

Abstract
Affinity purification approaches have been successful in isolating native complexes for proteomic characterization. Structural heterogeneity and a degree of compositional heterogeneity of a complex do not usually impede progress in conducting such studies. In contrast, a complex intended for structural characterization should be purified in a state that is both compositionally and structurally homogeneous as well as at a higher concentration than required for proteomics. Recently, there have been significant advances in the application of electron microscopy for structure determination of large macromolecular complexes. This has heightened interest in approaches to purify native complexes of sufficient quality and quantity for structural determination by electron microscopy. The Tandem Affinity Purification (TAP) method has been optimized to extract and purify an 18-subunit, ~ 0.8 MDa ribonucleoprotein assembly from budding yeast (Saccharomyces cerevisiae) suitable for negative stain and electron cryo microscopy. Herein is detailed the modifications made to the TAP method, the rationale for making these changes, and the approaches taken to assay for a compositionally and structurally homogeneous complex.
Keywords: Biochemistry, Issue 113, Electron microscopy, purification, complexes, Tandem Affinity Purification, ribonucleoprotein, splicing, structure, spliceosome, U1 snRNP, affinity chromatography
Introduction
Many major cellular processes are carried out by large protein and protein-RNA complexes1. A significant bottleneck to conducting biophysical and structural studies of such complexes is obtaining them of a suitable quality (i.e., homogeneity) and at an appropriate concentration. Isolating a complex from a native source has many advantages, including retaining relevant post-transcriptional and/or translational modifications of subunits and insuring proper complex assembly. However, large cellular complexes are often present in a cell at a low copy number and the purification must be highly efficient and occur under near physiological conditions to ensure complex integrity is maintained. Purifying a complex from a eukaryotic source is particularly challenging and can be financially prohibitive. Thus, strategies or methods that are efficient and yield a homogeneous complex are highly desired.
A strategy that has been successful in purifying native complexes from eukaryotic cells for their initial characterization is the Tandem Affinity Purification (TAP) method2,3. The TAP method was initially devised to purify a native protein from the budding yeast (S. cerevisiae) in complex with interacting factor(s)2. The TAP method utilized two tags, each tag fused in tandem to the same protein-coding gene sequence. The tags were selected so as to balance a need for tight and selective binding to an affinity resin with a desire to maintain near physiological solution conditions. This balance serves to preserve stable interaction(s) of the tagged protein with interacting factor(s) for post-purification characterization. The genomically incorporated TAP tag was placed at the end (C-terminal) of a protein-coding gene and consisted of a sequence coding for a Calmodulin Binding Peptide (CBP) followed by Protein A - an addition of just over 20 kDa to the tagged protein. CBP is short, 26 amino acids, and recognized by the ~ 17 kDa protein Calmodulin (CaM) in the presence of calcium with a KD on the order of 10-9 M 4. The Protein A tag is larger, consisting of two repeats of 58 residues with a short linker between the repeats. Each 58 amino acid repeat is recognized by immunoglobulin G (IgG) with a KD ~ 10-8 M 5. Between these two tags was incorporated a recognition site for TEV protease, an endopeptidase from the Tobacco Etch Virus6,7. As illustrated in Figure 1, in the first affinity step of the method the TAP tagged protein is bound to an IgG resin via the Protein A. The tagged protein is eluted by on column cleavage upon the addition of TEV protease, site-specifically cleaving between the two tags. This is a necessary step as the interaction of IgG and Protein A is very strong and can only be adequately perturbed under denaturing solution conditions. Lacking a Protein A tag, the protein is bound to CaM resin in the presence of calcium and eluted from this resin with addition of the metal ion chelator EGTA (ethylene glycol tetraacetic acid) (Figure 1).
Soon after the introduction of the TAP method, it was used in a large-scale study to generate a 'map' of complex interactions in S. cerevisiae8. Importantly, as a consequence of this effort an entire yeast-TAP Tagged Open Reading Frame (ORF) library as well as individual TAP tagged ORFs9 are available from a commercial source. Thus, one can obtain any yeast strain with a tagged protein for any yeast complex. The TAP method also spurred modifications or variations of the TAP tag, including: its use for purification of complexes from other eukaryotic as well as bacterial cells10,11; the design of a "split tag", wherein the Protein A and CBP are placed on different proteins12; and the tags changed, so as to accommodate the need of the investigator, such as sensitivity of the complex to Ca2+ or EGTA13.
Recent advances in both instrumentation and methodology have led to significant advances in the application of electron microscopy (EM) for structure determination, that have led to high, near atomic resolution images of macromolecular complexes14. The resolution obtainable of a complex by EM, however, remains contingent upon the quality of the complex under study. This study has utilized the TAP tag approach to purify from S. cerevisiae the U1 snRNP, an 18-subunit (~ 0.8 MDa) low copy number ribonucleoprotein complex that is part of the spliceosome15,16. A number of steps have been taken to purify this complex such that it is homogeneous and of an adequate concentration. Potential problems encountered at various stages of the purification are described and strategies taken to overcome challenges highlighted. By carefully assessing and optimizing steps in the purification, the U1 snRNP purified is of a quality and at a quantity suitable for negative stain and electron cryo microscopy (cryo-EM) studies. An optimized TAP method protocol for purification of native complexes for structural studies is described herein.
Protocol
Note: The following protocol was devised for purification of a complex from 4 L of cell culture, approximately 40 g wet weight of cells. Once prepared, all buffers should be stored at 4 °C and used within a month of their preparation. Reducing agent and protease inhibitors are only added to buffers just prior to use.
