS-Treated Mice with [18F]28c and [15O]Water
Subsequent microPET imaging in the LPS model of liver inflammation also clearly showed increased accumulation of [18F]28c in the liver of LPS-treated mice compared to that in sham controls (Figure 4A). Time–activity curves (TAC) confirmed higher tracer uptake in the liver of LPS pretreated mice compared with that in the sham controls (Figure 4B). Quantification of the liver region of interest (ROI) in the microPET scan from 100 to 120 min shows a 61% increase (P < 0.001, n = 4) (Figure 4C). These results were confirmed by euthanasia of the mice post-PET for an acute biodistribution study. The liver uptake of [18F]28c in the LPS-treated mouse at 2 h was 1.95-fold higher than that in the sham mice (12.5 vs 24.4% ID/gram, P < 0.001, n = 4). To further demonstrate that the increased uptake of [18F]28c in the liver was not attributed to changes in blood flow in the LPS-induced model of liver injury, additional microPET studies were performed with an injection of [15O]water 30 min prior to the injection of [18F]28c. [15O]Water is a widely used radiotracer for assessing blood flow.47,48 As shown in Figure 4D, the liver TAC for [15O]water in LPS-treated mice showed no significant change compared to that in the sham mice. This suggested that the increase in vivo uptake of [18F]28c in the liver of LPS-treated mice is the result of increased S1P1 expression and not increased blood flow.
Figure 4.
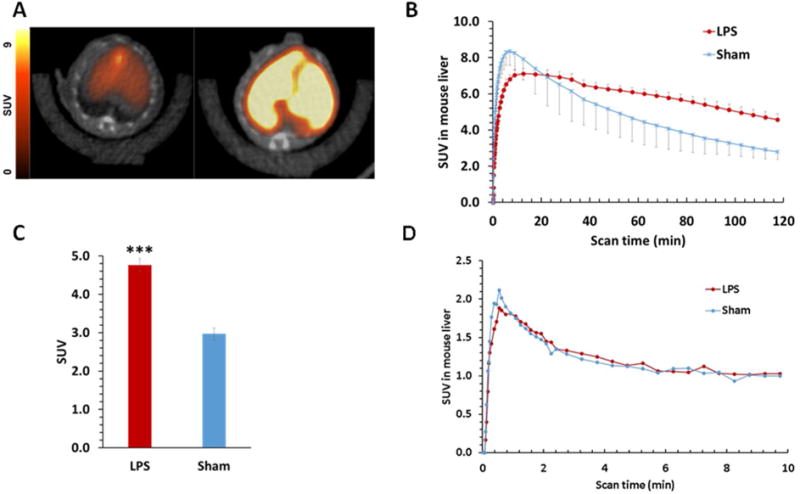
(A) Representative summed 120 min transverse PET/CT images of [18F]28c in a sham (left) and LPS-treated (right) mouse. (B) Liver time–activity curves (TAC) of [18F]28c standardized uptake values (SUV) in sham and LPS-treated mice. (C) Sham vs LPS-treated summed liver SUVs from 100 to 120 min postinjection. P < 0.001. (D) Liver TACs of [15O]H2O in sham and LPS-treated mice.
2.4.4. Biodistribution of [18F]28c and [99mTc]Mebrofenin in LPS-Treated Mice
To confirm that the uptake of [18F]28c was associated with S1P1 expression, we utilized a mouse model of LPS-induced liver injury and inflammation, in which inflammation caused by LPS is known to cause increased liver expression of S1P1 after ∼24 h.44,45 Biodistribution studies comparing [18F]28c uptake in saline-treated sham controls vs the LPS-treated mouse model demonstrated a large increase in liver uptake (Figure 5). In order to demonstrate that tracer retention in the liver was not due to impaired hepatobiliary clearance in the liver injury model, an additional study was carried out with this model using a liver imaging agent. [99mTc]Mebrofenin is used to detect if the hepatobiliary transport is impaired. Increased liver uptake of [99mTc]-mebrofenin indicates the potential for nonspecific retention of drugs (in this case, [18F]28c) in the liver due to impaired clearance, rather than uptake specific to the pathological increase in S1P1 at the molecular level.36,49,50,32,33 We hypothesized that increased liver uptake of [99mTc]mebrofenin in LPS-treated mice versus the sham control mice at 60 min would suggest that increased uptake of the S1P1 tracer resulted from a severely damaged liver; however, if the liver uptake of [99mTc]mebrofenin in LPS-treated mice versus the sham mice was similar at 60 min (as we observed), the increase should reflect an increase in S1P1 rather than impaired clearance.36,49 To confirm that this increase in uptake of [18F]28c resulted from the increase expression of S1P1 receptor response to liver injury, the biodistribution study of [99mTc]mebrofenin was performed in the LPS-induced liver injury models. Although increased [99mTc]mebrofenin was observed in the liver of LPS-treated mice at 30 min p.i. compared with sham mice, at 60 min p.i. there was no significant difference between the sham and LPS study groups. The uptake of [18F]28c showed a significant increase in the LPS-treated mice compared to that of the sham control mice at both 30 and 60 min (Figure 5). The biodistribution data demonstrated that the increased hepatobiliary uptake of [18F]28c in LPS-treated mice is mainly caused by the upregulation of S1P1 expression in the liver.
Figure 5.
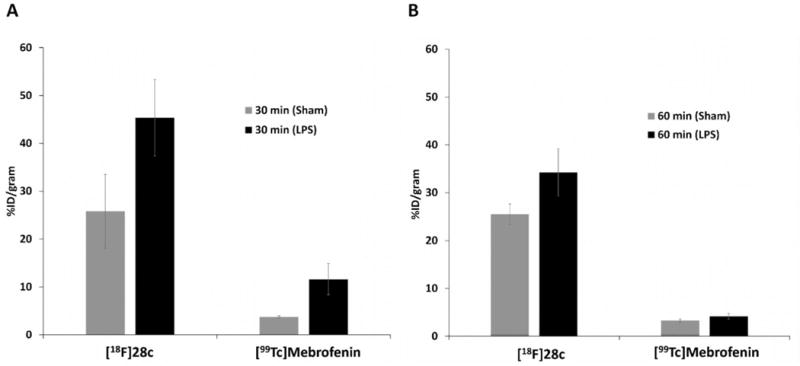
Liver uptake in LPS-treated and sham mice 30 min (A) and 60 min (B) postinjection of [18F]28c or [99mTc]mebrofenin.
3. CONCLUSIONS
We synthesized 14 fluorine-containing analogues based on two lead pharmacophores and determined their affinity for S1P1. Potent compounds (IC50 < 50 nM for S1P1) were subsequently screened for their S1P2 and S1P3 binding affinities. Seven compounds were found to have IC50 values <100 nM, while three were very potent with S1P1 IC50 values <10 nM. We explored the SAR studies on the oxadiazole containing pharmacophore by optimizing the lipophilic tail binding portion of the binding pocket. The oxadiazole 28c had a high potency (2.63 nM for S1P1) and selectivity (>100-fold for S1P1 versus S1P2/3), and was successfully radiolabeled in high yield and high specific activity. Biodistribution in normal rats showed no evidence of defluorination, so the mouse model of liver inflammation was used for subsequent biological evaluation of [18F]28c as a PET tracer for imaging S1P1. In vitro autoradiography and microPET imaging studies showed increased binding in vitro and in vivo retention of [18F]28c in the liver of LPS-treated vs control mice. IHC staining studies confirmed that S1P1 expression was increased in the liver of LPS-treated mice. Additional microPET imaging showed no difference in liver uptake of [15O]water between LPS-treated and control mice,37,38 further supporting the hypothesis that the higher uptake and retention of [18F]28c in the liver of LPS-treated mice was a reflection of increased S1P1 due to inflammation and not due to a change in blood flow. Parallel acute biodistribution studies in the LPS model were carried out with both [18F]28c and the liver imaging agent [99mTc]-mebrofenin.36 The absence of a significant difference in liver uptake of [99mTc]mebrofenin in LPS-treated vs control liver uptake 60 min postinjection suggested that the increased liver uptake of [18F]28c observed in the PET studies of LPS-treated mice was not the result of reduced hepatobiliary clearance due to liver injury. Together, these data indicate that uptake of [18F] 28c reflects the expression of S1P1 and suggest that [18F]28c is a S1P1 specific radiotracer with potential for use in quantifying S1P1 receptor expression in response to inflammation.
4. EXPERIMENTAL SECTION
4.1. Chemistry Materials and Methods
Unless otherwise indicated, all reactions were conducted in oven-dried (140 °C) glassware. Stainless steel syringes or cannulae that had been oven-dried (140 °C) and cooled under a nitrogen atmosphere or in a desiccator were used to transfer air- and moisture-sensitive liquids. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials, unless otherwise stated. Reactions were monitored by thin-layer chromatography (TLC) carried out on precoated glass plates of silica gel (0.25 mm) 60 F254 from EMD Chemicals Inc. Visualization was accomplished with ultraviolet light (UV 254 nm) or by shaking the plate in a sealed jar containing silica gel and iodine. Alternatively, plates were treated with one of the following solutions (this was accomplished by holding the edge of the TLC plate with forceps or tweezers and immersing the plate into a wide-mouth jar containing the desired staining solution) and carefully heating with a hot-air gun (450 °C) for approximately 1–2 min (note: excess stain was removed by resting the TLC on a paper towel prior to heating): 10% phosphomolybdic acid in ethanol, 1% potassium permanganate/7% potassium carbonate/0.5% sodium hydroxide aqueous solution, and/or anisaldehyde in ethanol with 10% sulfuric acid. Flash column chromatography was performed using Silia Flash P60 silica gel (40–63 μm) from Silicycle. All workup and purification procedures were carried out with reagent grade solvents in air.
1H NMR spectra were recorded on a Varian 400 MHz instrument. Chemical shifts are reported in parts per million (ppm) and are calibrated using residual undeuterated solvent as an internal reference (CDCl3, δ 7.26 ppm; MeOD-d4, δ 3.31 ppm; DMSO-d6, δ 2.50 ppm). Data are reported as follows: chemical shift, multiplicity, coupling constants (Hz), and integration. The following abbreviations or combinations thereof were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, pent = pentet, sext = sextet, sept = septet, m = multiplet, at = apparent triplet, aq = apparent quartet, and b = broad. 13C NMR spectra were recorded on a Varian instrument (100 MHz) spectrometer with complete proton decoupling. Chemical shifts are reported in ppm and are calibrated using a residual undeuterated solvent as an internal reference (CDCl3, δ 77.16 ppm; MeOD-d4, 49.00 ppm; and DMSO-d6, 39.52 ppm). Elemental compositions (C, H, and N) are within ±0.4% of the calculated values and were determined by Atlantic Microlab, Inc. Lipophilicity values as cLogD are reported as the calculated Log P value at pH 7.4 and were obtained using ACD/I-Lab version 7.0 (Advanced Chemistry Development, Inc. Toronto, Ontario, Canada). The biological activity of all of the final compounds was determined by an analytical HPLC method with purity ≥ 95%.
4.1.1. Synthesis. General Procedure A: Suzuki Coupling
To an oven-dried round-bottomeded flask equipped with a stir bar was added Pd(OAc)2 (2 mol %), 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPhos, 5 mol %), commercially available 4-bromo-3-(trifluoromethyl)benzoic acid (6) (1 equiv), the boronic acid (1.1 equiv), and a fluoride base (3 equiv). 1,4-Dioxane (0.37 M) and degassed H2O (0.37 M) were added to the reaction mixture, and the reaction was degassed by sparging with N2(g) for 10 min, at which time it was equipped with a condenser and placed in a preheated 110 °C oil-bath. The reaction mixture was stirred for the time indicated, cooled to room temperature (r.t.), and poured into a 1:1 mixture of ethyl acetate and 1 N HCl (aq). The quenched reaction mixture was stirred for 20 min, and the layers were then separated. The aqueous layer was extracted with ethyl acetate (×2). The combined organic layers were dried over MgSO4, concentrated in vacuo, and purified as specified.
General Experimental B: Amide Formation
To an oven-dried round-bottomeded flask equipped with a stir bar was added carboxylic acid (1 equiv) followed by CH2Cl2 (0.1 M). Five drops of N,N-dimethylformamide was added via pipet followed by the slow addition of oxalyl chloride (2.3 equiv) via a syringe. The reaction was then stirred at r.t. for 2 h, at which time it was concentrated in vacuo. The crude acid chloride was then dissolved in toluene (0.15 M), and 10% NaHCO3(aq) (0.3 M) was added. The aniline 9a or commercially available 2-amino-4-bromophenol (9b) (1 equiv) was added, and the reaction was stirred overnight. Upon completion of the reaction, the resulting precipitate was filtered and washed with H2O, toluene, and hexanes to give the desired amide.
General Experimental C: Benzoxazole Formation
To a round-bottomeded flask equipped with a stir bar was added the amide (1 equiv), toluene (0.1 M), and p-toluenesulfonic acid (2.1 equiv) or pyridinium p-toluenesulfonate (2.1 equiv). The reaction was equipped with a reflux condenser and lowered into a preheated 130 °C oil-bath. The reaction mixture was stirred for the specified time, cooled to r.t., diluted with EtOAc, and washed with sat. NaHCO3(aq) (×2), and brine. The organic layer was dried over MgSO4 and concentrated in vacuo to give the desired benzoxazole.
General Procedure D: Buchwald–Hartwig Coupling
To an oven-dried flask (pressure-vessel or Schlenk tube) equipped with a stir bar was added Pd2(dba)3·CHCl3 (2 mol %), XPhos (8 mol %), sodium tert-butoxide (1.4 equiv), and the aryl bromide (1 equiv). Toluene (0.5 M) was added followed by the amine (1.2 equiv), and the reaction mixture was degassed by sparging with N2(g) for 10 min. The reaction vessel was sealed with a Teflon screw-cap and lowered into a preheated 110 °C oil-bath. After 18–20 h, the reaction mixture was allowed to cool to r.t. and diluted with ethyl acetate. The reaction mixture was then concentrated in vacuo and purified as specified.
General Procedure E: Saponification
To a 16 × 150 mm test tube equipped with a stir bar was added the ester (1 equiv), THF (0.24 M), H2O (1.2 M), and lithium hydroxide (2 equiv). The reaction mixture was stirred at r.t. for 16–18 h, at which time it was acidified to pH 1 with 1 M HCl(aq). The reaction mixture was extracted with ethyl acetate (×4). The combined organic layers were washed with brine and concentrated in vacuo to give the desired products.
General Procedure F: Oxadiazole Formation
To an oven-dried round-bottomeded flask equipped with a stir bar was added the acid (1 equiv), EDC·HCl (1 equiv), HOBt (1 equiv), and DMF (0.8 M). The reaction mixture was stirred at r.t. for 30 min, at which time amidoxime (1 equiv) was added. A reflux condenser was attached, and the reaction vessel was placed in a preheated 120–140 °C oil-bath and stirred overnight (14–18 h). The reaction mixture was cooled to r.t. and diluted with EtOAc and water. The layers were separated, and the aqueous layer was extracted with EtOAc (×1). The combined organic layers were washed with water, 1 M HCl(aq), water, and brine, dried over MgSO4, and concentrated in vacuo to give the desired product.
General Procedure G: Reductive Amination
To a round-bottomeded flask equipped with a stir bar was added aldehyde (1 equiv), amine (1.05 equiv), methanol (0.07 M), and acetic acid (2 M). The reaction mixture was stirred for 30 min at which time sodium cyanoborohydride (0.5 equiv) was added as a solution in methanol (0.25 M). The reaction mixture was stirred for 1–2 h, at which time the solids were filtered and washed with methanol to give the desired product.
