

Abstract
The canonical set of amino acids leads to an exceptionally wide range of protein functionality. Nevertheless, the set of residues still imposes limitations on potential protein applications. The incorporation of noncanonical amino acids can enlarge this scope. There are two complementary approaches for the incorporation of noncanonical amino acids. For site-specific incorporation, in addition to the endogenous canonical translational machineries, an orthogonal aminoacyl-tRNA-synthetase-tRNA pair must be provided that does not interact with the canonical ones. Consequently, a codon that is not assigned to a canonical amino acid, usually a stop codon, is also required. This genetic code expansion enables the incorporation of a noncanonical amino acid at a single, given site within the protein. The here presented work describes residue-specific incorporation where the genetic code is reassigned within the endogenous translational system. The translation machinery accepts the noncanonical amino acid as a surrogate to incorporate it at canonically prescribed locations, i.e., all occurrences of a canonical amino acid in the protein are replaced by the noncanonical one. The incorporation of noncanonical amino acids can change the protein structure, causing considerably modified physical and chemical properties. Noncanonical amino acid analogs often act as cell growth inhibitors for expression hosts since they modify endogenous proteins, limiting in vivo protein production. In vivo incorporation of toxic noncanonical amino acids into proteins remains particularly challenging. Here, a cell-free approach for a complete replacement of L-arginine by the noncanonical amino acid L-canavanine is presented. It circumvents the inherent difficulties of in vivo expression. Additionally, a protocol to prepare target proteins for mass spectral analysis is included. It is shown that L-lysine can be replaced by L-hydroxy-lysine, albeit with lower efficiency. In principle, any noncanonical amino acid analog can be incorporated using the presented method as long as the endogenous in vitro translation system recognizes it.
Keywords: Molecular Biology, Issue 114, Cell-Free Expression, Noncanonical Amino Acid Analog, Residue-Specific Incorporation, L-Arginine Analog, L-Canavanine, Bioengineering, Synthetic Biology, Escherichia coli Cell Extract, Unnatural Amino Acid
Introduction
The genetic code is universal to the biosphere. It codes for a set of 20 canonical amino acids, which is sometimes extended by selenocysteine1 or pyrrolysine2. It is the ribosome that translates the genetic code with the help of tRNAs into chains of amino acids that fold into proteins. The functional groups of the canonical amino acids, in combination with posttranslational modifications, contribute to an exceptionally wide range of protein function3,4. In principle, functional limitations due to the limited set of canonical amino acids can be overcome by incorporating further, noncanonical amino acids (ncAAs) that enable new chemistries and new functionalities3,4.
There are two complementary approaches for the incorporation of ncAAs: the site- or the residue-specific incorporation. The former method entails considerable technical difficulties, since the canonical set of aminoacyl-tRNA-synthetases (aaRS) and tRNAs must be expanded by an orthogonal aaRS-tRNA pair that must not interact with the endogenous translation machinery. Based on careful engineering, this approach incorporates the ncAAs as single point mutations at the desired protein sites. Site-specific incorporation of ncAAs is genetically encoded by a codon that is not assigned to a canonical amino acid (cAA), usually a stop codon5-9. This method entails changes in function at a given site rather than across the entire protein10-13.
In contrast, residue-specific incorporation relies on erroneous recognition of the noncanonical amino acid by the canonical translation machinery. The incorporation occurs due to the lack of substrate specificity of the aaRS. The residue-specific incorporation of ncAAs, built on the work of Cohen and coworkers14, has led to important applications3,10, among them bio-orthogonal labeling15-17 of proteins or structure elucidation of proteins in X-ray crystallography18.
As natural aaRS generally prefer their cognate amino acid over an isostructural ncAA, efficient in vivo residue-specific incorporation usually requires an auxotrophic expression host not capable of synthesizing the canonical analog of the ncAA. The host cells are cultivated in growth medium that delivers only a low concentration of the analogous cAA. Its exhaustion in combination with the consecutive supplementation with the ncAA forces the expression host to incorporate the ncAA into the model protein at multiple, canonically prescribed sites. In contrast to the site-specific approach, this generally has a deep impact on the entire protein structure, leading to considerably modified physical and chemical properties of proteins19,20. However, most of the ncAAs are growth inhibitors for the expression host3, as they are incorporated into many other proteins besides those of interest during recombinant gene expression. This clearly limits the in vivo approach. The in vivo incorporation of amino acids that are toxic or have strong influence on the protein structure remains particularly challenging. However, these molecules are among the most promising to engineer proteins with extraordinary functions.
One example is the toxic, noncanonical, naturally occurring L-canavanine (Can), an analog of L-arginine (Arg). It affects and blocks Arg associated regulatory and catalytic reaction pathways, and its presence in the living cell can lead to immediate death3,21-23. Its incorporation into proteins at arginine positions can reduce protein stability21-23. Due to the resulting toxicity, expression of canavanine containing proteins in Escherichia coli (E. coli) and other common expression hosts remains a challenge. For these reasons, complete in vivo incorporation of Can at all Arg positions has appropriately been confirmed only once24, using an elaborated single-protein production system. However, Can has been proposed as an anti-cancer agent25-27, and as a stimulator for autoimmune diseases in humans28. Additionally, it is subject of various studies on its anti-metabolic, antibacterial, antifungal and antiviral properties25. These properties raise a demand for efficient and easy-to-perform methods to express Can containing proteins for pharmaceutic, medical and functional studies.
Although many problems that are connected to in vivo production can be circumvented using cell-free expression systems, in vitro residue-specific approaches have only been poorly explored. The cell-free residue-specific incorporation of an L-tryptophan analog29 and multiple ncAAs30 have been reported. These methods are based on the highly efficient T7 RNA polymerase. The T7 RNA polymerase entails bacteriophage-like transcription, thereby reducing genetic functionality in comparison to endogenous transcription.
The complete residue-specific incorporation of Can into a model protein at all Arg positions was recently reported31, using a cell-free expression system32. A slight modification of the same system enabled site-specific incorporation of different pyrrolysine analogs into a model protein via stop codon suppression33. The employed cell-free system31-33 is based on an all E. coli transcription-translation system. Nevertheless, it enables protein expression as efficiently as in current bacteriophage systems (0.5 - 1 mg/ml of recombinant protein)32, while retaining much of the original transcription-translation modularity.
In this work, a detailed protocol is provided on how the residue-specific incorporation of ncAAs can be realized, using this all E. coli cell-free system32. Additionally, further steps to prepare the expressed proteins for appropriate evaluation via HPLC-ESI mass spectroscopy are proposed. To expand the properties of this cell-free system, this work does not only refer to the published incorporation of Can31 but also presents new data related to the noncanonical L-lysine analog L-hydroxy-lysine.
The following protocol for the residue-specific incorporation of ncAAs is an adaptation of a protocol recently published in JoVE34. The latter protocol describes how to perform highly efficient cell-free expression with standard amino acids. Furthermore, it presents the preparation of the crude cell free extract, the amino acid solution, the energy stock solution and the energy buffer used in this approach. The following protocol focuses on modified steps in comparison to the previous protocol in order to enable the residue-specific incorporation of ncAAs. Calibrated pipets, low-binding pipette tips and micro-centrifuge tubes are recommended for the preparation. In the following, IUPAC abbreviations for the amino acids are used.
Protocol
Caution! Please consult all relevant material safety data sheets (MSDS) before use. Several of the used chemicals are acutely toxic. Personal protective equipment is required (eyeshield, dust mask, gloves, lab coat, full length pants, closed-toe shoes) as well as working in a fume hood.
1. Amino Acid Solution Preparation
- Stock solution preparation of the ncAA (168 mM) NOTE: The stock solution preparation of the ncAA is described for the Arg analog Can as an example. Accordingly adapt the values for other ncAAs.
- Place a 1.5 ml reaction tube onto a microbalance. Weigh out 46.1 mg of Can inside the reaction tube for the preparation of 1 ml of a 168 mM solution. Use a sterile microspatula. For a racemic mixture of the ncAA, double the concentration of the stock solution.
- Add 977 µl of sterile ddH2O. Thoroughly vortex until Can is in complete dissolution. NOTE: For a total solution volume of 1 ml, the physical volume of the dissolved amino acid has to be compensated. For any of the amino acids, estimate half of the solid mass in mg as the corresponding volume increase in µl (100 mg solid will take a volume of 50 µl in the solution)35. Most amino acids can be dissolved at this concentration. If not, reduce the concentration until complete dissolution.
- Directly use the ncAA stock solution for the preparation of the amino acid solutions in section 1.2 or flash freeze it in liquid nitrogen and store it at –20 °C. CAUTION! For safety, wear an eyeshield and Cryo-gloves to be protected from liquid nitrogen splashes.
- Preparation of the amino acid solutions NOTE: For the preparation of the amino acid solutions, use the amino acid sampler providing the L-isomers of the 20 cAAs in separate stock solutions (1.5 ml, buffered with HEPES/KOH, < 0.1% NaN3, pH 7.5), each at a concentration of 168 mM, except for L-Leucine (140 mM). For a homemade preparation of these stock solutions (buffered with KOH), follow this protocol35.
