

Abstract
Imaging of Förster resonance energy transfer (FRET) is a powerful tool for examining cell biology in real-time. Studies utilizing FRET commonly employ two-dimensional (2D) culture, which does not mimic the three-dimensional (3D) cellular microenvironment. A method to perform quenched emission FRET imaging using conventional widefield epifluorescence microscopy of cells within a 3D hydrogel environment is presented. Here an analysis method for ratiometric FRET probes that yields linear ratios over the probe activation range is described. Measurement of intracellular cyclic adenosine monophosphate (cAMP) levels is demonstrated in chondrocytes under forskolin stimulation using a probe for EPAC1 activation (ICUE1) and the ability to detect differences in cAMP signaling dependent on hydrogel material type, herein a photocrosslinking hydrogel (PC-gel, polyethylene glycol dimethacrylate) and a thermoresponsive hydrogel (TR-gel). Compared with 2D FRET methods, this method requires little additional work. Laboratories already utilizing FRET imaging in 2D can easily adopt this method to perform cellular studies in a 3D microenvironment. It can further be applied to high throughput drug screening in engineered 3D microtissues. Additionally, it is compatible with other forms of FRET imaging, such as anisotropy measurement and fluorescence lifetime imaging (FLIM), and with advanced microscopy platforms using confocal, pulsed, or modulated illumination.
Keywords: Molecular Biology, Issue 114, Microfluidic, microtissue, fluorescence, Förster, scaffold, polyethylene glycol, hydrogel, FRET, intracellular signaling, chondrocytes
Introduction
Cellular signaling is both complex and consequential for numerous fields, including pharmacology, tissue engineering, and regenerative medicine. Useful investigational tools are needed to further our understanding of cell biology and to develop optimal materials for tissue regeneration. Förster resonance energy transfer (FRET) imaging is an essential tool enabling analysis of receptor activation, molecular conformation, and supramolecular interactions (e.g., protein/DNA complexation) in live cells. FRET is a type of non-radiative energy transfer between fluorophores1. It has an efficiency that is inversely proportional to the sixth power of the distance between a donor fluorophore and acceptor fluorophore. Thus, it can provide information on the spatial distance between two different molecules or across regions of one molecule at the nanometer scale (typically 1 - 10 nm)2. The donor and acceptor fluorophores may be different molecule types (hetero-FRET), in which the donor has the higher energy emission (shorter wavelength), or of the same type (homo-FRET). FRET imaging can be performed on fixed or live cells, but in general fixed cells are used for imaging of molecular interactions at high spatial resolution when high speed confocal systems are unavailable. Live cells are used to study interactions and processes that are inhibited or altered by fixation, such as the real-time intracellular signaling response3.
Live cell studies that employ FRET imaging use two-dimensional (2D) cellular cultures in part because of simplicity and lower background (no emission from out of plane cells). However, 2D cultures also alter numerous cellular processes compared to the physiologic microenvironment and three-dimensional (3D) culture4,5. For example, native cell to cell and cell to extracellular matrix interactions are drastically altered in 2D culture leading to the easily observed changes in cell morphology and polarity6. Membrane microdomains (e.g., caveolae and lipid rafts) and receptor signaling are strongly regulated by the culture environment, in part because they bind cytoskeletal components7,8 and the cell shape and cytoskeletal structure are strongly regulated by the spatial culture environment9. Mechanotransduction is not only influenced by the 3D matrix, but by the more complex secondary loading conditions engendered in 3D environments compared to 2D cultures10. Finally, the permeability of 3D matrices is less than the culture medium, in general with decreased diffusion and increased binding of cell signaling molecules compared to 2D cultures. With 3D cultures one is able to create and observe a microenvironment more similar to the physiologic with respect to adhesive, chemical, and mechanical cues. Cell to cell interactions can be promoted and the adhesive properties can be controlled with the structure, composition, and architecture (patterning) of the 3D material11-13. The mechanical properties of the material can likewise be tailored by the composition and structure14,15. Culturing cells in hydrogels therefore permits FRET studies on primary cells that would otherwise de-differentiate or change in phenotype using conventional techniques, e.g., cells from articular cartilage. Thus, a method is needed to enable FRET assays in live 3D cultures.
Hydrogel scaffolds are ideal for 3D FRET based studies because they can be made optically transparent and tailored to control the microenvironment and to provide cellular cues. Hydrogels made from natural polymers have been used in cell culture for decades, including gelatin and fibrin16. Greater control over the cellular microenvironment can be achieved with chemical modification of these polymers and with the use of synthetic polymers17,18. Hydrogel mechanical stiffness and permeability can be controlled by changing their polymeric composition and mesh size (polymer and crosslink density)19,20. Further, hydrogels can be made bioactive through the incorporation of growth factors and cellular ligands. Because of these possibilities, the development of hydrogels for biosensing, drug delivery, and tissue engineering applications is a very active area of research21. 3D printing of hydrogels is also undergoing development for fabrication of micro-tissues and -organs15. Thus, FRET imaging of cells in hydrogels is not only useful as a means to approximate physiologic cellular behavior, but also as a tool to study and optimize engineered tissue growth.
Binary ratiometric FRET probes are very useful in studying cellular signaling and can be utilized in hydrogel cultures. For a partial list of published fluorescent and FRET probes, see the Cell Migration Consortium website22. The most common probe type contains two different fluorophores that are associated or linked by a recognition element(s) (analyte sensitive region). These regions undergo a conformational change, association, or disassociation upon detection of the analyte (e.g., binding of an ion or molecule, enzymatic cleavage of the region) that alters the distance between fluorophores and therefore the FRET magnitude (either higher or lower)23. A less common yet useful probe type relies on an analyte sensitive change in the spectral overlap of the two fluorophores, which alters FRET magnitude. "Single-chain" ratiometric probes utilize a fluorophore pair linked on a single molecule. "Dual-chain" probes utilize fluorophore pairs on separate molecules, but their expression can be coupled under the same expression cassette24. Unlike studies with single fluorophore expression (e.g., fluorescent fusion proteins), studies with ratiometric FRET probes do not require dual transfection to control for transfection efficiency or cell viability. Ratiometric FRET probes are relatively insensitive to transfection efficiency when expressed above a minimum threshold (concentration insensitive)23,25. They are insensitive to excitation source fluctuations and other intracellular environment factors (e.g., pH, ions) so long as both fluorophores are similarly responsive. In addition, their response is not subject to transcriptional delay, unlike fluorescent and bioluminescent promoter-reporter constructs26,27.
Ratiometric FRET probes are easily and frequently employed in conventional widefield epifluorescence microscope systems. Widefield microscopes are preferred over line scanning confocal systems for live cell imaging because of concerns over system cost, fluorophore bleaching, and capture of signal changes with sufficient temporal resolution. In addition, single cell analysis over a large population of cells is possible with a motorized stage and image capture over multiple fields of view. Widefield microscopy requires intensity based analysis of the hetero-FRET probe signals. Though intensity analysis does not require complex hardware like other FRET methods, more post-acquisition work is needed to correct for the effects of fluorophore behaviors and microscope/imaging system configurations28. The captured emission intensity of a fluorophore is a function of the excitation energy, fluorophore quantum yield, and the combined filter and camera/detector spectral sensitivity and linearity2. These may be dissimilar for the donor and acceptor and will require calibration for quantitative analysis. In addition, imaging of the acceptor emission is influenced by the spectral overlap of the fluorophores (excitation of acceptor by donor excitation light and bleed-through of donor emission into the acceptor emission spectra)2. "Single-chain" probes are internally normalized because their fluorophores are equimolar at the nanoscale, while the fluorophores of "dual-chain" probes may exist at different stoichiometries throughout a cell25,28. If the fluorophores of "single-chain" probes are of similar brightness (function of extinction coefficient and quantum yield), the effects of non-linear detector sensitivity are small and only correction for the background fluorescence and the coupled quantum yield/detector sensitivity is needed23. Thus "single-chain" ratiometric FRET probes are most easily implemented in widefield microscopy24.