4-(2-Fluoroethoxy)-3-(trifluoromethyl)benzoic Acid (7a)
To an oven-dried 500 mL round-bottomed flask equipped with a stir bar was added 4-fluoro-3-(trifluoromethyl)benzoic acid (8)(8.32 g, 40.0 mmol), 100 mL of DMSO, and 2-fluoroethanol (3.52 mL, 60.0 mmol). Sodium hydride (3.60 g, 90.0 mmol) was added portionwise, and the reaction mixture was stirred for 16 h. The reaction mixture was poured into water and acidified with 12 M HCl(aq) to pH 1. The resulting precipitate was filtered and washed with water and hexanes to give a tan solid (9.8 g, 97% yield). 1H NMR (400 MHz, DMSO-d6) δ = 12.18 (s, 1H), 8.18 (dd, J = 8.8 Hz, 2.0 Hz, 1H), 8.11 (d, J = 2.0 Hz, 1H), 7.39 (d, J = 8.8 Hz, 1H), 4.85–4.40 (m, 4H). 13C NMR (101 MHz, DMSO-d6) δ = 166.0, 159.44, 135.6, 128.0 (q, JC–F = 4.9 Hz), 123.2 (q, JC–F = 274 Hz), 123.2, 117.4 (q, JC–F = 31.1 Hz), 113.9, 81.8 (d, JC–F = 168 Hz), 68.6 (d, JC–F = 19.1 Hz), MP: 131–133 °C. HRMS (EI-TOF) m/z calcd for C10H7F4O3 [M – H] 251.0337. Found [M – H] 251.0289.
4′-Fluoro-2-(trifluoromethyl)-[1,1′-biphenyl]-4-carboxylic Acid (7b)
Following general experimental procedure A, Pd(OAc)2 (67 mg, 0.30 mmol), SPhos (306 mg, 0.75 mmol), 6 (4.0 g, 14.9 mmol), 4-fluorophenylboronic acid (2.29 g, 16.4 mmol), cesium fluoride (6.79 g, 44.7 mmol), 1,4-dioxane (40 mL), and degassed H2O (40 mL) were combined. The reaction mixture was stirred for 18 h, to give 3.0 g of a tan solid which was used without further purification. 1H NMR (400 MHz, DMSO-d6) δ = 13.56 (br s, 1H), 8.28 (s, 1H), 8.23 (d, J = 8 Hz, 1H), 7.57 (d, J = 8 Hz, 1H), 7.43–7.38 (m, 2H), 7.35–7.29 (m, 2H).
2′-Hydroxy-2-(trifluoromethyl)-[1,1′-biphenyl]-4-carboxylic Acid (7c)
Following general experimental procedure A, Pd(OAc)2 (84 mg, 0.37 mmol), SPhos (382 mg, 0.93 mmol), 6 (5.0 g, 18.6 mmol), 2-hydroxyphenylboronic acid (2.82 g, 20.4 mmol), cesium fluoride (8.48 g, 55.8 mmol), 1,4-dioxane (50 mL), and degassed H2O (50 mL) were combined. The reaction mixture was stirred for 20 h and purified on a silica gel column, eluting with 5% MeOH/CH2Cl2, to provide 5.2 g of a yellow semisolid. 1H NMR (400 MHz, DMSO-d6) δ = 9.54 (s, 1H), 8.25 (s, 1H), 8.18 (d, J = 8 Hz, 1H), 7.47 (d, J = 8 Hz, 1H), 7.23 (td, J = 8.8 Hz, 1.6 Hz, 1H), 7.04 (d, J = 7.2 Hz, 1H), 6.91 (d, J = 8.4 Hz, 1H), 6.85 (t, J = 7.2 Hz, 1H).
4′-Hydroxy-2-(trifluoromethyl)-[1,1′-biphenyl]-4-carboxylic Acid (7d)
Following general experimental procedure A, Pd(OAc)2 (67 mg, 0.30 mmol), SPhos (306 mg, 0.75 mmol), 6 (4.0 g, 14.9 mmol), 4-hydroxyphenylboronic acid (2.46 g, 17.8 mmol), cesium fluoride (6.79 g, 44.7 mmol), 1,4-dioxane (40 mL), and degassed H2O (40 mL). The reaction mixture was stirred for 20 h and purified on a silica gel column, eluting with 5% MeOH/CH2Cl2, to provide 4.2 g of a yellow semisolid. 1H NMR (400 MHz, DMSO-d6) δ = 9.73 (s, 1H), 8.25 (s, 1H), 8.18 (d, J = 8 Hz, 1H), 7.52 (d, J = 8 Hz, 1H), 7.16 (d, J = 8.4 Hz, 2H), 6.83(d, J = 8.8 Hz, 2H).
2,4′-Bis(trifluoromethyl)-[1,1′-biphenyl]-4-carboxylic Acid (7e)
Following general experimental procedure A, Pd(OAc)2 (42 mg, 0.19 mmol), SPhos (191 mg, 0.47 mmol), 6 (2.5 g, 9.3 mmol), 4-(trifluorofluoromethyl)phenylboronic acid (2.12 g, 11.2 mmol), potassium fluoride (4.24 g, 27.9 mmol), 1,4-dioxane (25 mL), and degassed H2O (25 mL). The reaction mixture was stirred for 22 h to give 2.35 g of a yellow solid, which was used without further purification. 1H NMR (400 MHz, DMSO-d6) δ = 12.67 (br s, 1H), 8.31 (d, J = 1.2 Hz, 1H), 8.26 (d, J = 8 Hz, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.61 (t, J = 7.6 Hz, 3H).
4-(6-Fluoropyridin-3-yl)-3-(trifluoromethyl)benzoic Acid (7f)
Following general experimental procedure A, Pd(OAc)2 (50 mg, 0.22 mmol), SPhos (230 mg, 0.56 mmol), 6 (3.0 g, 11.15 mmol), (6-fluoropyridin-3-yl)boronic acid (1.89 g, 13.4 mmol), cesium fluoride (5.08 g, 33.5 mmol), 30 mL of 1,4-dioxane, and 30 mL of degassed H2O. The reaction mixture was stirred for 18 h to give a yellow solid, which was used without further purification (3.0 g, 94% yield). 1H NMR (400 MHz, DMSO-d6) δ = 13.63 (br s, 1H), 8.31 (s, 1H), 8.30–8.24 (m, 2H), 8.09–7.98 (m, 1H), 7.66 (d, J = 7.6 Hz), 7.34 (dd, J = 8.4 Hz, 2.8 Hz, 1H).
3′-Methoxy-2-(trifluoromethyl)-[1,1′-biphenyl]-4-carboxylic Acid (7g)
Following general experimental procedure A, Pd(OAc)2 (50 mg, 0.22 mmol), SPhos (230 mg, 0.56 mmol), 6 (3.0 g, 11.15 mmol), 3-methoxyphenylboronic acid (2.04 g, 13.4 mmol), cesium fluoride (5.08 g, 33.5 mmol), 30 mL of 1,4-dioxane, and 30 mL of degassed H2O. The reaction mixture was stirred for 18 h, to give a yellow solid, which was used without further purification (3.12 g, 95% yield). 1H NMR (400 MHz, DMSO-d6) δ = 13.22 (br s, 1H), 8.28 (d, J = 1.2 Hz, 1H), 8.22 (dd, J = 8.0 Hz, 1.6 Hz, 1H), 7.57 (d, J = 8.0 Hz, 1H), 7.38 (t, J = 8.4 Hz, 1H), 7.06–7.01 (m, 1H), 6.93–6.86 (m, 2H), 3.78 (s, 3H).
Methyl 3-amino-4-hydroxybenzoate (9a)
To a 250 mL round-bottomed flask equipped with a stir bar was added methyl 4-hydroxy-3-nitrobenzoate (5.0 g, 25.4 mmol), 120 mL of EtOH, and 540 mg of 10% Pd/C. One balloon of H2(g) was bubbled through the reaction mixture, and then it was stirred under 1 atm of H2(g) overnight (16 h). At this time, the reaction was filtered through a Celite plug, washing with EtOH, and concentrated in vacuo to give a brown solid (4.17 g, 98% yield). 1H NMR (400 MHz, CDCl3) δ = 7.45 (d, J = 2.0 Hz, 1H), 7.42 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 7.75 (d, J = 8.4 Hz, 1H), 3.87 (s, 3H).
Methyl 3-(4-(2-fluoroethoxy)-3-(trifluoromethyl)benzamido)-4-hydroxybenzoate (10a)
To an oven-dried 100 mL of round-bottomed flask equipped with a stir bar was added acid 7a (2.50 g, 9.91 mmol), 50 mL of CH2Cl2, and 10 drops of DMF. Oxalyl chloride (1.68 mL, 19.8 mmol) was added carefully, and the reaction mixture was stirred for 2 h at r.t. The reaction was concentrated in vacuo and dissolved in 33 mL of CH2Cl2. Triethylamine (5.52 mL, 39.6 mmol) was added followed by DMAP (120 mg, 0.99 mmol) and aniline 9a (1.66 g, 9.91 mmol). The reaction mixture was stirred overnight and diluted with MTBE, at which point a precipitate had formed. The precipitate was filtered, washed with MTBE, and hexanes to give a white solid which was used in the next step without further purification. 1H NMR (400 MHz, DMSO-d6) δ = 10.75 (br s, 1H), 9.83 (s, 1H), 8.27 (app q, J = 2.4 Hz, 2H), 8.23 (d, J = 2.4 Hz, 1H), 7.70 (dd, J = 8.4 Hz, 2.4 Hz, 1H), 7.43 (d, J = 9.2 Hz, 1H), 7.02 (d, J = 8.4 Hz, 1H), 4.87–4.68 (m, 2H), 4.57–4.42 (m, 2H).
N-(5-Bromo-2-hydroxyphenyl)-4′-fluoro-2-(trifluoromethyl)-[1,1′-biphenyl]-4-carboxamide (10b)
Following general experimental procedure B, carboxylic acid 7b (720 mg, 2.53 mmol), 25.3 mL of CH2Cl2, oxalyl chloride (0.49 mL, 5.82 mmol), 17 mL of toluene, 8.5 mL of 10% NaHCO3(aq), and 9b (476 mg, 2.53 mmol) were combined to give amide 10b as a tan solid (1.11 g, 99% yield). 1H NMR (400 MHz, DMSO-d6) δ = 8.30 (s, 1H), 8.19 (d, J = 8 Hz, 1H), 7.99 (s, 1H), 7.51 (d, J = 8 Hz, 1H), 7.35–7.25 (m, 4H), 7.08 (d, J = 8 Hz, 1H), 6.76 (d, J = 8 Hz, 1H).
N-(5-Bromo-2-hydroxyphenyl)-2′-hydroxy-2-(trifluoromethyl)-[1,1′-biphenyl]-4-carboxamide (10c)
Following general experimental procedure B, carboxylic acid 7c (3.5 g, 12.4 mmol), 124 mL of CH2Cl2, oxalyl chloride (2.41 mL, 28.5 mmol), 82 mL of toluene, 41 mL of 10% NaHCO3(aq), and 9b (2.33 g, 12.4 mmol) were combined to give amide 10c as a brown solid (1.15 g, 20% yield). 1H NMR (400 MHz, DMSO-d6) δ = 8.32 (s, 1H), 8.19 (d, J = 8.0 Hz, 1H), 7.87 (d, J = 2.4 Hz, 1H), 7.48 (d, J = 7.6 Hz, 1H), 7.28–7.17 (m, 2H), 7.05 (d, J = 6.8 Hz, 1H), 6.92 (d, J = 8.0 Hz, 1H), 6.89–6.82 (m, 2H).
N-(5-Bromo-2-hydroxyphenyl)-4′-hydroxy-2-(trifluoromethyl)-[1,1′-biphenyl]-4-carboxamide (10d)
Following general experimental procedure B, carboxylic acid 7d (4.2 g, 14.9 mmol), 149 mL of CH2Cl2, oxalyl chloride (2.90 mL, 34.3 mmol), 100 mL of toluene, 50 mL of 10% NaHCO3(aq), and 9b (2.80 g, 14.9 mmol) were combined to give amide 10d as a brown solid which was used in the next step without further purification (3.4 g, 50% yield).
N-(5-Bromo-2-hydroxyphenyl)-2,4′-bis(trifluoromethyl)-[1,1′-biphenyl]-4-carboxamide (10e)
Following general experimental procedure B, acid 7e (2.27 g, 6.79 mmol), 68 mL of CH2Cl2, oxalyl chloride (1.21 mL, 14.3 mmol), 46 mL toluene, 23 mL of 10% NaHCO3(aq), and 9b (1.28 g, 6.79 mmol) were combined to give a light brown solid (2.81 g, 82% yield). 1H NMR (400 MHz, DMSO-d6) δ = 10.09 (br s, 1H), 8.42 (s, 1H), 8.29 (d, J = 8.0 Hz, 1H), 7.92–7.84 (m, 3H), 7.65–7.56 (m, 3H), 7.23 (dd, J = 8.8 Hz, 2.4 Hz, 1H), 6.89 (d, J = 8.8 Hz, 1H).
N-(5-Bromo-2-hydroxyphenyl)-4-(6-fluoropyridin-3-yl)-3-(trifluoromethyl)benzamide (10f)
Following general experimental procedure B, acid 7f (1.00 g, 3.51 mmol), 35 mL of CH2Cl2, oxalyl chloride (0.68 mL, 8.1 mmol), 24 mL of toluene, 12 mL of 10% NaHCO3(aq), and 9b (660 mg, 3.51 mmol) were combined to give a tan solid (1.0 g, 63% yield). 1H NMR (400 MHz, DMSO-d6) δ = 10.32 (br s, 1H), 8.40 (s, 1H), 8.35–8.18 (m, 2H), 8.03 (t, J = 8.4 Hz, 1H), 7.89 (d, J = 1.6 Hz, 1H), 7.64 (d, J = 7.6 Hz, 1H), 7.35 (d, J = 6.4 Hz, 1H), 7.19 (d, J = 8.4 Hz, 1H), 6.87 (d, J = 8.8 Hz, 1H).
Methyl 3-(4-(6-Fluoropyridin-3-yl)-3-(trifluoromethyl)-benzamido)-4-hydroxybenzoate (10g)
To an oven-dried 100 mL round-bottomed flask equipped with a stir bar was added acid 7f (1.50 g, 5.26 mmol), 53 mL of CH2Cl2, and 10 drops of DMF. Oxalyl chloride (0.89 mL, 10.5 mmol) was added carefully, and the reaction was stirred for 2 h, where upon it was concentrated in vacuo to give a yellow semisolid. The crude acid chloride was suspended in 26 mL of CH2Cl2, and triethylamine (2.93 mL, 21.0 mmol) was added, followed by DMAP (64 mg, 0.53 mmol). The reaction was stirred for 5 min at which time aniline 9a (880 mg, 5.26 mmol) was added. The reaction mixture was stirred overnight (18 h), quenched with 1 M HCl(aq), and diluted with CH2Cl2, causing a suspension to form in the organic layer. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (×3). The combined organic layers were diluted with hexanes and filtered to give a brown solid (1.02 g, 45% yield). 1H NMR (400 MHz, DMSO-d6) δ = 10.78 (br s, 1H), 10.05 (br s, 1H), 8.45 (s, 1H), 8.33 (d, J = 7.6 Hz, 1H), 8.29 (s, 2H), 8.05 (t, J = 6.4 Hz, 1H), 7.73 (d, J = 8.4 Hz, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 8.4 Hz, 1H), 7.04 (d, J = 8.4 Hz, 1H), 3.82 (s, 3H).
N-(5-Bromo-2-hydroxyphenyl)-3′-methoxy-2-(trifluoromethyl)-[1,1′-biphenyl]-4-carboxamide (10h)
Following general experimental procedure B, acid 7g (3.1 g, 10.5 mmol), 105 mL of CH2Cl2, oxalyl chloride (2.04 mL, 24.2 mmol), 70 mL of toluene, 35 mL of 10% NaHCO3(aq), and 9b (1.97 g, 10.5 mmol) were combined to give a brown solid which was used in the next step without further purification (3.96 g, 81% yield).
Methyl 2-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)benzo-[d]oxazole-5-carboxylate (11a)
Following general experimental procedure C, amide 10a (crude from previous step), 99 mL of toluene, and p-toluenesulfonic acid (3.77 g, 19.8 mmol) were combined to give a tan solid (1.1 g, 29% yield over two steps). 1H NMR (400 MHz, CDCl3) δ = 8.56–8.49 (m, 1H), 8.45 (d, J = 1.6 Hz, 1H), 8.41 (dd, J = 8.8 Hz, 2.4 Hz, 1H), 8.12 (dd, J = 8.4 Hz, 1.6 Hz, 1H), 7.62 (d, J = 8.4 Hz, 1H), 7.17 (d, J = 8.4 Hz, 1H), 4.93–4.75 (m, 2H), 4.48–4.37 (m, 2H), 3.97 (s, 3H).
