

Abstract
Myotonic dystrophy 1 (DM1) is a common form of muscular dystrophy. Although several animal models have been established for DM1, myoblast cell models are still important because they offer an efficient cellular alternative for studying cellular and molecular events. Though C2C12 myoblast cells have been widely used to study myogenesis, resistance to gene transfection, or viral transduction, hinders research in C2C12 cells. Here, we describe an optimized protocol that includes daily maintenance, transfection and transduction procedures to introduce genes into C2C12 myoblasts and the induction of myocyte differentiation. Collectively, these procedures enable best transfection/transduction efficiencies, as well as consistent differentiation outcomes. The protocol described in establishing DM1 myoblast cell models would benefit the study of myotonic dystrophy, as well as other muscular diseases.
Keywords: Medicine, Issue 113, Myotonic dystrophy, C2C12 myoblast, Celf1, plasmid transfection, retroviral transduction, lentiviral transduction, immunostaining
Introduction
Myotonic dystrophy (DM) is an autosomal dominant disease that affects multiple systems, most notably cardiac and skeletal muscles1. There are two subtypes of this disease, DM1 and DM2. DM1 is more common and has a more severe manifestation than DM22. The genetic mutation underlying DM1 is an expansion of CUG triplet repeats located in the 3' untranslated region (UTR) of DM protein kinase gene (DMPK)3. The CUG repeat number in unaffected individuals varies from 5 to 37. In contrast, it increases to more than 50, and sometimes up to thousands in DM1 patients4. As a result, RNA-binding proteins, such as muscleblind-like 1 (MBNL1), CUGBP, and Elav-like family 1 (Celf1), are misregulated. Due to the sequestration on the expanded CUG repeats, MBNL1 loses its ability to regulate alternative splicing5. Celf1, on the other hand, is up-regulated6,7. Overexpression of Celf1 is associated with muscle loss and weakness, which are not attributed to loss of MBNL1 function. Animal models simulating DM1-related alterations, including DMPK 3'-UTR CUG expansion, loss of MBNL1, and overexpression of Celf1, have been established. However, modeling DM1 in myoblasts offers an efficient alternative, especially for dissecting DM1-related cellular and molecular events.
C2C12 myoblast cell line was first isolated from injured C3H mouse muscle and widely used to study myogenic differentiation8,9. C2C12 cells rapidly proliferate in fetal bovine serum (FBS)-containing media and readily undergo differentiation when FBS is depleted. Yet, using this myoblast differentiation model presents two challenges: C2C12 cells are often resistant to gene transfection/viral transduction; and slight variations in cell handling and differentiation procedure can lead to marked changes in myotube formation.
Our lab routinely uses C2C12 myoblasts as a cell model and has developed protocols that effectively deliver genes by plasmid transfection, retroviral transduction, and lentiviral transduction into C2C12 cell line10. In the video, we demonstrate the optimized procedures for transfecting/transducing C2C12 cells and maintaining differentiation consistency in establishing DM1 myoblast models.
Protocol
1. C2C12 Cell Culture
Maintain C2C12 mouse myoblasts in a 100 mm plate in growth medium (Dulbecco's modified Eagle's medium (DMEM)) supplemented with 20% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin, and 2 mM L-glutamine. Allow C2C12 passaged cells to become approximately 50 - 60% confluent.
Discard the growth medium and wash C2C12 cells with 3 ml room temperature phosphate-buffered saline (PBS). Remove the PBS and add 500 µl 0.25% Trypsin-EDTA to detach the cells. Place the plate in a 37 °C, 5% CO2 incubator for 3 - 5 min.
Neutralize the trypsin by adding 3 ml growth medium. Pipette 6 - 8 times to suspend the cells. Add 1 ml of suspended cell to a new 100 mm plate and incubate at 37 °C, 5% CO2.
