

Abstract
RNA sequencing (RNAseq) is a versatile method that can be utilized to detect and characterize gene expression, mutations, gene fusions, and noncoding RNAs. Standard RNAseq requires 30 - 100 million sequencing reads and can include multiple RNA products such as mRNA and noncoding RNAs. We demonstrate how targeted RNAseq (capture) permits a focused study on selected RNA products using a desktop sequencer. RNAseq capture can characterize unannotated, low, or transiently expressed transcripts that may otherwise be missed using traditional RNAseq methods. Here we describe the extraction of RNA from cell lines, ribosomal RNA depletion, cDNA synthesis, preparation of barcoded libraries, hybridization and capture of targeted transcripts and multiplex sequencing on a desktop sequencer. We also outline the computational analysis pipeline, which includes quality control assessment, alignment, fusion detection, gene expression quantification and identification of single nucleotide variants. This assay allows for targeted transcript sequencing to characterize gene expression, gene fusions, and mutations.
Keywords: Molecular Biology, Issue 114, RNA sequencing, hybridization, capture, gene fusions, expression, targeted RNAseq
Introduction
Whole transcriptome or RNA sequencing (RNAseq) is an unbiased sequencing method to assess all RNA products. The goal of targeted RNAseq (Capture) is a focused evaluation of selected transcripts with increased sensitivity, dynamic range, reduced cost or scale, and increased throughput compared to standard RNAseq. Similar to standard RNAseq, targeted enrichment approaches can be used to evaluate gene expression, multiple RNA species such as mRNA, microRNA (miRNA), lncRNA1, other noncoding RNAs2, gene fusions3, and mutations4-6.
Capture involves hybridization of complementary oligonucleotides to enrich cDNA libraries for sequencing. The rationale for RNAseq Capture is similar to microarray approaches where complementary oligonucleotides or probes are hybridized to samples and then measured for relative abundance. For microarray technologies, expression is based on relative signal measured for transcripts binding to these probes. Microarrays are thus limited by range, potential background noise from non-specific binding, and cross-hybridization of probes. Furthermore, arrays have limited dynamic range for low and highly expressed transcripts compared to RNAseq1. Microarrays are widely utilized due to their reduced cost and high throughput capacity compared to RNAseq.
Here, we demonstrate a method for RNAseq Capture that offers a middle ground between RNAseq and microarray approaches for evaluating the transcriptome. RNAseq Capture has intermediate throughput, greater dynamic range and sensitivity, and is scaled for fast turnaround on desktop sequencers. RNAseq Capture also requires reduced computational resources in terms of storage space and data processing.
Protocol
Note: This protocol describes the simultaneous processing and analysis of four samples. This method is compatible with RNA isolated from cells, fresh frozen tissue and formalin-fixed paraffin-embedded tissue (FFPE). This protocol begins with 50 - 1,000 ng (250 ng recommended) of starting RNA input for each sample.
1. rRNA Depletion and Fragmentation of RNA Procedure
- rRNA Depletion
- Remove elute, prime, fragment mix, rRNA removal mix, rRNA binding buffer and resuspension buffer from -20 °C and thaw at room temperature. Remove elution buffer, rRNA removal beads and RNA/cDNA specific paramagnetic beads from 4 °C and bring to room temperature.
- Add 0.25 µg of RNA to PCR tube. Dilute the total RNA with nuclease-free ultra-pure water to a total final volume of 10 µl. Add 5 µl of rRNA binding buffer to each tube then add 5 µl of rRNA removal mix and gently pipette up and down to mix. Place tubes in thermal cycler with lid pre-heated to 100 °C and program thermal cycler to 68 °C for 5 min to denature the RNA.
- After RNA denaturation, remove tubes from thermal cycler and incubate at room temperature for 1 min. Vortex rRNA removal bead tube vigorously to resuspend the beads. Add 35 µl of rRNA removal beads to new tubes and transfer the RNA denaturation reaction (20 µl) to tubes containing rRNA removal beads.
- Adjust pipette to 45 µl and pipette quickly up and down 20x to mix. Incubate tubes at RT for 1 min. Then place tubes in magnetic stand at room temperature for 1 min. Transfer supernatant to newly labeled PCR tubes and place these tubes again in magnetic stand at room temperature for 1 min. This ensures that no beads are transferred. Transfer supernatant to newly labeled PCR tubes.
- Vortex RNA/cDNA specific paramagnetic beads and add 99 µl of beads to each tube. Gently pipette entire volume up and down 10x to mix. If starting with degraded-RNA, add 193 µl of well-mixed RNA/cDNA specific paramagnetic beads to each tube. Incubate at RT for 15 min. Then place tubes on the magnetic stand at room temperature for 5 min. Remove and discard the supernatants.
- Add 200 µl of freshly prepared 70% ethanol (EtOH). Keep tubes on magnetic stand and take care to not disturb the beads. Incubate at room temperature for 30 sec, then remove and discard supernatant. Allow tubes to stand at room temperature for 5 - 10 min to dry.
- Centrifuge thawed room temperature elution buffer at 600 × g for 5 sec. Add 11 µl of elution buffer to each tube and gently pipette up and down 10x to mix. Incubate tubes at RT for 2 min. Place tubes on magnetic stand at room temperature for 5 min. Transfer 8.5 µl of the supernatant to newly labeled PCR tubes.