1. Preparation of Whole Cell Extract for Tandem Affinity Purification
- Growth of S. cerevisiae Cells
- Streak the desired TAP tagged S. cerevisiae strain from storage (-80 °C) onto a yeast peptone dextrose (YPD) plate. Incubate for 3 - 5 days (30 °C), until yeast cells appear white and fluffy.
- Inoculate a streak of approximately 2 mm2 of the fresh cells from the plate into 10 ml YPD media in a 50 ml conical polypropylene centrifuge tube. Shake the tube at 180 revolutions per min (rpm) overnight (30m°C).
- Inoculate 2 ml of the overnight culture per liter of YPD media in a 2 L Erlenmeyer or conical flask. Shake at 180 rpm (30 °C). Monitor cell growth at an optical density of 600 nm (OD600) until late log phase, OD600 of 1.8 to 2, typically 20 - 24 hr.
- Harvesting S. cerevisiae Cells
- Harvest cells by centrifugation at 5,400 x g for 15 min (4 °C).
- Quickly decant the supernatant and keep cells at 4 °C or on ice.
- Suspend each liter of cell pellet with 2 ml chilled Lysis Buffer (10 mM Tris-HCl, pH 8.0; 300 mM NaCl; 10 mM KCl; 0.2 mM EDTA, pH 8.0; 5 mM imidazole, pH 8.0; 10% glycerol; 0.1% v/v NP-40). Suspend cells by swirling and/or repeat pipetting using a 10 ml serological pipet. Note: The importance of NP-40 as it pertains to complex quality and post-purification analysis is discussed below.
- Transfer the cell suspension into an empty 50 ml conical polypropylene centrifuge tube kept on ice. Wash each centrifuge bottle with an additional 2 ml of chilled Lysis Buffer and add to the cell suspension in the 50 ml tube.
- Determine the wet weight of cell pellet by weighing the tube and subtracting the weight of the empty tube as well as the added Lysis Buffer. Note: A liter of cell culture having an OD600 of 1.8 is approximately 10 g.
- Create a liquid nitrogen bath in a 50 ml conical polypropylene centrifuge tube. Draw suspended cells into a 5 ml syringe. Connect a 16 G needle to the syringe. Freeze the cell suspension by passing it through the syringe/needle to generate cell "droplets." Rate of freezing should be approximately 1 min/ml.
- Store frozen cell droplets (-80 °C) or proceed to lysis.
- Lysing Cells and Lysate Clarification
- Lyse the yeast cells using a coffee grinder. Lyse cells eight to nine times with 25 sec grinding bursts, while shaking the coffee grinder. Prior to use, pre-chill the coffee grinder with liquid nitrogen. Every two rounds of grinding, add a shallow layer of liquid nitrogen to the grinder and allow it to evaporate.
- Stir with a liquid nitrogen chilled spatula as needed to keep cells from clumping. Take caution to prevent thawing of cells, which can result in cell clumping. The cells should appear as a fine white powder following lysis. A maximum cell pellet from 4 L of yeast culture (~ 40 g) can be ground at a time. Note: Observations regarding method of lysis are discussed below.
- Assess the degree of cell lysis using a hemocytometer or alternative cell counting method. To provide an adequate analysis, take the following samples: (1) before lysis; (2) after four rounds of lysis; and (3) following lysis. To check these samples, take a small amount of cells with a liquid nitrogen chilled spatula and place in a 1.5 ml tube.
- Once cells are thawed, pipette 5 µl of cells and mix with 495 µl water. If 40 - 60% lysis of cells is observed, proceed to next step in the protocol. Note: Observations regarding effect of long periods of lysis are discussed below.
- Once adequate lysis is observed, store lysed cells (-80 °C) or proceed to next step.
- Prepare two 50 ml conical polypropylene centrifuge tubes each with 15 ml Lysis Buffer, including 1 mM phenylmethanesulfonyl fluoride (PMSF), 1 mM dithiothreitol (DTT), and a commercially available cocktail of protease inhibitors. Note: If significant proteolysis is observed, consider use of a protease deficient strain.
- Use a liquid nitrogen chilled spatula to scoop the frozen lysed cell powder into the prepared 50 ml tubes. Add the cell powder/solid incrementally to the Lysis Buffer, containing both a reductant and protease inhibitor(s).
- Rotate the 50 ml tubes gently at RT so as to thaw/dissolve the cells, but also to prevent bubbles from forming. As the cell powder/solid dissolves, add additional cell solid/powder.
- Fill each of the two 50 ml tubes until each has approximately 20 g of cells or all cells added. Continue thawing/dissolving until there are no frozen cell clumps observed in the 50 ml tubes. This should take approximately 50 min.
- Centrifuge the suspended cell lysate for 20 min at 25,000 x g (4 °C). Well-lysed cells may exhibit a small amount of black precipitate.
- Transfer the 50 ml of supernatant to polycarbonate ultra-centrifuge tubes (26.3 ml tubes used) and add 10 µl 200 mM PMSF to each full tube.
- Centrifuge for 1 hr at 100,000 x g (4°C). Four layers should be visible, from bottom to top: (1) a hard clear pellet, containing ribosomal complexes; (2) a soft lipid-rich pellet; (3) a large clear yellow-tinged layer containing most of the cell's soluble proteins and complexes; and (4) a 'scaly' top layer consisting of lipids.