- Thaw the stock solutions of the 20 cAAs (amino acid sampler or prepared according to 35) and of the ncAA (prepared in section 1.1) at RT.
- After thawing, frequently vortex the stock solutions to redissolve any precipitated amino acids. As some amino acids are harder to dissolve, incubate them in a heating block at 37 °C until complete dissolution. Cys may not fully dissolve. Place all amino acids on ice, except for Asn, Phe and Cys — keep these at RT to avoid precipitation.
- Use the following values to use one-seventh of the complete kit. NOTE: Scale down appropriately to work with smaller volumes and to save parts of the kit for further experiments. Scale up for incorporation of ncAAs into model proteins on a large scale. To prevent frequent thawing, which is likely to reduce the stability of the amino acids, aliquot the individual amino acid stock solutions into volumes of 200 µl. This 200 µl aliquot volume and the aliquot volumes used in step 1.2.4.1 account for losses due to pipetting.
- First, prepare an amino acid master mix solution that will be split in step 1.2.4.3 to finalize the preparation of 3 differently composed amino acid solutions (sections 1.2.5 - 1.2.7). In these solutions, concentrate all amino acids at 6 mM, except for Leu (5 mM).
- Transfer 1.4 ml of sterile ddH2O into a 15 ml centrifuge tube. Put it on ice. Add 175 µl of each amino acid stock solution. Add one after another, except for the stock solution of the cAA (e.g., Arg) to be replaced by the ncAA (e.g., Can). Thoroughly vortex after each addition and put the solution back on ice. NOTE: Leu is at 5 mM in the 3 differently composed amino acid solutions, compared to 6 mM for the other amino acids. The reduced concentration does not reduce the expression efficiency. Scaling up to 6 mM is suitable as well.
- Transfer the amino acid stock solutions in the following order to avoid precipitation: Ala, Arg, Asn, Asp, Gln, Glu, Gly, His, Ile, Lys, Met, Phe, Pro, Ser, Thr, Val, Trp, Tyr, Leu, and Cys. Remember to not add the stock solution of the cAA (e.g., Arg) that is analogous to the ncAA (e.g., Can). Finally, thoroughly vortex. Incubate at 37 °C to make the solution as clear as possible.
- Split this amino acid master mix solution into three equal volumes of 1.35 ml. Transfer each of the split volumes into 1.5 ml reaction tubes. Keep them on ice.
- Prepare an amino acid solution that consists of all 20 cAAs at a concentration of 6 mM each, except for Leu that is 5 mM. To the first volume of 1.35 ml, as a result of the split in step 1.2.4.3, add 50 µl of the 168 mM stock solution of the cAA (e.g., Arg) that is analogous to the ncAA (e.g., Can). Thoroughly vortex.
- Put back on ice. Aliquot this 1.4 ml solution in volumes of 16 µl into reaction tubes. Note that this solution volume approximately leads to 85 aliquots. Label these aliquots "+ cAA" (e.g., + Arg).
- Flash freeze the aliquots in liquid nitrogen and store at -80 °C. CAUTION! For safety, wear an eyeshield and Cryo-gloves to be protected from liquid nitrogen splashes.
- Prepare an amino acid solution that is composed of 19 cAAs except for the cAA (e.g., Arg) that is analogous to the ncAA (e.g., Can). Add each amino acid to a concentration of 6 mM, except for Leu (5 mM).
- Add 50 µl of sterile ddH2O to the second volume of 1.35 ml, as a result of the split in step 1.2.4.3. Thoroughly vortex and put back on ice. Aliquot this 1.4 ml solution in volumes of 16 µl into reaction tubes. Note that this solution volume approximately leads to 85 aliquots. Label these aliquots "- cAA" (e.g., - Arg).
- Flash freeze the aliquots in liquid nitrogen and store at -80 °C. CAUTION! For safety, wear an eyeshield and Cryo-gloves to be protected from nitrogen splashes.
- Prepare an amino acid mixture that contains 19 cAAs and the ncAA (e.g., Can) that substitutes the canonical one (e.g., Arg). Add each amino acid to a concentration of 6 mM, except for Leu (5 mM). To the last 1.35 ml volume, as a result of the split in step 1.2.4.3, add 50 µl of the 168 mM stock solution of the ncAA (e.g., Can). Label it "+ ncAA" (e.g., + Can). Thoroughly vortex and put back on ice.
- Aliquot this 1.4 ml solution in volumes of 16 µl into reaction tubes. Note that this solution volume approximately leads to 85 aliquots. Label these aliquots "+ ncAA" (e.g., + Can).
- Flash freeze the aliquots in liquid nitrogen and store at -80 °C. CAUTION! For safety, wear an eyeshield and Cryo-gloves to be protected from nitrogen splashes. NOTE: The 16 µl aliquot volumes used in the steps 1.2.5.1, 1.2.6.1 and 1.2.7.1 are slightly higher than required to account for losses due to pipetting.
2. Energy Buffer Preparation
NOTE: Each batch of crude extract is unique and requires optimized concentrations of Mg- and K-glutamate34. The crude extract aliquot volume depends on the protein concentration34. Use the provided calculation template (Supplemental Material 1) for different values. Find further instructions in Supplemental Material 1 figure legend, explaining how to employ this template.
Prepare and store at -80 °C the 14x energy solution and crude extract aliquots according to the unmodified protocol34. Calibrate the crude extract depending on the Mg- and K-glutamate concentrations for optimized expression efficiency34. NOTE: The final composition of 14x energy solution is: 700 mM HEPES (pH 8), 21 mM ATP, 21 mM GTP, 12.6 mM CTP, 12.6 mM UTP, 2.8 mg/ml tRNA, 3.64 mM CoA, 4.62 mM NAD, 10.5 mM cAMP, 0.95 mM folinic acid, 14 mM spermidine, 420 mM 3-PGA.
Thaw on ice the 100 mM Mg-glutamate stock solution, 3 M K-glutamate stock solution, 14x energy solution and 40% PEG-8000 to prepare the master mix. Keep them on ice.
Mix 9.18 µl of 100 mM Mg-glutamate stock solution, 3.06 µl of 3 M K-glutamate stock solution, 21.86 µl of 14x energy solution, 15.3 µl of 40% PEG-8000 and 1.6 µl sterile ddH2O in a reaction tube. Thoroughly vortex this master mix after each addition, and keep it on ice.
Aliquot the master mix (51 µl) into volumes of 16 µl (3 aliquots) into reaction tubes. Frequently vortex the master mix during aliquoting. Flash freeze the aliquots in liquid nitrogen. NOTE: The 16 µl aliquot volume as well as the master mix volume are slightly higher than required to account for losses due to pipetting.
Use a strainer to collect the energy buffer tubes. Store the tubes at -80 °C. CAUTION! Wear an eyeshield and Cryo-gloves to be protected from nitrogen splashes.
3. Preparation and Execution of Cell-free Reactions for Residue-specific Incorporation of ncAAs
- First, prepare the DNA vector solution in ddH2O.
- For highly efficient protein expression, use the expression vector pBEST-OR2-OR1-Pr-UTR1-deGFP-T50032. Clone the gene that codes for the model protein into this vector36,37. NOTE: Alternatively, use other promoters that are recognized by σ70 as well, but note that expression efficiency may be reduced.
- Transform37,38 the vector in E. coli strain KL 74032 (Yale CGCS#:4382), purify amplified vector DNA37,39 and quantify the concentration of the DNA solution40-42. Store the DNA solution at -20 °C or directly use it for the cell-fee reaction preparation (steps 3.4.1, 3.4.2 and 3.4.3).
Calibrate the cell-free expression efficiency depending on the used vector construct concentration according to the unmodified protocol34. Use the optimal concentration that leads to highest protein yields for the cell-free reaction preparation (steps 3.4.1, 3.4.2 and 3.4.3). NOTE: The preparation of cell-free reactions is exemplified using a 90 nM vector DNA stock solution that leads to a final vector concentration of 10 nM in the cell-free reaction and follows the above optimal values of Mg- and K-glutamate as well as the extract aliquot volume. Use the calculation template for different values.
Thaw on ice 3 crude extract aliquots each of 30 µl volume (prepared according to unmodified protocol34), 1 amino acid solution aliquot labeled "+ cAA" (e.g., + Arg), 1 amino acid solution aliquot labeled "- cAA" (e.g., - Arg) and 1 amino acid solution aliquot labeled "+ ncAA" (e.g., + Can) (prepared in sections 1.2.5 - 1.2.7), 3 energy buffers aliquots (prepared in section 2) and the vector DNA solution (prepared in section 3.1). NOTE: The crude extract is slightly viscous and it may contain air bubbles. Remove air bubbles by centrifuging at 10,000 x g for 30 sec at 4 °C. Put crude extract aliquots back on ice.