- Fragmentation of rRNA Depleted RNA
- Add 8.5 µl elute, 8.5 µl prime, and 8.5 µl fragment mix to each tube. Gently pipette up and down 10x to mix. Place tubes in thermal cycler with following program: pre-heated lid to 100 °C, 94 °C for 8 min, then 4 °C hold. NOTE: This step is designed to generate an average insert size of 155 bp. If average fragment size of RNA sample is lower than 200 bp, skip this step and proceed to First Strand cDNA Synthesis.
2. cDNA Synthesis
- Synthesize First Strand cDNA
- Remove first strand synthesis mix from -20 °C and thaw at room temperature.
- Pre-program the thermal cycler with the following settings: pre-heat lid option and set to 100 °C, then 25 °C for 10 min, 42 °C for 15 min, 70 °C for 15 min and hold at 4 °C. Save this program as "Synthesize 1st Strand."
- Centrifuge thawed-first strand synthesis mix tube at 600 × g for 5 sec. Mix 1 µl reverse transcriptase with 9 µl of first strand synthesis mix.
- Add 8 µl of first strand synthesis and reverse transcriptase mix to each tube, gently pipette up and down 6x to mix. Centrifuge tubes for 4 sec using a fixed speed mini tabletop centrifuge at 6.0 x g to bring liquid to bottom. Place tubes in the thermal cycler and select Synthesize 1st Strand. When the thermal cycler reaches 4 °C, remove tubes and proceed immediately to Synthesize Second Strand cDNA.
- Synthesize Second Strand cDNA
- Pre-heat thermal cycler to 16 °C with lid pre-heated to 30 °C. Thaw second strand master mix and re-suspension buffer on ice. In advance, remove the bottle of paramagnetic beads from 4 °C and let stand for at least 30 min to bring them to room temperature.
- Add 5 µl of resuspension buffer to each PCR tube. Centrifuge second strand master mix at 600 × g for 5 sec and add 20 µl to each PCR tube. Place tubes in pre-heated thermal cycler at 16 °C for 1 hr. When incubation is completed, remove from thermal cycler and allow tubes to come to room temperature.
- Vortex the paramagnetic beads until they are well dispersed. Add 90 µl of well-mixed paramagnetic beads to each tube. Gently pipette the entire volume up and down 10x to mix. Incubate tubes at room temperature for 15 min. Place tubes on the magnetic stand at room temperature for 5 min.
- Remove and discard 135 µl of supernatant then leave tubes on the magnetic stand to perform EtOH wash. Add 200 µl freshly prepared 80% EtOH to each tube without disturbing the beads, then incubate at room temperature for 30 sec. Remove and discard all of the supernatant from each. Repeat for a total of two 80% EtOH washes.
- Let tubes stand at room temperature for 5 - 10 min to dry on the magnetic stand. Centrifuge the thawed room temperature resuspension buffer at 600 × g for 5 sec. Remove PCR tubes from magnetic stand. Add 17.5 µl resuspension buffer to each PCR tube and gently pipette the entire volume up and down 10x to mix thoroughly. Incubate tubes for 2 min at room temperature.
- Place tubes on the magnetic stand at room temperature for 5 min, then transfer 15 µl supernatant (double stranded cDNA) to 0.2 ml PCR strip tubes. NOTE: This is a safe stopping point as cDNA can be stored at -20 °C for up to 7 days.
3. Library Preparation
- Adenylate 3' Ends
- Remove A-tailing mix from -20 °C and thaw at room temperature. Pre-program the thermal cycler with the following settings: choose the pre-heat lid option and set to 100 °C, then set at 37 °C for 30 min, 70 °C for 5 min and hold at 4 °C. Save this program as "ATAIL70."
- Add 2.5 µl of resuspension buffer to each tube. Then add 12.5 µl of thawed A-tailing mix. Gently pipette the entire volume up and down 10x to mix thoroughly. Place the tubes in the thermal cycler and select ATAIL70. When the thermal cycler temperature is at 4 °C, remove the PCR tubes from the thermal cycler and proceed immediately to ligate adapters.
- Ligate Adapters
- Remove appropriate RNA adapter tubes, stop ligation buffer and resuspension buffer from -20 °C and thaw at room temperature. Do not remove the ligation mix tube from -20 °C until instructed to do so in the protocol. Remove the bottle of paramagnetic beads from 4 °C and let stand for at least 30 min to bring to room temperature.
- Pre-heat the thermal cycler to 30 °C and choose the pre-heat lid option and set to 100 °C. Centrifuge the thawed RNA adapter tubes at 600 × g for 5 sec. Immediately before use, remove the ligation mix tube from -20 °C storage.
- Add 2.5 µl of resuspension buffer to each sample tube. Add 2.5 µl of ligation mix. Return the ligation mix tube to -20 °C storage immediately after use. Add 2.5 µl of the thawed RNA adapter index to each sample tube. Gently pipette the entire volume up and down 10x to mix thoroughly.
- Centrifuge tubes for 4 sec using a fixed speed mini tabletop centrifuge at 6.0 x g. Place tubes in the pre-heated thermal cycler. Close the lid and incubate at 30 °C for 10 min.
- Remove tubes from thermal cycler and add 5 µl of stop ligation buffer to each tube to inactivate the ligation. Gently pipette the entire volume up and down 10x to mix thoroughly.
- Repeat wash as described in 2.2.3 - 2.2.4, using 42 µl of mixed paramagnetic beads, and discarding 79.5 µl supernatant. With the tubes on the magnetic stand, let the samples air-dry at room temperature for 5 - 10 min.