This paper describes a straightforward technique to perform FRET imaging of live cells in 3D hydrogel cultures using a conventional widefield epifluorescent microscope. It can be easily adopted by laboratories already carrying out FRET experiments in 2D, as well laboratories interested in intercellular signaling in microtissues. Here we demonstrate the method with FRET imaging based measurement of cyclic adenosine monophosphate (cAMP) levels in live chondrocytes within photocrosslinking (PC-gel, polyethylene glycol dimethacrylate) and thermoresponsive (TR-gel, Matrigel) hydrogels. A ratiometric FRET probe is utilized based on EPAC1 (ICUE1) to detect cAMP levels in response to forskolin, a chemical agonist for adenylyl cyclase which catalyzes the conversion of ATP to cAMP. cAMP is one of the principal second messengers mediating G protein-coupled receptor signaling and it activates various factors, including downstream exchange proteins directly activated by cAMP (EPACs). The fluorophores move apart upon cAMP binding to the EPAC domain resulting in a decrease in FRET. Described here is the preparation of the hydrogel materials, cell transfection with the ICUE1 probe, and embedding of the cells in 3D hydrogels. The 3D FRET experiment procedure and the image analysis necessary to evaluate probe activity per cell are explained. We also discuss the inherent limitations of FRET analysis in 2D and 3D using widefield microscopy. The technique presented here is an improvement over the existing 2D methods since it enables analysis in a more physiological context and requires less image correction and calibration. Great utility lies in the fact that many transparent materials can be used while cell and transfection preferences can be maintained.
Protocol
1. Materials Preparation
Prepare polyethylene glycol dimethacrylate (PEGDM, 4,000 mw) by methacrylating polyethylene glycol according to methods described by Lin-Gibson et al.29. Alternatively, PEGDM may be purchased.
Synthesize the photoinitiator, lithium phenyl-2,4,6-trimethylbenzoylphosphinate (LAP), through a two-step process where first dimethyl phenylphosphonite is reacted with 2,4,6-trimethylbenzoyl choride via a Michaelis-Arbuzov reaction and then the lithium salt created by adding lithium bromide in 2-butanone, in accordance with Majima et al.30. Note: Alternatively, other photoinitiators may be purchased but LAP shows better biocompatibility and crosslinking efficiency31.
- Functionalize the glass coverslips to enable bonding of the hydrogel to the glass and thereby prevent movement during imaging.
- In a chemical hood with proper personal protective equipment, use 3-(Trimethoxysilyl)propyl methacrylate according to the manufacturer's protocol for PC-gels and epoxy silanes (3-Glycidoxypropyltrimethoxysilane) for TR-gels31. Note: Alternatively, precoated slides may be purchased. Note that the cover slip must be larger than the well of the PDMS dish in Step 3.8.
2. Cell Transfection
- In a tissue culture hood and using sterile techniques, thaw and plate the desired cells at sub-confluency (50 - 70%), herein bovine chondrocytes at 15,000 cells/cm2 in 6-well non-treated culture dishes. Note: Any relevant cell type may be used, including anchorage independent cells.
- Allow cells to recover for one day in an incubator at 37 °C. Before subsequent steps, remove medium, rinse with PBS, and add 2 ml phenol- and serum-free medium per well.
The next day for each well, add 100 µl of phenol- and serum-free medium as well as 3 µl transfection reagent to an autoclaved glass test tube. To mix, shake the tube lightly. Incubate at RT for 5 min. Limit any exposure to light.
Add 1 µg FRET probe DNA and shake lightly. Incubate the mixture at RT and away from light for 30 min. Note: In this experiment ICUE1 plasmid (courtesy of Dr. Jin Zhang33) was used. The fluorophores in ICUE1 are cyan fluorescent protein (CFP, donor) and yellow fluorescent protein (YFP, acceptor). The ratio of transfection reagent to plasmid DNA will need adjustment for optimal transfection of different cell types.
Distribute the prepared mixture (~104 µl) drop wise to each of the 6 wells containing cells plus 2 ml of phenol- and serum-free medium. Do not pipet up and down. Gently swirl the plate to mix.
Incubate at 37 °C for 48 hr. Note: Confluency inhibits transfection efficiency and alters endogenous expression of receptors.
3. Fabrication of PDMS Molds for Hydrogel Casting and Culture
- To prepare a large glass slide to cast the polydimethylsiloxane (PDMS) onto, clean the glass first with soap, rinse with a copious amount of water, and then dry with isopropanol followed by air.
- Treat the glass with a siliconizing reagent as per manufacturer's protocol so that the PDMS can be easily peeled off after curing. Rinse residual reagent off of the surface with toluene over a catchment container. Allow the surface to air dry completely before or use heat at 100 °C to shorten the waiting time.
- Select a hydrogel height (e.g., 1 mm) and calculate 1.2 times the volume of PDMS needed to cover the glass slide with PDMS of this thickness using the dimensions of the glass slide and hydrogel height. Note: PDMS does not shrink.
- Mix the PDMS kit components at a 10:1 ratio by weight in a beaker. Once it is mixed well, place the beaker under house vacuum to degas. Relieve the vacuum if bubbles begin to overflow the container and repeat. Note: Other hydrogel thicknesses may be used, but at the cost of increased background and increased opacity if centrifugation (step 6.2) is not used.
Wrap aluminum foil around the base and sides of the glass to contain the PDMS. Carefully pour the desired amount of degassed PDMS onto this glass surrounded by foil. Measure the PDMS height. Keep in mind that the height of the PDMS will be the maximum height of the hydrogel. If any bubbles are created when pouring, degas again or pop them with a spatula or needle.
Place the uncured PDMS on a level surface in an oven at 100 °C for 2 hr, or leave at RT for 24 hr.
Carefully remove the PDMS from the surface once it has cured. Punch out the desired shape (e.g., cylindrical punch of 3 mm) for the hydrogels. Note: Photocrosslinking will yield slightly narrower PC-gel cylinders than the mold diameter due to quenching of the reaction by molecular oxygen in the PDMS.
Sterilize the punched PDMS. For short term experiments, submerge PDMS in 70% ethanol for 1 hr.
Assemble the mold by fitting the base of the sterilized PDMS with a sterile glass coverslip of a thickness matched to the microscope objective correction. Set aside for use in Step 6.
Fabricate a larger PDMS dish using the methods above (with a well wider and taller than the hydrogel) to contain the hydrogel and medium. Accommodate a medium volume at least 10x more than the hydrogel to immerse the hydrogel and to minimize dilution of analyte.
4. Preparation of Hydrogel Precursor Solutions
- In a tissue culture hood and using sterile techniques, prepare the hydrogel precursor solutions appropriate for the study immediately before adding cells. Prepare PC-gel from the polymer precursor as follows.
- Mix PEGDM powder into phosphate buffered saline (PBS, pH 7.2) at 10% (w/v).
- Add the photoinitiator (e.g., LAP) at a final concentration of 0.005 - 0.01% (w/v) and mix by pipetting. Cover the solution with aluminum foil or shield from light as LAP is light sensitive. Note: Here the TR-gel reduced growth factor extracellular matrix is used as supplied by the manufacturer.
5. Cell Suspension in Hydrogel Solutions
In a tissue culture hood and using sterile techniques, aspirate the medium from the wells.
Add PBS to the wells and then remove to thoroughly rinse attached cell monolayers.
Dissociate the cells from the culture substrate using 2 to 3 ml per 25 cm2 of cell dissociation solution. Rock gently, then incubate at 37 °C for 10 - 20 min or until dissociated. Firmly tap dish if necessary to dislodge cells. Note: Alternate cell dissociation solutions are available with selection limited to those that do not impact the signaling pathway under analysis by cleaving receptors of interest.
After the cells are detached, add 2 ml of phenol- and serum-free medium, suspend the cells, and count cells with a hemocytometer. Note: Chemically defined serum free medium (e.g., supplemented with insulin, transferrin and selenium) may also be used.
Aliquot the desired number of cells based on the cell type into sterile vials and pellet (e.g., 2.0 x 106 cells per vial).
- Remove the supernatant, add the desired volume of hydrogel precursor and suspend the cells by pipetting. Use a volume of precursor that will yield 2 x 105 cells/cm2 in the xy plane when visualizing the z-axis of the hydrogel as collapsed (e.g., 1.0 ml per vial for a 1 mm thick hydrogel containing 2 x 106 cells/ml). Take care not to produce bubbles.
- Quickly move on to the next step to limit negative effects on cell viability34.
6. Hydrogel Preparation
In a tissue culture hood and using sterile techniques, add the cell-containing hydrogel precursor to the casting molds prepared in step 3 (e.g., 7.1 µl per 3 mm diameter x 1 mm high mold). Create an extra hydrogel, according to the same methods described, to use for microscope imaging system calibration and probe calibration (if the inherent probe efficiency is unknown).