2. C2C12 Cell Transfection/transduction and Selection
- C2C12 plasmid transfection
- 24 hr prior to transfection, plate 7.0 x 105 C2C12 cells into one well of a six-well plate for each transfection. Note: This leads to 50 - 60% confluence by the time of transfection.
- Allow the transfection reagent to come to room temperature prior to use.
- Add 2 µg of plasmid DNA (GFP-CUG5 or GFP-CUG200) into 200 µl pre-warmed serum free medium and vortex briefly. Add 6 µl transfection reagent to the medium and mix immediately for 30 sec using a vortex. Incubate the mixture for 30 min at room temperature.
- Change the media of C2C12 cells to 2.5 ml serum-free growth media. Add the transfection mixture to the cells after incubation. Return the plates to a 5% CO2, 37 °C incubator.
- Keep the cells in transfection mixture/serum-free growth media for 4 hr or up to overnight. Change media back to the growth media the next day or when cytotoxicity is apparent. Note: Cytotoxicity is assessed by microscopic inspection of the cells. Observe the changes in cell morphology and cell detachment. As long as there is no overt cytotoxicity, overnight incubation in serum-free growth media enhances transfection.
- Select the cells 48 hr after transfection by adding 1.2 - 1.6 mg/ml G418 for ~ 10 days until the control culture (transfected without the plasmids) does not have live cells. Change the media every two days with growth media supplemented with G418.
- Maintain the G418 resistant cells in growth medium and 5% CO2 at 37 °C for subsequent experiments.
- Retrovirus preparation and C2C12 retroviral transduction
- Prepare HEK 293 culture medium by supplementing DMEM high glucose with 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin, and 2 mM L-glutamine.
- Approximately 24 hr prior to transfection, plate 4.0 x 106 ecotropic HEK 293-based packaging cells in a 100 mm plate (80% confluence at transfection) containing cell culture medium.
- Allow the transfection reagent to come to room temperature prior to use.
- Mix 15 µg plasmid DNA (pMSCV-Puro or pMSCV-Celf1Flag-Puro) in 1 ml serum-free growth medium. Vortex briefly. Add 30 µl transfection reagent from step 2.2.2 into the DNA/serum-free growth medium tube, and mix well using vortex. Incubate the mixture at room temperature for 30 min.
- Add transfection mixture dropwise to the ecotropic HEK 293-based packaging cells. Gently swirl the plates and return them back to the 5% CO2, 37 °C incubator. After 24 hr, change the media to 10 ml pre-warmed fresh growth medium.
- Harvest the supernatant (containing retrovirus) 48 hr after transfection using a pipette and transfer the supernatant into a 50 ml centrifuge tube. Add 10 ml of new warm media to the plate and return it to the incubator. Keep the supernatant at 4 °C. Note: The media is added gently to the side of the well so as not to disrupt the cell monolayer.
- Harvest the second batch of supernatant 60 hr after transfection and pool it with the supernatant from 2.2.5. Centrifuge at 500 x g, 4 °C for 7 min to remove cellular debris.
- Transfer the supernatant into a new 50 ml centrifuge tube. Use retrovirus to transduce C2C12 cells at this point by proceeding to step 2.2.9 or aliquot and store the supernatant at -20 °C for future use.
- Thaw the retrovirus from 2.2.7 at room temperature, if a frozen stock is used here. Note: Avoid multiple freeze-thaw cycles.
- Plate C2C12 cells on the same day of transduction aiming for 30 - 40% of confluence.
- Discard the growth medium and wash C2C12 cells with 3 ml room temperature PBS. Remove the PBS, add 500 µl 0.25% Trypsin-EDTA, and incubate the plate in a 37 °C, 5% CO2 incubator for 3 - 5 min.
- Neutralize the trypsin by adding 3 ml growth medium. Pipette 6 - 8 times to suspend the cells. Count cells using a hemocytometer and add 8.0 x 105 C2C12 cells to a 60 mm plate.
- Add 0.5 ml retrovirus to infect one 60 mm plate of cells in 3 ml growth media. Return the plate to the incubator.