- Remove the PCR strip tubes from the magnetic stand and add 52.5 µl resuspension buffer to each tube. Gently pipette the entire volume up and down 10x to mix thoroughly. Incubate the tubes at room temperature for 2 min. Place the tubes on the magnetic stand at RT for 5 min or until the liquid is clear. Transfer 50 µl of supernatant from each tube to new 0.2 ml PCR strip tubes. Take care not to disturb the beads.
- Repeat wash as described in 2.2.3 - 2.2.4, using 50 µl of mixed paramagnetic beads, and discarding 95 µl supernatant. With the tubes on the magnetic stand, let the samples air-dry at RT for 5 - 10 min.
- Remove the PCR strip tubes from the magnetic stand and add 22.5 µl resuspension buffer to each tube. Gently pipette the entire volume up and down 10x to mix thoroughly.
- Incubate the tubes at RT for 2 min. Place the tubes on the magnetic stand at room temperature for 5 min or until the liquid is clear. Transfer 20 µl supernatant from each tube to a new 0.2 ml PCR strip tube. Take care not to disturb the beads. This is a safe stopping point and cDNA can be stored at -20 °C for up to 7 days.
4. Library Amplification
- Enrich DNA Fragments
- Remove the PCR master mix, PCR primer cocktail and resuspension buffer from -20 °C storage and thaw at room temperature. Remove the bottle of paramagnetic beads from 4 °C storage and let stand for at least 30 min to bring to room temperature.
- Pre-program the thermal cycler with the following settings: choose the pre-heat lid option and set to 100 °C, then set initial denaturation at 98 °C for 30 sec, 15 cycles of denaturation at 98 °C for 10 sec, annealing at 60 °C for 30 sec, and extension at 72 °C for 30 sec, one final extension cycle at 72 °C for 5 min and hold at 4 °C. Save this program as "PCR."
- Centrifuge the thawed PCR master mix and PCR primers tubes at 600 × g for 5 sec. Add 5 µl of thawed PCR primers to each sample tube. Add 25 µl of thawed PCR master mix to each sample tube. Gently pipette the entire volume up and down 10x to mix thoroughly.
- Place the capped PCR strip tubes in the pre-programmed thermal cycler. Close the lid and run PCR program. When PCR is completed, remove tubes from thermal cycler and keep on ice.
- Repeat wash as described in 2.2.3 - 2.2.4, using 50 µl of mixed paramagnetic beads, and discarding 95 µl supernatant. With the tubes on the magnetic stand, let the samples air-dry at room temperature for 5 - 10 min.
- Remove tubes from the magnetic stand and add 32.5 µl resuspension buffer to each sample tube. Gently pipette the entire volume up and down 10x to mix thoroughly. Incubate at RT for 2 min. Place the PCR tubes on the magnetic stand at room temperature for 5 min or until the liquid is clear. Then transfer 30 µl supernatant to fresh 0.2 ml PCR tubes.
- Perform quantification of cDNA using a fluorometer7 and determine cDNA quality using a capillary electrophoresis system8. NOTE: This is a safe stopping point and cDNA can be stored at -20 °C for up to 7 days. Note: This library is considered a RNAseq library. Subsequent steps lead to a Capture library.
5. Hybridization, Capture and Sequencing
- Multiplexed Hybridization
- Remove custom hydrated probes, Cot-1 DNA, universal blocking oligos, and adapter specific blocking oligos from -20 °C and thaw on ice.
- In a low bind 1.5 ml tube, combine 500 ng DNA (125 ng per sample when multiplexing 4 samples), 5 µl Cot-1 DNA (1 µg/µl), 1ul universal blocking oligos, 0.5 µl p7 (6 nucleotide) adapter specific blocking oligos (this amount may need to be adjusted depending on multiplex conditions) and 0.5 µl p7 (8 nucleotide) adapter specific blocking oligos (this amount may need to be adjusted depending on multiplex conditions).
- Place the sample tube in a vacuum concentrator with cap open facing the opposite direction of rotation. Dry contents at 45 °C for 20 min or until complete evaporation of the liquid.
- Resuspend dried content with 8.5 µl 2x hybridization buffer, 3.4 µl hybridization component A and 1.1 µl nuclease free water. Allow 10 min for resuspension and vortex every 2.5 min. Transfer resuspended-material to a 0.2 ml PCR tube and incubate at 95 °C for 10 min on a thermal cycler.
- Remove hybridization sample tube from thermal cycler and add 2 µl of custom probes resuspended at a concentration of 1.5 pmol/µl. Alternatively, add 4 µl of custom probes resuspended at a concentration of 0.75 pmol/µl. Incubate hybridization reaction overnight (16 - 24 hr) at 65 °C. NOTE: Probes purchased from various Vendors may be used and manufacturer's instructions should be followed. The duration of the hybridization step may also vary.
- Bead Preparation and Capture
- Remove bottle of streptavidin-coupled paramagnetic beads from 4 °C and equilibrate at room temperature for 30 min. Dilute 10x Wash Buffers (I, II, III, and Stringent) and 2.5x Bead Wash Buffer to create 1x working solutions.
- Aliquot 140 µl of 1x wash buffer I into to a fresh 1.5 ml tube. Heat entire amount of 1x stringent buffer and aliquot of 1x wash buffer I at 65 °C in a heat block for at least 2 hr.