- Prepare 3 differently composed cell-free reactions (each 90 µl final volume) by mixing crude extract (33.33%), energy buffer (16.67%), 1 of the 3 differently composed amino acid solution aliquots (16.67%) and vector DNA solution. Optionally, add further biomolecules (DNA, proteins, tRNA, etc.), but appropriately reduce the volume of ddH2O.
- Prepare the reference cell-free reaction (90 µl) expressing the unmodified model protein.
- Add the 15 µl of energy buffer, 15 µl of the amino acid solution aliquot labeled "+ cAA" (e.g., + Arg), 10 µl of 90 nM vector DNA solution and 20 µl of sterile ddH2O to the 30 µl of crude extract. Mix by pipetting up and down and gently vortex after the addition of each ingredient.
- Aliquot the 90 µl of cell-free reaction in 15 equal volumes of 6 µl. Transfer each of the 15 volumes into a separate reaction tube. Close the tubes and put them back on ice. Label all reaction tubes as "CFR(+cAA)" (e.g., CFR(+Arg)).
- Prepare the negative control cell-free reaction (90 µl), where neither the cAA (e.g., Arg) nor the noncanonical analog (e.g., Can) are added.
- Add the 15 µl of energy buffer, 15 µl of the amino acid solution aliquot labeled "- cAA" (e.g., - Arg), 10 µl of 90 nM vector DNA solution and 20 µl of sterile ddH2O to the 30 µl of crude extract. Mix by pipetting up and down and gently vortex after the addition of each ingredient.
- Aliquot the 90 µl of cell-free reaction in 15 equal volumes of 6 µl. Transfer each of the 15 volumes into a separate reaction tube. Close the tubes and put them back on ice. Label all reaction tubes as "CFR(-cAA)" (e.g., CFR(-Arg)).
- Prepare the cell-free reaction (90 µl) that is supposed to residue-specifically incorporate the ncAA (e.g., Can) into the expressed model protein.
- Add the 15 µl of energy buffer, 15 µl of the amino acid solution aliquot labeled "+ ncAA" (e.g., + Can), 10 µl of 90 nM DNA solution and 20 µl of sterile ddH2O to the 30 µl of crude extract. Mix by pipetting up and down and gently vortex after the addition of each ingredient. NOTE: The expression efficiency may be reduced in comparison to the expression with standard amino acids. If necessary, simply scale up the cell-free reaction volume.
- Aliquot the 90 µl of cell-free reaction in 15 equal volumes of 6 µl. Transfer each of the 15 volumes into a separate reaction tube. Close the tubes and put them back on ice. For increased reaction volumes, accordingly aliquot in further volumes of 6 µl. Label all reaction tubes as "CFR(+ncAA)" (e.g., CFR(+Can)).
Incubate all tubes at 29 °C O/N. NOTE: Only small reaction volumes enable adequate oxygen diffusion into the reaction that is crucial for highly efficient protein expression. Reaction volumes greater than 10 µl require active oxygenation through agitation34. For large volumes, split the reaction into volumes smaller than 15 µl.
After cell-free expression, pool all identically composed split cell-free reactions of 6 µl. First, pool all 15 split cell-free reactions of 6 µl labeled "CFR(+cAA)" (e.g., CFR(+Arg)). Then, pool all 15 split cell-free reactions of 6 µl labeled "CFR(-cAA)" (e.g., CFR(-Arg)). Finally, pool all 15 split cell-free reactions of 6 µl labeled "CFR(+ncAA)" (e.g., CFR(+Can)). For increased reaction volumes, accordingly pool further volumes of 6 µl.
Check the expression level of the model protein in all three pooled, differently composed cell-free reactions by performing denaturing SDS-PAGE43,44 (section 4.1). NOTE: Use this method for a preliminary evaluation of the incorporation experiment.
Prepare the cell-free expressed model proteins for an appropriate analysis via HPLC-ESI mass spectroscopy (section 4.4). First, purify45 them (section 4.2). Finally, exchange the buffer (section 4.3) to avoid high background noise during mass spectroscopy. NOTE: The sections 3.4.1, 3.4.2 and 3.4.3 lead to typical cell-free reaction conditions34: 8.9 - 9.9 mg/ml protein (from crude extract), 4.5 - 10.5 mM Mg-glutamate, 40 - 160 mM K-glutamate, 1 mM of each amino acid except leucine, 0.83 mM leucine, 50 mM HEPES, 1.5 mM ATP and GTP, 0.9 mM CTP and UTP, 0.2 mg/ml tRNA, 0.26 mM CoA, 0.33 mM NAD, 0.75 mM cAMP, 0.068 mM folinic acid, 1 mM spermidine, 30 mM 3-PGA, 2% PEG-8000 and 10 nM pBEST-OR2-OR1-Pr-UTR1-gene_of_model_protein-T500. If desired, a different procedure of cell-free reaction preparation can be conducted that leads to the reaction conditions above.
4. Preliminary Evaluation via SDS-PAGE43,44 and Preparation of the Cell-free Expressed Model Proteins for HPLC-ESI Mass Spectrometry
- SDS-PAGE of the cell-free reactions NOTE: Perform denaturating SDS-PAGE for a quick and preliminary analysis of the expressed proteins without any further purification or extraction, and execute the following steps.
- Thaw the protein standard on ice. Ensure that it consists of proteins with molecular weights similar to the expressed model protein for its localization on the gel (step 4.1.13). NOTE: Here, the used standard provides proteins over a wide range of molecular weights (myosin: 212 kDa, maltose-binding-protein-β-galactosidase: 158 kDa, β-galactosidase: 116 kDa, phosphorylase b: 97 kDa, serum albumin: 66 kDa, glutamic dehydrogenase: 56 kDa, maltose-binding-protein: 43 kDa, thioredoxin reductase: 35 kDa, triosephosphate isomerase: 27 kDa, trypsin inhibitor: 20 kDa, lysozyme: 14 kDa, aprotinin: 7 kDa, insulin A: 3 kDa, B chain: 2 kDa).
- Since cell-free reactions are slightly viscous due to the high protein concentration, dilute them with sterile ddH2O 5 - 10 times before SDS-PAGE to ensure appropriate gel migration and to avoid saturation after staining. In order not to waste too much sample, dilute 1 µl of cell-free reaction in 4 µl of sterile ddH2O. Prepare the dilution in reaction tubes, thoroughly vortex and shortly spin the liquid down with a mini centrifuge for 2 - 3 sec at 2,000 x g.
- Add 5 µl of 2x loading buffer to 5 µl of diluted cell-free reaction. Thoroughly vortex and spin down with a mini centrifuge for 2 - 3 sec at 2,000 x g. NOTE: Here, after addition, the biomolecules are dissolved in 62.5 mM Tris/Cl-, 10% glycerol, 2% SDS, and 0.0025% bromophenol blue at pH 6.8, a typical composition. Using other loading dyes that lead to slightly different dilution conditions may be suitable as well.
- Thoroughly vortex the protein standard and transfer 15 µl of it into a reaction tube. NOTE: Depending on gel size and for other protein standards, the recommended volume can differ from the above.
- Place the reaction tubes into a heating block. Check if the lids of the tubes are properly closed. Heat at 95 - 100 °C for 3 - 5 min. Do this to denature the proteins and to enable SDS-wrapping around the protein backbone. NOTE: Some protein standards must NOT be heated, see supplier instructions.
- Meanwhile, transfer the running buffer into the gel electrophoresis chamber. The content of 1x running buffer is 25 mM Tris, 192 mM glycine, 0.1% SDS at pH 8.3 (with HCl).
- Fix the precast 4 - 20% gradient Tris-Glycine-SDS gel (10 cm x 10 cm x 1 mm) to the electrophoresis chamber. Remove the comb and rinse the wells with a syringe loaded with running buffer. NOTE: The gel concentration strongly depends on the size and nature of the expressed model protein. The above concentration separates the E. coli crude extract proteins as well as low molecular weight model proteins. Self-cast gels are suitable as well.
- Remove the tubes from the heating block. Spin the liquid down with a mini centrifuge for 2 - 3 sec at 2,000 x g. Shortly vortex and repeat spinning down for 2 - 3 sec at 2,000 x g.
- Transfer the 15 µl of protein standard (24 - 48 µg protein) and 10 µl (11 - 22 µg protein) of each sample into the wells and start electrophoresis. Here, SDS-PAGE is performed at 125 V and 20 - 40 mA for approximately 90 min, a typical protocol.
- Carefully extract the gel from the cassette. Transfer it into the fixing solution (50% methanol, 10% acetic acid, 40% ddH2O) for 30 min. CAUTION! Methanol is toxic by inhalation and in contact with skin. Wear protective gloves and work under a fume hood.
- Transfer the gel into staining solution (0.025% Coomassie brilliant blue G-250, 10% acetic acid, 90% ddH2O). Stain for 60 min.
- Transfer the gel into destaining solution (10% acetic acid, 90% ddH2O) for 60 - 120 min. NOTE: Fixing, staining and destaining strongly depend on the size and nature of the expressed protein. This protocol applies to a wide range of proteins of rather small molecular weights46.
- Displace the gel onto a matte transparency on a white background that generates an appropriate contrast for the stained protein bands.
