

Abstract
The recent development of a high-throughput single-cell assay technique enables the screening of novel enzymes based on functional activities from a large-scale metagenomic library1. We previously proposed a genetic enzyme screening system (GESS) that uses dimethylphenol regulator activated by phenol or p-nitrophenol. Since a vast amount of natural enzymatic reactions produce these phenolic compounds from phenol deriving substrates, this single genetic screening system can be theoretically applied to screen over 200 different enzymes in the BRENDA database. Despite the general applicability of GESS, applying the screening process requires a specific procedure to reach the maximum flow cytometry signals. Here, we detail the developed screening process, which includes metagenome preprocessing with GESS and the operation of a flow cytometry sorter. Three different phenolic substrates (p-nitrophenyl acetate, p-nitrophenyl-β-D-cellobioside, and phenyl phosphate) with GESS were used to screen and to identify three different enzymes (lipase, cellulase, and alkaline phosphatase), respectively. The selected metagenomic enzyme activities were confirmed only with the flow cytometry but DNA sequencing and diverse in vitro analysis can be used for further gene identification.
Keywords: Molecular Biology, Issue 114, Genetic circuits, high-throughput screening, enzyme screening, metagenome, phenolic compound, dmpR
Introduction
A recently developed high-throughput single-cell assay technique allows novel enzymes to be screened from a large-scale genetic library based on their functional activities1. At the single cell level, proteins regulating transcription are employed to trigger reporter gene expression by sensing small molecules that are produced as a result of a target enzyme activity. One early approach involved the isolation of a phenol-degrading operon from Ralstonia eutropha E2 using the substrate-induced genetic expression screening (SIGEX) method, in which the substrate induces the expression of a reporter protein2. NhaR of Pseudomonas putida was used to select benzaldehyde dehydrogenase3, and LysG from Corynebacterium glutamicum was utilized for the high-throughput screening of a new L-lysine-producing strain from diverse mutant libraries4.
Previously, a genetic enzyme screening system (GESS) was proposed as a generally applicable screening platform5. This system uses the phenol-recognizing dimethylphenol regulator, DmpR, of P. putida. DmpR(E135K), and a mutant of DmpR, can also be employed in GESS (pNP-GESS) for the detection of p-nitrophenol (pNP). In the presence of target enzymes producing phenolic compounds, GESS in E. coli cells emits a fluorescence signal, enabling the rapid isolation of single cells using a fluorescence-activated cell sorter (FACS). But the expression of metagenomic enzyme appears to be weaker than that of conventional recombinant enzymes; therefore, GESS was designed to detect phenolic compounds with maximum sensitivity by investigating the combination of ribosomal binding site (RBS) and terminator sequences along with optimal operating condition5.
One of the fundamental advantages of GESS is that this single method theoretically allows the screening of over than 200 different types of enzymes in the BRENDA database (Table 1, http:// www.brenda-enzymes.info, 2013.7) by simply employing different substrates. It was shown that cellulase, lipase, and methyl parathion hydrolase (MPH) can be detected using pNP-GESS with appropriate substrates of p-nitrophenyl butyrate, p-nitrophenyl-cellotrioside, and methyl parathion, respectively5. Recently, it was proved that an alkaline phosphatase (AP), which is one of the novel enzymes identified using pNP-GESS, is the first thermolabile AP found in cold-adapted marine metagenomes6.
Here, details of the screening process is presented with pNP-GESS detecting the activities of three different types of enzymes- lipase, cellulase, and alkaline phosphatase -and rapidly identifying novel candidate enzymes from a metagenomic library5,6. The processes include metagenome preprocessing with pNP-GESS and operating a flow cytometry sorter. While the hits obtained will need to be sequenced for further identification, this protocol covers the procedure up to the steps of enzyme activity confirmation using flow cytometry.
Protocol
1. Preparing the Metagenomic Library with pNP-GESS
Construct a metagenomic library in E. coli with a fosmid vector using a fosmid library production kit according to the manufacturer's protocol 5.
Aliquot 100 µl of the library for storage at −70 °C, which is a source of metagenomic library cells. Note: The optical density of a sample measured at a wavelength of 600 nm (OD600) of this library stock is approximately 100.