- Perform all subsequent steps in a cold room (4 °C). Remove as much of the top 'scaly' lipid layer as possible using a 1 ml pipet and discard. Use a 10 ml serological pipet to recover most of the yellow, clear layer.
- Recover the last few ml of this layer using a 1 ml pipette to avoid disturbing the bottom two layers. From 40 g wet cell pellet, approximately 48 ml of the middle layer is typically recovered and 8 ml of lipid layer removed/discarded. Note: Column purification flow is greatly hindered by the presence of lipids, which also may reduce the quality of the complex, discussed below.
2. Column Purification step 1: IgG Chromatography
- Resin Equilibration and Binding
- Prepare a 300 µl IgG Sepharose slurry for 40 g of cell pellet. Cut the end of a 1 ml pipet tip to facilitate pipetting the slurry. Wash/equilibrate the IgG resin three times, each time with 5 ml IgGD150 Buffer (10 mM Tris-HCl, pH 8.0; 150 mM NaCl; 150 mM KCl; 1 mM MgCl2; 5 mM imidazole, pH 8; 0.1% v/v NP-40; 1 mM DTT; a protease inhibitor tablet per 50 ml volume).
- Spin to pellet at 160 x g (4°C) between the washes and remove supernatant.
- Rotate the IgG resin with the middle phase supernatant and two mini protease inhibitor tablets per 40 g of cells, using an appropriate rotator, for 2 hr (4 °C). This is the IgG batch solution.
- Prepare two 10 ml poly-prep columns for use by cutting the end of the column to produce a flat cut. To expedite the packing and washing of the column by gravity flow, load a maximum of 200 µl of packed IgG resin or ~ 25 ml of the IgG batch solution per 10 ml column.
- Pour the IgG batch solution into the column and allow to sediment by gravity. If sedimentation takes longer than 30 min, there most likely was lipid contamination when recovering the middle phase from the centrifuge tubes.
- Wash the packed column with four successive 10 ml volumes of IgGD150 Buffer.
- On Column TEV Cleavage
- Seal the bottom of column and add 1 ml of IgGD150 Buffer and 100 µl of TEV protease (0.6 mg/ml stock, mutant variant S219V of TEV produced in-house). Seal the top of the column and mix for 20 min (18 °C) using a thermal mixer, shaking at 750 rpm. Resuspend resin and mix again for 20 min, repeat once more for a total of 1 hr incubation. Note: It is critical to keep time of incubation to a minimum.
- Return the column to 4 °C and elute the protein/complex by gravity. Elute the dead-volume with an additional 200 μl of IgGD150 Buffer.
3. Column Purification Step 2: Calmodulin Affinity Chromatography
- Resin Equilibration and Binding
- Prepare 200 µl of Calmodulin affinity resin per 40 g of cell pellet. Wash the resin three times, each time with 5 ml Calmodulin Binding Buffer (10 mM Tris-HCl, pH 8.0; 150 mM NaCl; 10 mM KCl; 1 mM MgCl2; 5 mM imidazole, pH 8; 2 mM CaCl2; 0.1% v/v NP-40; 10 mM β-mercaptoethanol), centrifuging at 160 x g (4 °C) to pellet for each wash.
- To each 1 ml IgG eluate, add 3 volumes of Calmodulin Binding Buffer. To keep the calcium level constant, add CaCl2 and β-mercaptoethanol to account for the lack of these constituents in the IgG elution. Final concentrations should be 2 mM CaCl2 and 10 mM β-mercaptoethanol.
- Add the sample to the Calmodulin resin and bind for 1 hr (4 °C) using a tube rotator.
- Packing and Eluting from Calmodulin Resin
- Prepare a 2 ml poly-prep column per 100 µl Calmodulin slurry by cutting the end of the column to create a flat opening. Load no more than 100 µl Calmodulin slurry per column to minimize the amount of resin the sample comes in contact with during elution.
- Pack the column by gravity and wash the resin in the column three times with 5 ml Calmodulin Binding Buffer.
- Elute protein with addition of 200 µl Calmodulin Elution Buffer (10 mM Tris-HCl, pH 8.0; 150 mM NaCl; 10 mM KCl; 1 mM MgCl2; 5 mM imidazole, pH 8.0; 4 mM EGTA, pH 8.0; 0.08% v/v NP-40; 10 mM β-mercaptoethanol). Repeat six times the addition of 200 µl of Calmodulin Elution Buffer.
- For elutions 1 to 3, apply the volume to the column and collect the eluate immediately. For elution 4, seal the bottom of the column and incubate with elution buffer for 2.5 min and then unseal and allow to elute by gravity. For elution 5, incubate for 5 min and then unseal and elute. For elution 6, incubate for 10 min and then unseal and elute. Note: The integrity of the complex may vary between elutions/fractions (see representative results section).
- Dialyze elutions 2 to 6 individually overnight using an appropriate micro dialysis method. Dialyze the fractions against Dialysis Buffer (10 mM Tris-HCl, pH 8.0; 150 mM NaCl; 10 mM KCl; 1 mM MgCl2; 5 mM imidazole, pH 8; 10 mM β-mercaptoethanol). Note: NP-40 does not pass appreciably, if at all, through the dialysis membrane.
4. Post-Purification Analyses to Assess Complex Quality
- Native Gel and Silver Staining
- Prepare a 4 - 16% precast native gel for analysis of the complex as per the instructions from the manufacturer. Use an appropriate molecular weight protein standard and load 15 µl of each of the dialyzed Calmodulin elution/fractions onto the gel.