- Replace with 3 ml fresh media supplemented with selection antibiotic (puromycin 1 - 3 µg/ml) 48 hr after infection. Replace 3 ml medium with antibiotic every day for ~ 5 days until the untransduced C2C12 control culture dies out.
- Lentivirus preparation and C2C12 lentiviral transduction Note: Perform all the lentivirus-related procedures in Biosafety Level 2 cabinet and follow biological safety guidelines. Liquid waste containing the virus must be disinfected prior to disposal.
- Plate 4.0 x 106 293T cells in a 100 mm plate (~ 80% confluence at day of transfection) with HEK 293 cell culture medium (DMEM high glucose, supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin, and 2 mM L-glutamine) 24 hr before transfection.
- Warm the transfection reagent to room temperature. Mix the lentiviral packaging vectors (4 µg pMD2.G and 6 µg psPAX2) together with 8 µg lentiviral transfer vector (Celf1 shRNA or scrambled control shRNA) in 1 ml serum free cell culture medium. Vortex briefly.
- Add 36 µl pre-warmed transfection reagent to the DNA/DMEM tube and mix well by vortex. Incubate the mixture at room temperature for 30 min.
- Remove the 293T cells from the incubator and add the transfection mixture to the plate. Gently swirl the plate and return it back to 5% CO2, 37 °C incubator. After 24 hr, replace the transfection mixture with 10 ml fresh culture medium.
- Collect the supernatant containing lentivirus 48 hr after transfection and transfer it into a 50 ml centrifuge tube. Add 10 ml new warm media to the plate and return it to incubator. Store the supernatant at 4 °C.
- Collect the second batch of supernatant 60 hr after transfection and combine it with the supernatant from 2.3.5. Centrifuge briefly at 500 x g, 4 °C for 7 min.
- Transfer the supernatant into a fresh 50 ml centrifuge tube. If using fresh virus, proceed to step 2.3.9. For future use, store virus-containing supernatant at -20 °C in aliquots of 10 ml.
- Thaw the virus from 2.3.7 at room temperature before use, if a frozen stock is used. Note: Avoid multiple freeze-thaw cycles.
- Plate C2C12-CUG200 cells (obtained in Section 2.1) as described in 2.2.9 on the same day of transduction. Note: Aim for 30 - 40% of confluence.
- Add 0.5 ml virus to infect a 60 mm plate of C2C12-CUG200 and return the plate to the incubator.
- Replace the culture medium with fresh media supplemented with the selection antibiotic (puromycin 1 - 3 µg/ml) 72 hr after transduction. Refresh culture media everyday with the selection antibiotic for ~5 days until all untransduced C2C12-CUG200 control culture dies out.
- Use Western blot to verify Celf1 knockdown in GFP-CUG200 transfected cells.
3. C2C12 Cell Differentiation
- Plate 2 x 106 cells in one well of a 6-well plate in 2.5 ml growth media 24 hr before initiation of differentiation. Note: The plating density may be adjusted so that full confluence is reached in 24 hr.
- Rinse confluent cells with PBS and add 2.5 ml of low-serum differentiation medium (Dulbecco's modified Eagle's medium supplemented with 2% equine serum, 100 U/ml penicillin, 100 µg/ml streptomycin, 2 mM L-glutamine, and 1 µM insulin). Replace the medium every two days. Note: Keep stock insulin frozen and add it to the media at each medium change.
During C2C12 differentiation, collect samples for quantitative RT-PCR (see section 4) at the following time points: Day0, 1, 2, 4, and 6. Process the culture for immunostaining at day 6 - 8 (see section 5).
4. RNA Isolation and Quantitative RT-PCR
Use the manufacturer's protocol for RNA isolation. After isolation, resuspend the RNA pellet in 30 µl RNase-free water and pipet up and down several times. Note: The samples may proceed to Quantitative RT-PCR or can be stored at -80 °C.