5-Bromo-2-(4′-fluoro-2-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)-benzo[d]oxazole (11b)
Following general experimental procedure C, amide 10b (980 mg, 2.17 mmol), 21.7 mL of toluene, and p-toluenesulfonic acid (867 mg, 4.56 mmol) were combined to give oxazole 11b as a red solid (760 mg, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ = 8.52 (s, 1H), 8.48 (d, J = 8 Hz, 1H), 8.13 (d, J = 2 Hz, 1H), 7. 86 (d, J = 8.8 Hz, 1H), 7.69 (d, J = 8 Hz, 1H), 7.66 (dd, J = 8.8 Hz, 2 Hz, 1H), 7.50–7.41 (m, 2H), 7.35 (t, J = 8.8 Hz, 2H).
5-Bromo-2-(2′-(2-fluoroethoxy)-2-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)benzo[d]oxazole (11c)
Following general experimental procedure C, amide 10c (1.06 g, 2.34 mmol), 23.4 mL of toluene, and p-toluenesulfonic acid (0.94 g, 4.92 mmol) were combined to give the crude product which was applied to a silica gel column, eluted with 20% EtOAc/hexanes, to provide the phenol intermediate as a red solid (820 mg, 80% yield). 1H NMR (400 MHz, DMSO-d6) δ = 9.64 (s, 1H), 8.49 (s, 1H), 8.43 (d, J = 8.0 Hz, 1H), 8.13 (d, J = 1.6 Hz, 1H), 7.86 (d, J = 8.4 Hz, 1H), 7.65 (dd, J = 8.8 Hz, 2.0 Hz, 1H), 7.60 (d, J = 8.8 Hz, 1H), 7.26 (t, J = 7.6 1H), 7.09 (d, J = 7.2 Hz, 1H), 6.94 (d, J = 8.4 Hz, 1H), 6.87 (t, J = 7.2 Hz, 1H).
To an oven-dried 16 × 125 mm test tube equipped with a stir bar was added the phenol prepared above (290 mg, 0.67 mmol), cesium carbonate (437 mg, 1.34 mmol), and 2.2 mL of N,N-dimethylformamide. 2-Fluoroethyl 4-methylbenzenesulfonate (160 mg, 0.73 mmol) was added via syringe, and the reaction vessel was placed in a preheated 70 °C oil-bath and stirred for 4 h. The reaction mixture was cooled to r.t. and diluted with EtOAc and H2O, and the layers were separated. The organic layer was washed with H2O (×5) and brine, dried over sodium sulfate, and concentrated in vacuo to give a light brown solid, which was used without further purification (310 mg, 97% yield). 1H NMR (400 MHz, CDCl3) δ = 8.58 (s, 1H), 8.34 (d, J = 8.4 Hz, 1H), 7.89 (s, 1H), 7.53–7.43 (m, 3H), 7.38 (t, J = 7.6 Hz, 1H), 7.20 (d, J = 7.2 Hz, 1H), 7.04 (t, J = 7.6 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 4.52 (dt, J = 47.2 Hz, 4 Hz, 2H), 4.31–4.03 (m, 2H).
5-Bromo-2-(4′-(2-fluoroethoxy)-2-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)benzo[d]oxazole (11d)
Following general experimental procedure C, amide 10d (3.4 g, 7.52 mmol), 75 mL of toluene, and p-toluenesulfonic acid (3.0 g, 15.8 mmol) were combined to give the crude product, which was applied to a silica gel column, eluted with 20% EtOAc/hexanes, to provide the phenol intermediate as a red solid (1.21 g, 37% yield). 1H NMR (400 MHz, DMSO-d6) δ = 9.77 (s, 1H), 8.46 (s, 1H), 8.41 (d, J = 8 Hz, 1H), 8.10 (d, J = 1.6 Hz, 1H), 7.83 (d, J = 8.8 Hz, 1H), 7.63 (d, J = 8 Hz, 1H), 7.66 (dd, J = 8.8 Hz, 2 Hz, 1H), 7.20 (d, J = 8.4 2H), 7.86 (d, J = 8.4 Hz, 2H).
To an oven-dried 16 × 125 mm test tube equipped with a stir bar was added the phenol prepared above (217 mg, 0.50 mmol), cesium carbonate (326 mg, 1.0 mmol), and 1.7 mL of N,N-dimethylformamide. 2-Fluoroethyl 4-methylbenzenesulfonate (120 mg, 0.55 mmol) was added via a syringe, and the reaction vessel was placed in a preheated 70 °C oil-bath and stirred for 4 h. The reaction mixture was cooled to r.t. and diluted with EtOAc and H2O, and the layers were separated. The organic layer was washed with H2O (×5) and brine, dried over sodium sulfate, and concentrated in vacuo to give 232 mg of a light brown solid, which was used without further purification. 1H NMR (400 MHz, DMSO-d6) δ = 8.51 (s, 1H), 8.46 (d, J = 8 Hz, 1H), 8.13 (d, J = 2 Hz, 1H), 7.86 (d, J = 8.4 Hz, 1H), 7.70–7.62 (m, 2H), 7.34 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 8.8 Hz, 2H), 4.87–4.70 (m, 2H), 4.37–4.25 (m, 2H).
2-(2,4′-Bis(trifluoromethyl)-[1,1′-biphenyl]-4-yl)-5-bromobenzo-[d]oxazole (11e)
Following general experimental procedure C, amide 10e (2.70 g, 5.35 mmol), 54 mL of toluene, and p-toluenesulfonic acid monohydrate (2.14 g, 11.2 mmol) were combined to give a light red solid (2.39 g, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ = 8.55 (s, 1H), 8.52 (d, J = 8.0 Hz, 1H), 8.15 (d, J = 2.0 Hz, 1H), 7.92–7.85 (m, 3H), 7.75 (d, J = 8.4 Hz, 1H), 7.69–7.61 (m, 3H).
5-Bromo-2-(4-(6-fluoropyridin-3-yl)-3-(trifluoromethyl)phenyl)-benzo[d]oxazole (11f)
To an oven-dried 25 mL round-bottomed flask equipped with a stir bar was added amide 10f (935 mg, 2.05 mmol) and 8.2 mL of THF. Triphenylphosphine (1.19 g, 4.52 mmol) was added followed by diisopropylazodicarboxylate (0.89 mL, 4.52 mmol). The reaction mixture was stirred at r.t. for 22 h and concentrated in vacuo. The crude product was diluted with diethyl ether and filtered through a Celite plug, washing with ether. The ether mixture was concentrated in vacuo and applied to a silica gel column, eluting with 15% EtOAc/hexanes to give a pink solid (500 mg, 56% yield). 1H NMR (400 MHz, CDCl3) δ = 8.66 (d, J = 1.2 Hz, 1H), 8.46 (dd, J = 8.0 Hz, 1.6 Hz, 1H), 8.24 (d, J = 2.0 Hz, 1H), 7.82 (td, J = 8.0 Hz, 2.4 Hz, 1H), 7.55–7.47 (m, 3H), 7.04 (dd, J = 8.4 Hz, 2.8 Hz, 1H).
Methyl 2-(4-(6-Fluoropyridin-3-yl)-3-(trifluoromethyl)phenyl)-benzo[d]oxazole-5-carboxylate (11g)
Following general experimental procedure C, amide 10g (500 mg, 1.15 mmol), 11.5 mL of toluene, and pyridinium p-toluenesulfonate (607 mg, 2.42 mmol) were combined to give a yellow solid (415 mg, 87% yield). 1H NMR (400 MHz, CDCl3) δ = 8.64 (d, J = 1.2 Hz, 1H), 8.48–8.42 (m, 2H), 8.22 (d, J = 2.0 Hz, 1H), 8.13 (dd, J = 8.4 Hz, 1.6 Hz, 1H), 7.81 (td, J = 8.0 Hz, 2.4 Hz, 1H), 7.64 (d, J = 8.4 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.03 (dd, J = 8.4 Hz, 2.8 Hz, 1H), 3.96 (s, 3H).
5-Bromo-2-(3′-methoxy-2-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)-benzo[d]oxazole (11h)
Following general experimental procedure C, amide 10h (3.96 g, 8.49 mmol), 85 mL of toluene, and p-toluenesulfonic acid (3.39 g, 17.8 mmol) were combined to give a red solid (3.24 g, 85% yield). 1H NMR (400 MHz, CDCl3) δ = 8.61 (d, J = 1.6 Hz, 1H), 8.39 (dd, J = 8.0 Hz, 1.6 Hz, 1H), 7.96–7.93 (m, 1H), 7.57–7.49 (m, 3H), 7.36 (t, J = 7.6 Hz, 1H), 7.02–6.90 (m, 3H), 3.85 (s, 3H).
(2-(4-(6-Fluoropyridin-3-yl)-3-(trifluoromethyl)phenyl)benzo[d]-oxazol-5-yl)methanol (12a)
To a 25 mL round-bottomed flask equipped with a stir bar was added ester 11g (415 mg, 0.99 mmol) and 5 mL of THF. The reaction was cooled in a −78 °C cooling bath (CO2(s)/acetone), and DIBAl-H (2.99 mL, 2.99 mmol, 1.0 M in hexanes) was added slowly. The reaction was allowed to warm in the bath to −15 °C for 1 h, at which time TLC indicated consumption of the starting ester. The reaction was quenched with 2 mL of EtOAc and 5 mL of saturated Rochelle’s salt solution and stirred for 30 min. The reaction mixture was diluted with EtOAc and the layers separated. The aqueous layer was extracted with EtOAc (×2). The combined organic layers were washed with H2O, brine, dried over MgSO4, and concentrated in vacuo to give a white solid (350 mg, 91% yield). 1H NMR (400 MHz, CDCl3) δ = 8.69 (s, 1H), 8.48 (dd, J = 8.0 Hz, 1.6 Hz, 1H), 8.24 (d, J = 2.0 Hz, 1H), 7.86–7.78 (m, 2H), 7.62 (d, J = 8.4 Hz, 1H), 7.51 (d, J = 8.0 Hz, 1H), 7.45 (dd, J = 8.4 Hz, 1.6 Hz, 1H), 7.04 (dd, J = 8.4 Hz, 3.2 Hz, 1H), 4.86 (s, 2H).
(2-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)benzo[d]-oxazol-5-yl)methanol (12b)
To an oven-dried 50 mL round-bottomed flask equipped with a stir bar was added ester 11a (844 mg, 2.2 mmol) and 11 mL of 2-methyltetrahydrofuran. The reaction mixture was cooled to −78 °C (CO2(s)/acetone bath), and DIBAl-H (1.0 M solution in hexanes, 6.6 mL, 6.6 mmol) was added carefully. The reaction mixture was allowed to warm to −5 °C in the bath over 1 h, at which time TLC showed the consumption of the ester. The reaction was quenched with EtOAc and saturated Rochelle’s salt solution(aq), and stirred for 30 min when a precipitate had formed. The reaction mixture was filtered through a Celite plug, washing with EtOAc and water. The layers were separated, and the aqueous layer was extracted with EtOAc (×1). The combined organic layers were washed with water, brine, dried over MgSO4, and concentrated in vacuo to give a white solid (743 mg, 95% yield). 1H NMR (400 MHz, CDCl3) δ = 8.48 (d, J = 2.0 Hz, 1H), 8.37 (dd, J = 8.8 Hz, 2.0 Hz, 1H), 7.74 (s, 1H), 7.55 (d, J = 8.4 Hz, 1H), 7.40–7.35 (m, 1H), 7.14 (d, J = 8.8 Hz, 1H), 4.90–4.74 (m, 2H), 4.82 (s, 2H), 4.47–4.35 (m, 2H).
5-(Chloromethyl)-2-(4-(6-fluoropyridin-3-yl)-3-(trifluoromethyl)-phenyl)benzo[d]oxazole (13a)
To an oven-dried 10 mL round-bottomed flask equipped with a stir bar was added 0.11 mL of DMF, followed by cyanuric chloride (194 mg, 1.05 mmol). The reaction was stirred for 30 min, at which time TLC analysis showed the consumption of the cyanuric chloride. Then, 2.5 mL of CH2Cl2 was added, followed by alcohol 12a (388 mg, 1.00 mmol). The reaction was stirred for 1 h, at which time TLC analysis showed the consumption of the alcohol. The reaction was quenched with H2O and diluted with CH2Cl2. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (×1). The combined organic layers were washed with sat. sodium carbonate solution, 1 M HCl(aq), brine, dried over MgSO4, and concentrated in vacuo to give 398 mg of an off-white solid. 1H NMR (400 MHz, CDCl3) δ = 8.68 (s, 1H), 8.48 (d, J = 8.0 Hz, 1H), 8.24 (d, J = 2.0 Hz, 1H), 7.86–7.77 (m, 2H), 7.63 (d, J = 8.4 Hz, 1H), 7.52 (d, J = 8.0 Hz, 1H), 7.47 (dd, J = 8.4 Hz, 1.6 Hz, 1H), 7.04 (dd, J = 8.4 Hz, 2.4 Hz, 1H), 4.75 (s, 2H).
5-(Chloromethyl)-2-(4-(2-fluoroethoxy)-3-(trifluoromethyl)-phenyl)benzo[d]oxazole (13b)
To an oven-dried 50 mL round-bottomed flask equipped with a stir bar was added 0.23 mL of DMF and cyanuric chloride (218 mg, 1.18 mmol). The reaction mixture was stirred for 1 h at which time 3 mL of CH2Cl2 and alcohol 12b (400 mg, 1.13 mmol) were added. The reaction mixture was stirred for 18 h and diluted with CH2Cl2 and water. The layers were separated, and the aqueous layer was extracted with CH2Cl2 (×2). The combined organic layers were washed with sat. Na2CO3(aq), 1 M HCl (aq), and brine, dried over MgSO4, and concentrated in vacuo to give a tan solid (250 mg, 59% yield). 1H NMR (400 MHz, CDCl3) δ = 8.47 (s, 1H), 8.38 (dd, J = 8.8 Hz, 2.4 Hz, 1H), 7.76 (s, 1H), 7.56 (d, J = 8.4 Hz, 1H), 7.40 (dd, J = 8.4 Hz, 1.6 Hz, 1H), 7.15 (d, J = 8.8 Hz, 1H), 4.92–4.74 (m, 2H), 4.73 (s, 2H), 4.48–4.36 (m, 2H).
Methyl 1-((2-(4-(6-Fluoropyridin-3-yl)-3-(trifluoromethyl)phenyl)-benzo[d]oxazol-5-yl)methyl)azetidine-3-carboxy-late (14a)
To a 10 mL round-bottomed flask equipped with a stir bar was added chloride 13a (260 mg, 0.64 mmol), methyl azetidine-3-carboxylate hydrochloride (146 mg, 0.96 mmol), 2.1 mL of acetonitrile, and potassium carbonate (265 mg, 1.92 mmol). The reaction was stirred for 3 h, at which time no progress was observed by TLC. iPr2NEt (0.33 mL, 1.92 mmol) was added and the reaction stirred for 20 h at r.t. The reaction was diluted with water and EtOAc and the layers separated. The aqueous layer was extracted with EtOAc (×1). The combined organic layers were washed with water (×2), brine, dried over MgSO4, and concentrated in vacuo to give a yellow semisolid. The crude product was then purified on a silica gel column, eluting with EtOAc to give a white solid (110 mg, 35% yield). 1H NMR (400 MHz, CDCl3) δ = 8.63 (d, J = 1.2 Hz, 1H), 8.43 (dd, J = 8 Hz, 1.6 Hz, 1H), 8.21 (d, J = 2.0 Hz, 1H), 7.80 (td, J = 8.4 Hz, 2.4 Hz, 1H), 7.69 (d, J = 0.8 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.47 (d, J = 8.0 Hz, 1H), 7.33 (dd, J = 8.4 Hz, 1.6 Hz, 1H), 7.01 (dd, J = 8.4 Hz, 3.2 Hz, 1H), 3.73 (s, 2H), 3.70 (s, 3H), 3.57–3.52 (m, 2H), 3.40–3.30 (m, 3H).