- Electrophorese the gel at 150 V for approximately 3 hr (4 °C).
- Silver stain the gel to assess the quality of each of the six elutions. Alternatively, use equally sensitive commercial fluorescent stain(s). At this stage, the concentration of the complex is most likely below the level of detection for Coomassie blue staining.
- Additional Gel Analysis Methods
- To detect protein by Western blotting, resolve the complex by SDS or native PAGE and then probe the gel for the Calmodulin Binding Peptide (CBP) epitope using a TAP tag antibody as per the instructions from the manufacturer.
- Using a specific fluorescent stain(s), confirm the presence and integrity of protein and/or nucleic acid in a TAP purified complex. Take care that no spectral overlap is detected between these stains.
- Negative Stain Electron Microscopy Visualization
- Use continuous carbon grids glow-discharged at -15 mA for 30 sec, within 30 min of complex application.
- Apply 3 µl of TAP purified complex (typically at a concentration of 1 nM) to grid. Incubate for a minute. Blot from the side of the grid to remove excess buffer.
- Wash twice with 20 µl drops of deionized water. Side blot between washes.
- Apply grid to a 50 µl negative stain drop and incubate for a min. Staining with uranyl acetate (2%) or uranyl formate (0.75%) is recommended. Caution: Uranyl salts and solutions are weakly radioactive and contain heavy metal. These should be handled with care and all material that comes in contact with these solutions should be disposed of following institutional recommended waste guidelines.
- Apply grid to a fresh 50 µl negative stain drop, without blotting. Incubate for a min, then side blot, and finally air dry and visualize.
- Cryo Electron Microscopy (cryo-EM) Visualization
- Perform cryo-EM with TAP purified complex, though optimization may be necessary, according to manufacturer's protocol. Note: Presence of detergent at high concentrations may impede progress and is discussed below.
5. Complex storage
- Preparing Complex for Storage
- Concentrate the complex, if needed, using a centrifugal filter having an appropriate molecular weight cut-off for the complex. Spin at 14,000 x g for 1 min increments until desired concentration is reached. Note: NP-40 concentration will increase in concentration during concentrating, as it does not pass through the filter nor dialysis membrane.
- Flash freeze the complex in 30 µl aliquots in liquid nitrogen or dry ice cooled ethanol bath and then store at -80 °C.
Optional Post-Purification Removal of Detergent
Note: The presence of a detergent such as NP-40 and at a concentration above that of its critical micelle concentration may hinder or inhibit progress for some applications. The consequences of its removal from the protocol as well as replacement with other additives or co-solvents are discussed below. If no NP-40 is desired, an option is to use a commercial application for removal, for example Bio-beads.
Pre-equilibrate commercially available detergent absorbing beads in Dialysis Buffer. Mix five beads per 30 μl of complex (usually at 10 nM concentration).
Incubate equilibrated beads from Step 5.2.1 with TAP purified complex for 15 min on ice. Use complex within a few hours following the removal of detergent.
Representative Results
A modified TAP method was used to purify from S. cerevisiae the U1 snRNP, an 18-subunit ribonucleoprotein complex. An initial TAP purification of the complex following the published protocol2,3 yielded a complex that appeared heterogeneous, migrating as three bands on a silver stained native polyacrylamide gel (Figure 2A). Multiple rounds of optimization of the TAP method, yielded a complex that migrated as primarily a single band on a native gel indicative of a more homogeneous assembly (Figure 2A). Using fluorescent dyes to stain for both protein and nucleic acid, it was established that the purified complex had both biopolymers present (Figure 2B).
The purified complex was also analyzed by SDS PAGE and Western blotting using a TAP tag antibody (Figure 3A). Consistent with the native gel result, the complex purified according to the published protocol exhibited proteolysis of the TAP tagged protein (Snu71). The amount of proteolysis, however, was significantly reduced with modifications of the TAP method. Nearly all of the seventeen proteins in the U1 snRNP complex were resolved by SDS PAGE and positively identified by MALDI mass spectrometry (Figure 3B). As well as native and SDS PAGE, complex quality was assessed at different stages of the purification by negative stain electron microscopy (Figure 4). The monodispersed particles observed in Figure 4A and B but absent from 4C are examples of good sample for structural study. This analysis method provided critical insight as to the impact of changes to the TAP method.
The TAP method calls for the inclusion of the non-ionic detergent NP-40 and at a concentration above that of its critical micelle concentration (CMC). The robustness of the methods for assessing the quality of the complex described in the protocol are well demonstrated by an experiment where the zwitterionic surfactant CHAPS, glycerol or no co-solvent were substituted for NP-40 in all buffers (Figure 5). By native PAGE and negative stain EM, the U1 snRNP appears most homogeneous and stable in NP-40 with decreasing stability from CHAPS to glycerol to no co-solvent added. It might be that the yeast U1 snRNP has a hydrophobic face and the presence of NP-40 prevents aggregation by coating this surface. Unfortunately, NP-40 cannot be removed by dialysis and concentrating the complex will then concentrate the NP-40. While the higher NP-40 did not appear to perturb the complex, it did negatively impact the freezing of the particle in vitreous ice for cryo-EM and large micelle-like aggregates were observed. Detergent absorbing beads were used successfully to remove the majority of the NP-40 from the U1 snRNP sample without appearing to impact the complex structurally.