Use a RT qPCR kit for Quantitative RT-PCR10 and follow the manufacturer's instruction. Prepare the reaction mix according to Table 1.
| Component | Volume (µl) | Final Concentration |
| 2x reaction buffer | 10 | 1x |
| Forward primer | 0.5 | 200 nM |
| Reverse primer | 0.5 | 200 nM |
| Probe | 0.5 | 200 nM |
| Reverse Transcriptase & RNase Inhibitor | 0.1 | 0.25 U/ml |
| Template | 1 | 100 ng total RNA |
| RNase free water | 7.4 | NA |
| Total Mix | 20 |
Table 1. Quantitative RT-PCR Master Mix Preparation
Run a 20 µl RT-qPCR reaction using thermal profile Table 2.
| Reverse transcription step | 30 min, 48 °C |
| DNA polymerase activation/Reverse Transcriptase inactivation | 10 min, 95 °C |
| 40 Cycles | 15 sec, 95 °C |
| 1 min, 60 °C |
Table 2. Quantitative RT-PCR thermal profile
5. Immunostaining
Fix the monolayer culture in 2.5 ml freshly prepared 4% paraformaldehyde/PBS at room temperature for 10 min.
Permeabilize the samples in 2.5 ml 0.1% Triton X-100/PBS for 30 min at room temperature.
Transfer the samples to a humidified chamber. Block the samples in 2.5 ml blocking buffer (0.1% nonionic surfactant/PBS, 10% normal goat serum) for 30 min at 37 °C.
Discard blocking buffer and apply 800 µl primary antibody against Myosin Heavy Chain diluted in blocking buffer (1:100). Incubate in a humidified chamber overnight at room temperature.
Wash the plate 3 times (10 min each) with 2.5 ml wash buffer (0.1% polysorbate 20/PBS) on day 2.
Remove the wash buffer and apply 800 µl secondary antibody (goat anti-mouse IgG, 1:500 diluted in PBS). Incubate in a humidified chamber at room temperature for 1 hr.
Wash two times (10 min each) with 2.5 ml wash buffer.
Incubate with 800 µl DAPI (1 µg/ml, diluted in distilled deionized H2O) for 5 min.
Wash with 2.5 ml wash buffer again for 10 min.
Change the washing buffer to 2.5 ml PBS and use a fluorescent microscope to capture images.
Representative Results
C2C12 cells were transfected with GFP-CUG5 or GFP-CUG200. After drug-resistance selection, stable pools were established, which can be visualized by GFP expression (Figure 1A). Myotube formation in the differentiated myoblasts was detected by myosin heavy chain immunostaining10 (Figure 1B). The quantification of myotube formation demonstrated that fusion indices were decreased from 35.4 ± 4.1% to 2.6 ± 1.1% and myotube areas were decreased from 35.6 ± 2.2% to 2.7 ± 0.8% by abnormal CUG expansion in GFP-CUG200 (Figure 1C). The fusion index and the myotube area were counted via the total number of nuclei in myotubes (≥ 2 nuclei) divided by the total number of nuclei and the percentage of the total image area covered by myotubes, respectively. Real-time RT-PCR10 was conducted to analyze expression for a number of muscle differentiation related genes including MyoD, MyoG, Mef2c and Celf1. Compared to CUG5 cultures, CUG200 increased the expression of Celf1 mRNA in proliferating myoblasts at the beginning of differentiation. Congruent with previous reports, Celf1 upregulation was associated with CUG-expansion in myogenic dystrophy 1 (Figure 1D). Further, Western blotting10 consistently showed Celf1 protein level was elevated (Figure 1E), and CUG200 inhibited the expression of MyoD, MyoG and Mef2c during differentiation (Figure 1D). These results suggest that the CUG-expansion leads to myotube defects and impaired myoblast differentiation.