Centrifuge the prepared cell-containing hydrogel precursor prior to gelation/crosslinking to optimize imaging by confining cells to a single focal plane and minimizing out of plane signal (Figure 1). Adjust centrifugation time and force to the cell type and viscosity of the precursor (400 g for 4 min).
- Gel or crosslink the solution
- For the PC-gel hydrogel, irradiate the cell-containing precursor with a UV-A light source (λ = 365 nm) for an exposure time equal to 3.2 J/cm2 per millimeter thickness to photocrosslink. Note: Use a 1 mm minimum thickness in the calculation. Exposures should fall in the 1-5 min range. Use the minimum exposure time. Avoid over-irradiating the cells as this will decrease viability and bleach the CFP donor. However, exposure times less than 60 s are not recommended because high intensity irradiation leads to radical self-quenching and poor cell viability.
- For the TR-gel hydrogel, place the filled mold in a petri dish and move to an incubator to thermally gelate. Incubate at 37 °C for 30 min.
- After gelling or crosslinking the hydrogel, remove the PDMS hydrogel casting mold and replace it with the prepared larger PDMS dish (from step 3.5). Press the PDMS down onto the glass with a sterile spatula to adhere it.
- Place this PDMS dish with coverslip into a sterile container, such as a petri dish. Add phenol-free serum-free medium or chemically defined medium (serum free medium supplemented with insulin, transferrin and selenium (step 5.4)) to the well created with the PDMS dish and coverslip to keep the hydrogel immersed. Incubate at 37 °C for 30 min. Note: If using a specialized dish with a coverslip, then place the coverslip in that and similarly add medium.
- Alternately, perform this step the day before FRET imaging to allow the probes to recover as the irradiation wavelengths overlap with the CFP donor excitation spectrum.
7. 3D FRET Imaging
Remove the PDMS dish with the hydrogel from the petri dish, prepared in step 6.4, and secure it in an adjustable specimen holder for the XY microscope stage (Figure 2). Position the specimen holder on the microscope stage. Apply oil to the objective if required. Use an objective of at least 40X and of high numerical aperture (NA) (i.e., 1.3 NA) to capture the relatively weak fluorophore emissions.
- Configure the fields of view (FOVs) for image acquisition: Select cells for imaging using transmitted light imaging and verify transfection with fluorescence imaging. Choose at least 15 different cells.
- Select cells that are neither too bright nor too dim (which could otherwise lead to probe over-saturation or low sensitivity) and with the FRET ratio between 0.0 and 1.0 (which otherwise indicates damage or decay of the binary probe). Then switch off the light to limit the exposure.
- Select FOVs close to the center of the hydrogel and with the cells of interest within the relatively flat illumination field (See Figure 3B and Step 9.2 below).
- Configure the camera settings for the FRET image channels. Choose among a few cells near the center of the FOV to optimize the "optical configurations" (i.e., exposure and gain per image channel). Note: The nomenclature below varies depending on the software used to operate the microscope and camera. The microscope software, NIS-Elements, is used here for image capture and analysis.
- For FRET analysis of single chain probes, select two image channels per FOV: Quenched emission (QE), which is the donor emission under donor excitation and Acceptor emission under acceptor excitation (Aem). Note: For the ICUE1 CFP-YFP probe, a CFP excitation and emission filter pair for QE (418 - 442 ex, 458 - 482 em) and a YFP excitation and emission pair for Aem (490 - 510 ex, 520 - 550 em) are used.
- Set the exposure for QE and Aem to acquire the maximum signal intensity without saturation (and if using a CCD, with the minimal possible gain) to minimize background noise. Keep the optical configurations the same for both fluorophores and throughout the experiment, as well as among experimental groups to avoid biasing the results (i.e., excitation power, iris size, exposure, and camera gain) (Figure 3A).
- Acquire additional image channels to allow for more analysis options after acquisition (e.g., corrected sensitized emission (cSE) imaging (see Discussion)). For the ICUE1 CFP-YFP probe, acquire the excitation (Ex) and emission (Em) channels of Exdonor - Emdonor (QE, select CFP-CFP in the microscope software), Exdonor - Emacceptor (SE, select CFP-YFP), Exacceptor - Emacceptor (Aem, select YFP-YFP), and Exacceptor - Emdonor (YFP-CFP) configurations (See Figure 3A).
- Configure the capture rate for time-lapse image acquisition.
- Decrease the capture rate if limited by the hardware. For short term assays (e.g., 30 - 60 min), denote around 12 acquisitions per minute for each FOV (5 sec sampling rate) to capture signaling kinetics. Use the minimum sampling rate necessary to avoid significantly photobleaching the probe.
- Set the acquisition rate by typing the periodicity of the captures in the microscope software. For long term assays (e.g., 24 hr), begin with the high acquisition rate to capture the initial pulse response and then switch to a low acquisition rate, e.g., every 5 to 10 min.
Select "Run now" in the microscope software to initiate image acquisition and capture images for 5 min to establish a baseline for probe response at the designated FOVs.
After 5 min of image acquisition, pipet in the desired agonist or antagonist analyte (e.g., Forskolin at 100 nM), mix by pipetting, and continue imaging.
8. FRET Calibration
- System Calibration: Use the additional calibration hydrogel, fabricated as described in step 6, to acquire Aem images of at least 6 representative cells using the same FOV criteria, optical configurations, and camera settings used for FRET imaging above (Steps 7.1, 7.2, and 7.3). Note: Quantification of the quantum yield and spectral sensitivity (QS) for the combined probe and microscope imaging system is needed for "absolute" FRET analysis but not for "relative" FRET.
- Photobleach the acceptor fluorophore by selecting a 5 min long exposure to the excitation light at full intensity (excitation on Aem channel). Use a narrow aperture to bleach only the cells of interest. Verify that the Aem signal is at least 10% lower than the original signal by comparing the signal intensity histogram ("LUT" window) before and after photobleaching. Continue photobleaching if signal loss is less than 90%. Note: Ensure the donor fluorophore is not bleached during this procedure by using Aem excitation filters that do not overlap with the donor excitation spectrum.
- Acquire donor emission under donor excitation with the now bleached acceptor fluorophore (Dem) using the same optical configuration as for QE in step 7.3.
- Calculate QS per pixel as Dem divided by Aem after background correction, as in Section 9 below. QS = Dem/Aem
- Calculate the average QS among the cells from the cellular averages.
- Estimate the inherent FRET efficiency of the probe (Ei) using the calibration sample. Induce maximum FRET of the probe and calculate Ei = 1 - QE/(Aem x QS) as in section 9 below. Alternately, use Ei = cSE/Aem, or Ei calculated using a conventional endpoint assay for FRET efficiency with Ei = 1 - (QE/Dem). Note: Ei is needed to quantify the absolute fraction of activated probes (FAP, i.e., fraction of probes binding analyte), but not necessary for "relative" FRET analysis.
- Saturate probe response by stimulating the culture with an agonist of analyte production/activation for probes that increase in FRET upon interaction with analyte.
- Inhibit probe interaction with the analyte using an antagonist of signaling for probes that decrease in FRET upon binding the analyte. Note: In the case of the ICUE1 probe, suppress cAMP levels with an adenyl cyclase inhibitors such as 2′,5′-Dideoxyadenosine (specific) or 3-isobutyl-methyl-xanthine (non-specific).
9. FRET Ratio Calculation
- Draw and designate one region of interest (ROI) per FOV near the cells to define the background level (Figure 3B) in each FOV over time, but restrict the ROI to the area of relatively homogenous illumination over the center of the image (Figure 4A). See the discussion for an explanation of background correction options.
- With the microscope software: Select the arrow on the "Background ROI Icon" and select "Draw Elliptical Background ROI" (other shapes will work). Ensure that "Keep Updating Background Offset" is selected. Activate viewing of the background ROI by clicking the Background ROI icon. Alternately, use the "ROI Icon" and the ROI properties set as "Use as Background ROI" and "ROIs are different in each multipoint".
- With ImageJ: Use the toolbar to select the circular drawing tool. Then add the shape to the ROI manager via the menu "Analyze→ Tools→ ROI Manager". Set a different color for the background ROI in the ROI Manager if desired.
- For each FOV and image channel (e.g., QE and Aem), subtract the average value within the background ROI per time-point to create a corrected image per channel, e.g., sQEt,p = QEt,p - bQEt,p where t is the acquisition time, p is the FOV (i.e., xy position), and bQE and bAem are the scalar values of signal within the background ROI. Use the image arithmetic functions available in the software.