Thaw 100 µl of the stock metagenomic library on ice and inoculate in a 500 ml flask containing 50 ml Luria-Bertani (LB) and 12.5 µg/ml chloramphenicol followed by 37 °C incubation for 2 hr.
Harvest the cells in a 50 ml conical tube by centrifugation at 1,000 x g for 20 min at 4 °C.
Resuspend the pellet quickly in 50 ml ice-cold distilled water (DW) and centrifuge at 1,000 x g for 20 min at 4 °C again.
Resuspend the pellet in 50 µl ice-cold DW with 10% (v/v) glycerol. Use 50 µl of this cell aliquot for electroporation.
Place the mixture of electrocompetent cells 50 µl and pGESS(E135K)5 DNA (100ng) in an ice-cold electroporation cuvette and electroporate (1.8 kV/cm, 25 µF) the mixture.
Quickly add 1 ml super optimal broth with catabolite repression (SOC) medium and resuspend the cells gently.
Transfer the cells into 14 ml round-bottom tube using a pipette and incubate at 37 °C for 1 hr.
Spread 500 µl of the recovered cells on a LB 20 x 20 cm square plate containing 12.5 µg/ml chloramphenicol and 50 µg/ml ampicillin. Incubate plate at 30 °C for 12 hr.
Scrape the colonies using a cell scraper and collect cells into a 50 ml conical tube using ice-cold cell storage media.
Centrifuge at 1,000 x g for 20 min at 4 °C. Resuspend the pellet in 10 ml ice-cold cell storage media to reach an OD600 of 100.
Aliquot 20 µl of the cells for storage at −70 °C.
2. Removing False Positives from the Metagenomic Library
Thaw the stock metagenomic library cells (from Step 1.13) containing metagenomic DNA fosmid and pGESS(E135K) on ice.
Inoculate 10 µl of the cells in 2 ml LB containing 50 µg/ml ampicillin and 12.5 µg/ml chloramphenicol in a 14-ml round-bottom tube. Incubate at 37 °C with shaking at 200 rpm for 4 hr.
Meanwhile, turn on the FACS machine and open the default FACS software. Use the following settings: nozzle tip diameter, 70 µm; Forward Scatter Area (FSC-A) sensitivity, 300 V-logarithmic amplification; Side Scatter Area (SSC-A) sensitivity, 350 V-logarithmic amplification; Fluorescein Isothiocyanate Area (FITC-A) sensitivity, 450 V-logarithmic amplification; threshold parameter, FSC-A value 5. Note: Typical settings are given for FACS device mentioned in the Table of Materials. Adjust the settings for other FACS devices.
Dilute the metagenomic library cells from Step 2.2 by adding 5 µl of the sample to a 5-ml round bottom tube containing 1 ml PBS.
Place the diluted library sample onto the loading port of the FACS instrument and click on Load button on the Acquisition Dashboard of the FACS software.
Adjust the event rate to 1,000 - 1,500 events/sec by clicking and controlling the Flow Rate buttons on the dashboard.
Create log scaled FSC-A vs. log scaled SSC-A scatter plot on a global worksheet by clicking on the Dot Plot button in the tool bar. Adjust the scatter gate R1 to encompass the singlet events (bacterial population) using the Polygon Gate button in the tool bar.
Plot a histogram with cell count vs. log scaled FITC-A on the worksheet by clicking on Histogram button. Then, adjust the FITC voltage from the Cytometer Settings tab on the Inspector Control window such that the peak of the bell-shaped distribution is less than 102 of FITC-A.
Create a log scaled FSC-A vs. the log FITC-A plot by clicking on the Dot Plot button on the global worksheet. Set a sorting gate R2 on the plot using the Polygon Gate button on the tool bar so that the gate is located between +5% and -5% cells from the center of the distribution (total 10% cells around the peak of the bell-shaped curve).
Place a collection tube containing 1.2 ml LB containing 50 µg/ml ampicillin and 12.5 µg/ml chloramphenicol at the outlet of the FACS instrument and sort out 106 cells satisfying both the R1 and R2 gates.
Remove the collection tube, cap, and gently vortex.