Following the purification of the complex with the modifications detailed in the protocol, a complex suitable for both negative stain and cryo-EM was obtained, as evidenced by images of the lawn of particles and distinctive representative class averages (Figure 6).
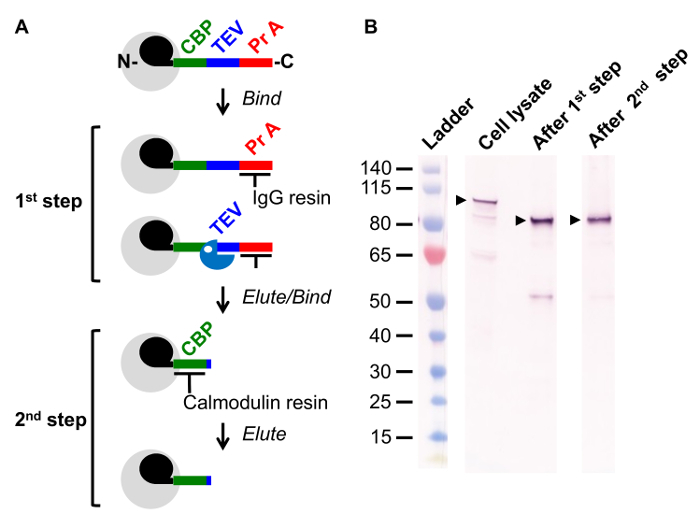 Figure 1. Overview of Tandem Affinity Purification (TAP) Method. (A) Schematic of the two step TAP method. Complex of interest (grey) includes a TAP tagged subunit (black; U1 snRNP subunit Snu71) genomically fused with a sequence comprised of a Calmodulin Binding Peptide (CBP, green), a recognition site for the site-specific protease TEV (Blue), and two IgG binding regions of Protein A (Pr A, Red). In the 1st step, cell lysate is incubated with an IgG resin and the complex is retained via its interaction with the Protein A sequence. The complex is released from the resin by TEV mediated cleavage. In the 2nd step, the complex is incubated with a Calmodulin resin, a Ca2+ dependent reaction. The complex is released via addition of the Ca2+ chelator EGTA. (B) Western blot following a TAP tagged protein through the purification steps illustrated in (A). Samples were taken of cell lysate, after the 1st step, and after the 2nd step of the TAP method. The reactive protein observed in cell lysate migrates faster after the 1st step, following TEV protease cleavage and thus loss of the Protein A sequence. Please click here to view a larger version of this figure.
Figure 1. Overview of Tandem Affinity Purification (TAP) Method. (A) Schematic of the two step TAP method. Complex of interest (grey) includes a TAP tagged subunit (black; U1 snRNP subunit Snu71) genomically fused with a sequence comprised of a Calmodulin Binding Peptide (CBP, green), a recognition site for the site-specific protease TEV (Blue), and two IgG binding regions of Protein A (Pr A, Red). In the 1st step, cell lysate is incubated with an IgG resin and the complex is retained via its interaction with the Protein A sequence. The complex is released from the resin by TEV mediated cleavage. In the 2nd step, the complex is incubated with a Calmodulin resin, a Ca2+ dependent reaction. The complex is released via addition of the Ca2+ chelator EGTA. (B) Western blot following a TAP tagged protein through the purification steps illustrated in (A). Samples were taken of cell lysate, after the 1st step, and after the 2nd step of the TAP method. The reactive protein observed in cell lysate migrates faster after the 1st step, following TEV protease cleavage and thus loss of the Protein A sequence. Please click here to view a larger version of this figure.
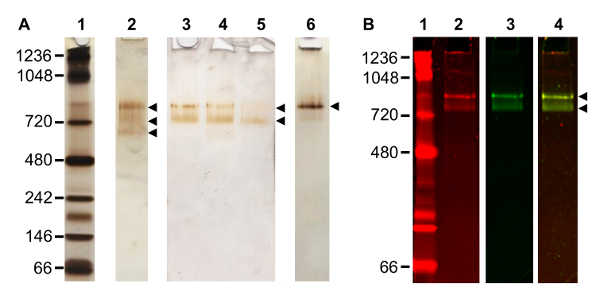 Figure 2. Optimization of the TAP Method. (A) Silver stained native gel of TAP purified complexes. Native PAGE of: protein standard (lane 1); complex purified according to original TAP method3 (lane 2), three bands observed (arrowheads); three consecutive elutions from the same TAP purification following alterations to buffer, two bands observed (arrowheads) (lanes 3 - 5); further changes to the TAP method, yielding primarily a single complex band migrating at ~ 800 kDa (lane 6). (B) Analysis of composition of a purified complex by staining a single gel lane with both protein and RNA reactive stains. Native PAGE of: protein standard (lane 1) stained with a protein fluorescent stain; complex imaged with protein fluorescent stain (lane 2), two bands observed; same lane in lane 2, now imaged with a nucleic acid fluorescent stain (lane 3), two bands observed; and fluorescent signal overlay of stained complex in lanes 2 and 3 (lane 4). Please click here to view a larger version of this figure.