- With the microscope software, use the "ROI Background Icon" and select "Subtract Background Using ROI". In the pop-up window, select "Every Image" under "Subtract background ROI on" and select "All Frames" under "Apply To". Note: A background ROI must be defined in every FOV for this function to work properly (e.g., the function does not properly subtract backgrounds for FOV with background ROIs if some FOV have undefined background ROIs).
- With ImageJ, use the "BG Subtraction from ROI" macro written by Michael Cammer (http://microscopynotes.com/imagej/macros/).
- Save the corrected time series of images by selecting "Save as" for the following analysis steps.
- Define a ROI around the cell using a binary mask for each cell under analysis in each FOV (Figure 4B). Use the Aem channel to threshold the image per FOV at the start of the experiment, identify the cell boundaries, and defined the ROIs. Save the ROIs.
- With the microscope software: For each frame (i.e., FOV), first use the menu "Binary→ Define Threshold" and select "apply to current frame". Adjust the lower threshold value to select the cells. Then use the "Binary→ Close" function to fill in holes in the binary mask if necessary. Next use the ROI icon to select "Copy Binary to ROI"; the binary layer is automatically deleted.
- Append ROIs for subsequent frames to the existing pool of ROIs. Delete any spurious ROIs. Next "right-click" on the ROIs and make sure their properties are "Use as Standard ROI" and "ROIs are different in each multipoint".
- With ImageJ: For each FOV, first use the menu "Image→ Adjust→ Threshold". Then use "Analyze→ Analyze Particles" with Show Outlines and Add to Manager selected. Use "Binary→ Close" to fill in "holes" in the binary mask if necessary. Delete spurious ROIs. Free plugins are available to automate this process.
- Calculate the QE based FRET ratio at each pixel, sQE/sAem for relative analysis (See Step 10) and EQE = sQE/(QS x sAem) for absolute analysis (see Step 10) with QS obtained in Step 8.1. Use floating point division of the images. See the discussion section for an explanation of the ratio derivations.
- With the microscope software: Use the menu "Image →Image Operations" and select "Floating Point".
- With ImageJ: Use either the "Process →Image Calculator" function or the "Calculator_Plus.java" plugin. Note: Alternately, the macro "pH_ratioMacro".txt" may be adapted to calculate the FRET ratios. It employs the background correction macro described above and is available at
- Export the average ratio per cell (i.e., per ROI) into a spreadsheet and graphing software.
- With the microscope software: Use either the ND export button in the "ROI Statistics" analysis control or the "Spreadsheet Export" button in the "Time Analysis" analysis control.
- With ImageJ: First select the menu "Analyze →Set Measurements" and make sure "Mean value" and "Standard deviation" are selected. Then use the ROI Manager and select the button "Measure". Save the generated results window.
10. FRET Ratio Analysis
- Calculate the average cellular FRET ratio over time from the average ratios per cell (exported in Step 9.4). Consider the baseline FRET ratios (the ratio before administering the analyte) in this calculation. Note: Spreadsheet and graphing software is used here to plot the baseline-subtracted average FRET ratios and their standard error of the mean (SEM) using a linear regression on the baseline as in Step 10.1.2 and 10.1.3 below (Figures 5 and 6).
- Relative Analysis (magnitude of response): Normalize each curve to a common reference sample (e.g., untreated sample).
- Absolute Analysis (time-course of response): Shift the curves to a common start point by normalizing each curve with its baseline FRET ratio.
- In the spreadsheet and graphing software, interleave the data (columns of average ratios per cell) with new columns, such that each original column is paired with a new column containing only ratio data for the baseline period (ratios before analyte added). Select "Insert Column" to insert an empty column after every column of data. Then copy the baseline data into the paired empty columns.
- In the spreadsheet and graphing software, select "Insert → New Analysis → Transform Remove baseline" and then set "Selected column" to "every other", select "assume the baseline is linear", and select "Difference" under "Calculation".
- Calculate the average cellular ratio and descriptive statistics. In the spreadsheet and graphing software, select "Insert → New Analysis → XY Analyses → Row Means with SD or SEM" and then select "Row means with SEM" in the pop-up window.
Representative Results
All hydrogel precursors, cells, and casting molds were prepared as described above. The cell laden precursor solutions were cast in PDMS molds and then centrifuged before gelation/photocrosslinking to immobilize cells near the coverslip and facilitate FRET imaging analysis (Figure 1). The hydrogels with attached coverslips were placed within prepared PDMS imaging wells for microscopic imaging (Figure 2). The optical configurations and image acquisition parameters were then set as shown in Figure 3A. Capture of all four FRET related image channels for CFP-YFP fluorophore pairs is demonstrated (see under "Optical Conf." in Figure 3), as is needed for non-binary probes. However in this work only two images are required for the single chain binary ICUE1 probe, QE = "CFP CFP" and Aem = "YFP YFP" (excitation emission). Multiple FOVs (xy positions) were selected in which several cells resided within the homogenous illumination area (Figure 3B). After time lapse acquisition, the signal data for the probe was exported. ROIs for background correction of channel signals and for cell analysis were designated using thresholded Aem images from the start of the experiments (Figure 4). Cells were selected from among the cell pool as those expressing sufficient (not too dim), but not too high (excessively bright) probe (Figure 4B). The QE and SE FRET ratios per pixel at each acquisition were calculated using floating-point division of the background subtracted image channels. The average FRET ratio per cell (i.e., per ROI) was calculated, normalized to the baseline signal prior to stimulation (i.e., a regression on the baseline subtracted from all time-points), and plotted with error bars as the standard error of the mean (SEM). Both the QE and cSE based FRET ratios detect the increased cAMP activity in chondrocytes under forskolin stimulation (100 nM, Figure 5). The QE FRET ratio was more sensitive to background correction than the cSE FRET ratio because the QE signal was lower due to the low basal level of cAMP activity in chondrocytes embedded within the hydrogels. Both hydrogel types (TR-gel and PC-gel) showed an increase in FRET ratio over time (Figure 6), indicating an increased cAMP signaling response to the addition of Forskolin. As indicated by the QE FRET ratio, chondrocytes in PC-gel hydrogels show a greater change in cAMP (shown) and a higher basal level of cAMP (EQE = 0.19 in PC-gel vs. EQE = 0.09 in TR-gel, not shown in baseline subtracted figure).
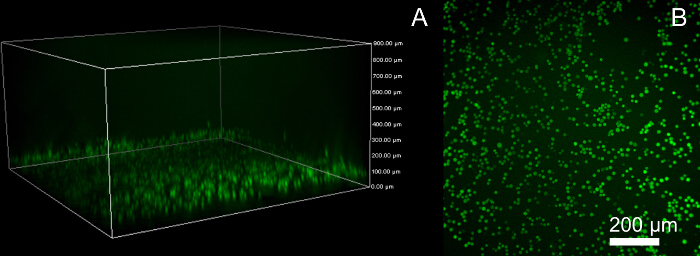 Figure 1:Cell Distribution in Photocrosslinked Hydrogel (PC-gel) A: The cell laden hydrogel precursor was centrifuged to maximize the number of cells at a focal plane near the coverslip. B: This increases the number of cells in the FOV and minimizes background signal from out of plane cells. (Scale bar = 200 µm) Please click here to view a larger version of this figure.
Figure 1:Cell Distribution in Photocrosslinked Hydrogel (PC-gel) A: The cell laden hydrogel precursor was centrifuged to maximize the number of cells at a focal plane near the coverslip. B: This increases the number of cells in the FOV and minimizes background signal from out of plane cells. (Scale bar = 200 µm) Please click here to view a larger version of this figure.
 Figure 2: Hydrogel in PDMS Dish The hydrogel was placed inside a tall PDMS well (10x volume of hydrogel) with a coverslip base for microscopic imaging and analyte stimulation. The PC-gel hydrogels are transparent and bonded to the coverslip so that they remain in place throughout the assay. The PDMS well can be made larger to contain 10x more medium than the hydrogel volume using a wider biopsy punch in addition to a thicker PDMS sheet. (Scale bar = 2 mm) Please click here to view a larger version of this figure.