To study the role of Celf1 in myoblast differentiation, pMSCV- Celf1Flag retroviral vector was constructed to transduce C2C12 cells.10 Puromycin-resistant clones were pooled for differentiation studies. Western blot results showed that the Flag-tagged Celf1 was only present in the pMSCV-Celf1Flag transduced cultures and the expression of Celf1 protein was upregulated (Figure 2A). The formation of myotubes in Celf1-overexpressing cells is rare when compared to the control cultures (Figure 2B), which suggests that the overexpression of Celf1 severely impairs myoblast differentiation.
To determine if Celf1 knockdown rescues the differentiation deficiency in CUG-expansion cells, Celf1 shRNA was delivered to GFP-CUG200 cells by lentiviral vectors. Double resistant clones (G418 and puromycin) were selected and pooled for differentiation studies. The level of endogenous Celf1 protein was markedly reduced in the presence of Celf1 shRNA (Figure 3A). After 6 days of differentiation, the effect of Celf1 shRNA on increasing myotube formation was evident (Figure 3B). Fusion indices and myotube areas were 16.1 ± 3.0% and 15.2 ± 1.2%, respectively, compared to 4.6 ± 0.8% and 5.1 ± 1.3% in control cultures (Figure 3C). Meanwhile, real-time RT-PCR results suggest that the expression of MyoD, MyoG and Mef2c was significantly increased in Celf1 shRNA cells (Figure 3D) supporting Celf1 knockdown rescued myocyte differentiation deficiency.
 Figure 1. CUG-Expansion Inhibits Myocyte Differentiation in C2C12 Cells. (A) C2C12 cells expressed GFP-CUG5 or GFP-CUG200 ubiquitously after selection. Scale bars are 100 µm. This figure has been modified from our previous paper10. (B) Myotubes were formed in GFP-CUG5 cells but less in GFP-CUG200 cells. Myotubes were visualized by myosin heavy chain (MF-20) immunostaining. Scale bars are 100 µm. (C) GFP-CUG200 cultures significantly reduced fusion index and myotube areas. The error bars represent standard deviations. (D) Real-time RT-PCR analysis of Celf1, transcription factors MyoD, MyoG, and Mef2c expression during myoblast differentiation. mRNA levels were plotted after the GAPDH normalized. The error bars represent standard deviations. Student's T test was performed to compare the tests to controls. n > 3, *P < 0.05. (E) GFP-CUG200 transfection increased Celf1 protein expression. ß-actin is shown as the loading control. Please click here to view a larger version of this figure.
Figure 1. CUG-Expansion Inhibits Myocyte Differentiation in C2C12 Cells. (A) C2C12 cells expressed GFP-CUG5 or GFP-CUG200 ubiquitously after selection. Scale bars are 100 µm. This figure has been modified from our previous paper10. (B) Myotubes were formed in GFP-CUG5 cells but less in GFP-CUG200 cells. Myotubes were visualized by myosin heavy chain (MF-20) immunostaining. Scale bars are 100 µm. (C) GFP-CUG200 cultures significantly reduced fusion index and myotube areas. The error bars represent standard deviations. (D) Real-time RT-PCR analysis of Celf1, transcription factors MyoD, MyoG, and Mef2c expression during myoblast differentiation. mRNA levels were plotted after the GAPDH normalized. The error bars represent standard deviations. Student's T test was performed to compare the tests to controls. n > 3, *P < 0.05. (E) GFP-CUG200 transfection increased Celf1 protein expression. ß-actin is shown as the loading control. Please click here to view a larger version of this figure.