3. Metagenomic Enzyme Screening
Incubate the sorted cells (from Step 2.11) at 37 °C with shaking at 200 rpm until the OD600 reaches 0.5.
Add 1 μl copy induction solution to amplify the intracellular fosmid copy number. Incubate the cells for an additional 3 hr at 37 °C with shaking at 250 rpm.
- To prepare cells for sorting, add 0.5 ml of the cultured cells and an appropriate substrate (p-nitrophenyl acetate, p-nitrophenyl-β-D-cellobioside, or phenyl phosphate) into a 14-ml round-bottom tube at a final concentration of 100 µM.
- For a control sample, add 0.5 ml of the cultured cells from Step 3.2 into a 14-ml round-bottom tube. Add the same volume of PBS as the substrate volume in Step 3.3.
Incubate the two samples at 37 °C with shaking at 200 rpm for 3 hr.
Meanwhile, prepare the FACS machine with the same configurations as Step 2.3.
Add 5 µl of the cells (for sorting) and control cells to two 5 ml round-bottom tubes containing 1 ml PBS, respectively.
Place the tube containing control cells onto the loading port of the FACS and click on Load button on Acquisition Dashboard of the FACS software. Adjust the event Rate to 1,000 - 3,000 events/sec by controlling Flow Rate buttons on the dashboard.
Create log scaled FSC-A vs. log scaled SSC-A scatter plot by clicking on Dot Plot button on the global worksheet of the software and adjust the scatter gate R1 to encompass the singlet events (bacterial population) using the Polygon Gate button on the tool bar.
Create log scaled FSC-A vs. log scaled FITC-A scatter plot by clicking on the Dot Plot button on the worksheet and set a sorting gate R2 on the plot using the Polygon Gate button on the tool bar so that less than 0.1 % of negative cells are detected within the R2 gate.
Replace the control sample tube with the sorting sample tube, and adjust the event rate to 1,000 - 3,000 events/sec by controlling the Flow Rate buttons on the dashboard.
Place a collection tube containing 0.5 ml LB at the outlet of the FACS instrument and sort out 104 cells satisfying both the R1 and R2 gates. Note: The sorting criterion can range from top 0.1% to 5% of FITC-A in the R2 gate. In this protocol, top 1% cells were collected as positives.
Remove the collection tube, cap, and gently vortex.
Spread the 0.5 ml collected samples on two agar plates (90 mm Petri dishes) containing LB, 50 µg/ml ampicillin, 12.5 µg/ml chloramphenicol and incubate the plate at 37 °C overnight.
If necessary, perform additional rounds of sorting for FITC-A enrichment by repeating Steps 3.1-3.14.
4. Hit Selection and Enzyme Activity Confirmation
- Flow cytometry analysis
- From the incubated plate at Step 3.14, select a colony showing no fluorescence using a colony picker under the observation of 488 nm wavelength of a LED illuminator.
- Inoculate the selected colony in a 14-ml round-bottomed tube containing 2 ml LB containing 50 µg/ml ampicillin and 12.5 µg/ml chloramphenicol at 37 °C with shaking at 200 rpm until its OD600 reaches 0.5.
- Add 2 μl copy induction solution to amplify the intracellular fosmid copy number. Incubate the cells for an additional 3 hr at 37 °C with shaking at 250 rpm.
- Prepare a 14 ml round-bottom tube and add 1 ml of the cultured cells (Step 4.1.3). Add an appropriate substrate (p-nitrophenyl acetate, p-nitrophenyl-β-D-cellobioside, or phenyl phosphate) at a final concentration of 100 µM.
- For a control, add the 1 ml cultured cells from Step 4.1.3 into a 14 ml round-bottom tube and add the same volume of PBS as the substrate volume in Step 4.1.4.
- Incubate the two samples at 37 °C with shaking at 200 rpm for 3 hr.
- Meanwhile, prepare the FACS machine with the same configurations as Step 2.3.
- Add 5 µl of the control and substrate treated cells to 5 ml round-bottom tubes containing 1 ml PBS, respectively.