Figure 2: Hydrogel in PDMS Dish The hydrogel was placed inside a tall PDMS well (10x volume of hydrogel) with a coverslip base for microscopic imaging and analyte stimulation. The PC-gel hydrogels are transparent and bonded to the coverslip so that they remain in place throughout the assay. The PDMS well can be made larger to contain 10x more medium than the hydrogel volume using a wider biopsy punch in addition to a thicker PDMS sheet. (Scale bar = 2 mm) Please click here to view a larger version of this figure.
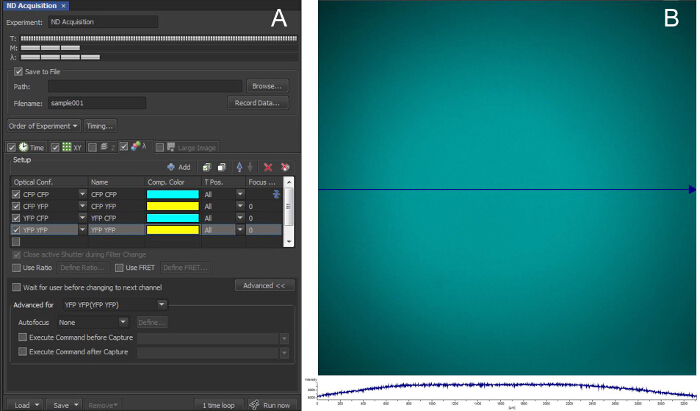 Figure 3:Acquisition Configuration for FRET Imaging A: The acquisition was configured for image capture every 7 sec at multiple locations. For QE FRET ratio calculation, only two image channels are needed for the ICUE1 probe used here (binary single chain probe), QE = "CFP CFP" and Aem = "YFP YFP" (excitation emission) under the "Optical Conf." column in the λ tab of the microscope software. B: FOVs ("XY" positions) were selected in which several cells resided within the homogenous illumination area. The plot below the image shows the illumination intensity along the blue arrowed line that traverses through the center of the FOV. Please click here to view a larger version of this figure.
Figure 3:Acquisition Configuration for FRET Imaging A: The acquisition was configured for image capture every 7 sec at multiple locations. For QE FRET ratio calculation, only two image channels are needed for the ICUE1 probe used here (binary single chain probe), QE = "CFP CFP" and Aem = "YFP YFP" (excitation emission) under the "Optical Conf." column in the λ tab of the microscope software. B: FOVs ("XY" positions) were selected in which several cells resided within the homogenous illumination area. The plot below the image shows the illumination intensity along the blue arrowed line that traverses through the center of the FOV. Please click here to view a larger version of this figure.
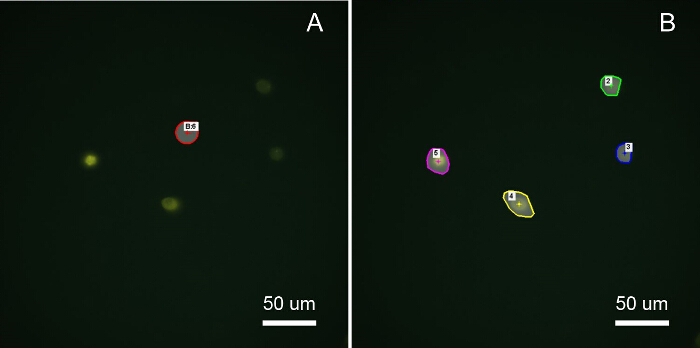 Figure 4: Selection of ROIs for FRET Imaging Analysis A: A region of interest (ROI) free of cells was selected to correct for background signal on each channel. An image of a cell-free hydrogel may instead be used for background subtraction if cell density or migration is high, or if a shading mask is not used when cells do not lie within a homogeneous illumination field. B: The cell ROIs were selected using image thresholding and binary masking of the Aem channel. Selecting a ROI larger than the cells will adversely affect calculation of the average FRET ratio per cell due to inclusion of extracellular ratios generated from background noise. Calculation of the cellular FRET ratio using the average of each channel per cell is not recommended as this underestimates the ratio. (Scale bar = 50 µm) Please click here to view a larger version of this figure.
Figure 4: Selection of ROIs for FRET Imaging Analysis A: A region of interest (ROI) free of cells was selected to correct for background signal on each channel. An image of a cell-free hydrogel may instead be used for background subtraction if cell density or migration is high, or if a shading mask is not used when cells do not lie within a homogeneous illumination field. B: The cell ROIs were selected using image thresholding and binary masking of the Aem channel. Selecting a ROI larger than the cells will adversely affect calculation of the average FRET ratio per cell due to inclusion of extracellular ratios generated from background noise. Calculation of the cellular FRET ratio using the average of each channel per cell is not recommended as this underestimates the ratio. (Scale bar = 50 µm) Please click here to view a larger version of this figure.
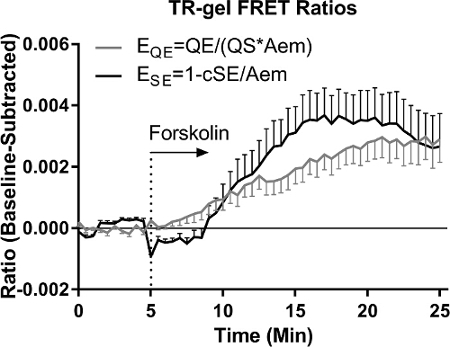 Figure 5: Comparison of FRET Ratios in Thermoresponsive Hydrogel (TR-gel) Both QE and SE based FRET ratios detect the increased cAMP activity in chondrocytes under forskolin stimulation. Forskolin is an adenyl cyclase agonist which induces cAMP production. FRET decreases when the ICUE1 probe binds cAMP, and thus the QE FRET ratio (EQE = sQE/(sAem x QS)) increases. Conversely, the SE FRET ratio decreases and is thus plotted as ESE = 1 - cSE/sAem. In hydrogel embedded chondrocytes, the QE FRET ratio is more sensitive to background correction than the SE FRET ratio because the QE signal is low due to the low basal cAMP activity. Error bars = SEM, n = 25. Please click here to view a larger version of this figure.
Figure 5: Comparison of FRET Ratios in Thermoresponsive Hydrogel (TR-gel) Both QE and SE based FRET ratios detect the increased cAMP activity in chondrocytes under forskolin stimulation. Forskolin is an adenyl cyclase agonist which induces cAMP production. FRET decreases when the ICUE1 probe binds cAMP, and thus the QE FRET ratio (EQE = sQE/(sAem x QS)) increases. Conversely, the SE FRET ratio decreases and is thus plotted as ESE = 1 - cSE/sAem. In hydrogel embedded chondrocytes, the QE FRET ratio is more sensitive to background correction than the SE FRET ratio because the QE signal is low due to the low basal cAMP activity. Error bars = SEM, n = 25. Please click here to view a larger version of this figure.
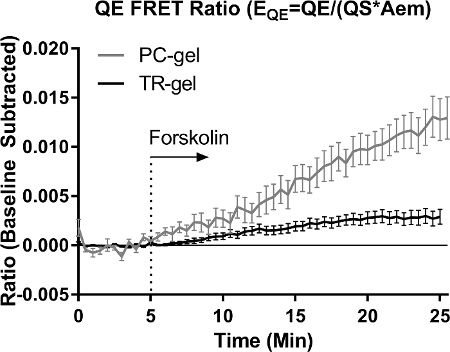 Figure 6:Comparison of FRET Ratio in Different Hydrogels The chondrocytes in both hydrogel types responded to forskolin stimulation with an increase in cAMP activity as demonstrated by the increased QE FRET ratio over time. The hydrogel type impacts the cellular response through several effects, including differences in permeability to forskolin and in basal cellular cAMP activity (EQE = 0.19 in PC-gel vs. EQE = 0.09 in TR-gel, not shown). Error bars = SEM, n for TR-gel = 25, n for PC-gel = 15. Please click here to view a larger version of this figure.
Figure 6:Comparison of FRET Ratio in Different Hydrogels The chondrocytes in both hydrogel types responded to forskolin stimulation with an increase in cAMP activity as demonstrated by the increased QE FRET ratio over time. The hydrogel type impacts the cellular response through several effects, including differences in permeability to forskolin and in basal cellular cAMP activity (EQE = 0.19 in PC-gel vs. EQE = 0.09 in TR-gel, not shown). Error bars = SEM, n for TR-gel = 25, n for PC-gel = 15. Please click here to view a larger version of this figure.