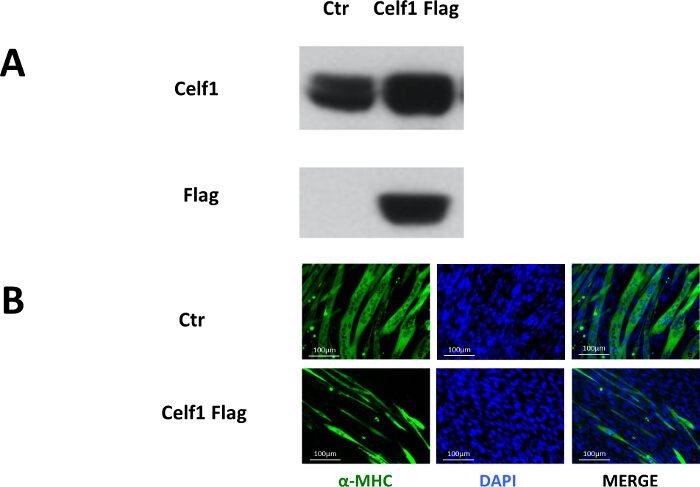 Figure 2. Overexpression of Celf1 Impairs Myoblast Differentiation. (A) Proof of exogenous Celf1Flag expression in C2C12 cells by Western blot. (B) Myotube formation was impaired in Celf1Flag-overexpressing C2C12 myoblasts. Myotubes were visualized by MF-20 immunostaining. Scale bars are 100 µm. Please click here to view a larger version of this figure.
Figure 2. Overexpression of Celf1 Impairs Myoblast Differentiation. (A) Proof of exogenous Celf1Flag expression in C2C12 cells by Western blot. (B) Myotube formation was impaired in Celf1Flag-overexpressing C2C12 myoblasts. Myotubes were visualized by MF-20 immunostaining. Scale bars are 100 µm. Please click here to view a larger version of this figure.
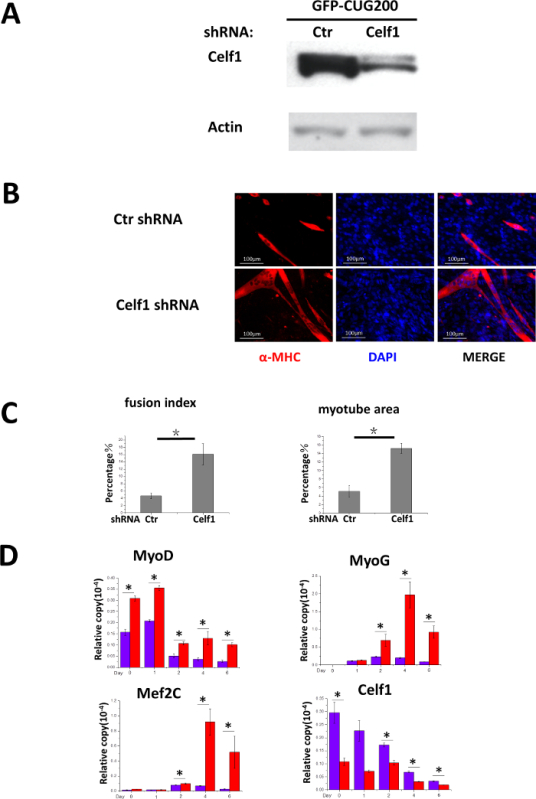 Figure 3.Knockdown of Celf1 Partially Rescues CUG-Expansion-Induced Myoblast Differentiation Deficiency. (A) Proof of Celf1 knockdown by Western blot. (B) Celf1 shRNA induced myotube formation in GFP-CUG200 myoblasts. Myotubes were visualized by MF-20 immunostaining. Scale bars are 100 µm. (C) Celf1 shRNA-rescued cells that had increased fusion index and myotube areas. The error bars represent standard deviations. (D) Real-time RT-PCR analysis of Celf1, transcription factors MyoD, MyoG, and Mef2c expression during differentiation. mRNA level was plotted after normalizing to that of GAPDH. The error bars represent standard deviations. Student's T test was performed to compare the tests to control. n > 3, *P < 0.05. Please click here to view a larger version of this figure.