Figure 2. Optimization of the TAP Method. (A) Silver stained native gel of TAP purified complexes. Native PAGE of: protein standard (lane 1); complex purified according to original TAP method3 (lane 2), three bands observed (arrowheads); three consecutive elutions from the same TAP purification following alterations to buffer, two bands observed (arrowheads) (lanes 3 - 5); further changes to the TAP method, yielding primarily a single complex band migrating at ~ 800 kDa (lane 6). (B) Analysis of composition of a purified complex by staining a single gel lane with both protein and RNA reactive stains. Native PAGE of: protein standard (lane 1) stained with a protein fluorescent stain; complex imaged with protein fluorescent stain (lane 2), two bands observed; same lane in lane 2, now imaged with a nucleic acid fluorescent stain (lane 3), two bands observed; and fluorescent signal overlay of stained complex in lanes 2 and 3 (lane 4). Please click here to view a larger version of this figure.
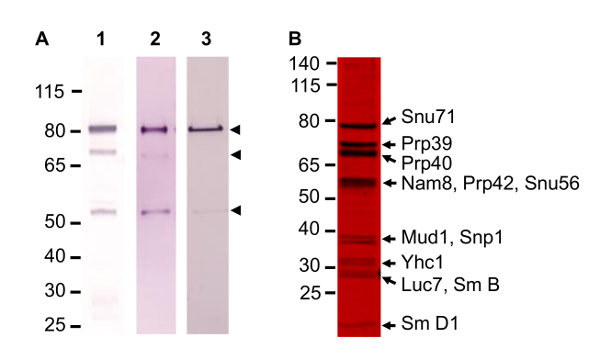 Figure 3. Optimization of TAP Method. (A) Western blot analysis of improvement in complex quality, following TAP tagged subunit Snu71 probed with α-TAP antibody. Several proteolyzed bands are visible initially (lane 1) but improvements reduce the amount of lower bands considerably (lanes 2 and 3). (B) A representative SDS gel stained with a fluorescent protein gel stain. Gel resolves 12 of the 17 protein subunits of the complex. Identity of protein in the bands was established by mass spectrometry. Please click here to view a larger version of this figure.
Figure 3. Optimization of TAP Method. (A) Western blot analysis of improvement in complex quality, following TAP tagged subunit Snu71 probed with α-TAP antibody. Several proteolyzed bands are visible initially (lane 1) but improvements reduce the amount of lower bands considerably (lanes 2 and 3). (B) A representative SDS gel stained with a fluorescent protein gel stain. Gel resolves 12 of the 17 protein subunits of the complex. Identity of protein in the bands was established by mass spectrometry. Please click here to view a larger version of this figure.
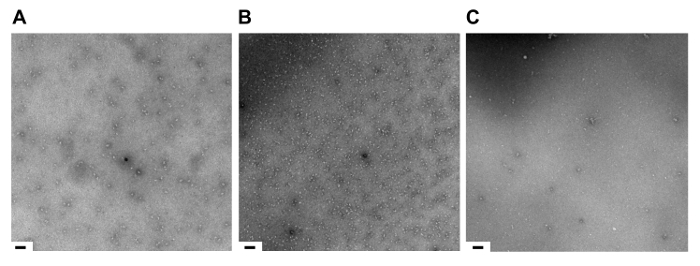 Figure 4. Sample Quality at Stages in the TAP Method Assessed by Negative Stain Electron Microscopy. Assessment by negative stain electron microscopy of complex after: (A) the 1st step of TAP; (B) the 2nd step of TAP; (C) complex destabilized. Scale bar, 125 nm. Please click here to view a larger version of this figure.
Figure 4. Sample Quality at Stages in the TAP Method Assessed by Negative Stain Electron Microscopy. Assessment by negative stain electron microscopy of complex after: (A) the 1st step of TAP; (B) the 2nd step of TAP; (C) complex destabilized. Scale bar, 125 nm. Please click here to view a larger version of this figure.
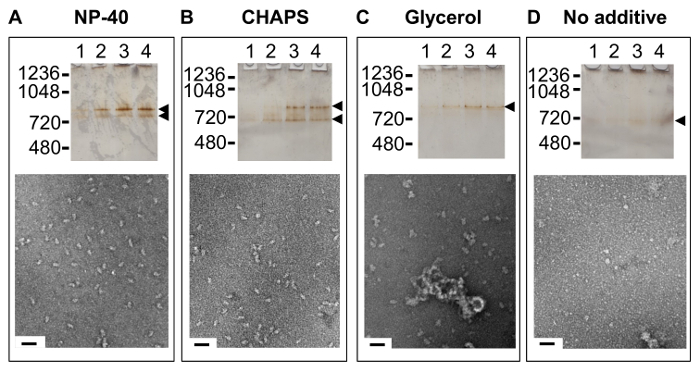 Figure 5. Impact on Sample Quality of Additives Present in TAP Method Buffers. Lanes 1 - 4 are the first four elutions of the final or 2nd step of TAP purification of complex purified in the presence of: (A) the non-ionic detergent nonyl phenoxypolyethoxylethanol (NP-40), analyzed by native PAGE (top) and negative stain EM (bottom); (B) the zwitterionic detergent 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), analyzed by native PAGE (top) and negative stain EM (bottom); (C) glycerol, analyzed by native PAGE (top) and negative stain EM (bottom); and (D) no additive, analyzed by native PAGE (top) and negative stain EM (bottom). Scale bar, 50 nm. Please click here to view a larger version of this figure.