Discussion
This work demonstrates how FRET imaging can be used to analyze cellular signaling in 3D hydrogels. While prior studies have demonstrated FRET imaging of cells seeded on hydrogel substrates35-37, of extracellular interactions with hydrogels38, and of molecules trapped in hydrogels39-41, this is the first publication to describe FRET imaging based intracellular analysis of cells embedded within 3D hydrogels. The work solves several implementation issues when translating FRET imaging from 2D to 3D. First, high numerical aperture objectives are needed for FRET imaging to gather sufficient emission signal, but they limit the depth of field. The cells here are allowed to slightly settle via centrifuging to increase the number of cells in a focal plane near the glass to compensate for this. Second, 3D hydrogel scaffolds may drift across the field of view during imaging which complicates analysis. The hydrogels here are bonded to the coverslip so that they remain fixed throughout the experiment. Third, selection of appropriate hydrogel compositions for 3D FRET is essential because hydrogels may hinder visualization of the cells. Here transparent hydrogels of PC-gel and TR-gel are used. However, gathering the cells with centrifugation to a focal plane near the coverslip can be used to image cells in hydrogels with higher diffraction.The choice of hydrogel depends on the microenvironment conditions that must be simulated, which can be found in a review on hydrogels42. Fourth, an alternate calculation is employed here for the FRET ratio of the single chain ICUE1 probe (i.e., EQE = QE/cAem) to minimize the number of image channels required for analysis and thereby minimize photobleaching. The average FRET ratio per cell is calculated as the average of the ratios per pixel within a cell to minimize underestimation of the actual FRET ratio. Finally, a linear ratiometric analysis and means to calculate the activated fraction of single chain binary probes is described.
There are several critical aspects of carrying out successful FRET experiments using widefield microscopy. With respect to hardware, the camera must be sensitive enough to resolve small signal changes, leaving newer scientific CMOS cameras and electron multiplying CCDs better suited than conventional CCDs. To best analyze the spatial distribution of the FRET ratio, the camera must at minimum possess twice the spatial resolution of the point spread function of the objective (Nyquist criterion). This makes dual-view systems less suited to the task. More crucial is to select an appropriate exposure and image acquisition rate for the given FRET pair so as to minimize photobleaching of the fluorophores. A decreased acquisition rate is recommended as experiment duration increases. The use of rapid filter wheels increases acquisition rate and the number of FOVs for analysis while avoiding the image registration issues with dual-view and turret filter cubes. The inhomogeneous illumination field complicates correction for background fluorescence that arises from sample autofluorescence and camera system noise. Some hydrogels, particularly those containing collagenous proteins are highly autofluorescent43,44. The FRET ratios are underestimated without background correction. Here we employ a simple background correction method that relies on the relatively flat illumination field which can often be configured with epi-fluorescence illumination over the center of the image (Figure 3B, Step 7.2). This method therefore restricts cells of interest to those within this illumination field. The background correction described here does not account for cellular autofluorescence in the wavelengths read for the binary probe. In such a case, unlabeled cells (no probe transfection) should be imaged under the same conditions and background subtracted. A mix of labeled and unlabeled cells may be used and the unlabeled cells selected for the background ROI. Alternate methods which provide for cell analysis over the entire image field include use of a shading mask, multiple background ROIs, or identical samples without cells and a protocol is available upon request.
Specific to 3D FRET imaging, probe response is highly sensitive to the hydrogel permeability to the analyte (Figure 6). Therefore the same hydrogel regions are compared across experimental treatments, preferably at a point of geometry symmetry such as near the scaffold center as used here. Alternatively, microfluidic chambers/bioreactors may be employed to rapidly and uniformly perfuse the hydrogel with analyte solution45. A FRET signal may not be detected (signal to noise ratio is too low) when the hydrogel's autofluorescence is too high; a thinner hydrogel or different probe (different fluorophores) should be used. With respect to 3D FRET imaging in photocrosslinked hydrogels, use of minimum irradiation exposure and wavelengths outside the excitation spectrums of the probe fluorophores is recommended. LAP may be used with a visible light source at 405 nm to further minimize potential cellular damage31,46. However, using LAP with 365 nm irradiation is recommended because this minimizes probe bleaching. 365 nm lies at the tail end of the CFP donor excitation spectrum, whereas 405 nm lies at the peak excitation. Alternatively, the probe expression may be allowed to recover for one day. Use of binary probes minimizes issues of sensitivity to differences in expression levels and in molecular diffusion of the donor and acceptor fluorophores. Non-binary probes may be used, but with SE instead of QE imaging and additional correction for spectral bleed-through of excitation and emission spectra (see below). Furthermore, the low commercial availability and the complexity in designing FRET probes may be a hindrance. The most critical factor for successful experiments remains proper correction and analysis of the fluorescence signals.
An alternate calculation of the FRET ratio is utilized for several additional reasons besides minimization of photobleaching. For binary single-chain probes, the commonly reported ratio is acceptor emission under donor excitation (sensitized emission, SE) to donor emission under donor excitation (quenched emission, QE), i.e., FRET ratio = SE/QE25,47,48. SE/QE is non-linear relative to changes in FRET efficiency21,22,47. Namely, the ratio has an exponentially non-linear relationship to inherent FRET efficiency of the probe (Ei)(Donius, A. E.,Taboas, J. M. unpublished work. (2015)), with the ratio most linear for Ei less than approximately 15% . SE/QE will also underestimate the fraction of activated probes (FAP, i.e., fraction of probes binding analyte). Therefore, analysis of SE/QE requires a cumbersome equilibrium dose response study and curve fit. Fortunately, the ratios of SE or QE to the acceptor emission under acceptor excitation (Aem) are linear with respect to Ei and the FAP, i.e., the FRET ratios SE/Aem and QE/Aem. SE/Aem is often used for binary probes and only requires end-point calibration (at no probe activity and full probe activity), but basal cellular signaling makes this difficult. To eliminate the need for endpoint calibration, SE can be corrected (cSE) for spectral overlap of donor and acceptor excitation and emission spectrums (spectral bleed-through) and for the quantum yield and spectral sensitivity (QS) of the combined probe fluorophores and microscope imaging system. The corrected SE FRET ratio based on cSE defines the observed FRET efficiency of the probes within each pixel of an image, i.e., ESE = cSE/Aem. Commercial and free software is available to correct SE for these effects and the reader is referred to the work of Chen and Periasamy 2006 for a detailed explanation of the methods50. However, calculating ESE requires capture of three image channels per time-point (SE, QE, Aem). Therefore, in this work we also employ an alternate FRET ratio EQE = 1 - QE/cAem, which requires capture of only two image channels (QE and Aem). Calculating EQE from these channels only requires correcting Aem for QS (cAem = Aem x QS). QS = Dem/Aem is easily estimated using two images from the one calibration sample, where Dem is the donor emission under donor excitation with acceptor bleached. The FAP at any instant can be calculated by comparing ESE or EQE to the Ei of the probe.
Ultimately, FRET ratio selection (ESE = cSE/Aem vs. EQE = QE/cAem) should be based on consideration of the probe type and the channels with best signal. Both approaches include background noise correction of images per frame as described above to account for changes in autofluorescence over time. They assume no overlap of Aem excitation light into the Dem excitation spectrum, which is often the case due to probe design and microscope filter configuration. Single-chain probes can use either and selection is based on best probe signal (brightness) to camera noise and to background signal on SE and QE channels. For the ICUE1 probe and experimental conditions used here (cell and hydrogel types), SE has a stronger signal than QE. Dual-chain probes require use of ESE due to non-equimolar distribution of donor and acceptor fluorophores. For probes that decrease in FRET upon binding analyte such as ICUE1, FRET efficiency can be "inverted" to present a positive signaling change upon binding the analyte as we do here, with ESE = 1 - cSE/Aem or EQE = QE/(Aem x QS).
Analysis of the FAP is sensitive to proper correction of the FRET ratios and to the estimate of Ei. It can be estimated with the same calibration sample used for QS and a conventional endpoint FRET efficiency assay as Ei = 1-(QE/Dem) (QE image followed by acceptor photobleaching and a Dem image)28, but care must be taken to minimize accidental bleaching of the donor. We describe a modified version where the estimate of Dem based on Aem adjusted for QS is substituted, i.e., Ei = 1 - (QE/(Aem x QS)). Alternately, Ei = cSE/Aem may be used. Estimation of Ei in living cells requires that all probes be driven to full FRET. This is difficult to achieve in practice for probes, such as ICUE1 that decrease in FRET upon binding the analyte. An in vitro based calculation of Ei using cell lysates and purified analyte is best, but outside the scope of this work. Here we recommend using 2′,5′-Dideoxyadenosine to decrease cAMP in the cells. However, a relative FAP may be calculated using an Ei estimate derived from the baseline level of signaling. For these reasons, studies most often report the FRET ratio (as done here) or a relative FAP based on endpoint calibration, where the signaling baseline before adding agonist is the lower bound and the signal after adding a second agonist that saturates signaling is the upper bound. Forskolin is often used as a control agonist to saturate cAMP signal. Alternately, a cAMP analog may be used such as 8-Bromoadenosine 3',5'-cyclic monophosphate. Positive controls used at the completion of studies to identify potential changes in the signaling pathway among experimental treatments are recommended.