Figure 3.Knockdown of Celf1 Partially Rescues CUG-Expansion-Induced Myoblast Differentiation Deficiency. (A) Proof of Celf1 knockdown by Western blot. (B) Celf1 shRNA induced myotube formation in GFP-CUG200 myoblasts. Myotubes were visualized by MF-20 immunostaining. Scale bars are 100 µm. (C) Celf1 shRNA-rescued cells that had increased fusion index and myotube areas. The error bars represent standard deviations. (D) Real-time RT-PCR analysis of Celf1, transcription factors MyoD, MyoG, and Mef2c expression during differentiation. mRNA level was plotted after normalizing to that of GAPDH. The error bars represent standard deviations. Student's T test was performed to compare the tests to control. n > 3, *P < 0.05. Please click here to view a larger version of this figure.
Discussion
C2C12 cell line has been used as a model to study myogenesis11-14. These cells retain a fibroblast-like look, proliferate rapidly in media containing 20% fetal bovine serum and readily differentiate in media containing 2% equine serum15. The fast growth and differentiation are advantageous characteristics in a myogenesis cell model. Here, we demonstrate the use of plasmid, retroviral, and lentiviral vectors to introduce cDNA, 3'-UTR, and shRNA into C2C12 cells. The critical points for transfection/transduction and maintenance of consistency in differentiation are highlighted below.
The cell density in daily cell maintenance is critical. These cells need to be cultured below 50 - 70% confluence. Due to the short population doubling time, high confluence (> 70%) C2C12 culture spontaneously differentiates when left overnight. While the fraction of spontaneously differentiated cells may be small, the presence of differentiated cells will contribute to inconsistencies in subsequent differentiation experiments. Additionally, freshly thawed C2C12 cells that have low passage number are preferred. For differentiation, cells are plated at pre-determined numbers so that they reach full confluence after 24 hr. Insulin is kept frozen in small aliquots and is added each time the differentiation medium is changed. These measures improve differentiation outcomes.
Serum free medium enhances C2C12 cell transfection. The cells can be incubated in transfection mixture/serum free media overnight. In our lab, we achieve up to 20% transfection rate, accompanied by slight signs of cytotoxicity. The concentration of drug used to select C2C12 cell is higher, 1.2 to 1.6 mg/ml G418 or 1 to 3 µg/ml puromycin, and the duration of the selection is longer than most other cell lines. Due to potential toxicity under high drug concentration and the extended incubation period, lot-to-lot variation in differentiation potential may arise. It is crucial to adjust the selection scheme (concentration and duration) based on cell appearance and to treat the experiment and control groups together.
We have successfully introduced GFP-CUG200 into C2C12 cells that reproduce 3'-UTR CUG expansion in DM1. Introducing Celf1 into C2C12 cells and Celf1 shRNA into GFP-CUG200 C2C12 cells has allowed the evaluation of cellular and molecular events triggered by Celf1 upregulation and exploration of targeting Celf1 to help DM1 myoblast differentiation, respectively. In summary, the C2C12 myoblast model presented here benefits the investigation of myotonic dystrophy as well as general muscle cell biology and myogenic differentiation. The limitation of this model is that the findings in cell lines may not completely extend to organisms. The findings in the current models are often advanced to isolating fresh myoblasts from animals and studying their differentiation. Ultimately we substantiate myoblast findings in organisms.
The unique strength of this study is the multi-tier gene manipulation in C2C12 myoblasts, which enables modeling the sequential genetic/pathological events in DM1. Previously, the RNA toxicity effect of DMPK CUG expansion was documented in myoblasts11-14, while the pathological roles of Celf1 were separately studied in mouse models16,17. Modeling these sequential genetic/pathological events is technically challenging and time consuming if performed in animal models. As demonstrated in this study, the combined use of fluorescent and drug resistant markers allows the modeling of additional molecular events in myotonic dystrophy. The models established in this study would be useful in dissecting the detailed mechanisms in DM1 pathogenesis. They would also be valuable in screening for drugs that target different steps of DM1 pathogenesis. The strategies and procedures of this study may shed light on establishing other muscular disease models in myoblasts.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Drs. Tom Cooper from the Baylor College of Medicine, Mani S. Mahadevan from the University of Wisconsin-Madison, and Didier Trono from the University of Geneva for reagents. This work is supported by a University of Houston startup fund (YL), American Heart Association grant (YL, 11SDG5260033), and the National Natural Science Foundation of China (XP, 81460047).