Methyl 1-((2-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-benzo[d]oxazol-5-yl)methyl)azetidine-3-carboxylate (14b)
To a 10 mL round-bottomed flask equipped with a stir bar was added chloride 13b (187 mg, 0.5 mmol), methyl azetidine-3-carboxylate hydrochloride (114 mg, 0.75 mmol), 1.7 mL of MeCN, and DIPEA (0.26 mL, 1.5 mmol). The reaction was placed in a preheated 60 °C oil-bath and stirred overnight (∼18 h). The reaction mixture was allowed to cool to r.t. and diluted with water and EtOAc. The layers were separated and the aqueous layer extracted with EtOAc (×2). The combined organic layers were washed with water (×3), brine, dried over MgSO4, and concentrated in vacuo to give a tan solid (150 mg, 66% yield). 1H NMR (400 MHz, CDCl3) δ = 8.41 (s, 1H), 8.29 (d, J = 8.8 Hz, 1H), 7.60 (s, 1H), 7.44 (d, J = 8.4 Hz, 1H), 7.23 (d, J = 8.0 Hz, 1H), 7.07 (d, J = 8.8 Hz, 1H), 4.85–4.67 (m, 2H), 4.40–4.28 (m, 2H), 3.75–3.65 (m, 4H), 3.55–3.45 (m, 2H), 3.36–3.25 (m, 2H).
1-((2-(4-(6-Fluoropyridin-3-yl)-3-(trifluoromethyl)phenyl)benzo-[d]oxazol-5-yl)methyl)azetidine-3-carboxylic Acid (15a)
To a 13 × 100 mm test tube equipped with a stir bar was added ester 14a (70 mg, 0.14 mmol), 1.1 mL of THF, and 0.21 mL of 2 M LiOH(aq). The reaction was stirred overnight at r.t. and quenched with 1 M HCl(aq) to give a white precipitate. The precipitate was removed by filtration and dried under reduced pressure to provide 30 mg of the product. 1H NMR (400 MHz, MeOD-d4) δ = 8.69 (s, 1H), 8.57 (dd, J = 8.0 Hz, 2.0 Hz, 1H), 8.23 (d, J = 2.0 Hz, 1H), 8.07–7.97 (m, 2H), 7.86 (d, J = 8.4 Hz, 1H), 7.71 (d, J = 8.0 Hz, 1H), 7.66 (d, J = 8.4 Hz, 1H), 7.22 (dd, J = 8.4 Hz, 2.8 Hz, 1H), 4.63 (d, J = 22.4 Hz, 2H), 4.48 (t, J = 10.4 Hz, 1H), 4.39–4.32 (m, 3H), 3.83–3.71 (m, 1H).
1-((2-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)benzo[d]-oxazol-5-yl)methyl)azetidine-3-carboxylic Acid (15b)
To a 10 mL round-bottomed flask equipped with a stir bar and containing ester 14b (120 mg, 0.27 mmol) was added 2.1 mL of THF and 0.4 mL of 2 M LiOH(aq). The reaction mixture was stirred overnight (18 h) and quenched with 1 M HCl(aq). The reaction mixture was neutralized with sat. NaHCO3(aq), which caused the formation of a precipitate. This precipitate was filtered and washed with Et2O to give a white solid (41 mg, 35% yield). 1H NMR (400 MHz, MeOD-d4) δ = 8.50–8.43 (m, 2H), 7.90 (s, 1H), 7.80 (d, J = 8.4 Hz, 1H), 7.54 (d, J = 8.4 Hz, 1H), 7.45 (d, J = 9.6 Hz, 1H), 4.86–4.71 (m, 2H), 4.56 (d, J = 8.0 Hz, 2H), 4.54–4.25 (m, 6H), 3.72 (pent, J = 9.2 Hz, 1H).
5-(2-(4-(6-Fluoropyridin-3-yl)-3-(trifluoromethyl)phenyl)benzo-[d]oxazol-5-yl)thiophene-2-carboxylic Acid (16)
To an oven-dried 25 mL Schlenk tube equipped with a stir bar was added palladium acetate (2.2 mg, 0.01 mmol), bromide 11f (219 mg, 0.5 mmol), 2-carboxythiophene-5-boronic acid (95 mg, 0.55 mmol), cesium fluoride (228 mg, 1.5 mmol), 1.35 mL of 1,4-dioxane, and 1.35 mL of degassed H2O. The reaction vessel was equipped with a cold finger, placed in a preheated 110 °C oil-bath, and heated overnight (∼18 h). The reaction was cooled to r.t. and poured into a 1:1 mixture of EtOAc/1 M HCl(aq). The layers were separated, and the aqueous layer was extracted with EtOAc (×3). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated in vacuo to give a brown solid (223 mg, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ = 13.19 (br s, 1H), 8.57 (s, 1H), 8.54 (d, J = 8.0 Hz, 1H), 8.31 (d, J = 2.4 Hz, 1H), 8.26 (d, J = 2.0 Hz, 1H), 8.09 (td, J = 8.0 Hz, 2.4 Hz, 1H), 7.95 (d, J = 8.8 Hz, 1H), 7.86 (dd, J = 8.8 Hz, 2.0 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.75 (d, J = 3.6 Hz, 1H), 7.68 (d, J = 4.0 Hz, 1H), 7.37 (dd, J = 8.8 Hz, 2.8 Hz, 1H).
Methyl 1-(2-(4′-Fluoro-2-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)-benzo[d]oxazol-5-yl)piperidine-4-carboxylate (17a)
Following general experimental procedure D, Pd2(dba)3·CHCl3 (10 mg, 0.01 mmol), XPhos (19 mg, 0.040 mmol), sodium tert-butoxide (67 mg, 0.70 mmol), oxazole 11b (218 mg, 0.5 mmol), toluene (1 mL), and methylpiperidine-4-carboxylate (81 μL, 0.60 mmol) were combined to give the crude product which was applied to a silica gel column, eluting with 5–15% ethyl acetate in hexanes to give ester 17a as a yellow solid (165 mg, 66% yield). 1H NMR (400 MHz, CDCl3) δ = 8.59 (s, 1H), 8.36 (d, J = 8.0 Hz, 1H), 7.47 (d, J = 6.0 Hz, 1H), 7.44 (d, J = 4.4 Hz, 1H), 7.37–7.27 (m, 3H), 7.11 (t, J = 8.4 Hz, 2H), 7.07 (dd, J = 8.8 Hz, 2.4 Hz, 1H), 3.71 (s, 3H), 3.61 (dt, J = 8.8 Hz, 3.2 Hz, 2H), 2.82 (td, J = 12 Hz, 2.0 Hz, 2H), 2.47 (tt, J = 11.2 Hz, 4 Hz, 1H), 2.13–2.02 (m, 2H), 2.01–1.87 (m, 2H).
Methyl 1-(2-(2′-(2-Fluoroethoxy)-2-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)benzo[d]oxazol-5-yl)piperidine-4-carboxylate (17b)
Following general experimental procedure D, Pd2(dba)3·CHCl3 (6.2 mg, 0.006 mmol), XPhos (11.4 mg, 0.024 mmol), sodium tert-butoxide (40 mg, 0.42 mmol), oxazole 11c (144 mg, 0.30 mmol), toluene (0.6 mL), and methylpiperidine-4-carboxylate (48 μL, 0.36 mmol) were combined to give the crude product which was applied to a silica gel column, eluting with 20% ethyl acetate in hexanes to give ester 17b as a yellow solid (90 mg, 56% yield). 1H NMR (400 MHz, CDCl3) δ = 8.60 (s, 1H), 8.37 (d, J = 8.0 Hz, 1H), 7.47 (dd, J = 9.2 Hz, 4.0 Hz, 2H), 7.39 (t, J = 7.6 Hz, 1H), 7.32 (br s, 1H), 7.21 (d, J = 7.2 Hz, 1H), 7.13–7.01 (m, 2H), 6.97 (d, J = 8.4 Hz, 1H), 4.54 (dt, J = 47.6 Hz, 4.0 Hz, 2H), 4.29–4.04 (m, 2H), 3.72 (s, 3H), 3.62 (d, J = 12.4 Hz, 2H), 2.84 (t, J = 11.2 Hz, 2H), 2.53–2.44 (m, 1H), 2.14–2.04 (m, 2H), 2.03–1.85 (m, 2H).
Methyl 1-(2-(4′-(2-Fluoroethoxy)-2-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)benzo[d]oxazol-5-yl)piperidine-4-carboxy-late (17c)
Following general experimental procedure D, Pd2(dba)3·CHCl3 (7.2 mg, 0.007 mmol), XPhos (13 mg, 0.028 mmol), sodium tert-butoxide (47 mg, 0.49 mmol), oxazole 11d (168 mg, 0.35 mmol), toluene (0.7 mL), and methylpiperidine-4-carboxylate (57 μL, 0.42 mmol) were combined to give the crude product, applied to a silica gel column, eluting with 20% ethyl acetate in hexanes to give ester 17c as a yellow solid (95 mg, 50% yield). 1H NMR (400 MHz, CDCl3) δ = 8.59 (s, 1H), 8.36 (d, J = 8.0 Hz, 1H), 7.47 (d, J = 6.0 Hz, 1H), 7.44 (d, J = 4.4 Hz, 1H), 7.37–7.27 (m, 3H), 7.11 (t, J = 8.4 Hz, 2H), 7.07 (dd, J = 8.8 Hz, 2.4 Hz, 1H), 3.71 (s, 3H), 3.61 (dt, J = 8.8 Hz, 3.2 Hz, 2H), 2.82 (td, J = 12 Hz, 2.0 Hz, 2H), 2.47 (tt, J = 11.2 Hz, 4 Hz, 1H), 2.13–2.02 (m, 2H), 2.01–1.87 (m, 2H).
Methyl 1-(2-(2,4′-Bis(trifluoromethyl)-[1,1′-biphenyl]-4-yl)benzo-[d]oxazol-5-yl)piperidine-4-carboxylate (17d)
Following general experimental procedure D, Pd2(dba)3·CHCl3 (21 mg, 0.02 mmol), XPhos (38 mg, 0.08 mmol), sodium tert-butoxide (135 mg, 1.4 mmol), bromide 11e (486 mg, 1.0 mmol), and 2 mL of toluene were combined to provide the crude product. This was applied to a silica gel column, eluting with 20% EtOAc/hexanes to provide a yellow powder (265 mg, 48% yield). 1H NMR (400 MHz, CDCl3) δ = 8.63 (s, 1H), 8.41 (d, J = 8.0 Hz, 1H), 7.70 (d, J = 8.0 Hz, 2H), 7.52–7.45 (m, 4H), 7.31 (d, J = 2.4 Hz, 1H), 7.10 (dd, J = 8.8 Hz, 2.0 Hz, 1H), 3.73 (s, 3H), 3.63 (dt, J = 12.4 Hz, 3.2 Hz, 2H), 2.84 (td, J = 11.6 Hz, 2.4 Hz, 2H), 2.47 (tt, J = 10.8 Hz, 4.0 Hz, 1H), 2.13–2.04 (m, 2H), 2.02–1.87 (m, 2H).
Methyl 1-(2-(3′-Methoxy-2-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)-benzo[d]oxazol-5-yl)piperidine-4-carboxylate (17e)
Following general experimental procedure D, Pd2(dba)3·CHCl3 (21 mg, 0.02 mmol), XPhos (38 mg, 0.08 mmol), sodium tert-butoxide (135 mg, 1.4 mmol), bromide 11h (447 mg, 1.0 mmol), and 2 mL of toluene were combined to provide the crude product. This was applied to a silica gel column, eluting with 20% EtOAc/hexanes to provide a yellow powder (93 mg, 18% yield). 1H NMR (400 MHz, CDCl3) δ = 8.60 (s, 1H), 8.36 (d, J = 8.0 Hz, 1H), 7.49 (t, J = 8.0 Hz, 2H), 7.38–7.29 (m, 2H), 7.08 (d, J = 8.4 Hz, 1H), 7.00–6.87 (m, 3H), 3.84 (s, 3H), 3.72 (s, 3H), 3.62 (d, J = 12.4 Hz, 2H), 2.84 (t, J = 10.8 Hz, 2H), 2.53–2.42 (m, 1H), 2.13–2.04 (m, 2H), 2.03–1.86 (m, 2H).
1-(2-(4′-Fluoro-2-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)benzo[d]-oxazol-5-yl)piperidine-4-carboxylic Acid (18a)
Following general experimental procedure E, ester 17a (150 mg, 0.30 mmol), 1.25 mL of tetrahydrofuran, 0.25 mL of H2O, and lithium hydroxide (14 mg, 0.60 mmol) were combined to give carboxylic acid 18a as an off-white pearly solid (129 mg, 89% yield). 1H NMR (400 MHz, MeOD-d4) δ = 8.67 (s, 1H), 8.54 (d, J = 8.0 Hz, 1H), 8.18 (d, J = 2.0 Hz, 1H), 7.98 (d, J = 9.2 Hz, 1H), 7.78 (dd, J = 8.8 Hz, 2.4 Hz, 1H), 7.65 (d, J = 8 Hz, 1H), 7.41 (dd, J = 8.4 Hz, 5.2 Hz, 2H), 7.22 (t, J = 8.8 Hz, 2H), 3.93–3.74 (m, 4 H), 2.93–2.82 (m, 1H), 2.41 (dd, J = 15.2 Hz, 3.6 Hz, 2H), 2.32–2.16 (m, 2H).
1-(2-(2′-(2-Fluoroethoxy)-2-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)benzo[d]oxazol-5-yl)piperidine-4-carboxylic Acid (18b)
Following general experimental procedure E, ester 17b (90 mg, 0.17 mmol), 0.70 mL of tetrahydrofuran, 0.13 mL of H2O, and lithium hydroxide (8.0 mg, 0.332 mmol were combined to give carboxylic acid 18b as an off-white pearly solid (82 mg, 94% yield). 1H NMR (400 MHz, MeOD-d4) δ = 8.52 (s, 1H), 8.35 (d, J = 8.0 Hz, 1H), 7.64 (d, J = 8.4 Hz, 1H), 7.56 (s, 1H), 7.48 (d, J = 8.0 Hz, 1H), 7.45–7.31 (m, 2H), 7.16 (d, J = 7.2 Hz, 1H), 7.08 (d, J = 8.4 Hz, 1H), 7.03 (t, J = 7.2 Hz, 1H), 4.51 (d, J = 48 Hz, 2H), 3.31–3.05 (m, 2H), 3.68 (d, J = 12 Hz, 2H), 3.14 (t, J = 10.4 Hz, 2H), 2.59 (t, J = 10.4 Hz, 1H), 2.26–2.10 (m, 2H), 2.09–1.92 (m, 2H).
1-(2-(4′-(2-Fluoroethoxy)-2-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)benzo[d]oxazol-5-yl)piperidine-4-carboxylic Acid (18c)
Following general experimental procedure E, ester 17c (90 mg, 0.166 mmol), 0.7 mL of tetrahydrofuran, 0.13 mL of H2O, and lithium hydroxide (8.0 mg, 0.332 mmol) were combined to give carboxylic acid 18c as a light tan solid (87 mg, 99% yield). 1H NMR (400 MHz, MeOD-d4) δ = 8.64 (s, 1H), 8.50 (d, J = 7.6 Hz, 1H), 8.29 (s, 1H), 7.96 (d, J = 8.8 Hz, 1H), 7.83 (dd, J = 8.8 Hz, 2.0 Hz, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.32 (d, J = 8.4 Hz, 2H), 7.06 (d, J = 8.8 Hz, 2H), 4.77 (dt, J = 48 Hz, 4 Hz, 2H), 4.29 (dt, J = 28.8 Hz, 3.6 Hz, 2H), 3.90–3.74 (m, 4H), 2.93–2.83 (m, 1H), 2.49–2.35 (m, 2H), 2.35–2.20 (m, 2H).
1-(2-(2,4′-Bis(trifluoromethyl)-[1,1′-biphenyl]-4-yl)benzo[d]-oxazol-5-yl)piperidine-4-carboxylic Acid (18d)
Following general experimental procedure E, ester 17d (102 mg, 0.19 mmol), 0.79 mL of THF, and 0.16 mL of H2O, and LiOH (8.9 mg, 0.37 mmol) were combined to give an off-white solid. (101 mg, 99% yield). 1H NMR (400 MHz, MeOD-d4) δ = 8.70 (s, 1H), 8.58 (d, J = 8.0 Hz, 1H), 8.28 (d, J = 2.0 Hz, 1H), 7.99 (d, J = 9.2 Hz, 1H), 7.89 (dd, J = 8.8 Hz, 2.4 Hz, 1H), 7.80 (d, J = 8.4 Hz, 2H), 7.69 (d, J = 8.0 Hz, 1H), 7.60 (d, J = 8.0 Hz, 2H), 3.92–3.81 (m, 4H), 2.96–2.85 (m, 1H), 2.47–2.25 (m, 4H).