- Aliquot 100 µl of streptavidin-coupled paramagnetic beads per capture into a 1.5 ml tube. Place on magnet and discard supernatant. Add 200 µl bead wash buffer per 100 µl beads and vortex for 10 sec. Place on magnet for 2 - 5 min or until supernatant is clear. Once supernatant is clear, discard it and repeat once more for a total of two washes.
- After removal of bead wash buffer, add equal volume bead wash buffer as initial starting volume (i.e., 100 µl for one capture). Resuspend and transfer to a 0.2 ml PCR tube. Place tube in magnetic rack for 2 - 5 min or until supernatant is clear. Discard supernatant.
- With both the hybridization sample and beads in the thermal cycler at 65 °C, transfer the hybridization mix to the bead tube and pipette up and down 10x to mix. Incubate at 65 °C for 45 min, vortex and spin down sample for 4 sec using a fixed speed (6.0 x g) table-top mini centrifuge.
- Bead Wash
- Remove capture tube from thermal cycler and add 100 µl pre-heated 1x Wash Buffer I to the tube and vortex for 10 sec to mix. Transfer the mixture to a fresh low bind 1.5 ml tube. Place the tube in a magnetic separation rack and allow 2 - 5 min for separation or until supernatant is clear. Discard supernatant.
- Add 200 µl preheated 1x stringent wash buffer and pipette up and down 10 times to mix. Incubate at 65 °C for 5 min. Place the tube in the magnetic separation rack and allow 2 - 3 min for separation or until supernatant is clear. Discard supernatant. Repeat stringent wash once more for a total of two washes.
- Add 200 µl room temperature 1x wash buffer I and vortex for 2 min to mix. Place the tube in the magnetic separation rack and allowing 2 - 5 min for separation or until supernatant is clear. Discard supernatant.
- Add 200 µl room temperature 1x wash buffer II and vortex for 1 min to mix. Place the tube in the magnetic separation rack and allowing 2 - 5 min for separation or until supernatant is clear. Discard supernatant.
- Add 200 µl room temperature 1x wash buffer III and vortex for 30 sec to mix. Place the tube in the magnetic separation rack allowing 2 - 5 min for separation or until supernatant is clear. Discard supernatant.
- Remove the tube from the magnetic separation rack and add 20 µl nuclease-free water to resuspend the beads. Mix thoroughly by pipetting up and down 10 times.
- Post Capture PCR Amplification
- Remove paramagnetic beads from 4 °C and equilibrate at room temperature for 30 min.
- Remove the hot start PCR Ready Mix (2x) and PCR Primer Mix from -20 °C and thaw at RT and then place on ice. Prepare library amplification Master Mix by combining 27.5 µl of 2x hot start PCR Ready Mix with 2.75 µl of PCR Primer 1 and 2.75 µl of PCR primer 2 (these volumes are for 1 hybridization library plus 10% excess).
- Setup the reaction in PCR tube by adding 20 µl of beads plus captured DNA with 30 µl of library amplification master mix for a total volume of 50 µl. Cap tube properly and vortex to mix. Centrifuge tubes for 4 sec using a fixed speed mini table-top centrifuge at 6.0 x g.
- Set up the following PCR program: choose the pre-heat lid option and set to 100 °C, then set initial denaturation at 98 °C for 45 sec, 10 - 12 cycles of denaturation at 98 °C for 15 sec, annealing at 65 °C for 30 sec and extension at 72 °C for 60 sec, one final extension cycle at 72 °C for 60 sec and hold at 4 °C.
- Remove sample from thermocycler and add 75 µl paramagnetic beads. Mix well and incubate at RT for 15 min.
- Place tubes on magnet at room temperature for 2 - 3 min and then remove supernatant. Wash beads on magnet by adding 200 µl 80% Ethanol, incubating for 30 sec then removing supernatant. Repeat for a total of two 80% washes.
- Incubate at RT for 5 - 10 min to allow beads to dry. Do not over dry to cracking. Resuspend beads in 22 µl of Tris-EDTA pH 8.0 (1x TE Solution) and allow 3 min for elution. Place sample on magnet for 3 - 5 min then transfer 20 μl of eluted product to a fresh low-bind 1.5 ml tube, ensuring no beads are carried over.
- Perform quantification of captured cDNA using fluorometer7 and determine captured cDNA quality using a capillary electrophoresis system 8.
- Desktop Sequencer Loading Procedure 9
- Dilute captured cDNA library to a final concentration of 4 nM using 10 mM Tris-Cl pH 8.5 with 0.1% Tween 20. Thaw 10 N NaOH and Hybridization buffer on ice. Approximately 30 min before use, thaw desktop sequencer v2 reagent kit box 1 in RT water. Do not fill above MAX FILL LINE 10. Please note this is a sequencing platform specific procedure and may vary per manufacturer's instructions.
- Prepare 1ml of 0.2 N NaOH by combining 20 µl 10 N NaOH with 980 µl nuclease free water in a microcentrifuge tube (always prepare fresh). Dilute the PhiX library (library control) to 4 nM by combining 2 µl of 10 nM library control with 3 µl 10mM Tris-Cl pH 8.5 with 0.1% Tween 20.
- Denature final library and library control by combining 5 µl of 4nM library with 5 µl of 0.2 N NaOH and vortex briefly to mix. Centrifuge tubes for 4 sec using a fixed speed mini table-top centrifuge at 6.0 x g. Incubate at RT for 5 min to denature libraries.