- Purification of the His-tagged45 model proteins from the cell-free reactions NOTE: For protein purification, several methods exist that deliver similar results. This protocol purifies cell-free expressed model proteins that have a C-terminal polyhistidine-tag (His-tag). It uses a purification kit suitable for proteins that are expressed in the small reaction volumes of cell-free protein expression. It is identical for the purification of native or modified model proteins. Thus, it is generally described on the basis of one single cell-free reaction.
- For an appropriate HPLC-ESI mass spectroscopic analysis, extract and purify the model proteins from the cell-free reaction and execute the following steps.
- Mix equal volumes of 90 - 150 µl of His-binding buffer and 90 - 150 µl of cell-free reaction. Pipette up and down, followed by gentle vortexing. NOTE: Using His-binding buffer is recommended. However, cell-free reaction medium can be a starting material as long as the model protein is soluble, pH is between 7.5 - 8, imidazole/His concentration is < 10 mM, concentration of strong reducing agents is < 15 mM and no metal-chelating agents are present. If your mixture volume exceeds 300 µl, split it into equal volumes and aliquot theses volumes into different reaction tubes for the following purification steps.
- Prepare the column system. Thoroughly vortex the stock solution of the His-affinity gel until the gel resin is completely dissolved. Transfer 250 µl of gel resin into the column. Use a 1 ml pipette tip for the viscous gel resin. Place the column into a collection tube.
- Centrifuge column/collection tube for 5 - 10 sec at 13,000 - 15,000 x g. Ensure that the gel resin is completely drained. If not, extend the centrifugation time by additional 5 - 10 sec, but pay attention that the affinity gel is not over-dried. Accordingly extend the centrifugation time in steps 4.2.1.5, 4.2.1.7 and 4.2.1.9. NOTE: The gel resin is completely drained if it becomes stiff and no supernatant remains on top.
- Transfer 150 - 300 µl of the cell-free reaction/His-binding buffer to the column. If the mixture volume exceeds the recommended volume, split over several spin columns. Resuspend the gel resin by frequent tapping and gentle vortexing during an incubation time of at least 2 min. For volumes greater than 200 µl, incubate for additional 1 - 2 min. NOTE: Sufficient incubation time is crucial for binding of the model proteins to the gel resin.
- Centrifuge column/collection tube for 5 - 10 sec at 13,000 - 15,000 x g. Discard the flow-through and place the column back into the collection tube.
- Add 250 µl of His-wash buffer. Resuspend the gel resin by tapping and gentle vortexing.
- Centrifuge column/collection tube for 5 - 10 sec at 13,000 - 15,000 x g. Discard the flow-through and place the column back into the collection tube. Repeat step 4.2.1.6 and centrifuge again for 5 - 10 sec at 13,000 - 15,000 x g.
- Place the column into a standard 1.5 ml reaction tube. Add 150 µl of elution buffer and resuspend the gel resin by tapping and gentle vortexing. Reduce the volume to 100 µl for higher purified model protein concentrations after elution in the next step 4.2.1.9. NOTE: The total abundance of purified model protein may be reduced due to possible incomplete elution of all gel resin-bound proteins, if the elution buffer volume is reduced.
- Centrifuge column/reaction tube for 5 - 10 sec at 13,000 - 15,000 x g to dilute the purified protein in the elution buffer. If split before, pool all solutions into one stock solution.
- Before executing the buffer exchange, determine the protein concentration using standard methods, e.g., the Bradford assay47,48. NOTE: For an appropriate HPLC-ESI mass spectroscopy (section 4.4), use optimal protein concentrations that are usually about 0.5 mg/ml with required volumes of 20 - 50 µl. If the concentration is below 0.2 mg/ml, concentrate the protein solution after the buffer exchange using a spinning vacuum concentrator. In this case, first read section 4.3.8 to appropriately adapt the exchanging buffer.
- Buffer exchange for HPLC-ESI mass spectroscopy NOTE: Exchange the His-tag elution buffer to avoid high background noise in mass spectroscopic analysis due to high concentration of imidazole (> 150 mM) and other salts such as NaH2PO4 (> 300 mM) or NaCl (> 50 mM)49. The following buffer exchange protocol uses a prehydrated gel filtration spin column system. It is identically executed for native or modified model proteins. Thus, it is generally described on the basis of one single cell-free reaction. Note that there are other easy-to-perform buffer exchange methods, e.g., mini-dialysis cartridges.
- Prepare 100 ml of protein storage buffer (50 mM Tris-Cl pH 8, 100 mM NaCl, and 10% glycerol). Weigh out into an autoclavable 100 ml bottle 605.7 mg Tris-Base, 584.4 mg NaCl and add 10 ml of glycerol. Fill up to approximately 80 ml and adjust the pH value to 8 by adding NaOH. Steadily check the pH value with a pH meter. Finally, fill up to the 100 ml mark. NOTE: Use this buffer to stabilize a wide range of model proteins and not to disturb mass spectroscopic analysis, as long as HPLC is performed prior to the mass spectroscopic measurement. However, depending on the nature of the expressed model protein, use other buffers that might be better suitable. If your target protein is not stable at pH 8, adjust the pH accordingly. Do not use higher glycerol concentrations.
- Warm up the gel filtration spin columns for at least 15 min to RT.
- Resuspend the prehydrated gel by gentle tapping or vortexing and remove air bubbles.
- First remove the bottom cap, and then take the top cap away. Place the column into a wash tube (at least 2 ml). Transfer the column/wash tube into the centrifuge. Each column possesses an orientation mark. Centrifuge at 1,000 x g for 2 min to remove the gel storage buffer. Discard the flow-through. NOTE: Pay attention to proceed in this order. Ensure that the column has the same orientation in all further centrifugation steps.
- Add up to 400 µl of the protein storage buffer. Centrifuge for 2 min at 1,000 x g. Repeat this to completely load the protein storage buffer into gel. Carefully add up to 100 µl of the solution of the purified model protein. Pipette directly onto the center of the gel bed. NOTE: Solution volumes of purified proteins, particularly of modified proteins, might be higher. Split such solutions over several buffer exchange columns. Proteins must have molecular weights higher than 5 kDa for this kind of buffer exchange.
- Place the column into a collection tube and centrifuge for 2 min at 1,000 x g. NOTE: This step leads to the dilution of the purified model protein in the protein storage buffer. If split before, pool all solutions into one stock solution.
- Flash freeze the protein solution in liquid nitrogen and store at -80 °C or directly analyze it via HPLC-ESI mass spectroscopy50 (section 4.4). CAUTION! Wear an eyeshield and Cryo-gloves to be protected from nitrogen splashes.
- Perform the following steps of the protocol to concentrate the protein solution using a spinning vacuum concentrator that carefully evaporates the solvent51. NOTE: These steps are required if the protein concentration is too low for HPLC-ESI mass spectroscopy (< 0.2 mg/ml). The solution volume and model protein concentration is roughly the same before and after buffer exchange. The concentration process is described by means of the following example: After His-tag purification, the model protein is dissolved in 100 µl His elution buffer (step 4.2.1.9) and has a concentration of 0.07 mg/ml (step 4.2.1.10).
- To ensure that the protein concentration and the solution volume are after evaporation at least 0.2 mg/ml and 20 µl, respectively, dilute, before buffer exchange, the protein storage buffer prepared in step 4.3.1 at least threefold but at most fivefold. Equally evaporate in the following at least two third (66.67 µl) or at most four fifth (80 µl). NOTE: Dilution of the protein storage buffer before the buffer exchange is crucial to take into account that concentration values of this buffer (step 4.3.1) are still realized in spite of evaporation. After dilution of the protein storage buffer, first perform the buffer exchange (steps 4.3.2 - 4.3.6) before executing the following steps 4.3.8.2 - 4.3.8.4.
- After buffer exchange, put the complete 100 µl of model protein solution in an open tube into the spinning vacuum concentrator. NOTE: The model protein is dissolved in the diluted protein storage buffer. Incline it towards the rotation center to avoid liquid-loss during rotation. Ensure that an open blank of the same volume of H2O is symmetrically placed to balance the spinning system.
- Close the lid of the concentrator and start rotation. To assure protein stability, concentrate at RT or at low temperatures up to 30 °C. NOTE: The used concentrator automatically accelerates to 170 x g and establishes the vacuum. In the following it accelerates to its maximum speed of 240 x g.
- Frequently interrupt the concentration process to survey the liquid volume. Stop the concentration, when the remaining solution volume is between 20 µl and 33 µl. NOTE: Accordingly adapt the steps 4.3.8.1 - 4.3.8.4 for other solution volumes (step 4.2.1.9) and other protein concentrations (step 4.2.1.10). Flash freeze the concentrated protein solution in liquid nitrogen and store at -80 °C, or directly analyze it via HPLC-ESI mass spectroscopy50. CAUTION! Wear an eyeshield and Cryo-gloves to be protected from nitrogen splashes.