- Place the tube containing control cells on the loading port of the FACS device and click on Load button on Acquisition Dashboard of the FACS software. Adjust the event rate to 1,000 - 3,000 events/sec using Flow Rate buttons on the dashboard.
- Click on Acquisition button to measure FITC-A.
- Replace the control sample tube with the substrate treated sample tube. Adjust the event rate to 1,000 - 3,000 events/sec using Flow Rate buttons on the dashboard.
- Click on Acquisition button to measure FITC-A.
- Compare the fluorescence of the two groups of cells by plotting a histogram of cell count vs. the log scaled FITC-A.
- Other assays to confirm enzyme activity
- For further identification of the selected enzyme candidates, extract fosmid DNA using standard extraction procedures from the commercial extraction kits and analyze the nucleotide sequence, or test the in vitro enzyme activity6,7.
Representative Results
The three phenolic substrates were examined to identify novel metagenomic enzymes from a metagenome library of ocean-tidal flat sediments in Taean, South Korea by following the proposed protocol. For the library construction, average 30 - 40 kb metagenome sequences were inserted into fosmids, which are based on the E. coli F factor replicon and presented as a single copy in a cell. Note that fosmids have been widely used for constructing complex genomic libraries due to their stable propagation8.
Figure 1 shows a brief flowchart of the screening protocol including the removal of false positives with negative sorting (Figure 1 A) followed by positive hit selection with R1 and R2 sorting gates (Figure 1 B). The hits were evaluated by comparing the fluorescence of the samples with and without substrates (Figure 1 C). In case of p-nitrophenyl acetate mediated screening, five candidates of lipase were confirmed where the fluorescence of substrate treated cells were higher than that of control cells (Figure 2 A). In spite of the broad distributions of the three candidates of cellulase in Figure 2 B, they also showed clear differences of average FITC intensity of the populations. Four phosphatase candidates also showed high fluorescence level compared to the control cells (Figure 2 C). The more fluorescence difference between negative and the candidate cells the stronger enzyme activity can be deduced since the fluorescence reporter of pNP-GESS shows quantitative responses to the phenol concentrations5.
In this protocol, only the fluorescence differences were confirmed using a flow cytometry but various identification assays can be applied as mentioned in Step 4.2. Figure 3 demonstrates the example of AP identification6. A selected clone among forty-seven highly fluorescent colonies from the screening was sequenced and an ORF of the candidate AP was identified. The ORF inserted into a plasmid vector was confirmed to have AP activity with flow cytometry. In Figure 3, the hit shows stronger fluorescence signal than the cells carrying empty plasmid (negative). Further analysis of sequence and in vitro enzymatic activity discovered that this AP was highly similar to other known APs in its enzymatic characteristics except high catalytic activity at lower temperatures (35 °C) and a remarkable thermal instability (65 °C) within 15 min6. In addition, the screened AP was comparable to commercially available APs in removing terminal phosphates from cohesive and blunt ends of linearized plasmid DNA6 .
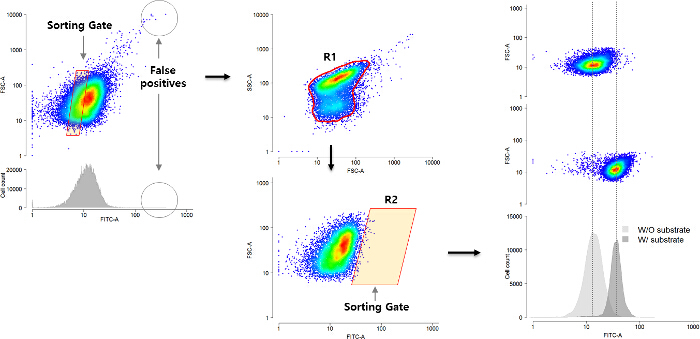 Figure 1: Multi Enzyme Screening Process. (A) Removal of false-positive cells showing fluorescence without substrate. The sorting gate is located in the middle of the cell population density so that the gate includes 10% of the total cells. (B) Sorting high fluorescent (FITC-A) cells (positive) satisfying R1 and R2 gates in the presence of an appropriate substrate. (C) After the selection and single cell isolation, the activity of the candidate enzymes was examined with and without the substrate (denoted as W/ and W/O substrates, respectively). The clear signal difference between the two samples implies the fosmid vector of the cells contains target genetic information of interest. Please click here to view a larger version of this figure.