- Add 990 µl of pre-chilled hybridization buffer to the tubes containing 10 µl of denatured libraries. This results in a 20 pM library. Mark the denatured 20 pM library with the date and can be stored for up to 3 weeks at -20 °C.
- Mix 375 µl of 20 pM library control with 225 µl of pre-chilled hybridization buffer and diluted library control to result in a 12.5 pM library control. Invert several times to mix the solution.
- Combine 594 µl of denatured final library with 6 µl of 12.5 pM denatured library control and vortex to mix. Set the combined sample library and library control aside on ice until samples are ready to load into the desktop sequencer reagent cartridge.
6. Data Analysis
- Sequence Quality Assessment
- Calculate the quality of the raw sequence data (fastq files) using sequence quality assessment11. NOTE: This step helps to assess the data before it is further subjected to downstream analysis. The software runs with in-built parameters and produces a set of metrics for each fastq file.
- Alignment
- Align sequence reads (fastq files) to the reference human genome hg19 and transcriptome using Tophat2 12 (version 2.0.10) while providing known transcripts as a GTF file. The output is in the form of a binary alignment format called the BAM file.
- Perform post processing steps including sorting and indexing using Samtools13 (version 0.1.19) on the BAM file. Perform duplicate marking, reordering SAM, insert size calculation and adding or replacing read groups using Picard tools14 (version 1.84).
- RNAseq Quality Assessment
- Compute a series of quality control metrics for RNAseq data using RNAseq quality assessment. The input to this software is a BAM file 15 from the Tophat2 alignment. The output is a HTML file that lists total read count, duplicates, mapped read percentage and rRNA percentage, etc., among others.
- Variant Calling
- Use STAR (version 2.4.0)16 for alignment and then call single nucleotide variants using GATK's (Version 3.3-0) HaplotypeCaller17.Follow GATK BAM post-processing steps and filtering criteria to flag and remove false positives from the output.
- Gene Expression
- Calculate gene expression using Cufflinks software (version 2.1.1) from Tuxedo suite18. NOTE: The input is a BAM file from Tophat2 alignment tool. The output is produced at the isoform, gene and transcript level, where expression is calculated as FPKM (Fragments per Kilobase per Million Mapped Reads).
- Fusion Calling
- Call fusions from each sample using ChimeraScan19 (version 0.4.5), Tophat Fusion20 custom and TRUP21. Annotate the fusions for domains using Oncofuse22 (version 1.0.9b2).
Representative Results
A schematic highlighting key steps in RNAseq Capture is shown in Figure 1. Four cancer cell lines with known mutations were used to demonstrate the effectiveness of the RNAseq Capture technique (K562 with ABL1 fusion, LC2 with RET fusion, EOL1 with PDGFRalpha fusion and RT-4 with FGFR3 fusion). The four samples were pooled together and sequenced with 2x 100 bp reads on a desktop sequencer, which generates FASTQ files. FASTQ files were run through an RNAseq analysis pipeline, which includes five main components: 1) quality control assessment, 2) alignment to human transcriptome, 3) gene expression quantification, 4) fusion calling, and 5) variant calling. The alignment file (BAM) is used to call single nucleotide variants and calculate gene expression. Fusions are called using fusion callers, such as TopHat Fusion (performing their own alignment) and the output is annotated using fusion detection software.
Comparison of gene expression from RNAseq and capture demonstrates enrichment of targeted transcripts by 10 to 1,000-fold using the capture method (Figure 2A). Additionally, Figure 2B shows an increase in the percent of reads mapping to the targeted transcript regions using capture compared to RNAseq. Assessment of quality control measures is represented in Figure 3. Capture and RNAseq perform equally in terms of alignment to the transcriptome (3A, 94% vs. 93%) and mean insert size (3B, 174 bp vs. 162 bp). Using the capture method, a higher percentage of exonic regions are sequenced (3C, 77% vs. 60%), and conversely a lower percentage of intronic regions are sequenced (3D, 4% vs. 20%). Total read counts per sample are depicted in 3E, and as expected, RNAseq generated over 50-fold more reads than capture. Finally, the percentage of rRNA sequences present in each sample was lower using the capture method when compared to RNAseq (3F, 4% vs. 15%).
Fusion detection output shown in Table 1 is generated with normalized fusion supporting reads. Capture RNAseq was successful in detecting fusions for all four cell lines. Comparison of single nucleotide variants called in overlapping regions of capture and RNAseq is displayed in Figure 4. This demonstrates a high concordance of variants between Capture and RNAseq within the target region.
 Figure 1. Schematic of RNAseq Capture Steps. In this experimental demonstration, RNA is first depleted of ribosomal RNA, followed by chemical fragmentation and synthesis of complementary DNA (cDNA) using reverse transcriptase. Next, the cDNA is polyadenylated and ligated on both ends to platform-specific adaptors to generate a library. Only cDNA libraries with proper adaptors are then amplified by PCR. Libraries are then hybridized to custom oligonucleotide probes and captured using magnetic beads. This small amount of captured library must be amplified a second time to have enough for next generation sequencing. Multiple libraries can then be sequenced in parallel. Sequencing data is analyzed for RNA events of interest such as gene fusions, expression or mutations. Please click here to view a larger version of this figure.