- HPLC-ESI mass spectrometric analysis 50 of model proteins
- Perform HPLC separation of 5 - 15 µl protein solution (prepared in section 4.3) on a C5 reverse-phase column (3 µm, 100 x 2.1 mm) with a gradient of 20% - 90% acetonitrile and 0.1% of formic acid over 25 min and subsequent ESI mass spectrometry with detection on a time-of-flight mass analysator in the range of 300 - 3,000 m/z. NOTE: Depending on the nature of the expressed model protein, use other solvents or columns that might be better suitable.
- Deconvolute measured mass spectra using appropriate software52 to calculate zero-charge masses.
Representative Results
This protocol guides through the cell-free residue-specific incorporation of ncAAs into model proteins. It proposes SDS-PAGE for a preliminary evaluation of the incorporation experiment and further steps to prepare the model proteins for an appropriate HPLC-ESI mass spectroscopic analysis.
Here, representative results of the cell-free residue-specific incorporation of the Arg analog Can, as well as the Lys analog L-hydroxy-lysine (Hyl) are presented. The different amino acid solutions, the energy buffer, the vector DNA coding for the model protein and cell-free reactions are prepared as described above. The reference cell-free reaction is provided with the amino acid solution consisting of the 20 cAAs. For each experiment, a negative control cell-free reaction is supplied with the amino acid solution that lacks the canonical analog of the ncAA in question. For each approach, one cell-free reaction expresses the model protein in the presence of the amino acid solution, where the cAA is substituted by the noncanonical analog. His-tag purification, buffer exchange and HPLC-ESI mass spectrometric analysis are executed according to the above described protocol.
The model protein is the C-terminal His-tagged deGFP32, a truncated version of EGFP53. Its one letter amino acid sequence can be found in (Supplemental Material 2). This model protein contains 6 Arg and 18 Lys positions, respectively. The expression vector is pBEST-OR2-OR1-Pr-UTR1-deGFP-T500.
In the case of complete incorporation of the ncAA, one can assume that the negative control reaction does not express deGFP, since one of the 20 cAAs is missing. Contrary, deGFP must be detectable in the other two reactions: the native one in the reference cell-free reaction and the modified protein in the cell-free reaction that is provided with the ncAA.
Figure 1A shows the preliminary SDS-PAGE evaluation of the Can incorporation experiment. The reference cell-free reaction has the highest deGFP expression level. In the cell-free reaction that is provided with Can, deGFP is expressed at slightly lower concentration. No deGFP expression can be detected in the negative control. This SDS-PAGE result is a good indication for a successful incorporation of Can into the target protein deGFP.
To prove the hypothesized complete incorporation of Can into deGFP, both purified model proteins, visualized in Figure 1B, are analyzed through HPLC-ESI mass spectroscopy. Figure 1C shows the deconvoluted mass spectra of the purified deGFP molecules. The deconvoluted mass of deGFP that is expressed in the reference cell-free reaction is 26,192.8 Da. For deGFP expressed in the Can containing cell-free reaction a mass of 26202.5 Da appears. The expected masses for the native deGFP6Arg and the modified deGFP6Can with Arg being fully replaced by Can are 26,193 Da and 26,204 Da, respectively. The mass difference of 1.5 Da for deGFP6Can is within the error of spectrum deconvolution. Thus, the full incorporation of Can into deGFP at all 6 Arg positions is confirmed.
The two peaks of reduced intensity correspond to the native deGFP6Arg and modified deGFP6Can that did not attain their mature fluorophore. The fluorophore is autocatalytically generated by elimination of a H2O molecule, followed by oxidation. This leads to a mass increased by 20 Da if this process does not proceed.
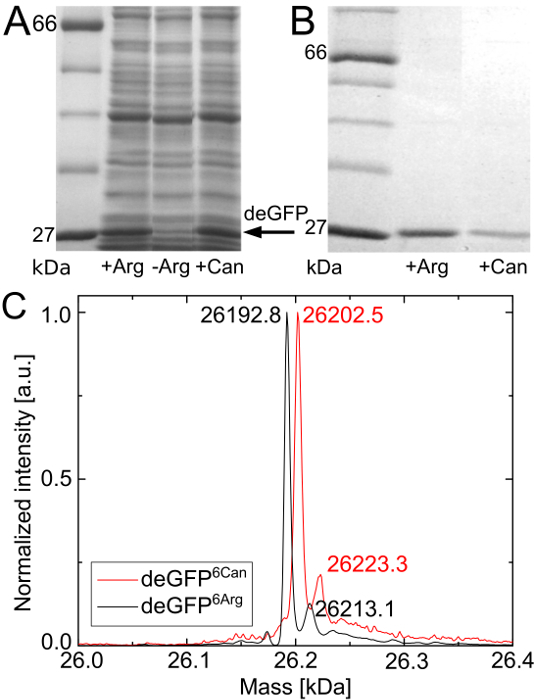 Figure 1.SDS-PAGE evaluation of the Can incorporation experiment and HPLC-ESI mass spectroscopy of the cell-free expressed and purified deGFP molecules. (A) Preliminary evaluation of the Can incorporation experiment using SDS-PAGE. From left to right: Protein standard, reference cell-free reaction, negative control and cell-free reaction providing Can instead of Arg. (B) SDS-PAGE after His-tag purification and buffer exchange of the expressed deGFP molecules. From left to right: Protein standard, purified deGFP from the reference reaction, purified deGFP from the Can containing reaction. (C) Confirmation of full incorporation of Can by HPLC-ESI mass spectroscopy. The expected masses of the native deGFP and the modified deGFP with Arg fully replaced by Can are 26,193 Da and 26,204 Da, respectively. Each spectrum is normalized to its highest intensity (counts). Peak positions are indicated in Da. For visualization purposes, the gel lanes are extracted from the gel pictures, joined together, are converted into gray scale format, size is optimized and contrast as well as brightness are enhanced. Original gel lanes are presented in Supplemental Material 3. Please click here to view a larger version of this figure.
Figure 1.SDS-PAGE evaluation of the Can incorporation experiment and HPLC-ESI mass spectroscopy of the cell-free expressed and purified deGFP molecules. (A) Preliminary evaluation of the Can incorporation experiment using SDS-PAGE. From left to right: Protein standard, reference cell-free reaction, negative control and cell-free reaction providing Can instead of Arg. (B) SDS-PAGE after His-tag purification and buffer exchange of the expressed deGFP molecules. From left to right: Protein standard, purified deGFP from the reference reaction, purified deGFP from the Can containing reaction. (C) Confirmation of full incorporation of Can by HPLC-ESI mass spectroscopy. The expected masses of the native deGFP and the modified deGFP with Arg fully replaced by Can are 26,193 Da and 26,204 Da, respectively. Each spectrum is normalized to its highest intensity (counts). Peak positions are indicated in Da. For visualization purposes, the gel lanes are extracted from the gel pictures, joined together, are converted into gray scale format, size is optimized and contrast as well as brightness are enhanced. Original gel lanes are presented in Supplemental Material 3. Please click here to view a larger version of this figure.
Figure 2A shows the preliminary SDS-PAGE evaluation of the Hyl incorporation experiment. In the cell-free reaction that is provided with Hyl, deGFP is expressed. Contrary to the first experiment, a weak deGFP band can be observed in the negative control reaction. This might be due to Lys residues in the cell-free reactions. This enables a faint deGFP expression in the negative control reaction, where neither Lys nor Hyl are added.
For HPLC-ESI mass spectroscopy, the deGFP molecules of the cell-free reaction provided with Hyl are purified and the buffer is exchanged (Figure 2B).
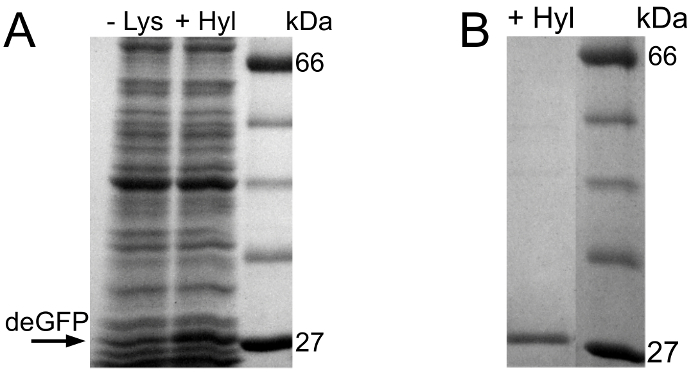 Figure 2.SDS-PAGE evaluation of the Hyl incorporation experiment. (A) From left to right: Negative control cell-free reaction, cell-free reaction containing Hyl and protein standard. (B) SDS-PAGE after His-tag purification and buffer exchange of the expressed deGFP molecules. From left to right: Purified deGFP from the Hyl containing reaction and protein standard. For visualization purposes, the gel lanes are extracted from the gel pictures, joined together, are converted into gray scale format, size is optimized and contrast as well as brightness are enhanced. Original gel lanes are presented in Supplemental Material 3. Please click here to view a larger version of this figure.