1-(2-(3′-Methoxy-2-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)benzo-[d]oxazol-5-yl)piperidine-4-carboxylic Acid (18e)
Following general experimental procedure E, ester 17e (92 mg, 0.18 mmol), 0.9 mL of THF, and 0.2 mL of H2O, and LiOH (8.6 mg, 0.36 mmol) were combined to give an off-white solid (72 mg, 81% yield). 1H NMR (400 MHz, MeOD-d4) δ = 8.62 (s, 1H), 8.47 (d, J = 8.0 Hz, 1H), 7.91 (s, 1H), 7.85 (d, J = 8.4 Hz, 1H), 7.65–7.56 (m, 2H), 7.37 (t, J = 8.0 Hz, 1H), 7.02 (dd, J = 8.4 Hz, 2.8 Hz, 1H), 6.95–6.89 (m, 2H), 3.83 (s, 3H), 3.84–3.73 (m, 2H), 3.58–3.43 (m, 2H), 2.76 (br s, 1H), 2.35–2.25 (m, 2H), 2.21–2.16 (m, 2H).
N-(Cyanomethyl)benzamide (19)
To an oven-dried 250 mL round-bottomed flask equipped with a stir bar was added amino-acetonitrile hydrochloride (8.0 g, 86.5 mmol). Pyridine (50 mL) was carefully added dropwise via an addition funnel. Benzoyl chloride (10.5 mL, 90 mmol) was added via an addition funnel dropwise over 30 min, and the reaction was stirred overnight (20 h). Then, 70 mL of H2O was added to dissolve the pyridinium hydrochloride and precipitate the product. The new precipitate was filtered, washed with H2O, dried, and recrystallized from 95% ethanol to give a white crystalline solid (9.5 g, 69% yield). 1H NMR (400 MHz, DMSO-d6) δ = 9.22 (t, J = 5.2 Hz, 1H), 7.86 (dd, J = 8.0 Hz, 0.8 Hz, 2H), 7.58 (tt, J = 7.2 Hz, 1.2 Hz, 1H), 7.50 (t, J = 7.6 Hz, 2H), 4.31 (d, J = 5.6 Hz, 2H).
N-((1H-Tetrazol-5-yl)methyl)benzamide (20)
To a 100 mL round-bottomed flask equipped with a stir bar was added nitrile 19 (5.9 g, 36.8 mmol), 32 mL of DMF, sodium azide (2.51 g, 38.7 mmol), and ammonium chloride (2.17 g, 40.5 mmol). The reaction vessel was lowered into a preheated 125 °C oil-bath and stirred overnight (18 h). The reaction was cooled to r.t. and diluted with 85 mL of 2 M HCl(aq) causing the product to precipitate. The precipitate was filtered, washed with copious amounts of H2O, and air-dried to give a white fluffy solid (6.89 g, 92% yield). 1H NMR (400 MHz, DMSO-d6) δ = 9.27 (t, J = 5.6 Hz, 1H), 7.89 (dd, J = 8.4 Hz, 1.2 Hz, 2H), 7.57 (tt, J = 7.2 Hz, 2.4 Hz, 1H), 7.49 (t, J = 7.2 Hz, 2H), 4.76 (d, J = 5.6 Hz, 2H).
(1H-Tetrazol-5-yl)methanamine Hydrochloride (21)
To a 250 mL round-bottomed flask equipped with a stir bar was added amide 20 (6.89 g, 33.9 mmol) and 50.6 mL of 12 M HCl(aq). The reaction was equipped with a reflux condenser, placed in a preheated 110 °C oil-bath, and stirred at reflux overnight (18 h). The reaction was cooled to r.t., then to 0 °C causing the formation of a precipitate. The reaction mixture was filtered, and the filtrate was washed with Et2O (×2) and concentrated in vacuo to give a white solid. This solid was triturated with ethanol and dried under vacuum to give a pale yellow solid (3.59 g, 78% yield). 1H NMR (400 MHz, D2O) δ = 4.59 (s, 2H).
Methyl 4-(2-fluoroethoxy)benzoate (23)
To a 500 mL round-bottomed flask equipped with a stir bar was added methyl 4-hydroxybenzoate (22) (5.0 g, 32.9 mmol), potassium carbonate (13.6 g, 98.6 mmol), and 219 mL of acetone. The reaction vessel was heated to reflux in a preheated 70 °C oil-bath, and stirred for 30 min. 1-Bromo-2-fluoroethane (7.34 mL, 98.6 mmol) was added via syringe, and the reaction mixture was stirred for 20 h. The reaction mixture was cooled to r.t. and concentrated in vacuo. The resulting solid was dissolved in EtOAc and H2O. The layers were separated, and the aqueous layer was extracted with EtOAc (×1). The combined organic layers were washed with brine, dried over MgSO4, and concentrated in vacuo to give a white solid (6.25 g, 96% yield). 1H NMR (400 MHz, CDCl3) δ = 8.00 (d, J = 8.8 Hz, 2H), 6.94 (d, J = 9.2 Hz, 2H), 4.78 (ddd, J = 47.2 Hz, 5.6 Hz, 4.0 Hz, 2H), 4.27 (ddd, J = 27.6 Hz, 5.6 Hz, 4.4 Hz, 2H), 3.89 (s, 3H).
4-(2-Fluoroethoxy)benzoic Acid (24)
To a 500 mL round-bottomed flask equipped with a stir bar was added ester 23 (5.84 g, 29.5 mmol), followed by 98 mL of methanol, and 49 mL of H2O. NaOH (4.71 g, 118 mmol) was added, and the reaction mixture was stirred for 16 h at r.t. The reaction was acidified with conc HCl(aq) to pH 1 and diluted with H2O. The resulting precipitate was filtered, washed with H2O and hexanes, and air-dried to give a white solid (5.41 g, 99%). 1H NMR (400 MHz, CDCl3) δ = 8.05 (d, J = 9.2 Hz, 2H), 6.97 (d, J = 8.8 Hz, 2H), 4.86–4.72 (m, 2H), 4.34–4.23 (m, 2H).
N′-Hydroxy-4-(hydroxymethyl)benzimidamide (25).33
To a 500 mL round-bottomed flask equipped with a stir bar was added 4-(hydroxymethyl)benzonitrile (13.3 g, 100 mmol), hydroxyamine hydrochloride (11.1 g, 160 mmol), sodium bicarbonate (26.9 g, 320 mmol), and 167 mL of methanol. The reaction was equipped with a reflux condenser and lowered into a preheated 70 °C oil-bath. The reaction mixture was stirred for 5 h and then cooled to r.t. The precipitate was filtered and washed with methanol. The filtrate was concentrated in vacuo to give a white solid (16.5 g, 99% yield). 1H NMR (400 MHz, MeOD-d4) data match the literature values.
(4-(5-(4-Ethoxyphenyl)-1,2,4-oxadiazol-3-yl)phenyl)methanol (26a).33
Following general experimental procedure F, 4-ethoxybenzoic acid (2.49 g, 15.0 mmol), EDC·HCl (2.88 g, 15.0 mmol), HOBt (2.03 g, 15.0 mmol), 19 mL of DMF, and amidine 25 (2.49 g, 15.0 mmol) were combined to give an off-white solid (2.2 g, 50% yield). 1H NMR (400 MHz, CDCl3) δ = 8.18–8.11 (m, 4H), 7.50 (d, J = 8.0 Hz, 2H), 7.02 (d, J = 8.8 Hz, 2H), 4.79 (s, 2H), 4.13 (q, J = 7.2 Hz, 2H), 1.47 (t, J = 6.8 Hz, 3H).
(4-(5-(4-(2-Fluoroethoxy)phenyl)-1,2,4-oxadiazol-3-yl)phenyl)-methanol (26b)
Following general experimental procedure F, carboxcyclic acid 24 (2.76 g, 15.0 mmol), EDC·HCl (2.88 g, 15.0 mmol), HOBt (2.03 g, 15.0 mmol), 19 mL of DMF, and amidine 25 (2.49 g, 15.0 mmol) were combined to provide the crude product, which was used without further purification for the next step.
(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)phenyl)methanol (26c)
Following general experimental procedure F, acid 7a (3.78 g, 15.0 mmol), EDC·HCl (2.88 g, 15.0 mmol), HOBt (2.54 g, 15.0 mmol), 19 mL of DMF, and amidine 25 (2.49 g, 15.0 mmol) were combined to give an off-white solid (3.12 g, 55% yield). 1H NMR (400 MHz, CDCl3) δ = 8.45 (d, J = 2.0 Hz, 1H), 8.33 (dd, J = 8.8 Hz, 2.0 Hz, 1H), 8.13 (d, J = 8.0 Hz, 2H), 7.50 (d, J = 8.0 Hz, 2H), 7.16 (d, J = 8.4 Hz, 1H), 4.90–4.74 (m, 4H), 4.48–4.35 (m, 2H). 13C NMR (101 MHz, CDCl3) δ = 174.4, 168.9, 159.7, 144.4, 133.4, 127.8, 127.8 (q, JC–F = 5.2 Hz), 127.3, 122.9, (q, JC–F = 274 Hz), 120.4 (q, JC–F = 32.1 Hz), 117.2, 113.5, 81.4 (d, JC–F = 173 Hz), 68.5 (d, JC–F = 21.1 Hz), 64.9. MP: 146–147 °C. HRMS (ESI) calcd for C10H14F4N2O3 [M + H+] 383.1013. Found [M + H+] 383.1011.
(4-(5-(4-Methoxy-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)phenyl)methanol (26d)
To a 100 mL round-bottomed flask equipped with a stir bar was added 4-methoxy-3-(trifluoromethyl)-benzoic acid (2.2 g, 10.0 mmol), HOBt (0.39 g, 2.0 mmol), TBTU (3.21 g, 10.0 mmol), 20 mL of DMF, and DIPEA (5.23 mL, 30.0 mmol). The reaction was stirred at r.t. for 30 min, and N′-hydroxy-4-(hydroxymethyl)benzimidamide (1.66 g, 10.0 mmol) was added. The reaction was stirred at r.t. for 1 h, heated in a 120 °C oil-bath for 3 h, and cooled to r.t. The reaction mixture was diluted with EtOAc and water. The layers were separated, and the aqueous layer was extracted with EtOAc (×1). The combined organic layers were washed with 1 M HCl(aq), water, sat. NaHCO3(aq), water (×3), and brine, dried over MgSO4, and concentrated in vacuo to give a tan solid (2.6 g, 74% yield). 1H NMR (400 MHz, CDCl3) δ = 8.45 (d, J = 2.0 Hz, 1H), 8.35 (dd, J = 8.8 Hz, 2.0 Hz, 1H), 8.15 (d, J = 8.0 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H), 7.16 (d, J = 8.8 Hz, 1H), 4.80 (s, 2H), 4.02 (s, 3H).
4-(5-(4-Ethoxyphenyl)-1,2,4-oxadiazol-3-yl)benzaldehyde (27a).33
To an oven-dried 100 mL round-bottomed flask equipped with a stir bar was added 32 mL of CH2Cl2 and dimethyl sulfoxide (1.27 mL, 17.9 mmol). The reaction mixture was cooled in a −78 °C bath (CO2(s)/acetone). Oxalyl chloride (1.03 mL, 12.1 mmol) was added carefully, and the reaction stirred for 30 min. Alcohol 26a (1.71 g, 5.77 mmol) was added, and the reaction mixture was stirred 30 min. Triethylamine (6.43 mL, 46.2 mmol) was added, and the reaction was allowed to warm to r.t. over 2 h. The reaction mixture was concentrated in vacuo and partitioned between EtOAc and 1 M HCl(aq). The layers were separated, and the aqueous layer was extracted with EtOAc (×1). The combined organic layers were washed with sat. NaHCO3(aq), water, brine, dried over MgSO4, and concentrated in vacuo to give an off-white solid (930 mg, 55% yield). 1H NMR (400 MHz, CDCl3) δ = 10.11 (s, 1H), 8.34 (d, J = 8.4 Hz, 2H), 8.15 (d, J = 8.8 Hz, 2H), 8.02 (d, J = 8.4 Hz, 2H), 7.03 (d, J = 8.8 Hz, 2H), 4.14 (q, J = 6.8 Hz, 2H), 1.47 (t, J = 7.2 Hz, 3H).
4-(5-(4-(2-Fluoroethoxy)phenyl)-1,2,4-oxadiazol-3-yl)-benzaldehyde (27b)
To an oven-dried 250 mL round-bottomed flask equipped with a stir bar was added 53 mL of CH2Cl2 and dimethyl sulfoxide (2.1 mL, 29.6 mmol). The reaction mixture was cooled in a −78 °C bath (CO2(s)/acetone). Oxalyl chloride (1.7 mL, 20.0 mmol) was added carefully and the reaction stirred 30 min. Alcohol 26b (3.00 g, 9.54 mmol) was added, and the reaction mixture was stirred 30 min. Triethylamine (10.6 mL, 76.3 mmol) was added, and the reaction was allowed to warm to r.t. over 2 h. The reaction mixture was concentrated in vacuo and partitioned between EtOAc and 1 M HCl(aq). The layers were separated, and the aqueous layer was extracted with EtOAc (×1). The combined organic layers were washed with sat. NaHCO3(aq), water, and brine, dried over MgSO4, and concentrated in vacuo to give an off-white solid (1.31 g, 44% yield). 1H NMR (400 MHz, CDCl3) δ = 10.11 (s, 1H), 8.34 (d, J = 8.4 Hz, 2H), 8.18 (d, J = 9.2 Hz, 2H), 8.02 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 8.8 Hz, 2H), 4.89–4.70 (m, 2H), 4.38–4.25 (m, 2H).
4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzaldehyde (27c)
To an oven-dried 100 mL round-bottomed flask equipped with a stir bar was added 29 mL of CH2Cl2 and DMSO (1.15 mL, 16.2 mmol). The reaction mixture was cooled to −78 °C (acetone/CO2(s)), and oxalyl chloride (0.93 mL, 11.0 mmol) was added carefully. The reaction mixture was stirred for 30 min at which time alcohol 26c (2.0 g, 5.23 mmol) was added. The reaction mixture was stirred for 30 min at which time Et3N (5.83 mL, 41.8 mmol) was added. The cooling bath was removed, and the reaction was allowed to warm to r.t. over 2 h. The reaction mixture was concentrated in vacuo and partitioned between EtOAc and 1 M HCl(aq). The layers were separated, and the aqueous layer was extracted with EtOAc (×2). The combined organic layers were washed with 1 M HCl(aq), sat. NaHCO3(aq), brine, dried over MgSO4, concentrated in vacuo, and triturated with MTBE to give a pale-yellow solid (1.75 g, 88% yield). 1H NMR (400 MHz, CDCl3) δ = 10.11 (s, 1H), 8.47 (d, J = 1.6 Hz, 1H), 8.39–8.31 (m, 3H), 8.02 (d, J = 8.4 Hz, 2H), 7.19 (d, J = 8.8 Hz, 1H), 4.93–4.75 (m 2H), 4.49–4.35 (m, 2H). 13C NMR (101 MHz, CDCl3) δ = 191.7, 174.9, 168.3, 159.9, 138.2, 133.5, 132.2, 130.2, 128.3, 127.9 (q, JC–F = 5.4 Hz), 122.9 (q, JC–F = 274 Hz), 120.5 (q, JC–F = 32.3 Hz), 116.9, 113.6, 81.4 (d, JC–F = 173 Hz), 68.6 (d, JC–F = 21.1 Hz). MP: 149–150 °C. HRMS (ESI) calcd for C18H13F4N2O3 [M + H+] 381.0857. Found [M + H+] 381.0857.