Calculation of the average signaling response per cell requires consideration of several factors. First, the average FRET ratio should be calculated as the average of the ratios at each pixel within a cell, i.e., (∑(QE⁄cAem))/(number of pixels), instead of the ratio of the average QE and Aem in a cell, i.e., (∑QE)⁄(∑cAem). Though the latter does not require careful masking as background noise is low, it severely underestimates the true average FRET ratio. However, pixel ratios require floating-point calculation and careful binary masking of the cell area to define the ROIs and avoid inclusion of spurious ratios generated from background noise outside the cell. The entire cell area must be quantified to avoid biasing results on cell shape. The masking may need to be redefined over time depending on changes in cell shape and migration. Alternately, ROIs larger than the cell may be used if exclusion criteria are defined to remove pixel ratios from QE and Aem signals that fall well below cellular levels and are near background levels. The described analysis methods detail the basic steps used in this work for FRET analysis without specialized software/plugins. Microscope manufacturers and other vendors sell add-on modules for their microscope software, which support automated ratiometric and FRET analysis. Likewise, the public domain ImageJ software has several plugins available for particle and FRET analysis, such as RiFRET. Second, the pixel-based average FRET ratio underestimates the true average ratio of the 3D cellular structure because a 2D image is used. The 2D signals represent and average along the z-axis. Calculation of the 3D spatial distribution of FRET ratios in live cells is not possible without high speed 3D imaging (e.g., using spinning disk confocal microscopy). This is a problem for both 2D and 3D cultures, but has a larger effect in 3D as more of the cellular structure exists out of plane. This is of great concern for probes that localize to cellular structures, such as the plasma membrane EPAC1 probe PM-ICUE. See Spiering et al. 2013 for suggestions to correct for cell thickness effects on ratio calculations24. Analysis of the distribution of Aem can be used to detect if probe accumulates in regions of the cell and imaging plane adjusted. This will also help to interpret the spatial distribution of FRET ratios and to eliminate spurious results, e.g., identifying probe saturation (i.e., a low Aem region will have low probe concentration and may exhibit probe saturation and high FRET ratio). Third, different FRET baseline levels may indicate a difference in transfection efficiency, probe expression, or basal signaling between experimental groups. "Relative Analysis" (Step 10.1.1) helps remove drift in the FRET ratio over time if present. It facilitates comparison of the relative magnitude of responses between samples, but impedes comparison to the time-course of response for the reference sample (control plot is a flat line). "Absolute Analysis" (Step 10.1.2) facilitates comparison of the rate of response across samples, but hampers comparison of the magnitude of response because each line is scaled by a dissimilar constant. Studies often use a linear regression on the baseline to correct for drift in the FRET ratio over time.
The described 3D FRET method is a useful and practical way to fabricate and perform time-lapse imaging of cell laden 3D hydrogels, which can be applied to other probes and imaging modalities. Other FRET system besides single chain binary probes will require more complex correction of intensity based signals, as discussed above. Alternately, fluorescence lifetime imaging of FRET (FLIM-FRET) may be used to calculate FRET efficiency via a change in emission lifetime of the probe donor. Unlike intensity based FRET, FLIM-FRET is insensitive to background noise, spectral bleed-through, quantum efficiency, and detector spectral sensitivity2. However, FLIM systems are expensive, complex, and uncommon, and work best with fluorophores with single exponent decay and no FLIM run-off 49. The described method may also be used with more advanced microscope platforms (e.g., FRET-TIRF, fluorescence anisotropy, and spectral correlation FRET). Applying this method to 3D imaging with high speed confocal and multiphoton microscopy will facilitate analysis of the sub-cellular distribution of signaling response and increase the accuracy of signaling analysis. This 3D FRET method will enable advanced cell biology studies in simulated 3D cellular microenvironments. As such, it can be readily applied to pharmacology and regenerative medicine needs, including studying intercellular signaling and screening drug response in engineered hydrogel-based microtissues.
Disclosures
The authors have no conflicts to declare.
Acknowledgments
The authors acknowledge the financial support of the School of Dental Medicine at the University of Pittsburgh, the National Institutes of Health award K01 AR062598, and grant P30 DE030740. The authors also thank Dr. Jin Zhang for the ICUE1 plasmid, Wayne Rasband for developing ImageJ and Michael Cammer for the ImageJ macro.
References
- Jares-Erijman EA, Jovin TM. Imaging molecular interactions in living cells by FRET microscopy. Curr. Opin. Chem. Biol. 2006;10(5):409–416. doi: 10.1016/j.cbpa.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Wallrabe H, Periasamy A. Imaging protein molecules using FRET and FLIM microscopy. Curr. Opin. Biotechnol. 2005;16(1):19–27. doi: 10.1016/j.copbio.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Nuber S, Hoffmann C. Fluorescence/bioluminescence resonance energy transfer techniques to study G-protein-coupled receptor activation and signaling. Pharmacol Rev. 2012;64(2):299–336. doi: 10.1124/pr.110.004309. [DOI] [PubMed] [Google Scholar]
- Baker BM, Chen CS. Deconstructing the third dimension - how 3D culture microenvironments alter cellular cues. J. Cell Sci. 2012;125(13):3015–3024. doi: 10.1242/jcs.079509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbitt MW, Anseth KS. Hydrogels as extracellular matrix mimics for 3D cell culture. Biotechnol. Bioeng. 2009;103(4):655–663. doi: 10.1002/bit.22361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mseka T, Bamburg JR, Cramer LP. ADF/cofilin family proteins control formation of oriented actin-filament bundles in the cell body to trigger fibroblast polarization. J. Cell Sci. 2007;120(24):4332–4344. doi: 10.1242/jcs.017640. [DOI] [PubMed] [Google Scholar]
- Holowka D, Sheets ED, Baird B. Interactions between Fc(epsilon)RI and lipid raft components are regulated by the actin cytoskeleton. J. Cell Sci. 2000;113 (Pt 6):1009–1019. doi: 10.1242/jcs.113.6.1009. [DOI] [PubMed] [Google Scholar]
- Alenghat FJ, Ingber DE. Mechanotransduction: all signals point to cytoskeleton, matrix, and integrins. Sci Signaling. 2002. [DOI] [PubMed]
- Guilak F, et al. Control of stem cell fate by physical interactions with the extracellular matrix. Cell stem cell. 2009;5(1):17–26. doi: 10.1016/j.stem.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polacheck WJ, Li R, Uzel SG, Kamm RD. Microfluidic platforms for mechanobiology. Lab Chip. 2013;13(12):2252–2267. doi: 10.1039/c3lc41393d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdick JA, Anseth KS. Photoencapsulation of osteoblasts in injectable RGD-modified PEG hydrogels for bone tissue engineering. Biomaterials. 2002;23(22):4315–4323. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- Benoit DSW, Schwartz MP, Durney AR, Anseth KS. Small functional groups for controlled differentiation of hydrogel-encapsulated human mesenchymal stem cells. Nat. Mater. 2008;7(10):816–823. doi: 10.1038/nmat2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang VL, et al. Fabrication of 3D hepatic tissues by additive photopatterning of cellular hydrogels. FASEB J. 2007;21(3):790–801. doi: 10.1096/fj.06-7117com. [DOI] [PubMed] [Google Scholar]