Figure 1. Schematic of RNAseq Capture Steps. In this experimental demonstration, RNA is first depleted of ribosomal RNA, followed by chemical fragmentation and synthesis of complementary DNA (cDNA) using reverse transcriptase. Next, the cDNA is polyadenylated and ligated on both ends to platform-specific adaptors to generate a library. Only cDNA libraries with proper adaptors are then amplified by PCR. Libraries are then hybridized to custom oligonucleotide probes and captured using magnetic beads. This small amount of captured library must be amplified a second time to have enough for next generation sequencing. Multiple libraries can then be sequenced in parallel. Sequencing data is analyzed for RNA events of interest such as gene fusions, expression or mutations. Please click here to view a larger version of this figure.
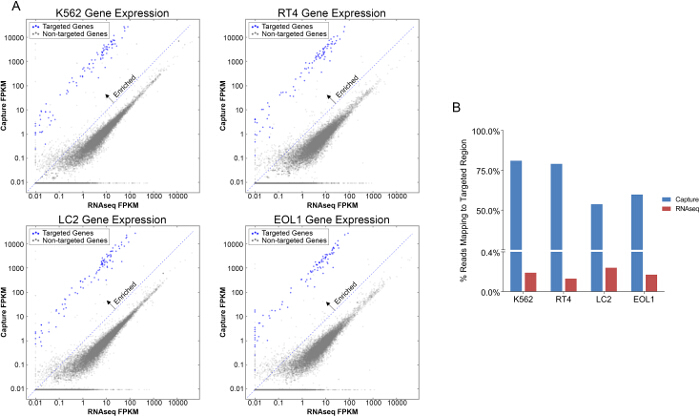 Figure 2. Comparison of Targeted Genes in Capture versus RNAseq. A, Gene expression comparison between Capture and RNAseq in four cancer cell lines K562, LC2, EOL1 and RT-4 measured by reads per kilobase per million mapped reads (FPKM)(Log scale). Targeted genes of interest are enriched (blue) compared to non-targeted genes (grey). B, Percentage of reads mapping to targeted region is increased in Capture versus RNAseq libraries in four cancer cell lines. Please click here to view a larger version of this figure.
Figure 2. Comparison of Targeted Genes in Capture versus RNAseq. A, Gene expression comparison between Capture and RNAseq in four cancer cell lines K562, LC2, EOL1 and RT-4 measured by reads per kilobase per million mapped reads (FPKM)(Log scale). Targeted genes of interest are enriched (blue) compared to non-targeted genes (grey). B, Percentage of reads mapping to targeted region is increased in Capture versus RNAseq libraries in four cancer cell lines. Please click here to view a larger version of this figure.
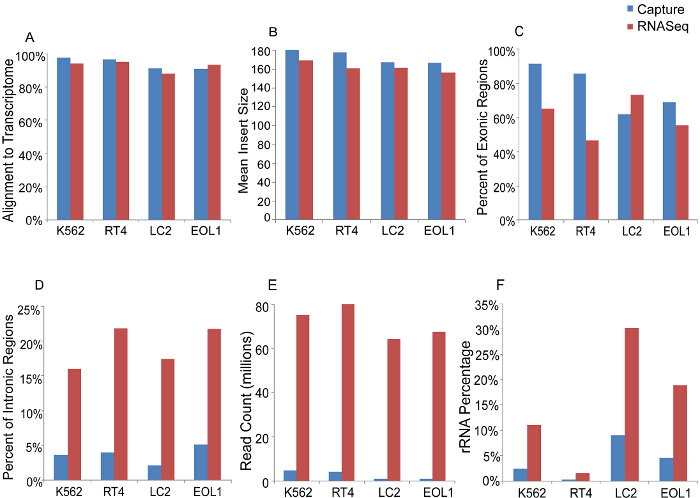 Figure 3. Sequencing Metrics of Capture versus RNAseq in Four Representative Cancer Cell Lines. A, Percentage of reads mapping to the transcriptome, B, Mean insert size of libraries. C, Percentage of reads in exonic regions. D, Percentage of reads in intronic regions. E, Total sequencing reads. F, Percentage of reads mapping to ribosomal RNA. Please click here to view a larger version of this figure.
Figure 3. Sequencing Metrics of Capture versus RNAseq in Four Representative Cancer Cell Lines. A, Percentage of reads mapping to the transcriptome, B, Mean insert size of libraries. C, Percentage of reads in exonic regions. D, Percentage of reads in intronic regions. E, Total sequencing reads. F, Percentage of reads mapping to ribosomal RNA. Please click here to view a larger version of this figure.
| Cell Line | Fusion | Library Type | Total Reads | On Target Reads | Normalized Fusion Supporting Reads (NFSR) | ||
| TophatFusion | ChimeraScan | TRUP | |||||
| K562 | BCR-ABL | RNAseq | 150,300,482 | 279,438 | 0 | 438 | 0 |
| Capture | 9,341,148 | 7,566,087 | 598 | 343 | 0 | ||
| LC2 | CCDC6-RET | RNAseq | 128,861,790 | 307,566 | 0 | 97 | 0 |
| Capture | 12,320,692 | 10,314,284 | 71 | 44 | 6 | ||
| EOL1 | FIP1L1-PDGFRA | RNAseq | 135,321,406 | 225,222 | 0 | 0 | 170 |
| Capture | 9,317,418 | 7,680,818 | 143 | 0 | 7 | ||
| RT4 | FGFR3-TACC3 | RNAseq | 161,350,024 | 208,741 | 0 | 131 | 469 |
| Capture | 8,305,950 | 6,563,574 | 358 | 88 | 34 |
Table 1. Fusion Detection for Capture versus RNAseq of K562, LC2, EOL1 and RT-4. This Table displays four cancer cell lines and three different fusion detection algorithms, TopHat2, ChimeraScan, and TRUP utilized in this demonstration. This table demonstrates the ability to detect fusions with Capture using less than 10 million total reads compared to greater than 60 million reads utilized for RNAseq. Fusion supporting reads were calculated by dividing fusion supporting reads by kinase reads, multiplied by one million.