4-(5-(4-Methoxy-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)-benzaldehyde (27d)
To an oven-dried 100 mL round-bottomed flask equipped with a stir bar was added 32 mL of CH2Cl2 and DMSO (1.26 mL, 17.7 mmol). The reaction mixture was cooled to −78 °C (acetone/CO2(s)), and oxalyl chloride (1.01 mL, 12.0 mmol) was added carefully. The reaction mixture was stirred for 30 min at which time alcohol 26d (2.0 g, 5.71 mmol) was added. The reaction mixture was stirred for 30 min at which time Et3N (6.37 mL, 45.7 mmol) was added. The cooling bath was removed, and the reaction was allowed to warm to r.t. over 2 h. The reaction mixture was concentrated in vacuo and partitioned between EtOAc and 1 M HCl(aq). The layers were separated, and the aqueous layer was extracted with EtOAc (×2). The combined organic layers were washed with 1 M HCl(aq), sat. NaHCO3(aq), brine, dried over MgSO4, and concentrated in vacuo to give a pale-yellow solid (1.51 g, 76% yield). 1H NMR (400 MHz, CDCl3) δ = 10.11 (s, 1H), 8.46 (s, 1H), 8.40–8.32 (m, 3H), 8.03 (d, J = 8.0 Hz, 2H), 7.19 (d, J = 8.8 Hz, 1H), 4.04 (s, 3H).
1-(4-(5-(4-Ethoxyphenyl)-1,2,4-oxadiazol-3-yl)benzyl)azetidine-3-carboxylic Acid (28a).33
Following general experimental procedure G, aldehyde 27a (500 mg, 1.70 mmol), azetidine-3-carboxylic acid (180 mg, 1.79 mmol), 24 mL of methanol, 0.85 mL of acetic acid, and NaBH3CN (53 mg, 0.85 mmol) in 3.4 mL of MeOH were combined to give a white solid (331 mg, 51% yield). 1H NMR (400 MHz, DMSO-d6) δ = 8.11 (d, J = 8.8 Hz, 2H), 8.01 (d, J = 8.0 Hz, 2H), 7.47 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 8.8 Hz, 2H), 4.16 (q, J = 7.2 Hz, 2H), 3.64 (s, 2H), 3.45–3.38 (m, 1H), 3.28–3.15 (m, 4H), 1.37 (t, J = 6.8 Hz, 3H).
1-(4-(5-(4-(2-Fluoroethoxy)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)-azetidine-3-carboxylic Acid (28b)
Following general experimental procedure G, aldehyde 27b (500 mg, 1.60 mmol), azetidine-3-carboxylic acid (170 mg, 1.68 mmol), 23 mL of methanol, 0.8 mL of acetic acid, and NaBH3CN (50 mg, 0.80 mmol) in 3.2 mL of methanol were combined to give 400 mg of 85% pure acetate salt. 1H NMR (400 MHz, MeOD-d4) δ = 8.21 (d, J = 8.0 Hz, 2H), 8.18 (d, J = 8.8 Hz, 2H), 7.62 (d, J = 8.0 Hz, 2H), 7.19 (d, J = 8.8 Hz, 2H), 4.85–4.69 (m, 2H), 4.42–4.30 (m, 2H), 4.37 (s, 2H), 4.19–4.09 (m, 4H), 3.41 (pent, J = 8.4 Hz, 1H), 1.96 (s, 3H).
1-(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)azetidine-3-carboxylic Acid (28c)
Following general experimental procedure G, aldehyde 27c (500 mg, 1.31 mmol), azetidine-3-carboxylic acid (139 mg, 1.38 mmol), 19 mL of MeOH, 0.70 mL of AcOH, and NaBH3CN (41 mg, 0.70 mmol) in 2.6 mL of MeOH were combined to give an off-white solid (317 mg, 52% yield). 1H NMR (400 MHz, MeOD-d4) δ = 8.45–8.38 (m, 2H), 8.23 (d, J = 8.0 Hz, 2H), 7.64 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 9.6 Hz, 1H), 4.87–4.71 (m, 2H), 4.54–4.43 (m, 2H), 4.43 (s, 2H), 4.25–4.15 (m, 4H), 3.44 (pent, J = 8.8 Hz, 1H). 13C NMR (101 MHz, MeOD-d4) δ = 166.3, 160.5, 151.8, 137.2, 125.3, 119.0, 118.8, 118.7, 118.6 (q, JC–F = 5.45 Hz), 117.3, 114.9 (q, JC–F = 273 Hz), 111.3 (q, JC–F = 32.0 Hz), 108.4, 105.8, 105.8, 73.1 (d, JC–F = 70.4 Hz), 60.7, 60.5, 55.2 (d, JC–F = 11.0 Hz), 55.1. MP: 146–148 °C. Anal. Calcd for C22H19F4N3O4·2H2O: C, 52.70; H, 4.62; N, 8.38. Found: C, 52.93; H, 4.46; N, 8.37. HRMS (ESI) calcd for C22H20F4N3O4 [M + H+] 466.1384. Found [M + H+] 466.1383.
1-(4-(5-(4-(2-Fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)piperidine-4-carboxylic Acid (28d)
Following general experimental procedure G, aldehyde 27c (380 mg, 1.0 mmol), piperidine-4-carboxylic acid (136 mg, 1.05 mmol), 14.3 mL of MeOH, 0.5 mL of AcOH, and NaBH3CN (31 mg, 0.5 mmol) in 2 mL of methanol were combined to give a white solid (193 mg, 39% yield). 1H NMR (400 MHz, DMSO-d6) δ = 8.45 (dd, J = 8.8 Hz, 1.6 Hz, 1H), 8.33 (d, J = 1.6 Hz, 1H), 8.05 (d, J = 8.0 Hz, 2H), 7.58 (d, J = 8.8 Hz, 1H), 7.51 (d, J = 8.0 Hz, 2H), 4.75–4.50 (m, 4H), 3.54 (s, 2H), 3.25–1.51 (m, 9H).
N-((1H-Tetrazol-5-yl)methyl)-1-(4-(5-(4-(2-fluoroethoxy)-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)phenyl)methanamine (28e)
Following general experimental procedure G, aldehyde 27c (380 mg, 1.0 mmol), 21 (142 mg, 1.05 mmol), 14.3 mL of MeOH, 0.5 mL of AcOH, and NaBH3CN (31 mg, 0.5 mmol) in 2 mL of methanol were combined. The reaction mixture was stirred for 3 h and diluted with MTBE to give a precipitate. The precipitate was filtered to give an off-white solid (145 mg, 31% yield). 1H NMR (400 MHz, MeOD-d4) δ = 8.46–8.42 (m, 2H), 8.24 (d, J = 8.4 Hz, 2H), 7.70 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 8.4 Hz, 1H), 4.83–4.72 (m, 2H), 4.54–4.44 (m, 2H), 4.50 (s, 2H), 4.40 (s, 2H).
1-(4-(5-(4-Methoxy-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)azetidine-3-carboxylic Acid (28f)
Following general experimental procedure G, aldehyde 27d (500 mg, 1.44 mmol), azetidine-3-carboxylic acid (152 mg, 1.51 mmol), 21 mL of methanol, 0.72 mL of acetic acid, and NaBH3CN (45 mg, 0.72 mmol) in 2.9 mL of methanol were combined to give a white solid (300 mg, 48% yield). 1H NMR (400 MHz, DMSO-d6) δ = 8.43 (d, J = 8.4 Hz, 1H), 8.30 (s, 1H), 8.02 (d, J = 7.2 Hz, 2H), 7.60–7.42 (m, 3H), 4.03 (s, 3H), 3.64 (br s, 2H), 3.23 (br s, 3H).
4-(3-(4-(Hydroxymethyl)phenyl)-1,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenol (30)
To a 100 mL round-bottomed flask equipped with a stir bar was added 4-hydroxy-3-(trifluoromethyl)-benzoic acid (29) (2.06 g, 10.0 mmol), HOBt (0.39 g, 2.0 mmol), TBTU (3.21 g, 10.0 mmol), 20 mL of DMF, and DIPEA (5.23 mL, 30.0 mmol). The reaction was stirred at r.t. for 30 min, and 25 (1.66 g, 10.0 mmol) was added. The reaction was stirred at r.t. for 1 h, heated in a 120 °C oil-bath for 3 h, and cooled to r.t. The reaction mixture was diluted with EtOAc and water. The layers were separated, and the aqueous layer was extracted with EtOAc (×1). The combined organic layers were washed with 1 M HCl(aq), water, sat. NaHCO3(aq), water (×3), and brine, dried over MgSO4, and concentrated in vacuo to give a tan solid (1.7 g, 51% yield). 1H NMR (400 MHz, DMSO-d6) δ = 11.83 (br s, 1H), 8.29–8.22 (m, 2H), 8.04 (d, J = 8.0 Hz, 2H), 7.52 (d, J = 8.0 Hz, 2H), 7.27 (d, J = 8.4 Hz, 1H), 5.38 (br s, 1H), 4.60 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ = 174.3, 168.1, 160.0, 146.4, 133.5, 126.9, 126.8 (q, JC–F = 5.6 Hz), 124.5, 123.3 (q, JC–F = 274 Hz), 118.2, 118.1, 116.4 (q, JC–F = 31.0 Hz), 114.0, 62.5. MP: >200 °C. HRMS (ESI, m/z) calcd for C16H11F3N2O3 [M + H+] 337.0795. Found [M + H+] 337.0789.
4-(5-(4-Hydroxy-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)-benzaldehyde (31)
To a 250 mL round-bottomed flask equipped with a stir bar was added alcohol 30 (1.0 g, 2.97 mmol), 59 mL of 1,4-dioxane, and 10 g of MnO2. A reflux condenser was added, then the reaction was heated in an 80 °C oil-bath and stirred 4 h at which point TLC showed consumption of the alcohol. The reaction mixture was cooled to r.t. and filtered through a Celite plug, washing with EtOAc. The filtrate was concentrated in vacuo to give a tan solid (620 mg, 62% yield). 1H NMR (400 MHz, DMSO-d6) δ = 10.11 (s, 1H), 8.34–8.22 (m, 4H), 8.10 (d, J = 8.0 Hz, 2H), 7.24 (d, J = 8.4 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ = 192.7, 174.9, 167.5, 160.7, 138.0, 133.7, 131.2, 130.2, 127.8, 126.9 (q, JC–F = 5.66 Hz), 123.3 (q, JC–F = 274 Hz), 118.3, 116.4 (q, JC–F = 30.8), 113.3. MP: >200 °C. HRMS (ESI, m/z) calcd for C16H8F3N2O3 [M – H] 333.0493. Found [M – H] 333.0400.
Methyl 1-(4-(5-(4-hydroxy-3-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-3-yl)benzyl)azetidine-3-carboxylate (32)
To a 25 mL round-bottomed flask equipped with a stir bar was added aldehyde 31 (300 mg, 0.90 mmol), 4.1 mL of CH2Cl2, methyl azetidine-3-carboxylate hydrochloride (204 mg, 1.35 mmol), AcOH (0.21 mL, 3.6 mmol), DIPEA (0.24 mL, 1.35 mmol), and 4.1 mL of MeOH. The reaction mixture was stirred for 30 min, and sodium cyanoborohydride (57 mg, 0.90 mmol) was added. The reaction mixture was stirred for 2 h at which time TLC showed the consumption of the aldehyde. The reaction mixture was concentrated in vacuo, dissolved in EtOAc, and quenched with sat. NaHCO3(aq). The layers were separated, and the aqueous layer was extracted with EtOAc (×2). The combined organic layers were washed with water and brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified on a silica gel column eluted with 75–100% EtOAc/hexanes to give a white crystalline solid (193 mg, 49% yield). 1H NMR (400 MHz, MeOD-d4) δ = 8.14 (d, J = 1.2 Hz, 1H), 8.00 (d, J = 8.8 Hz, 1.6 Hz, 1H), 7.91 (d, J = 8.0 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 8.8 Hz, 1H), 3.72–3.62 (m, 5H), 3.56 (t, J = 8.0 Hz, 2H), 3.45 (t, J = 6.8 Hz, 2H), 3.34 (pent, J = 7.6 Hz, 1H). 13C NMR (101 MHz, MeOD-d4) δ = 174.8, 173.1, 168.1, 161.4, 139.6, 132.7, 129.0, 127.1, 126.9 (q, JC–F = 5.15 Hz), 126.0, 123.5 (q, JC–F = 273 Hz), 117.8, 117.4 (q, JC–F = 31.1 Hz), 113.5, 61.8, 56.1, 51.2, 33.4. MP: 125 °C (decomposition). HRMS (ESI, m/z) calcd for C22H18F3N3O4 [M + H+] 434.1322. Found [M + H+] 434.1320.
2-(4-(3-(4-Formylphenyl)-1,2,4-oxadiazol-5-yl)-2-(trifluoromethyl)phenoxy)ethyl 4-methylbenzenesulfonate (33)
To a 50 mL round-bottomed flask equipped with a stir bar was added phenol 31 (450 mg, 1.35 mmol), ethylene 1,2-bis(tosylate) (748 mg, 2.02 mmol), potassium carbonate (933 mg, 6.75 mmol), and 6.8 mL of acetonitrile. The reaction vessel was equipped with a reflux condenser placed in a preheated 90 °C oil-bath. The reaction mixture was stirred overnight (∼18 h), cooled to r.t., and diluted with EtOAc and water. The layers were separated, and the aqueous layer was extracted with EtOAc (×3). The combined organic layers were washed with brine, dried over MgSO4, and concentrated in vacuo. The product was purified by silica-gel chromatography to give an off-white solid (210 mg, 29% yield). 1H NMR (400 MHz, CDCl3) δ = 10.07 (s, 1H), 8.37 (d, J = 1.6 Hz), 8.34–8.24 (m, 3H), 7.98 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.4 Hz, 2H), 7.09 (d, J = 8.8 Hz, 1H), 4.46–4.33 (m 4H), 2.43 (s, 3H). 13C NMR (100 MHz, CDCl3) δ = 191.7, 174.7, 168.2, 159.4, 145.3, 138.2, 133.5, 132.4, 132.1, 130.1, 130.0, 128.2, 128.0, 127.7 (q, JC–F = 5.35 Hz), 122.7, (q, JC–F = 274 Hz), 120.2, (q, JC–F = 32.1 Hz), 116.9, 113.5, 67.4, 66.7, 21.7. MP: 125 °C (decomposition). HRMS (ESI, m/z) calcd for C25H20F3N2O6S [M + H+] 533.0989. Found [M + H+] 533.0990.
Methyl 1-(4-(5-(4-(2-(tosyloxy)ethoxy)-3-(trifluoromethyl)-phenyl)-1,2,4-oxadiazol-3-yl)benzyl)azetidine-3-carboxylate (34)
To a 25 mL round-bottomed flask equipped with a stir bar was added aldehyde 33 (200 mg, 0.38 mmol), methyl 3-azetidinecarboxylate hydrochloride (61 mg, 0.40 mmol), 1.9 mL of 1,2-dichloroethane, and 6.3 mL of methanol. Diisopropylethylamine (0.07 mL, 0.41 mmol) was added to the reaction mixture, and it was stirred for 1 h at r.t. The reaction mixture was concentrated in vacuo, suspended in 1.9 mL of 1,2-dichloroethane, at which time sodium triacetoxyborohydride (250 mg, 1.18 mmol) and acetic acid (0.02 mL, 0.38 mmol) were added. The reaction mixture was stirred overnight (∼18 h), diluted with EtOAc, and quenched with sat. NaHCO3(aq). The layers were separated, and the aqueous layer extracted with EtOAc (×3). The combined organic layers were washed with water and brine, dried over MgSO4, and concentrated in vacuo. The crude product was purified on a silica gel column eluted with 2–3% MeOH/CH2Cl2 to give a clear oil (110 mg, 41% yield). 1H NMR (400 MHz, CDCl3) δ = 8.40 (s, 1H), 8.29 (d, J = 8.8 Hz, 1H), 8.08 (d, J = 6.8 Hz, 2H), 7.79 (d, J = 7.2 Hz), 7.41 (d, J = 5.6 Hz, 2H), 7.33 (d, J = 5.6 Hz, 2H), 7.07 (dd, J = 8.4 Hz, 2.4 Hz, 1H), 4.45–4.32 (m 4H), 3.74–3.64 (m, 5H), 3.60–3.50 (m, 2H), 3.42–3.28 (m, 3H), 2.45–2.38 (m, 3H). 13C NMR (100 MHz, CDCl3) δ = 174.1, 173.5, 168.8, 159.0, 145.2, 141.2, 133.3, 132.3, 129.9, 128.9, 127.9, 127.6 (q, JC–F = 5.35 Hz), 127.6, 125.5, 122.6, (q, JC–F = 274 Hz), 119.7 (q, JC–F = 32.2 Hz), 117.2, 113.2, 67.2, 66.5, 63.1, 56.9, 52.0, 33.9, 21.6. HRMS (ESI, m/z) calcd for C30H29F3N3O7S [M + H+] 632.1673. Found [M + H+] 632.1669.