Figure 2.SDS-PAGE evaluation of the Hyl incorporation experiment. (A) From left to right: Negative control cell-free reaction, cell-free reaction containing Hyl and protein standard. (B) SDS-PAGE after His-tag purification and buffer exchange of the expressed deGFP molecules. From left to right: Purified deGFP from the Hyl containing reaction and protein standard. For visualization purposes, the gel lanes are extracted from the gel pictures, joined together, are converted into gray scale format, size is optimized and contrast as well as brightness are enhanced. Original gel lanes are presented in Supplemental Material 3. Please click here to view a larger version of this figure.
Figure 3 shows the deconvoluted mass spectrum of purified deGFP molecules. Figure 3A confirms the hypothesis of already present Lys residues in the cell-free reactions. The predominant peak of the spectrum corresponds to the native deGFP (expected mass: 26,193 Da). Again, deGFP molecules of a 20 Da higher mass that did not develop their fluorophore can be detected. The Lys residues are preferentially loaded onto the tRNALys by lysyl-tRNA-synthetase leading to a high expression level of the native deGFP18Lys.
The mass difference between Hyl and Lys is 16 Da. Due to the presence of Hyl that is in competition to the Lys residues all possible deGFP species are generated (deGFP18Hyl, deGFP17Hly+1Lys, …, deGFP16Hyl+2Lys) (Figure 3B). Admittedly, the peak of deGFP1Hyl+17Lys overlaps with the peak of the native deGFP that did not produce its fluorophore (Figure 3A) and the mass of some peaks differs more than 2 Da from the expected mass. These mass differences can be attributed to high noise due to low amounts of the deGFP species. However, Hyl is generally incorporated by the cell-free system. Further improvements have to be done to abolish Lys residues in the cell-free reactions.
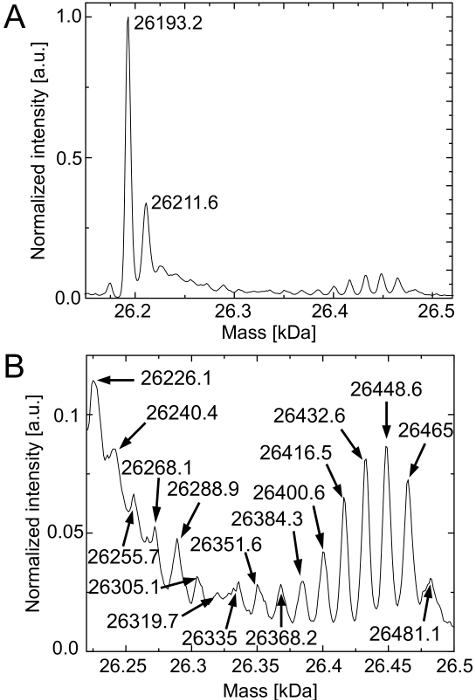 Figure 3.HPLC-ESI mass spectroscopy of the purified deGFP molecules of the cell-free reaction containing Hyl. (A) The native deGFP18Lys is predominantly detected (expected masses: with fluorophore 26,193 Da, without mature fluorophore 26,213 Da). (B) Magnification reveals the existence of all possible deGFP species (deGFP18Hyl, deGFP17Hly+1Lys, deGFP16Hyl+2Lys,…, deGFP1Hyl+17Lys). Their expected masses are 26,193 Da + N x 16 Da (N = 1, …, 18). The spectrum is normalized to its highest intensity (counts). Peak positions are indicated in Da. Please click here to view a larger version of this figure.
Figure 3.HPLC-ESI mass spectroscopy of the purified deGFP molecules of the cell-free reaction containing Hyl. (A) The native deGFP18Lys is predominantly detected (expected masses: with fluorophore 26,193 Da, without mature fluorophore 26,213 Da). (B) Magnification reveals the existence of all possible deGFP species (deGFP18Hyl, deGFP17Hly+1Lys, deGFP16Hyl+2Lys,…, deGFP1Hyl+17Lys). Their expected masses are 26,193 Da + N x 16 Da (N = 1, …, 18). The spectrum is normalized to its highest intensity (counts). Peak positions are indicated in Da. Please click here to view a larger version of this figure.
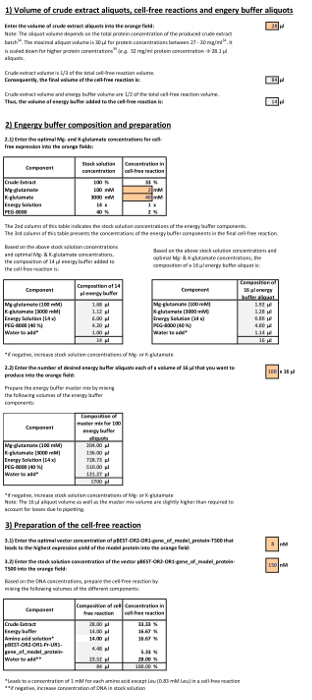
Supplemental Material 1. Calculation template. In section 2, the preparation of the energy buffer master mix is exemplified using crude extract aliquots of 30 µl and optimal Mg- and K-glutamate concentrations of respectively 3 mM and 30 mM. This example leads to a master mix volume that yields 3 energy buffer aliquots. In section 3, the preparation of the cell-free reaction is exemplified using a 90 nM DNA stock solution leading to an optimal vector DNA concentration of 10 nM in the cell-free reaction. Please click here to download this file.
For an appropriate traceability, the use of the calculation template is exemplified by insertion of these typical values that differ from the above example: Crude extract volume: 28 µl, optimal Mg-glutamate: 2 mM, optimal K-glutamate: 40 mM, number of desired buffer aliquots: 100, optimal vector concentration in cell-free reaction: 8 nM, DNA vector stock solution: 150 nM.
First enter 28 µl as crude extract volume into the orange field of the first template section. Then, enter into the second template section 2 mM and 40 mM as optimal Mg- and K-glutamate concentrations into the orange fields. Taking into account the optimal Mg- and K-concentrations, the composition of a 15 µl energy buffer, as well as a corresponding, scaled up 16 µl aliquot is calculated. Below, accordingly enter 100 as desired number of energy buffer aliquots (16 µl). The template adapts the volumes of the different buffer components for the 1,700 µl master mix as follows: 204 µl of 100 mM Mg-glutamate stock solution, 136 µl of 3 M K-glutamate stock solution, 728.73 µl of 14x energy solution, 510 µl of 40% PEG-8000 and 121.27 µl sterile ddH2O. Finally, in the third template section, enter 8 nM and 150 nM as optimal vector concentration in the cell-free reaction and respectively, vector DNA stock solution concentration. The template adapts the volumes of the different components that must be added to the 28 µl of crude extract to finalize the preparation of a 90 µl cell-free reaction as follows: 15 µl of energy buffer, 15 µl of one of the 3 differently composed amino acid solutions, 4.80 µl of vector DNA solution (150 nM), and 27.20 µl of sterile ddH2O.
 Supplemental Material 2. One letter amino acid sequence of the model protein deGFP. This model protein contains 6 Arg and 18 Lys positions. Please click here to view a larger version of this figure.
Supplemental Material 2. One letter amino acid sequence of the model protein deGFP. This model protein contains 6 Arg and 18 Lys positions. Please click here to view a larger version of this figure.
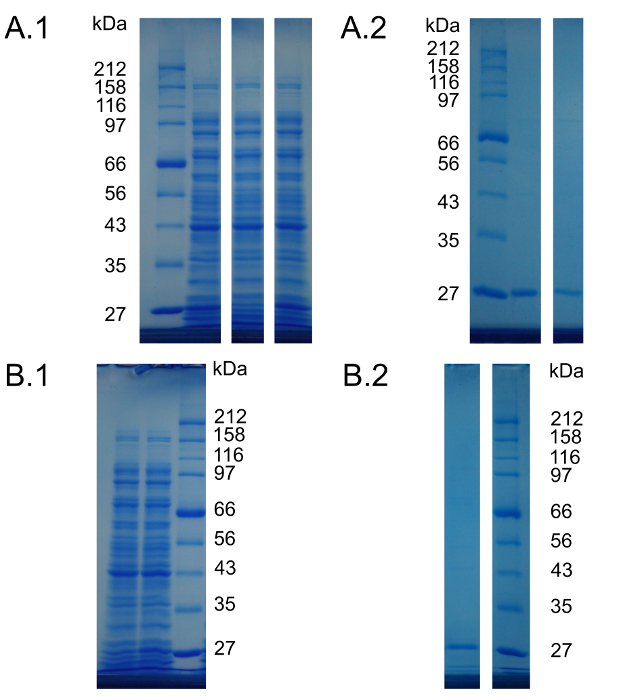 Supplemental Material 3. Full length and unmodified gel lanes that correspond to the gel pictures presented Figure 1 and 2. The gel lanes presented in each individual subfigure are extracted from the same SDS polyacrylamide gel. In Figure 1 and 2 these lanes were joined together for presentation purposes. Molecular weights of the protein standard bands are indicated beside the figures. (A.1) Uncropped gel lanes of Figure 1A. From left to right: Protein standard, reference cell-free reaction, negative control and cell-free reaction providing Can instead of Arg. (A.2) Uncropped gel lanes of Figure 1B. From left to right: Protein standard, purified deGFP from the reference reaction, purified deGFP from the Can containing reaction. (B.1) Uncropped gel lanes of Figure 2A. From left to right: Negative control cell-free reaction, cell-free reaction containing Hyl and protein standard. (B.2) Uncropped gel lanes of Figure 2B. From left to right: Purified deGFP from the Hyl containing reaction and protein standard. Please click here to view a larger version of this figure.