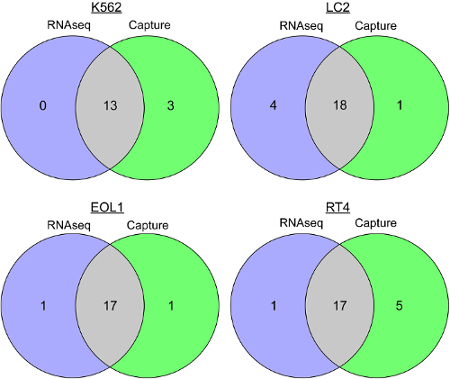 Figure 4. SNV calling for Capture versus RNAseq. These Venn diagrams show the number of Single Nucleotide Variants (SNVs) that were detected by Capture and RNAseq for each of four cell lines (K562, LC2, EOL1 and RT-4). This illustrates high concordance of SNVs between Capture and RNAseq within targeted-region: K562 (81.3%), LC2 (78.3%), EOL1 (89.5%) and RT-4 (73.9%). Please click here to view a larger version of this figure.
Figure 4. SNV calling for Capture versus RNAseq. These Venn diagrams show the number of Single Nucleotide Variants (SNVs) that were detected by Capture and RNAseq for each of four cell lines (K562, LC2, EOL1 and RT-4). This illustrates high concordance of SNVs between Capture and RNAseq within targeted-region: K562 (81.3%), LC2 (78.3%), EOL1 (89.5%) and RT-4 (73.9%). Please click here to view a larger version of this figure.
Discussion
RNAseq Capture is an intermediate strategy between RNAseq and microarray approaches for evaluating a selected part of the transcriptome. The advantages of Capture include reduced cost, rapid turnaround time on a desktop sequencer, high throughput, and detection of genomic alterations. The method can be adapted to characterize non-coding RNAs23, detect single nucleotide variants4-6, examine RNA splicing, and to identify gene fusions or structural rearrangements24. Further, this approach can be applied to clinical or processed samples that have undergone fixation with formalin and embedded in paraffin blocks24,25.
There are several significant benefits of RNAseq capture as compared to microarray, real-time quantitative PCR, Sanger sequencing and DNA sequencing. Microarray is limited by high background due to cross-hybridization and non-specific binding of probes. Quantification of genes with low expression is restricted due to background noise, while highly expressed gene measurements are affected by signal saturation1. Compared to RNAseq capture, real-time PCR proves difficult to reproduce. Additionally, RNAseq allows for detection of novel transcripts, requires less starting input material and can detect alternative splicing26.In contrast to Sanger sequencing, RNAseq allows for higher throughput and analysis of low expressed miRNA. Sanger sequencing has proved to be a valuable tool for verification of fusions with known exon-exon junctions and somatic DNA mutations, however identification of novel fusions is hindered by requirements of a priori candidate breakpoint. DNA sequencing is not cost efficient, requires larger storage space for data, and is incapable of detection of post-transcriptional modifications.
There are several critical steps involved in RNAseq Capture. First, to improve yield of library products from the RNA/cDNA specific paramagnetic beads and paramagnetic beads during washes, be cautious not to over dry the beads, which will lead to loss of yield. Also, do not under-dry the beads, ensure all ethanol is removed from the sample tubes, as ethanol can reduce cDNA yield. Second, the hybridization of cDNA libraries with complementary probes is dependent on consistent temperature, we recommend warming Wash Buffer I and Stringent Buffer to 65 °C for at least two hr in advance. Further, after hybridization it is essential to maintain 65 °C during the binding and wash steps. The probes used here were designed for the exons of genes of interest for drug development including kinases, genes involved in common rearrangements such as transcription factors, and house keeping genes. Moreover, gene content is customizable and capture panel sizes can vary. Further, as new information on genomic regions arises, additional probes can be designed and added to the existing capture panel.
Evaluation of alignment metrics, specifically on-target rate, provides information on how well the targeted region was enriched. A low on-target rate may be due to a failed hybridization and capture, whereby the desired target region was not captured and enriched. In this case, a re-hybridization and capture of the library set must be performed. A low on-target rate may also be due to failure to deplete rRNA, which can be confirmed by calculating the percentage of rRNA in the samples. High rRNA percentage within the sample will require re-preparation of the sample beginning with rRNA depletion. Additionally, if library concentration falls below the requirements for hybridization and capture, it would be advisable to optimize the amount of starting input for the sample type and quality (range: 50-1,000 ng).
While there are several advantages for targeted RNAseq applications, there are also limitations to consider. Samples with poor RNA quality based on RIN or degree of fragmentation may not yield quality libraries for sequencing. Several groups have demonstrated success with formalin fixed paraffin-embedded samples, however there are samples that will not pass for sequencing24,25,27. Further, since RNAseq Capture focuses on known transcripts, it loses the benefits of unbiased RNAseq for novel or unannotated transcripts. In addition, for SNP detection, RNAseq methods can only detect mutations in expressed transcripts.