4.2. In Vitro Screening
4.2.1. In Vitro Functional Assay (EC50)
The functional assay of the test compounds was carried out according to a published procedure.51 Briefly, S1P1-TANGO-U2OS cells were suspended at a concentration of 312,500 cells/mL in assay medium (Freestyle Expression Medium without supplements). Thirty-two microliters of the cell suspension was seeded in each well of a black, clear-bottomed 384-well plate, followed by 48 h of incubation at 37 °C in 5% CO2 and 95% relative humidity. After incubation, 100 nL of the test compound in DMSO was added to sample wells, while DMSO alone (0.5% final concentration) was added to control wells. Plates were then incubated at 37 °C in 5% CO2 for 5 h. After incubation, 8 μL/well of the LiveBLAzer FRET substrate mixture, prepared according to the manufacturer’s protocol and containing 10 mM Probenicid, was added to all wells. After 2 h of incubation at r.t. in the dark, plates were read on an EnVision plate reader (PerkinElmer Lifesciences, Turku, Finland) at an excitation wavelength of 405 nm and emission wavelengths of 460 and 535 nm.
4.2.2. In Vitro Binding Assay (IC50)
The potency of the test compounds for S1P receptors was determined according to our published procedure.34 In brief, test compounds dissolved in DMSO were preincubated for 30 min at r.t. in assay buffer (50 mM HEPES-Na (pH 7.5), 5 mM MgCl2, 1 mM CaCl2, and 0.5% fatty acid-free BSA) with cell membranes expressing recombinant human S1P1–3 (S1P1 cell membranes were purchased from ChanTest Corp, Cleveland, OH, and S1P2 and S1P3 cell membranes were from Chemicon/Millipore Inc., Billerica, MA). Diluted [32P]S1P, prepared in-house, was added to give a final volume of 150 μL containing 0.1–0.2 nM [32P]S1P with 1–2 μg of membrane protein in each well of a 96-well plate. Samples were incubated for 60 min at r.t., and filter-bound radioactivity was measured by Cherenkov counting on a Beckman LS 3801 scintillation counter. Nonspecific binding was determined from samples incubated with an additional 1 μM cold S1P.
4.3. Radiochemistry
[18F]Fluoride was produced by 18O(p,n)18F reaction through proton irradiation of enriched 18O water (95%) using Washington University’s RDS111 cyclotron (Siemens/CTI Molecular Imaging, Knoxville, TN). [18F]Fluoride was first passed through an ion-exchange resin and then eluted with 0.02 M potassium carbonate (K2CO3) solution. A sample of ∼200 mCi [18F]/fluoride was added to a reaction vessel containing Kryptofix 222 (5–6 mg), and the solution was evaporated under nitrogen (oil-bath temperature 110 °C). Acetonitrile (3 × 1.0 mL) was added and evaporated to ensure complete removal of water. After all of the water was removed, 1 mg of 34 was added as a solution in 200 μL of acetonitrile. The reaction tube was capped, briefly mixed, and placed in a preheated 110 °C oil-bath. The reaction mixture was incubated at 110 °C for 15 min, shaking occasionally. The reaction vessel was removed from the oil-bath, at which time 300 μL of EtOH and 200 μL of NaOH(aq) were added. The reaction mixture was heated in an 80 °C preheated oil-bath for 5 min, shaking occasionally. The reaction vessel was removed from the oil-bath, quenched with 400 μL of 1 M HCl(aq) and diluted with 1.9 mL of the HPLC mobile phase (38% MeCN in 0.1 M ammonium formate buffer, pH 4.5), passed through an alumina Neutral Sep-Pak Plus cartridge. The crude product was then loaded onto an Agilent SB-C18 semipreparative HPLC column (250 mm × 10 mm) with a UV detector set at 254 nm. The HPLC system used a 5 mL injection loop. With the above-mentioned mobile phase at 4.0 mL/min flow rate. The retention time of the product was approximately 18 min. After the HPLC collection was diluted with ∼50 mL of sterile water, the product was trapped on a C18 Sep-Pak Plus cartridge. The trapped product was eluted with ethanol (0.3 mL), and this portion was discarded. The Sep-Pak was sequentially eluted with ethanol (0.3 mL), which was followed by 2.7 mL of 0.9% saline. After sterile filtration into a glass vial, the final product was ready for quality control (QC) analysis and animal studies. An aliquot of sample was assayed by an analytical HPLC system (Agilent Zorbax, SB-C18 column, 250 × 4.6 mm), UV at 254 nm; the mobile phase consists of acetonitrile/0.1 M, pH 4.5, ammonium formate buffer (50/50, v/v). At this condition, the retention time for [18F]28c was approximately 5.1 min at a flow rate of 1.0 mL/min. The radioactive dose was authenticated by co-injection with the corresponding nonradioactive compound 28c onto an analytical HPLC system. The radiochemical purity was >98%, the labeling yield was 25.7 ± 4.6% (n = 10, decay corrected to start of synthesis), and the specific activity was 1.43 ± 0.12 Ci/μmol (decay corrected to the end of synthesis). The entire procedure took about 90 min.
4.4. Biological Evaluation in Rodents
All animal experiments were conducted under Washington University Animal Studies Committee IACUC-approved protocols in accordance with the US National Research Council’s Guide for the Care and Use of Laboratory Animals. Adult male Sprague–Dawley rats (Charles River, Inc.) were used to determine the biodistribution in normal rodents. Adult male C57BL/6 mice (Charles River Inc., Frederick, MD) were used for the mouse model of LPS-induced liver injury and inflammation to demonstrate specificity of the S1P1 tracer.50,52–54 LPS-treated mice received an intraperitoneal injection of LPS from E. coli 055:B5 (Sigma-Aldrich, St. Louis, MO, USA) in saline (3 mg/mL) at 15 mg/kg (5 mL/kg) dose 24 h prior to use, and sham mice received a saline injection.
4.4.1. Biodistribution in Normal Rats
The biodistribution of [18F] 28c in normal rodents was determined using adult male SD rats (230–270 g). Rats were anesthetized with isoflurane (2–3% in oxygen) and injected in the tail vein with ∼50 μCi (1.85 MBq) of [18F]28c in 5% ethanol/saline. At the appropriate time, rats were again anesthetized and were euthanized at 5, 30, 60, and 120 min postinjection (n = 4 for each group). Tissues of interest including blood, heart, lung, muscle, fat, pancreas, spleen, bone, thymus, brain, kidney, and liver were collected, weighed, and counted with a dilution of the injectate on an automated well counter (Beckman Gamma 8000 well counter). Uptake in Table 3 is reported as decay-corrected percent injected dose per gram (% ID/g).
4.4.2. In Vitro Autoradiography Study of LPS-Treated Mouse Liver
Livers from sham and LPS-treated mice were collected and snap frozen. Twenty micrometer sequential sections were cut with a Microm cryotome and thaw mounted on glass slides. The slides were then incubated for 60 min with ∼0.1 nM [18F]28c at r.t. Blocking studies were performed using adjacent sections incubated with an additional 10 μM of either S1P or SEW2871. Following the incubation, the slides were washed and exposed to the storage phosphor screen in an imaging cassette for 12 h in −80 °C in the dark. The distribution of radioactivity was visualized with a Fuji Bio-Imaging Analyzer FLA-7000 (Fuji Photo Film, Tokyo, Japan).
4.4.3. IHC Staining of S1P1 Expression in Mouse Liver
Liver samples were fixed in 10% formalin immediately, then embedded in paraffin and cut into 5 μm sections. Sections were deparaffinized in xylene and rehydrated through a graded alcohol series to water. Endogenous peroxidase activity was quenched with 3% H2O2 in methanol for 5 min. Slides were incubated in blocking buffer (10% normal goat serum in PBS) for 30 min before the incubation with a 1:50 dilution of a rabbit antimouse S1P1 antibody (Santa Cruz biotechnology, Santa Cruz, CA) overnight at 4 °C. The primary antibody binding was detected using an anti–rabbit HRP-DAB staining kit (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions. A Nikon E600 microscope coupled with a charge-coupled device camera was used to obtain all photomicrographs.
4.4.4. MicroPET Imaging of LPS-Treated Mice with [18F]28c and [15O]Water
MicroPET studies were performed using adult male C57BL/6 mice (18–23 g) ∼24 h after pretreatment with either saline (n = 4) or LPS (n = 3), and PET scans were conducted under light anesthesia (2–3% isoflurane) delivered by a nose cone. Mice were secured using a custom-designed acrylic restraining device and placed in transaxial position with their whole bodies inside the field of the window of the Inveon PET/CT system. Following a transmission scan and a CT for anatomical coregistration, animals received a bolus injection. [18F]28c (∼300 μCi (11.1 MBq), in 10% ethanol/saline) was injected i.v. and a microPET dynamic scan acquired from 0–120 min.
[15O]Water MicroPET Study: Small-animal microPET imaging using [15O]water serves as a noninvasive method to measure liver blood flow in rodents.48 A microPET scan was performed ∼30 min before the [18F] scan on an Inveon PET/CT system (Siemens Inc., Erlangen, Germany). Following a transmission scan and a CT for anatomical coregistration, animals received a bolus injection (30–55 MBq) of [15O]water via the tail vein, immediately followed by a 10 min scan (1 × 3 s, 6 × 2 s, 5 × 5 s, 11 × 10 s, 5 × 30 s, 5 × 60 s).
MicroPET Data Processing: Image reconstructions were performed using microPET Manager 2.3.3.6, ASIPro 6.3.3.0 (Siemens Inc., Erlangen, Germany). The list-mode data of the emission scans were reframed into a dynamic sequence. The data were reconstructed per time frame employing an iterative reconstruction algorithm and corrected for decay, random coincidences, scatter, and attenuation. Three-dimensional ROI of the mouse liver was drawn according to a standard mouse anatomy atlas. Averaged regional radioactivity was obtained and expressed as dimensionless standardized uptake values (SUVs). The parameter standardized uptake value (SUV) is defined as [tissue activity concentration (MBq/g) × body weight (g)/injected dose (MBq)]. For statistical analysis, two-tailed Student’s t test was applied, and significant difference is defined as a P value <0.05.
4.4.5. Biodistribution of [18F]28c and [99mTc]Mebrofenin in LPS-Treated Mice
The acute biodistribution studies in sham and LPS-treated mice, as described above, was carried out to determine tracer uptake in a rodent model with increased S1P1. Approximately 24 h after treatment, one set of sham and LPS-treated mice was injected in the tail vein with ∼20 μCi (0.74 MBq) of [18F]28c in 10% ethanol/saline. A separate set of sham and LPS-treated mice were injected with ∼4 μCi (148 KBq) of [99mTc]mebrofenin (Cardinal Health Nuclear Pharmacy Services, Overland, MO) under 2–3% isoflurane/oxygen anesthesia. Animals (n = 4 per each group for each tracer) were euthanized at 30 or 60 min postinjection. Tissues of interest were collected, weighed, and counted with a dilution of the injectate on an automated well counter (Beckman Gamma 8000 well counter). Uptake is reported as background- and decay-corrected percent injected dose per gram (% ID/g).
Supplementary Material
Acknowledgments
We thank Ms. Lynne Jones for many helpful discussions. This research work was directly supported by the Department of Energy (DOE, No. DESC0008432 and No. DESC0012737), and the National Institutes of Health through the National Institute of Neurological Disorders and Stroke (NINDS, R01NS075527), and the National Institute of Mental Health (NIMH, No. MH092797). During the process of accomplishing this work, we also used the NIH/NIGMS Biomedical Mass Spectrometry Resource at Washington University in St. Louis, MO, which is supported by National Institutes of Health/National Institute of General Medical Sciences Grant No. 8P41GM103422. We are grateful to the Washington University Cyclotron Facility and its personnel for [18F]fluoride production. We thank the Department of Chemistry staff and the Washington University High Resolution NMR Facility for assistance with NMR spectra; purchase of the 400 MHz NMR instrument was partially supported by Grant S10 RR027207 from the NIH Shared Instrument Grant program. We also thank the Elvie L. Taylor Histology Core Facility of Washington University School of Medicine for sample embedding, and the MIR Preclinical PET-CT facility of the Washington University School of Medicine, which was funded in part through the National Institute of Health HEI grant (S10 RR025097).
ABBREVIATIONS
- % ID/g
percent injected dose per gram
- EDC·HCl
N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride
- ROI
region of interest
- SD
standard deviation
- S1P
sphingosine 1-phosphate
- S1P1–5
sphingosine 1-phosphate receptor 1–5
- SPhos
2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl
- SUV
standardized uptake value
- TAC
time–activity curve
- [99mTc]-mebrofenin
[99mTc]-N-(3-bromo-2,4,6-trimethyacetanilide) iminodiacetic acid
- XPhos
2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.6b00390.
Molecular formula strings of the final products (CSV)
Notes The authors declare no competing financial interest.