- Zhu J, Marchant RE. Design properties of hydrogel tissue-engineering scaffolds. Expert Rev. Med. Devices. 2011;8(5):607–626. doi: 10.1586/erd.11.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiet T, Vandenhaute M, Schelfhout J, Van Vlierberghe S, Dubruel P. A review of trends and limitations in hydrogel-rapid prototyping for tissue engineering. Biomaterials. 2012;33(26):6020–6041. doi: 10.1016/j.biomaterials.2012.04.050. [DOI] [PubMed] [Google Scholar]
- Kumbar SG, Laurencin CT, Deng M. Natural and Synthetic Biomedical Polymers. Elsevier; 2014. [Google Scholar]
- Kirschner CM, Anseth KS. Hydrogels in Healthcare: From Static to Dynamic Material Microenvironments. Acta Mater. 2013;61(3):931–944. doi: 10.1016/j.actamat.2012.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guvendiren M, Burdick JA. Engineering synthetic hydrogel microenvironments to instruct stem cells. Curr Opin Biotechnol. 2013;24(5):841–846. doi: 10.1016/j.copbio.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulrez SKH, Al-Assaf S, Phillips GO. In: Progress in Molecular and Environmental Bioengineering - From Analysis and Modeling to Technology Applications, Ch. 5. Capri A, editor. Europe: InTech; 2011. pp. 117–149. [Google Scholar]
- Iza M, Stoianovici G, Viora L, Grossiord JL, Couarraze G. Hydrogels of poly(ethylene glycol): mechanical characterization and release of a model drug. J. Controlled Release. 1998;52(1-2):41–51. doi: 10.1016/s0168-3659(97)00191-0. [DOI] [PubMed] [Google Scholar]
- Deligkaris K, Tadele TS, Olthuis W, van den Berg A. Hydrogel-based devices for biomedical applications. Sens. Actuators, B. 2010;147(2):765–774. [Google Scholar]
- Axelrod D, Koppel DE, Schlessinger J, Elson E, Webb WW. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 1976;16(9):1055–1069. doi: 10.1016/S0006-3495(76)85755-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Lin W, Zheng K, Zhu S. FRET-Based Small-Molecule Fluorescent Probes: Rational Design and Bioimaging Applications. Acc. Chem. Res. 2013;46(7):1462–1473. doi: 10.1021/ar300273v. [DOI] [PubMed] [Google Scholar]
- Spiering D, Bravo-Cordero JJ, Moshfegh Y, Miskolci V, Hodgson L. Quantitative ratiometric imaging of FRET-biosensors in living cells. Methods Cell Biol. 2013;114:593–609. doi: 10.1016/B978-0-12-407761-4.00025-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K. Ch 42, Nano/Micro Biotechnology. In: Nagamune IE, editor. Advances in Biochemical Engineering Biotechnology. Vol. 119. Berlin Heidelberg: Springer; 2010. pp. 63–78. [Google Scholar]
- Van Dyk TK, et al. Rapid and sensitive pollutant detection by induction of heat shock gene-bioluminescence gene fusions. Appl Environ Microbiol. 1994;60(5):1414–1420. doi: 10.1128/aem.60.5.1414-1420.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M, Tsai SL, Chen W. Microbial biosensors: engineered microorganisms as the sensing machinery. Sensors. 2013;13(5):5777–5795. doi: 10.3390/s130505777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piston DW, Kremers GJ. Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem Sci. 2007;32(9):407–414. doi: 10.1016/j.tibs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- Lin-Gibson S, et al. Synthesis and characterization of PEG dimethacrylates and their hydrogels. Biomacromolecules. 2004;5(4):1280–1287. doi: 10.1021/bm0498777. [DOI] [PubMed] [Google Scholar]
- Majima T, Schnabel W, Weber W. Phenyl-2,4,6-Trimethylbenzoylphosphinates as Water-Soluble Photoinitiators - Generation and Reactivity of O=P(C6h5)(O-) Radical-Anions. Makromol Chem. 1991;192(10):2307–2315. [Google Scholar]
- Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Photoinitiated polymerization of PEG-diacrylate with lithium phenyl-2,4,6-trimethylbenzoylphosphinate: polymerization rate and cytocompatibility. Biomaterials. 2009;30(35):6702–6707. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Ellis AV, Voelcker NH. Recent developments in PDMS surface modification for microfluidic devices. Electrophoresis. 2010;31(1):2–16. doi: 10.1002/elps.200900475. [DOI] [PubMed] [Google Scholar]
- DiPilato LM, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci U S A. 2004;101(47):16513–16518. doi: 10.1073/pnas.0405973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Occhetta P, et al. Fabrication of 3D cell-laden hydrogel microstructures through photo-mold patterning. Biofabrication. 2013;5(3):035002. doi: 10.1088/1758-5082/5/3/035002. [DOI] [PubMed] [Google Scholar]
- Kubow KE, et al. Crosslinking of cell-derived 3D scaffolds up-regulates the stretching and unfolding of new extracellular matrix assembled by reseeded cells. Integr. Biol. 2009;1(11-12):635–648. doi: 10.1039/b914996a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legant WR, Chen CS, Vogel V. Force-induced fibronectin assembly and matrix remodeling in a 3D microtissue model of tissue morphogenesis. Integr. Biol. 2012;4(10):1164–1174. doi: 10.1039/c2ib20059g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron AR, Frith JE, Gomez GA, Yap AS, Cooper-White JJ. The effect of time-dependent deformation of viscoelastic hydrogels on myogenic induction and Rac1 activity in mesenchymal stem cells. Biomaterials. 2014;35(6):1857–1868. doi: 10.1016/j.biomaterials.2013.11.023. [DOI] [PubMed] [Google Scholar]
- Kong HJ, Polte TR, Alsberg E, Mooney DJ. FRET measurements of cell-traction forces and nano-scale clustering of adhesion ligands varied by substrate stiffness. Proc Natl Acad Sci U S A. 2005;102(12):4300–4305. doi: 10.1073/pnas.0405873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poehler E, et al. Microchamber arrays with an integrated long luminescence lifetime pH sensor. Anal. Biochem. 2015. pp. 1–9. [DOI] [PubMed]
- Son KJ, Shin DS, Kwa T, Gao Y, Revzin A. Micropatterned Sensing Hydrogels Integrated with Reconfigurable Microfluidics for Detecting Protease Release from Cells. Anal. Chem. 2013;85(24):11893–11901. doi: 10.1021/ac402660z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aimetti AA, Tibbitt MW, Anseth KS. Human Neutrophil Elastase Responsive Delivery from Poly(ethylene glycol) Hydrogels. Biomacromolecules. 2009;10(6):1484–1489. doi: 10.1021/bm9000926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieshaber SE, Jha AK, Farran AJE, Jia X. In: Biomaterials for Tissue Engineering Applications: A Review of the Past and Future Trends, Ch 2. Burdick JA, Mauck RL, editors. Springer-Verlag; 2011. pp. 9–46. [Google Scholar]
- Chiu YC, Brey E, Pérez-Luna V. A Study of the Intrinsic Autofluorescence of Poly (ethylene glycol)-co (L -Lactic acid) Diacrylate. J. Fluoresc. 2012;22(3):907–913. doi: 10.1007/s10895-011-1029-6. [DOI] [PubMed] [Google Scholar]
- Dewitt DD, Kaszuba SN, Thompson DM, Stegemann JP. Collagen I-Matrigel Scaffolds for Enhanced Schwann Cell Survival and Control of Three-Dimensional Cell Morphology. Tissue Eng., Part A. 2009;15(10):2785–2793. doi: 10.1089/ten.TEA.2008.0406. [DOI] [PubMed] [Google Scholar]
- Taboas JM, Tuan RS, Hudson SD. Bioreactor device, and method and system for fabricating tissues in the bioreactor device. PCT/US2006/028417. United States patent. 2014
- Fedorovich NE, et al. The effect of photopolymerization on stem cells embedded in hydrogels. Biomaterials. 2009;30(3):344–353. doi: 10.1016/j.biomaterials.2008.09.037. [DOI] [PubMed] [Google Scholar]
- Vandame P, et al. Optimization of ERK activity biosensors for both ratiometric and lifetime FRET measurements. Sensors. 2013;14(1):1140–1154. doi: 10.3390/s140101140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponsioen B, et al. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 2004;5(12):1176–1180. doi: 10.1038/sj.embor.7400290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarenbeek JB, Goedhart J, Hink MA, Gadella TW, Jalink K. A mTurquoise-based cAMP sensor for both FLIM and ratiometric read-out has improved dynamic range. PLoS ONE. 2011;6(4):e19170. doi: 10.1371/journal.pone.0019170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Periasamy A. Intensity range based quantitative FRET data analysis to localize protein molecules in live cell nuclei. J. Fluoresc. 2006;16(1):95–104. doi: 10.1007/s10895-005-0024-1. [DOI] [PubMed] [Google Scholar]