Supplemental Material 3. Full length and unmodified gel lanes that correspond to the gel pictures presented Figure 1 and 2. The gel lanes presented in each individual subfigure are extracted from the same SDS polyacrylamide gel. In Figure 1 and 2 these lanes were joined together for presentation purposes. Molecular weights of the protein standard bands are indicated beside the figures. (A.1) Uncropped gel lanes of Figure 1A. From left to right: Protein standard, reference cell-free reaction, negative control and cell-free reaction providing Can instead of Arg. (A.2) Uncropped gel lanes of Figure 1B. From left to right: Protein standard, purified deGFP from the reference reaction, purified deGFP from the Can containing reaction. (B.1) Uncropped gel lanes of Figure 2A. From left to right: Negative control cell-free reaction, cell-free reaction containing Hyl and protein standard. (B.2) Uncropped gel lanes of Figure 2B. From left to right: Purified deGFP from the Hyl containing reaction and protein standard. Please click here to view a larger version of this figure.
Discussion
An-easy-to use cell-free expression system as a viable strategy to residue-specifically incorporate ncAAs into proteins, is presented. To this end, the crude extract is supplemented with vector DNA coding for the protein of interest, the energy buffer and the corresponding amino acids. Note that the crude extract aliquot volume depends on the crude extract protein concentration34. The cell-free expression efficiency is optimized depending on the vector DNA construction concentration. The volumes of the energy buffer components vary as function of optimized Mg- and K-glutamate concentrations in order to enable high yields of the cell-free expressed model protein.
A preliminary evaluation of the incorporation experiment can be obtained by performing SDS-PAGE of the unpurified cell-free reaction medium. For a more detailed analysis, HPLC-ESI mass spectroscopy is proposed as a means to check for complete, residue-specific incorporation of the ncAA. As a preparation for the latter, spin column systems are used to enable His-tag purification and buffer exchange with the small volumes that we use in this protocol.
Including HPLC-ESI mass spectroscopy, the entire protocol can be performed within 2 days. It does not include any particularly critical steps. However, concentration optimizations of Mg- and K-glutamate as well as of vector DNA are crucial in order to express high yields of the model protein. The use of the highly efficient expression vector pBEST-OR2-Or1-Pr-UTR1-gene_of_model_protein-T500 is strongly recommended. Elution of His-tagged proteins is usually due to high concentration of imidazole (> 150 mM) and other salts such as NaH2PO4 (> 300 mM) or NaCl (> 50 mM) that generate high background noise in mass spectroscopic analysis49. Exchange of such elution buffers with a suitable protein storage buffer stabilizes the model protein and drastically reduces background noise during mass spectroscopic analysis.
As a result, Can replaces Arg at all six positions within the model protein. In the expression system, no Arg residues can be detected. This simplifies the residue-specific incorporation of Arg analogs compared to other expression systems that require further depletion strategies29,30. The presented cell-free approach circumvents the inherent limitations of in vivo approaches that are due to Can toxicity, or the strong dependency on mRNA sequence in single protein production strategies24,31. Contrary to the employed in vitro system, in vivo cleavage of Can to homoserine and hydroxyguanidine does occur31.
However, the cell-free system retains a sufficient amount of Lys to compete with analogs such as Hyl. The HPLC-ESI mass spectroscopic analysis shows that the model protein contains both, the canonical as well as the noncanonical analog in different proportions. The residue-specific incorporation of Lys is possible in general, but for complete substitution, further depletion strategies, or specially engineered aaRS and tRNA optimized for the recognition of ncAAs need to be developed.
We achieved excellent yields of cell-free expressed, modified model proteins by adding the ncAA at the same concentration as the canonical ones. The incorporation efficiency depends on the nature of the ncAA to be incorporated. Even higher yields might still be realizable by optimizing the concentration of the ncAA.
The presented results demonstrate the applicability of the employed system for the residue-specific incorporation of ncAAs as long as they are accepted by the canonical endogenous translational system. For the residue-specific incorporation of specific ncAAs, one further needs to check if the residues of the analogous cAA disturb the expression system.
Cell-free transcription-translation systems can be engineered from different organisms to respond to different demands54. The all E. coli transcription-translation machineries of the here presented cell-free system enable the usage of bacteriophage and E. coli promoters, and they can act in parallel or consecutively in cascades55. The general applicability and usability make the method a potent tool for further research in amino acid toxicity and therapeutic application.
Disclosures
The authors have nothing to disclose.
Acknowledgments
E.G. Worst and A. Ott acknowledge financial support by the Deutsche Forschungsgemeinschaft (DFG) within the collaborative research center SFB 1027 as well as Saarland University. E.G. Worst, A. Ott and V. Noireaux further acknowledge financial aid by the Human Frontiers Science Program Organization (HFSPO). The authors thank Tobias Baumann and Stefan Oehm (Institute of Chemistry, Technische Universität Berlin) for critical reading.
References
- Böck A, et al. Selenocysteine: the 21st amino acid. Mol. Microbiol. 1991;5(3):515–520. doi: 10.1111/j.1365-2958.1991.tb00722.x. [DOI] [PubMed] [Google Scholar]
- Srinivasan G, James CM, Krzycki JA. Pyrrolysine Encoded by UAG in Archaea: Charging of a UAG-Decoding Specialized tRNA. Science. 2002;296(5572):1459–1462. doi: 10.1126/science.1069588. [DOI] [PubMed] [Google Scholar]
- Budisa N. Prolegomena to Future Experimental Efforts on Genetic Code Engineering by Expanding Its Amino Acid Repertoire. Angew. Chem. Int. Ed. Engl. 2004;43:6426–6463. doi: 10.1002/anie.200300646. [DOI] [PubMed] [Google Scholar]
- Wang L, Xie J, Schultz PG. Expanding the Genetic Code. Annu. Rev. Biophys. Biomol. Struct. 2006;35:225–249. doi: 10.1146/annurev.biophys.35.101105.121507. [DOI] [PubMed] [Google Scholar]
- Chin JW. Expanding and Reprogramming the Genetic Code of Cells and Animals. Annu. Rev. Biochem. 2014;83:379–408. doi: 10.1146/annurev-biochem-060713-035737. [DOI] [PubMed] [Google Scholar]
- Neumann H. Rewiring translation - Genetic code expansion and its applications. FEBS Lett. 2012;586(15):2057–2064. doi: 10.1016/j.febslet.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Liu CC, Schultz PG. Adding New Chemistries to the Genetic Code. Annu. Rev. Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- Goerke AR, Swartz JR. High-Level Cell-Free Synthesis Yields of Proteins Containing Site-Specific Non-Natural Amino Acids. Biotechnol. Bioeng. 2009;102(2):400–416. doi: 10.1002/bit.22070. [DOI] [PubMed] [Google Scholar]
- Albayrak C, Swartz JR. Cell-free co-production of an orthogonal transfer RNA activates efficient site-specific non-natural amino acid incorporation. Nucleic Acids Res. 2013;41(11):5949–5963. doi: 10.1093/nar/gkt226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JA, Lu YY, Van Deventer JA, Tirrell DA. Residue-specific incorporation of non-canonical amino acids into proteins: recent developments and applications. Curr. Opin. Chem. Biol. 2010;14(6):774–780. doi: 10.1016/j.cbpa.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiu X, Puskar NL, Shanata JAP, Lester HA, Dougherty DA. Nicotine binding to brain receptors requires a strong cation-pi interaction. Nature. 2009;458(7237):534–537. doi: 10.1038/nature07768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünewald J, et al. Mechanistic studies of the immunochemical termination of self-tolerance with unnatural amino acids. Proc. Natl. Acad. Sci. USA. 2009;106(11):4337–4342. doi: 10.1073/pnas.0900507106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikić I, Lemke EA. Genetic code expansion enabled site-specific dual-color protein labeling: superresolution microscopy and beyond. Curr. Opin. Chem. Bio. 2015;28:164–173. doi: 10.1016/j.cbpa.2015.07.021. [DOI] [PubMed] [Google Scholar]