References
- 1.O’Sullivan C, Dev KK. The structure and function of the S1P1 receptor. Trends Pharmacol Sci. 2013;34:401–412. doi: 10.1016/j.tips.2013.05.002. [DOI] [PubMed] [Google Scholar]
- 2.Rosen H, Germana Sanna M, Gonzalez-Cabrera PJ, Roberts E. The Organization of the Sphingosine 1-Phosphate Signaling System. In: Oldstone MBA, Rosen H, editors. Sphingosine-1-Phosphate Signaling in Immunology and Infectious Diseases. Vol. 378. Springer International Publishing; Cham, Switzerland: 2014. pp. 1–21. 2014/04/15. [DOI] [PubMed] [Google Scholar]
- 3.Billich A, Bornancin F, Devay P, Mechtcheriakova D, Urtz N, Baumruker T. Phosphorylation of the immunomodulatory drug FTY720 by sphingosine kinases. J Biol Chem. 2003;278:47408–47415. doi: 10.1074/jbc.M307687200. [DOI] [PubMed] [Google Scholar]
- 4.Mandala S, Hajdu R, Bergstrom J, Quackenbush E, Xie J, Milligan J, Thornton R, Shei GJ, Card D, Keohane C, Rosenbach M, Hale J, Lynch CL, Rupprecht K, Parsons W, Rosen H. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–349. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 5.Brinkmann V, Billich A, Baumruker T, Heining P, Schmouder R, Francis G, Aradhye S, Burtin P. Fingolimod (FTY720) Discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discovery. 2010;9:883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 6.Forrest M, Sun SY, Hajdu R, Bergstrom J, Card D, Doherty G, Hale J, Keohane C, Meyers C, Milligan J, Mills S, Nomura N, Rosen H, Rosenbach M, Shei GJ, Singer II, Tian M, West S, White V, Xie J, Proia RL, Mandala S. Immune cell regulation and cardiovascular effects of sphingosine 1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypes. J Pharmacol Exp Ther. 2004;309:758–768. doi: 10.1124/jpet.103.062828. [DOI] [PubMed] [Google Scholar]
- 7.Fryer RM, Muthukumarana A, Harrison PC, Nodop Mazurek S, Chen RR, Harrington KE, Dinallo RM, Horan JC, Patnaude L, Modis LK, Reinhart GA. The clinically-tested S1P receptor agonists, FTY720 and BAF312, demonstrate subtype-specific bradycardia (S1P1) and hypertension (S1P3) in rat. PLoS One. 2012;7:e52985. doi: 10.1371/journal.pone.0052985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gergely P, Nuesslein-Hildesheim B, Guerini D, Brinkmann V, Traebert M, Bruns C, Pan S, Gray NS, Hinterding K, Cooke NG, Groenewegen A, Vitaliti A, Sing T, Luttringer O, Yang J, Gardin A, Wang N, Crumb WJ, Jr, Saltzman M, Rosenberg M, Wallstrom E. The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br J Pharmacol. 2012;167:1035–1047. doi: 10.1111/j.1476-5381.2012.02061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murakami A, Takasugi H, Ohnuma S, Koide Y, Sakurai A, Takeda S, Hasegawa T, Sasamori J, Konno T, Hayashi K, Watanabe Y, Mori K, Sato Y, Takahashi A, Mochizuki N, Takakura N. Sphingosine 1-phosphate (S1P) regulates vascular contraction via S1P3 receptor: Investigation based on a new S1P3 receptor antagonist. Mol Pharmacol. 2010;77:704–713. doi: 10.1124/mol.109.061481. [DOI] [PubMed] [Google Scholar]
- 10.Li MH, Sanchez T, Pappalardo A, Lynch KR, Hla T, Ferrer F. Induction of antiproliferative connective tissue growth factor expression in Wilms’ tumor cells by sphingosine-1-phosphate receptor 2. Mol Cancer Res. 2008;6:1649–1656. doi: 10.1158/1541-7786.MCR-07-2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Plano D, Amin S, Sharma AK. Importance of sphingosine kinase (SphK) as a target in developing cancer therapeutics and recent developments in the synthesis of novel SphK inhibitors. J Med Chem. 2014;57:5509–5524. doi: 10.1021/jm4011687. [DOI] [PubMed] [Google Scholar]
- 12.Ishii M, Egen JG, Klauschen F, Meier-Schellersheim M, Saeki Y, Vacher J, Proia RL, Germain RN. Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature. 2009;458:524–528. doi: 10.1038/nature07713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cross AH, Klein RS, Piccio L. Rituximab combination therapy in relapsing multiple sclerosis. Ther Adv Neurol Disord. 2012;5:311–319. doi: 10.1177/1756285612461165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong J, Wang H, Wu G, Zhao J, Zhang L, Zuo L, Zhu W, Gong J, Li Y, Gu L, Li J. Oral treatment with SEW2871, a sphingosine-1-phosphate type 1 receptor agonist, ameliorates experimental colitis in interleukin-10 gene deficient mice. Clin Exp Immunol. 2014;177:94–101. doi: 10.1111/cei.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McCandless EE, Budde M, Lees JR, Dorsey D, Lyng E, Klein RS. IL-1R signaling within the central nervous system regulates CXCL12 expression at the blood-brain barrier and disease severity during experimental autoimmune encephalomyelitis. J Immunol. 2009;183:613–620. doi: 10.4049/jimmunol.0802258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moberly JB, Rohatagi S, Zahir H, Hsu C, Noveck RJ, Truitt KE. Pharmacological modulation of peripheral T and B lymphocytes by a selective sphingosine 1-phosphate receptor-1 modulator. J Clin Pharmacol. 2012;52:996–1006. doi: 10.1177/0091270011408728. [DOI] [PubMed] [Google Scholar]
- 17.Graler MH. Targeting sphingosine 1-phosphate (S1P) levels and S1P receptor functions for therapeutic immune interventions. Cell Physiol Biochem. 2010;26:79–86. doi: 10.1159/000315108. [DOI] [PubMed] [Google Scholar]
- 18.Holman DW, Klein RS, Ransohoff RM. The blood-brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta, Mol Basis Dis. 2011;1812:220–230. doi: 10.1016/j.bbadis.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohno T, Hasegawa C, Nakade S, Kitagawa J, Honda N, Ogawa M. The prediction of human response to ONO-4641, a sphingosine 1-phosphate receptor modulator, from preclinical data based on pharmacokinetic-pharmacodynamic modeling. Biopharm Drug Dispos. 2010;31:396–406. doi: 10.1002/bdd.719. [DOI] [PubMed] [Google Scholar]
- 20.McCandless EE, Piccio L, Woerner BM, Schmidt RE, Rubin JB, Cross AH, Klein RS. Pathological expression of CXCL12 at the blood-brain barrier correlates with severity of multiple sclerosis. Am J Pathol. 2008;172:799–808. doi: 10.2353/ajpath.2008.070918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Z, Sims CR, Patil NK, Gokden N, Mayeux PR. Pharmacologic targeting of sphingosine-1-phosphate receptor 1 improves the renal microcirculation during sepsis in the mouse. J Pharmacol Exp Ther. 2015;352:61–66. doi: 10.1124/jpet.114.219394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong DF, Tauscher J, Grunder G. The role of imaging in proof of concept for CNS drug discovery and development. Neuropsychopharmacology. 2009;34:187–203. doi: 10.1038/npp.2008.166. [DOI] [PubMed] [Google Scholar]
- 23.Eckelman WC, Kilbourn MR, Mathis CA. Discussion of targeting proteins in vivo: in vitro guidelines. Nucl Med Biol. 2006;33:449–451. doi: 10.1016/j.nucmedbio.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 24.Eckelman WC, Mathis CA. Targeting proteins in vivo: in vitro guidelines. Nucl Med Biol. 2006;33:161–164. doi: 10.1016/j.nucmedbio.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 25.Briard E, Orain D, Beerli C, Billich A, Streiff M, Bigaud M, Auberson YP. BZM055, an iodinated radiotracer candidate for PET and SPECT imaging of myelin and FTY720 brain distribution. ChemMedChem. 2011;6:667–677. doi: 10.1002/cmdc.201000477. [DOI] [PubMed] [Google Scholar]
- 26.Prasad VP, Wagner S, Keul P, Hermann S, Levkau B, Schäfers M, Haufe G. Synthesis of fluorinated analogues of sphingosine-1-phosphate antagonists as potential radiotracers for molecular imaging using positron emission tomography. Bioorg Med Chem. 2014;22:5168–5181. doi: 10.1016/j.bmc.2014.08.009. [DOI] [PubMed] [Google Scholar]
- 27.Shaikh RS, Schilson SS, Wagner S, Hermann S, Keul P, Levkau B, Schafers M, Haufe G. Synthesis and evaluation of fluorinated fingolimod (FTY720) analogues for sphingosine-1-phosphate receptor molecular imaging by positron emission tomography. J Med Chem. 2015;58:3471–3484. doi: 10.1021/jm502021d. [DOI] [PubMed] [Google Scholar]
- 28.Yue X, Jin H, Liu H, Rosenberg AJ, Klein RS, Tu Z. A potent and selective C-11 labeled PET tracer for imaging sphingosine-1-phosphate receptor 2 in the CNS demonstrates sexually dimorphic expression. Org Biomol Chem. 2015;13:7928–7939. doi: 10.1039/c5ob00951k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin H, Yang H, Liu H, Zhang Y, Zhang X, Rosenberg AJ, Liu Y, Lapi SE, Tu Z. A promising carbon-11-labeled sphingosine-1-phosphate receptor 1-specific PET tracer for imaging vascular injury. J Nucl Cardiol. doi: 10.1007/s12350-015-0391-1. [Online early access] Published Online: Feb 02, 2016. [DOI] [PubMed] [Google Scholar]
- 30.Liu H, Jin H, Yue X, Luo Z, Liu C, Rosenberg AJ, Tu Z. PET imaging study of S1PR1 expression in a rat model of multiple sclerosis. Mol Imaging Biol. doi: 10.1007/s11307-016-0944-y. [Online early access] Published Online: Mar 14, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng G, Meng Q, Liu Q, Xu X, Xu Q, Ren F, Guo TB, Lu H, Xiang JN, Elliott JD, Lin X. Identification of benzoxazole analogs as novel, S1P3 sparing S1P1 agonists. Bioorg Med Chem Lett. 2012;22:3973–3977. doi: 10.1016/j.bmcl.2012.04.095. [DOI] [PubMed] [Google Scholar]
- 32.Quattropani A, Sauer WH, Crosignani S, Dorbais J, Gerber P, Gonzalez J, Marin D, Muzerelle M, Beltran F, Nichols A, Georgi K, Schneider M, Vitte PA, Eligert V, Novo-Perez L, Hantson J, Nock S, Carboni S, de Souza AL, Arrighi JF, Boschert U, Bombrun A. Pharmacophore-based design of novel oxadiazoles as selective sphingosine-1-phosphate (S1P) receptor agonists with in vivo efficacy. ChemMedChem. 2015;10:688–714. doi: 10.1002/cmdc.201402557. [DOI] [PubMed] [Google Scholar]
- 33.Li Z, Chen W, Hale JJ, Lynch CL, Mills SG, Hajdu R, Keohane CA, Rosenbach MJ, Milligan JA, Shei GJ, Chrebet G, Parent SA, Bergstrom J, Card D, Forrest M, Quackenbush EJ, Wickham LA, Vargas H, Evans RM, Rosen H, Mandala S. Discovery of potent 3,5-diphenyl-1,2,4-oxadiazole sphingosine-1-phosphate-1 (S1P1) receptor agonists with exceptional selectivity against S1P2 and S1P3. J Med Chem. 2005;48:6169–6173. doi: 10.1021/jm0503244. [DOI] [PubMed] [Google Scholar]
- 34.Rosenberg AJ, Liu H, Tu Z. A practical process for the preparation of [32P]S1P and binding assay for S1P receptor ligands. Appl Radiat Isot. 2015;102:5–9. doi: 10.1016/j.apradiso.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanna MG, Liao J, Jo E, Alfonso C, Ahn MY, Peterson MS, Webb B, Lefebvre S, Chun J, Gray N, Rosen H. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J Biol Chem. 2004;279:13839–13848. doi: 10.1074/jbc.M311743200. [DOI] [PubMed] [Google Scholar]
- 36.Chen DL, Zhou D, Chu W, Herrbrich P, Engle JT, Griffin E, Jones LA, Rothfuss JM, Geraci M, Hotchkiss RS, Mach RH. Radiolabeled isatin binding to caspase-3 activation induced by anti-Fas antibody. Nucl Med Biol. 2012;39:137–144. doi: 10.1016/j.nucmedbio.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sic H, Kraus H, Madl J, Flittner KA, von Munchow AL, Pieper K, Rizzi M, Kienzler AK, Ayata K, Rauer S, Kleuser B, Salzer U, Burger M, Zirlik K, Lougaris V, Plebani A, Romer W, Loeffler C, Scaramuzza S, Villa A, Noguchi E, Grimbacher B, Eibel H. Sphingosine-1-phosphate receptors control B-cell migration through signaling components associated with primary immunodeficiencies, chronic lymphocytic leukemia, and multiple sclerosis. J Allergy Clin Immunol. 2014;134:420–428. doi: 10.1016/j.jaci.2014.01.037. [DOI] [PubMed] [Google Scholar]
- 38.Cavone L, Felici R, Lapucci A, Buonvicino D, Pratesi S, Muzzi M, Hakiki B, Maggi L, Peruzzi B, Caporale R, Annunziato F, Amato MP, Chiarugi A. Dysregulation of sphingosine 1 phosphate receptor-1 (S1P1) signaling and regulatory lymphocyte-dependent immunosuppression in a model of post-fingolimod MS rebound. Brain, Behav, Immun. 2015;50:78–86. doi: 10.1016/j.bbi.2015.06.019. [DOI] [PubMed] [Google Scholar]
- 39.Poulain RF, Tartar AL, Déprez BtP. Parallel synthesis of 1,2,4-oxadiazoles from carboxylic acids using an improved, uronium-based, activation. Tetrahedron Lett. 2001;42:1495–1498. [Google Scholar]
- 40.Waterhouse RN. Determination of lipophilicity and its use as a predictor of blood-brain barrier penetration of molecular imaging agents. Mol Imaging Biol. 2003;5:376–389. doi: 10.1016/j.mibio.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 41.Komiya T, Sato K, Shioya H, Inagaki Y, Hagiya H, Kozaki R, Imai M, Takada Y, Maeda T, Kurata H, Kurono M, Suzuki R, Otsuki K, Habashita H, Nakade S. Efficacy and immunomodulatory actions of ONO-4641, a novel selective agonist for sphingosine 1-phosphate receptors 1 and 5, in preclinical models of multiple sclerosis. Clin Exp Immunol. 2013;171:54–62. doi: 10.1111/j.1365-2249.2012.04669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gibson RE. In Vitro Approaches to Site-Specific Imaging Agents. In: Welch MJ, Eckelman WC, editors. Targeted Molecular Imaging. CRC Press Taylor & Francis Group; Boca Raton, FL: 2012. pp. 3–15. [Google Scholar]
- 43.Hanson MA, Roth CB, Jo E, Griffith MT, Scott FL, Reinhart G, Desale H, Clemons B, Cahalan SM, Schuerer SC, Sanna MG, Han GW, Kuhn P, Rosen H, Stevens RC. Crystal structure of a lipid G protein-coupled receptor. Science. 2012;335:851–855. doi: 10.1126/science.1215904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JG. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med. 2004;169:1245–1251. doi: 10.1164/rccm.200309-1258OC. [DOI] [PubMed] [Google Scholar]
- 45.Kono M, Tucker AE, Tran J, Bergner JB, Turner EM, Proia RL. Sphingosine-1-phosphate receptor 1 reporter mice reveal receptor activation sites in vivo. J Clin Invest. 2014;124:2076–2086. doi: 10.1172/JCI71194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gonzalez-Cabrera PJ, Jo E, Sanna MG, Brown S, Leaf N, Marsolais D, Schaeffer MT, Chapman J, Cameron M, Guerrero M, Roberts E, Rosen H. Full pharmacological efficacy of a novel S1P1 agonist that does not require S1P-like headgroup interactions. Mol Pharmacol. 2008;74:1308–1318. doi: 10.1124/mol.108.049783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tomlinson RE, Silva MJ, Shoghi KI. Quantification of skeletal blood flow and fluoride metabolism in rats using PET in a preclinical stress fracture model. Mol Imaging Biol. 2012;14:348–354. doi: 10.1007/s11307-011-0505-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shiomi S, Iwata Y, Sasaki N, Morikawa H, Tamori A, Habu D, Takeda T, Nishiguchi S, Kuroki T, Ochi H. Assessment of hepatic blood flow by PET with 15O water: Correlation between per-rectal portal scintigraphy with 99Tcm-pertechnetate and scintigraphy with 99Tcm-GSA. Nucl Med Commun. 2000;21:533–538. doi: 10.1097/00006231-200006000-00006. [DOI] [PubMed] [Google Scholar]
- 49.Ghibellini G, Leslie EM, Pollack GM, Brouwer KL. Use of Tc-99m mebrofenin as a clinical probe to assess altered hepatobiliary transport: Integration of in vitro, pharmacokinetic modeling, and simulation studies. Pharm Res. 2008;25:1851–1860. doi: 10.1007/s11095-008-9597-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Joseph B, Bhargava KK, Tronco GG, Palestro CJ, Gupta S. Systemic and local release of inflammatory cytokines regulates hepatobiliary excretion of 99mTc-mebrofenin. Nucl Med Commun. 2008;29:336–344. doi: 10.1097/MNM.0b013e3282f81460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schurer SC, Brown SJ, Gonzalez-Cabrera PJ, Schaeffer MT, Chapman J, Jo E, Chase P, Spicer T, Hodder P, Rosen H. Ligand-binding pocket shape differences between sphingosine 1-phosphate (S1P) receptors S1P1 and S1P3 determine efficiency of chemical probe identification by ultrahigh-throughput screening. ACS Chem Biol. 2008;3:486–498. doi: 10.1021/cb800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583–594. doi: 10.1016/j.jhep.2013.03.033. [DOI] [PubMed] [Google Scholar]
- 53.Schroder M, Richter C, Juan MH, Maltusch K, Giegold O, Quintini G, Pfeilschifter JM, Huwiler A, Radeke HH. The sphingosine kinase 1 and S1P1 axis specifically counteracts LPS-induced IL-12p70 production in immune cells of the spleen. Mol Immunol. 2011;48:1139–1148. doi: 10.1016/j.molimm.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 54.Zhou D, Lee H, Rothfuss JM, Chen DL, Ponde DE, Welch MJ, Mach RH. Design and synthesis of 2-amino-4-methylpyridine analogues as inhibitors for inducible nitric oxide synthase and in vivo evaluation of [18F]6-(2-fluoropropyl)-4-methylpyridin-2-amine as a potential PET tracer for inducible nitric oxide synthase. J Med Chem. 2009;52:2443–2453. doi: 10.1021/jm801556h. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.