References
- Harper PS. Myotonic dystrophy. 3rd edn. W. B. Saunders; 2001. [Google Scholar]
- Timchenko L. Molecular mechanisms of muscle atrophy in myotonic dystrophies. Int J Biochem Cell Biol. 2013;45(10):2280–2287. doi: 10.1016/j.biocel.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook JD, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell. 1992;68(4):799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- Chau A, Kalsotra A. Developmental insights into the pathology of and therapeutic strategies for DM1: Back to the basics. Dev Dyn. 2015;244(3):377–390. doi: 10.1002/dvdy.24240. [DOI] [PubMed] [Google Scholar]
- Ho TH, et al. Muscleblind proteins regulate alternative splicing. EMBO J. 2004;23(15):3103–3112. doi: 10.1038/sj.emboj.7600300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol Cell. 2007;28(1):68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsotra A, et al. The Mef2 transcription network is disrupted in myotonic dystrophy heart tissue, dramatically altering miRNA and mRNA expression. Cell Rep. 2014;6(2):336–345. doi: 10.1016/j.celrep.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature. 1977;270(5639):725–727. doi: 10.1038/270725a0. [DOI] [PubMed] [Google Scholar]
- Blau HM, Chiu CP, Webster C. Cytoplasmic activation of human nuclear genes in stable heterocaryons. Cell. 1983;32(4):1171–1180. doi: 10.1016/0092-8674(83)90300-8. [DOI] [PubMed] [Google Scholar]
- Peng X, et al. Celf1 regulates cell cycle and is partially responsible for defective myoblast differentiation in myotonic dystrophy RNA toxicity. Biochim Biophys Acta. 2015;1852(7):1490–1497. doi: 10.1016/j.bbadis.2015.04.010. [DOI] [PubMed] [Google Scholar]
- Amack JD, Mahadevan MS. The myotonic dystrophy expanded CUG repeat tract is necessary but not sufficient to disrupt C2C12 myoblast differentiation. Hum Mol Genet. 2001;10(18):1879–1887. doi: 10.1093/hmg/10.18.1879. [DOI] [PubMed] [Google Scholar]
- Amack JD, Paguio AP, Mahadevan MS. Cis and trans effects of the myotonic dystrophy (DM) mutation in a cell culture model. Hum Mol Genet. 1999;8(11):1975–1984. doi: 10.1093/hmg/8.11.1975. [DOI] [PubMed] [Google Scholar]
- Bhagavati S, Shafiq SA, Xu W. (CTG)n repeats markedly inhibit differentiation of the C2C12 myoblast cell line: implications for congenital myotonic dystrophy. Biochim Biophys Acta. 1999;1453(2):221–229. doi: 10.1016/s0925-4439(98)00104-5. [DOI] [PubMed] [Google Scholar]
- Amack JD, Mahadevan MS. Myogenic defects in myotonic dystrophy. Dev Biol. 2004;265(2):294–301. doi: 10.1016/j.ydbio.2003.07.021. [DOI] [PubMed] [Google Scholar]
- Emerson CP, Sweeney HL. Methods in muscle biology. Vol. 52. Academic Press; 1997. [Google Scholar]
- Ward AJ, Rimer M, Killian JM, Dowling JJ, Cooper TA. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum Mol Genet. 2010;19(18):3614–3622. doi: 10.1093/hmg/ddq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshelev M, Sarma S, Price RE, Wehrens XHT, Cooper TA. Heart-specific overexpression of CUGBP1 reproduces functional and molecular abnormalities of myotonic dystrophy type 1. Hum Mol Genet. 2010;19(6):1066–1075. doi: 10.1093/hmg/ddp570. [DOI] [PMC free article] [PubMed] [Google Scholar]