- Munier R, Cohen GN. Incorporation d'analogues structuraux d'aminoacides dans les protéines bactériennes. Biochim. Biophys. Acta. 1956;21(3):592–593. doi: 10.1016/0006-3002(56)90207-4. [DOI] [PubMed] [Google Scholar]
- Lepthien S, Merkel L, Budisa N. In Vivo Double and Triple Labeling of Proteins Using Synthetic Amino Acids. Angew. Chem. Int. Ed. 2010;49(32):5446–5450. doi: 10.1002/anie.201000439. [DOI] [PubMed] [Google Scholar]
- Dieterich DC, Link AJ, Graumann J, Tirrell DA, Schuman EM. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT) Proc. Natl. Acad. Sci. USA. 2006;103(25):9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich DC, et al. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat. Neurosci. 2011;13(7):897–905. doi: 10.1038/nn.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson WA, Horton JR, LeMaster DM. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional structure. EMBO J. 1990;9(5):1665–1672. doi: 10.1002/j.1460-2075.1990.tb08287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoesl MG, et al. Lipase Congeners Designed by Genetic Code Engineering. ChemCatChem. 2011;3(1):213–221. [Google Scholar]
- Bae JH, et al. Expansion of the Genetic Code Enables Design of a Novel "Gold" Class of Green Fluorescent Proteins. J. Mol. Biol. 2003;328(5):1071–1081. doi: 10.1016/s0022-2836(03)00364-4. [DOI] [PubMed] [Google Scholar]
- Schachtele CF, Rogers P. Canavanine death in Escherichia coli. J. Mol. Biol. 1965;14(2):474–489. doi: 10.1016/s0022-2836(65)80197-8. [DOI] [PubMed] [Google Scholar]
- Rosenthal GA. The biological effects and mode of action of L-canavanine, a structural analogue of L-arginine. Q. Rev. Biol. 1977;52(2):155–178. doi: 10.1086/409853. [DOI] [PubMed] [Google Scholar]
- Rosenthal GA, Dahlman DL. Incorporation of L-Canavanine into Proteins and the Expression of Its Antimetabolic Effects. J. Agric. Food Chem. 1991;39(5):987–990. [Google Scholar]
- Ishida Y, Park JH, Mao L, Yamaguchi Y, Inouye M. Replacement of All Arginine Residues with Canavanine in MazF-bs mRNA Interferase Changes Its Specificity. J. Biol. Chem. 2013;288(11):7564–7571. doi: 10.1074/jbc.M112.434969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DA, Rosenthal GA, Gold DV, Dickey K. Growth Inhibition of a Rat Colon Tumor by L-Canavanine. Cancer Res. 1986;46(6):2898–2903. [PubMed] [Google Scholar]
- Bence AK, Worthen DR, Adams VR, Crooks PA. The antiproliferative and immunotoxic effects of L-canavanine and L-canaline. Anticancer Drugs. 2002;13(3):313–320. doi: 10.1097/00001813-200203000-00013. [DOI] [PubMed] [Google Scholar]
- Bence AK, Crooks PA. The Mechanism of L-Canavanine Cytotoxicity: Arginyl tRNA Synthetase as a Novel Target for Anticancer Drug Discovery. J. Enzyme Inhib. Med. Chem. 2003;18(5):383–394. doi: 10.1080/1475636031000152277. [DOI] [PubMed] [Google Scholar]
- Akaogi J, et al. Role of non-protein amino acid L-canavanine in autoimmunity. Autoimmun. Rev. 2006;5(6):429–435. doi: 10.1016/j.autrev.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Singh-Blom A, Hughes RA, Ellington AD. An amino acid depleted cell-free protein synthesis system for the incorporation of non-canonical amino acid analogs into proteins. J. Biotechnol. 2014;178:12–22. doi: 10.1016/j.jbiotec.2014.02.009. [DOI] [PubMed] [Google Scholar]
- Oh S-J, Lee K-H, Kim H-C, Catherine C, Yun H, Kim D-M. Translational Incorporation of Multiple Unnatural Amino Acids in a Cell-free Protein Synthesis System. Bioprocess. Eng. 2014;19(3):426–432. [Google Scholar]
- Worst EG, Exner MP, De Simone A, Schenkelberger M, Noireaux V, Budisa N, Ott A. Cell-free expression with the toxic amino acid canavanine. Bioorg. Med. Chem. Lett. 2015;25(17):3658–3660. doi: 10.1016/j.bmcl.2015.06.045. [DOI] [PubMed] [Google Scholar]
- Shin J, Noireaux V. Efficient cell-free expression with the endogenous E. Coli RNA polymerase and sigma factor 70. J. Biol. Eng. 2010;4(8) doi: 10.1186/1754-1611-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemla Y, Ozer E, Schlesinger O, Noireaux V, Alfonta L. Genetically expanded cell-free protein synthesis using endogenous pyrrolysyl orthogonal translation system. Biotechnol. Bioeng. 2015;112(8):1663–1672. doi: 10.1002/bit.25587. [DOI] [PubMed] [Google Scholar]
- Sun ZZ, Hayes CA, Shin J, Caschera F, Murray RM, Noireaux V. Protocols for Implementing an Escherichia coli Based TX-TL Cell-Free Expression System for Synthetic Biology. J. Vis. Exp. 2013. p. e50762. [DOI] [PMC free article] [PubMed]
- Caschera F, Noireaux V. Preparation of amino acid mixtures for cell-free expression systems. Biotechniques. 2015;58(1):40–43. doi: 10.2144/000114249. [DOI] [PubMed] [Google Scholar]
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Cambridge, MA: JoVE; 2015. Molecular Cloning. Available from: http://www.jove.com/science-education/5074/molecular-cloning. [Google Scholar]
- Green MR, Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2012. [Google Scholar]
- Froger A, Hall JE. Transformation of Plasmid DNA into E. coli Using the Heat Shock Method. J. Vis. Exp. 2007. p. e253. [DOI] [PMC free article] [PubMed]
- Zhang S, Cahalan MD. Purifying Plasmid DNA from Bacterial Colonies Using the Qiagen Miniprep Kit. J. Vis. Exp. 2007. p. e247. [DOI] [PMC free article] [PubMed]
- Moreno LA, Cox KL. Quantification of dsDNA using the Hitachi F-7000 Fluorescence Spectrophotometer and PicoGreen Dye. J. Vis. Exp. 2010. p. e2465. [DOI] [PMC free article] [PubMed]
- Desjardins P, Conklin D. NanoDrop Microvolume Quantitation of Nucleic Acids. J. Vis. Exp. 2010. p. e2699. [DOI] [PMC free article] [PubMed]
- Sukumaran S. Concentration Determination of Nucleic Acids and Proteins Using the Micro-volume Bio-spec Nano Spectrophotometer. J. Vis. Exp. 2011. p. e2699. [DOI] [PMC free article] [PubMed]
- Laemmli UK. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Cambridge, MA: JoVE; 2015. Separating Protein with SDS-PAGE. Available from: http://www.jove.com/science-education/5058/separating-protein-with-sds-page. [Google Scholar]
- Hochuli E, Bannwarth W, Döbeli H, Gentz R, Stüber D. Genetic Approach to Facilitate Purification of Recombinant Proteins with a Novel Metal Chelate Adsorbent. Nature Biotechnology. 1988;6(11):1321–1325. [Google Scholar]
- Schägger H, Aquila H, Von Jagow G. Coomassie blue-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for direct visualization of polypeptides during electrophoresis. Anal. Biochem. 1988;173(1):201–205. doi: 10.1016/0003-2697(88)90179-0. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A Rapid Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Ernst O, Zor T. Linearization of the Bradford Protein Assay. J. Vis. Exp. 2010. p. e1918. [DOI] [PMC free article] [PubMed]
- Hurst R, Kobs G, Johnson T. Mass Spectrometric Analysis of MagneHis Purified Proteins. Promega Corporation Web site. 2003. [Cited 2015 Oct 12]. http://www.promega.de/resources/pubhub/enotes/mass-spectrometric-analysis-of-magnehis-purified-proteins/
- Banerjee S, Mazumdar S. Electrospray Ionization Mass Spectrometry: A Technique to Access the Information beyond the Molecular Weight of the Analyte. Int. J. Anal. Chem. 2012;2012:1–40. doi: 10.1155/2012/282574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara K, Nomura SM. Condensation of an additive-free cell extract to mimic the conditions of live cells. PLoS One. 2013;8(1):e54155. doi: 10.1371/journal.pone.0054155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Marshall AG. A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J. Am. Soc. Mass. Spectrom. 1998;9(3):225–233. doi: 10.1016/S1044-0305(97)00284-5. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang G, Ngo N, Zhao X, Kain SR, Huang CC. Deletions of the Aequorea victoria Green Fluorescent Protein Define the Minimal Domain Required for Fluorescence. J. Biol. Chem. 1997;272(45):28545–28549. doi: 10.1074/jbc.272.45.28545. [DOI] [PubMed] [Google Scholar]
- Gagoski D, Polinkovsky ME, Mureev S, Kunert A, Johnston W, Gambin Y, Alexandrov K. Performance benchmarking of four cell-free protein expression systems. Biotechnol. Bioeng. 2016;113(2):292–300. doi: 10.1002/bit.25814. [DOI] [PubMed] [Google Scholar]
- Shin J, Noireaux V. An E. coli Cell-Free Expression Toolbox: Application to Synthetic Gene Circuits and Artificial Cells. ACS Synth. Biol. 2012;1(1):29–41. doi: 10.1021/sb200016s. [DOI] [PubMed] [Google Scholar]