Figure 5. Impact on Sample Quality of Additives Present in TAP Method Buffers. Lanes 1 - 4 are the first four elutions of the final or 2nd step of TAP purification of complex purified in the presence of: (A) the non-ionic detergent nonyl phenoxypolyethoxylethanol (NP-40), analyzed by native PAGE (top) and negative stain EM (bottom); (B) the zwitterionic detergent 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), analyzed by native PAGE (top) and negative stain EM (bottom); (C) glycerol, analyzed by native PAGE (top) and negative stain EM (bottom); and (D) no additive, analyzed by native PAGE (top) and negative stain EM (bottom). Scale bar, 50 nm. Please click here to view a larger version of this figure.
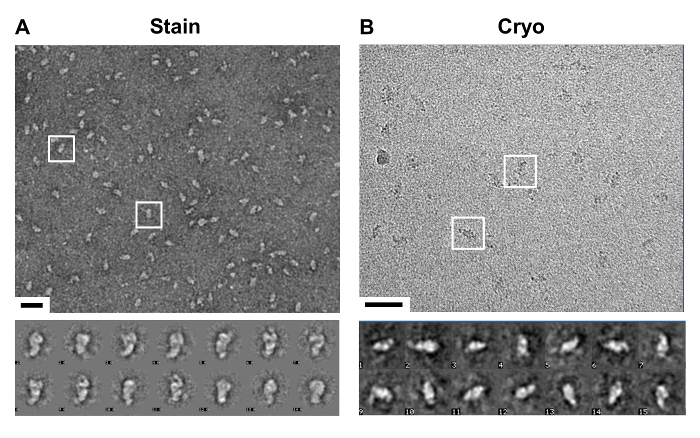 Figure 6. Visualization of TAP Method Purified Complex. (A) Top, a representative image of a negative stain electron microscopy (EM) micrograph. Observed is a lawn of similarly shaped particles, two are boxed, purified by the optimized TAP method. Bottom, a selection of class averages taken of the particles that highlight particle distinguishing features. Scale bar, 50 nm. (B) Top, a representative image of a cryo-EM micrograph. Observed are regions of higher contrast representative of particles, see two such regions boxed. Bottom, class averages of selected particles frozen in vitreous ice. Scale bar, 50 nm. Please click here to view a larger version of this figure.
Figure 6. Visualization of TAP Method Purified Complex. (A) Top, a representative image of a negative stain electron microscopy (EM) micrograph. Observed is a lawn of similarly shaped particles, two are boxed, purified by the optimized TAP method. Bottom, a selection of class averages taken of the particles that highlight particle distinguishing features. Scale bar, 50 nm. (B) Top, a representative image of a cryo-EM micrograph. Observed are regions of higher contrast representative of particles, see two such regions boxed. Bottom, class averages of selected particles frozen in vitreous ice. Scale bar, 50 nm. Please click here to view a larger version of this figure.
Discussion
The TAP method utilizes two tags that balance a need for tight and selective binding to an affinity resin with a desire to maintain near physiological solution conditions. This balance serves to preserve stable interaction(s) of the tagged protein with interacting factor(s) for post-purification characterization. In addition, individual TAP tagged ORFs are available from a commercial source, so that one can obtain any yeast strain with a tagged protein for any yeast complex. Preserving the integrity of a complex and the availability of a resource to test use of different tagged protein subunits in a complex for purification are two advantages for utilizing the TAP method for purifying a native complex. However, the original TAP method protocol was not devised to ensure that the complex purified is compositionally and structurally homogeneous. The TAP method has therefore been modified, as detailed above, to achieve this goal.
Various steps in TAP method were assessed to establish the optimal approach and conditions to purify a compositional and structural homogeneous complex. An early critical step is cell lysis. This step requires a balance between one's desire for a high yield and thus maximum lysis, while insuring that the complex obtained is of high quality, i.e., homogeneous. Different approaches to lysing yeast cells were explored, including using a bead-beater, high sheer fluid processor, and ball mill. Use of the less expensive coffee grinder was less efficient than the alternative methods (40 - 60% lysis), but the complex isolated and purified appeared monodisperse while several of these approaches appeared to either perturb particle integrity or cause a degree of aggregation. In the end, a significant number of cells were not lysed so as to ensure high quality complex was obtained. While 40 - 60% lysis is ideal for the yeast U1 snRNP, optimization is suggested when applying this protocol to a new biological complex.
It was critical to identify the steps in the method that could hasten purification to ensure that complex integrity was not compromised by cellular proteases, subunit dissociation as a result of dilution, and extended incubation at 18 °C during TEV protease cleavage. One simple step to achieve time efficient purification was to ensure that lipids did not carry over from the early lysate clarification, since lipids impede flow during gravity purification. Other steps included identifying an optimal balance of the amount resin and column size/dimensions to achieve efficient purification and a maximum flow rate. A particularly critical step was to reduce time of on column cleavage by TEV protease. The TEV protease used was purified in house to ensure it was protease/nuclease free and at a high concentration17. Significant amounts of TEV protease were used to obtain complete cleavage in under an hour.
In initial TAP purifications proteolysis was observed. Proteolysis during purification can be limited by working efficiently, adding a battery of inhibitors, and including in the purification high salt washes. A broad range of protease inhibitors supplied over the first half of the purification greatly improved the stability of the TAP tagged protein and complex. In addition, proteolysis was minimized by increasing the monovalent concentration of 300 to 500 mM during the cell lysis and IgG steps of purification. Even with the above modifications to the TAP method, successive eluted fractions off of the CBP affinity resin varied in their degree of compositional homogeneity. Thus, selective pooling of fractions from the second, CBP chromatography step, is warranted.