Figure 1: Multi Enzyme Screening Process. (A) Removal of false-positive cells showing fluorescence without substrate. The sorting gate is located in the middle of the cell population density so that the gate includes 10% of the total cells. (B) Sorting high fluorescent (FITC-A) cells (positive) satisfying R1 and R2 gates in the presence of an appropriate substrate. (C) After the selection and single cell isolation, the activity of the candidate enzymes was examined with and without the substrate (denoted as W/ and W/O substrates, respectively). The clear signal difference between the two samples implies the fosmid vector of the cells contains target genetic information of interest. Please click here to view a larger version of this figure.
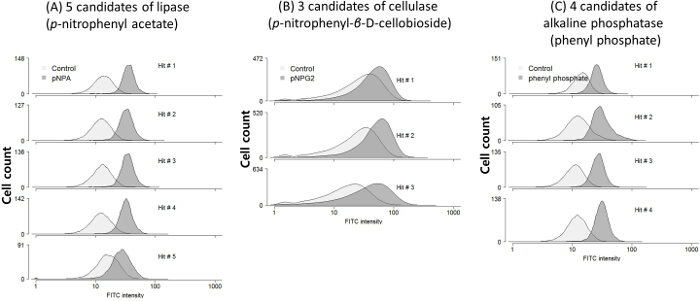 Figure 2:Enzyme Activity Evaluation Using Flow Cytometry. Metagenomic enzyme candidates screened by pNP-GESS with three different substrates (A) p-nitrophenyl acetate (pNPA), (B) p-nitrophenyl-β-D-cellobioside (pNPG2), and (C) phenyl phosphate. All the candidates show clear signal difference between cells with and without substrate. Controls are the cells without their corresponding substrate treatment. Please click here to view a larger version of this figure.
Figure 2:Enzyme Activity Evaluation Using Flow Cytometry. Metagenomic enzyme candidates screened by pNP-GESS with three different substrates (A) p-nitrophenyl acetate (pNPA), (B) p-nitrophenyl-β-D-cellobioside (pNPG2), and (C) phenyl phosphate. All the candidates show clear signal difference between cells with and without substrate. Controls are the cells without their corresponding substrate treatment. Please click here to view a larger version of this figure.
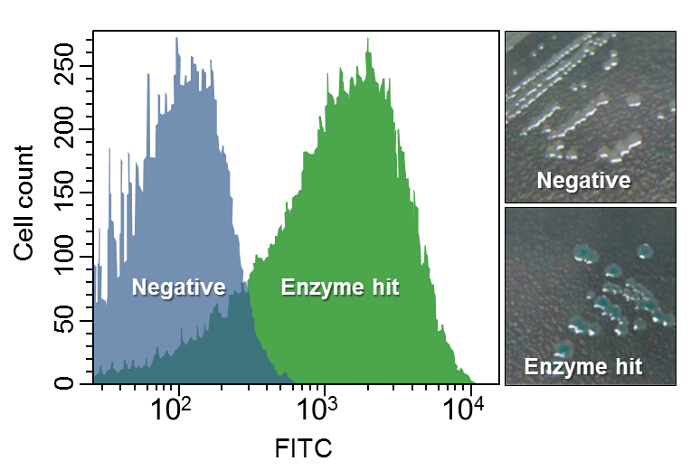 Figure 3:Alkaline Phosphatase Identification. Left panel shows clear fluorescence difference between the cells containing the enzyme hit and an empty plasmid vector (Negative). On the right, phosphatase assay of cells harboring the phosphatase candidate gene in a plasmid vector on LB agar containing 5-bromo-4-chloro-3-indolyl phosphate as a colorimetric substrate is shown. Blue colonies indicate that the selected cells contain the metagenomic gene encoding a phosphatase. See 6 for more details. Please click here to view a larger version of this figure.