Future opportunities of RNAseq Capture include research and clinical applications. Recent discovery of thousands of long non-coding RNA and their role in biology will require focused characterization. In the clinic, RNAseq Capture may extend beyond research testing and translate into clinical assays to characterize human disease such as cancer, infectious disease, and non-invasive testing. In conjunction with genomic sequencing approaches, RNAseq Capture can be integrated to study and characterize the expressed genome.
Disclosures
S.R. receives funding from Novartis and Ariad Pharmaceuticals for conduct of clinical trials. S.R. immediate family members own stock in Johnson and Johnson.
Acknowledgments
We give special thanks to Ezra Lyon, Eliot Zhu, Michele Wing, Esko Kautto and Eric Samorodnitsky for technical support. We would also like to thank Jenny Badillo for her administrative support for our team. We acknowledge the Ohio Supercomputer Center (OSC) for providing disk space, processing capacity, and support to run our analyses. We thank the Comprehensive Cancer Center (CCC) at The Ohio State University Wexner Medical Center for their administrative support of this work. S.R. and Team are supported by the American Cancer Society (MRSG-12-194-01-TBG), a Prostate Cancer Foundation Young Investigator Award, NHGRI (UM1HG006508-01A1), Fore Cancer Research Foundation, American Lung Association, and Pelotonia.
References
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer TR, et al. Targeted sequencing for gene discovery and quantification using RNA CaptureSeq. Nat Protoc. 2014;9:989–1009. doi: 10.1038/nprot.2014.058. [DOI] [PubMed] [Google Scholar]
- Maher CA, et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc Natl Acad Sci U S A. 2009;106:12353–12358. doi: 10.1073/pnas.0904720106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskol R, Ramaswami G, Li JB. Reliable identification of genomic variants from RNA-seq data. Am J Hum Genet. 2013;93:641–651. doi: 10.1016/j.ajhg.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn EM, et al. Development of strategies for SNP detection in RNA-seq data: application to lymphoblastoid cell lines and evaluation using 1000 Genomes data. PLoS One. 2013;8:e58815. doi: 10.1371/journal.pone.0058815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, et al. The eSNV-detect: a computational system to identify expressed single nucleotide variants from transcriptome sequencing data. Nucleic Acids Res. 2014;42:e172. doi: 10.1093/nar/gku1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Invitrogen. Qubit dsDNA HS Assay Kit Manual. 2010. Available from: http://www.science.smith.edu/cmbs/documents/QubitdsDNAHSAssay.pdf.
- Agilent. Agilent D1000 ScreenTape System Quick Guide. 2013. Available from: http://www.agilent.com/cs/library/usermanuals/Public/G2964-90032_ScreenTape_D1000_QG.pdf.
- Illumina. Preparing Libraries for Sequencing on the MiSeq®. 2013. Available from: https://support.illumina.com/content/dam/illumina-support/documents/documentation/system_documentation/miseq/preparing-libraries-for-sequencing-on-miseq-15039740-d.pdf.
- Illumina. MiSeq® Reagent Kit v2 Reagen Preparation Guide. 2012. Available from: https://support.illumina.com/downloads/miseq_reagent_kit_reagent_preparation_guide.html.
- Andrews S. FastQC. 2015. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Kim D, et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute. Picard. 2014. Available from: http://picard.sourceforge.net/
- DeLuca DS, et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics. 2012;28:1530–1532. doi: 10.1093/bioinformatics/bts196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;11:11.10.1–11.10.33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer MK, Chinnaiyan AM, Maher CA. ChimeraScan: a tool for identifying chimeric transcription in sequencing data. Bioinformatics. 2011;27:2903–2904. doi: 10.1093/bioinformatics/btr467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011;12:R72. doi: 10.1186/gb-2011-12-8-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Cuesta L, et al. Identification of novel fusion genes in lung cancer using breakpoint assembly of transcriptome sequencing data. Genome Biol. 2015;16:7. doi: 10.1186/s13059-014-0558-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shugay M, Ortiz de Mendibil , Vizmanos I, L J, Novo FJ. Oncofuse: a computational framework for the prediction of the oncogenic potential of gene fusions. Bioinformatics. 2013;29:2539–2546. doi: 10.1093/bioinformatics/btt445. [DOI] [PubMed] [Google Scholar]
- Clark MB, et al. Quantitative gene profiling of long noncoding RNAs with targeted RNA sequencing. Nat Methods. 2015;12:339–342. doi: 10.1038/nmeth.3321. [DOI] [PubMed] [Google Scholar]
- Cieslik M, et al. The use of exome capture RNA-seq for highly degraded RNA with application to clinical cancer sequencing. Genome Res. 2015. [DOI] [PMC free article] [PubMed]
- Cabanski CR, et al. cDNA hybrid capture improves transcriptome analysis on low-input and archived samples. J Mol Diagn. 2014;16:440–451. doi: 10.1016/j.jmoldx.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa C, Gimenez-Capitan A, Karachaliou N, Rosell R. Comprehensive molecular screening: from the RT-PCR to the RNA-seq. Transl Lung Cancer Res. 2013;2:87–91. doi: 10.3978/j.issn.2218-6751.2013.02.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, et al. Comparison of RNA-Seq by poly (A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genomics. 2014;15:419. doi: 10.1186/1471-2164-15-419. [DOI] [PMC free article] [PubMed] [Google Scholar]