An aspect of TAP method that received particular focus was the importance of the non-ionic detergent NP-40 to complex purification and at a concentration above its CMC. The presence of NP-40 at a concentration of 0.08% or higher had an overall beneficial effect on complex integrity and the final yield. The apparent beneficial effect(s) of NP-40 may be due in part to reducing non-specific interactions between resin and the complex. While NP-40 appears to have a beneficial effect during the purification, it could be removed post-purification without causing complex aggregation.
Finally, critical to optimizing the TAP method for structural study were several complementary assays used to assess the integrity and homogeneity of the complex. These included light scattering, SDS and native PAGE probed using the TAP antibody by Western blot and/or stained with fluorescent dyes, as well as negative stain electron microscopy. The changes made to the TAP method detailed herein, yielded a homogeneous complex at sufficient quantities for electron microscopy. Additional changes may need to be made to the TAP method for other complexes, in particular a determination of the importance of NP-40.
In summary, the TAP purification method can be used to successfully purify macromolecular complexes from a eukaryotic source of suitable quality for structural study. We have established a suitable approach for purification of the S. cerevisiae U1 snRNP and detail the steps taken as well as the rationale for many of these changes. For other complexes, we suggest that the investigator similarly explore the steps in the protocol, which may yield adequate amounts and quality of complex. The steps that should be examined include cell lysis, salt and additive type and concentrations including detergent, and sensitivity of the complex to calcium and metal chelator EGTA. Further, it is critical for long-term studies that the complex can be frozen and thawed without perturbing structure. Importantly, one should have several alternative ways to assay the structure of the complex during and following purification. Native and SDS PAGE, negative stain EM, and light scattering have served us well in our investigation of how best to purify a homogeneous complex using the TAP method.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors are grateful for the support and advice of Nikolaus Grigorieff. We thank Anna Loveland, Axel Brilot, Chen Xu, and Mike Rigney for helpful discussions and EM guidance. This work was funded by the National Science Foundation, Award No. 1157892. The Brandeis EM facility is supported by National Institutes of Health grant P01 GM62580.
References
- Gavin AC, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415(6868):141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nature Biotechnology. 1999;17(10):1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- Puig O, et al. The tandem affinity purification (TAP) method: A general procedure of protein complex purification. Methods. 2001;24(3):218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- Vaillancourt P, Zheng CF, Hoang DQ, Breister L. Affinity purification of recombinant proteins fused to calmodulin or to calmodulin-binding peptides. Methods Enzymol. 2000;326:340–362. doi: 10.1016/s0076-6879(00)26064-3. [DOI] [PubMed] [Google Scholar]
- Braisted AC, Wells JA. Minimizing a binding domain from protein A. Proc Natl Acad Sci U S A. 1996;93(12):5688–5692. doi: 10.1073/pnas.93.12.5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapust RB, et al. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14(12):993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- Nallamsetty S, et al. Efficient site-specific processing of fusion proteins by tobacco vein mottling virus protease in vivo and in vitro. Protein Expr Purif. 2004;38(1):108–115. doi: 10.1016/j.pep.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, et al. Global analysis of protein expression in yeast. Nature. 2003;425(6959):737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Howson R, et al. Construction, verification and experimental use of two epitope-tagged collections of budding yeast strains. Comp Funct Genomics. 2005;6(1-2):2–16. doi: 10.1002/cfg.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DM, Du M, Guo X, Siu KW, McDermott JC. Tandem affinity purification of protein complexes from mammalian cells. Biotechniques. 2002;33(2):267–268. doi: 10.2144/02332bm02. [DOI] [PubMed] [Google Scholar]
- Gully D, Moinier D, Loiseau L, Bouveret E. New partners of acyl carrier protein detected in Escherichia coli by tandem affinity purification. FEBS Lett. 2003;548(1-3):90–96. doi: 10.1016/s0014-5793(03)00746-4. [DOI] [PubMed] [Google Scholar]
- Tharun S. Purification and analysis of the decapping activator Lsm1p-7p-Pat1p complex from yeast. Methods Enzymol. 2008;448:41–55. doi: 10.1016/S0076-6879(08)02603-7. [DOI] [PubMed] [Google Scholar]
- Xu X, Song Y, Li Y, Chang J, Zhang H, An L. The tandem affinity purification method: an efficient system for protein complex purification and protein interaction identification. Protein Expr Purif. 2010;72(2):149–156. doi: 10.1016/j.pep.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Grigorieff N, Penczek PA, Walz T. A Primer to Single-Particle Cryo-Electron Microscopy. Cell. 2015;161(3):438–449. doi: 10.1016/j.cell.2015.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk A, et al. A comprehensive biochemical and genetic analysis of the yeast U1 snRNP reveals five novel proteins. RNA. 1998;4(4):374–393. [PMC free article] [PubMed] [Google Scholar]
- Neubauer G, Gottschalk A, Fabrizio P, Seraphin B, Luhrmann R, Mann M. Identification of the proteins of the yeast U1 small nuclear ribonucleoprotein complex by mass spectrometry. Proc Natl Acad Sci U S A. 1997;94(2):385–390. doi: 10.1073/pnas.94.2.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucast LJ, Batey RT, Doudna JA. Large-scale purification of a stable form of recombinant tobacco etch virus protease. Biotechniques. 2001;30(3):544–546. doi: 10.2144/01303st06. [DOI] [PubMed] [Google Scholar]