Figure 3:Alkaline Phosphatase Identification. Left panel shows clear fluorescence difference between the cells containing the enzyme hit and an empty plasmid vector (Negative). On the right, phosphatase assay of cells harboring the phosphatase candidate gene in a plasmid vector on LB agar containing 5-bromo-4-chloro-3-indolyl phosphate as a colorimetric substrate is shown. Blue colonies indicate that the selected cells contain the metagenomic gene encoding a phosphatase. See 6 for more details. Please click here to view a larger version of this figure.
| EC Number | Description | No. of p-nitrophenol producing enzymes | No. of phenol producing enzymes | |
| EC 1 | 1.3 | Acting on the CH-CH group of donors | - | 1 |
| Oxidoreductases | 1.11 | Peroxygenase | 2 | - |
| 1.14 | Acting on paired donors, with incorporation or | - | 2 | |
| reduction of molecular oxygen | ||||
| EC 2 | 2.3 | Acyltransferases | 1 | - |
| Transferases | 2.4 | Glycosyltransferases | 8 | - |
| 2.5 | Transferring alkyl or aryl groups, other than methyl groups | 1 | 1 | |
| 2.7 | Transferring phosphorus-containing groups | 1 | 1 | |
| 2.8 | Transferring sulfur-containing groups | 3 | 1 | |
| EC 3 | 3.1 | Act on ester bonds | 67 | 16 |
| Hydrolases | 3.2 | Glycosylases | 75 | 21 |
| 3.4 | Act on peptide bonds - peptidase | 26 | 4 | |
| 3.5 | Act on carbon-nitrogen bonds, other than peptide bonds | 5 | 3 | |
| 3.6 | Act on acid anhydrides | 4 | - | |
| 3.7 | Act on carbon-carbon bonds | 2 | - | |
| EC 4 | 4.1 | Carbon-carbon lyases | - | 3 |
| Lyases | 4.2 | Carbon-oxygen lyases | 1 | - |
| 4.3 | Carbon-nitrogen lyases | 1 | - | |
| Subtotal number of enzymes | 197 | 53 | ||
| Total number of enzymes (excluding overlapped enzymes) | 211 |
Table 1: The Number of Enzymes that Produce p-nitrophenol or Phenol from the BRENDA Database.
Discussion
Increasing production efficiency of biocatalysts is a key for the success of bio-chemical based industry9 and metagenome is considered one of the best natural enzyme source. In this sense, it is essential to screening novel enzymes from the metagenome where majority of the genetic resources have not been explored10. Several screening methods have been developed which directly detect enzyme products using transcriptional activators11, 12 but these techniques require specific metabolite-responsive transcriptional system. On the other hand, GESS is broadly applicable depending on the use of phenol or pNP tagged substrates. Although most of the reported enzymes producing phenol or pNP compounds in BRENDA database (Table 1), (including the three examples in this work) belong to the hydrolase (EC number 3) class enzymes, exploring more diverse phenolic substrates and even designing an artificial substrate subjecting to target enzyme activity, as alternatives of natural substrates, could significantly extend the applicability of the screening system. Note that choosing an appropriate phenol or pNP tagged substrate is one of the crucial steps of pNP-GESS application since the membrane permeability and cellular effect of the substrate considerably influence the screening protocol. Here, we show that industrially important three different types of enzymes can be identified in a high-throughput manner using the single screening system.
The approach of using FACS, in addition to the high-throughput screening of metagenomic enzymes, can be also applied to engineering transcriptional regulators (TRs) for altered TR specificity. One example is a study in which an AraC variant that detected mevalonate instead of arabinose was isolated using FACS, and then employed to screen cells producing mevalonate13. Also, the alteration of the specificity of TRs has allowed for further engineering of enzyme activity. Auto-regulated TR, PcaU, was engineered to detect 3,4-dihydroxybenzoate (34DHB) and then applied to screen for dehydroshikimate dehydratase enzymes (AsbF) at their maximum activity of converting endogenous dehydroshikimate to 34DHB14. The basic concept of these studies is similar to that of pNP-GESS, since pNP-GESS uses a DmpR variant to detect pNP and shows high-throughput performance in screening from a large-scale genetic library. However, specificity engineering may cause unintended false positives among the hits, as it is possible to trigger reporter expression by other inducers as a result of broadened specificity. Therefore, it would be desirable to investigate the orthogonality of TR response before the TR is applied. On the other hand, increasing the sensitivity and dynamic range of the TR could compensate for false positive generation. For example, the sensitivity of GESS was improved by inserting optimized RBS and terminator sequences into the system. False positives of the screening system possibly come from malfunction of the genetic circuit, unintended phenolic chemicals activating the TR, heterogeneous cell growth, and free diffusion of phenol or pNP. The negative selection processes of Steps 2.1 - 2.9 were expected to remove false positive cells releasing fluorescence without substrate treatment. These false positive cells can be also reduced by maximizing the signal to noise ratio of the genetic circuit. Along with the RBS and terminator engineering, the use of M9-glucose media significantly improved the signal (7-fold higher than LB media) in pNP-GESS performance5. But, in this protocol, LB was used as a compromise between the unknown metagenomic gene expression and GESS signal intensity. Investigating more delicate conditions of pNP-GESS cell growth and substrate effect in M9-glucose enables to improve the signal intensity. Tight regulation of reaction time of the substrates, in Step 3.5, is also important to prevent false positives triggered by phenol or pNP diffusion.
GESS enables the conversion of enzyme function into transcription-mediated reporter protein expression at a single-cell level but the indirect identification via reporter protein could be one of the innate limitations. So, in vitro enzyme assay and LC/GC-MS analysis of a product need to be followed for definite identification of a putative enzyme candidate6,7. Also it is possible to use GESS without the need for expensive FACS analysis by attaching alternative reporters such as antibiotic resistance genes or chromogenic enzymes with fluorescent reporters. For further improvement of GESS applications, it is necessary to establish a method to characterize enzyme activity and identify the enzymes in a high-throughput manner. Along with other high-throughput technologies, such as next-generation sequencing, GESS will be a widely used strategy for protein and enzyme catalyst engineering in the biology-based industry.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This research was supported by grants from the Intelligent Synthetic Biology Center of Global Frontier Project (2011-0031944), the Next-Generation Biogreen 21 Program (PJ009524), NRF-2015M3D3A1A01064875 and the KRIBB Research Initiative Program.
References
- Eggeling L, Bott M, Marienhagen J. Novel screening methods-biosensors. Curr. Opin. Biotech. 2015;35:30–36. doi: 10.1016/j.copbio.2014.12.021. [DOI] [PubMed] [Google Scholar]
- Uchiyama T, Abe T, Ikemura T, Watanabe K. Substrate-induced gene-expression screening of environmental metagenome libraries for isolation of catabolic genes. Nat. Biotechnol. 2005;23(1):88–93. doi: 10.1038/nbt1048. [DOI] [PubMed] [Google Scholar]
- Van Sint Fiet S, van Beilen JB, Witholt B. Selection of biocatalysts for chemical synthesis. Proc. Natl. Acad. Sci. U. S. A. 2006;103(6):1693–1698. doi: 10.1073/pnas.0504733102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder S, et al. A high-throughput approach to identify genomic variants of bacterial metabolite producers at the single-cell level. Genome Biol. 2012;13(5):R40. doi: 10.1186/gb-2012-13-5-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, et al. Toward a generalized and High-throughput Enzyme Screening System Based on Artificial Genetic Circuits. ACS Synthetic Biology. 2014;3(3):163–171. doi: 10.1021/sb400112u. [DOI] [PubMed] [Google Scholar]
- Lee DH, et al. A novel psychrophilic alkaline phosphatase from the metagenome of tidal flat sediments. BMC Biotechnology. 2015;15(1) doi: 10.1186/s12896-015-0115-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnecke F, et al. Metagenomic and functional analysis of hindgut microbiota of a wood-feeding higher termite. Nature. 2007;450(7169):560–565. doi: 10.1038/nature06269. [DOI] [PubMed] [Google Scholar]
- Kim UJ, Shizuya H, de Jong PJ, Birren B, Simon MI. Stable propagation of cosmid sized human DNA inserts in an F factor based vector. Nucleic Acids Research. 1992;20(5):1083–1085. doi: 10.1093/nar/20.5.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemli S, Ayadi-Zouari D, Hlima HB, Bejar S. Biocatalysts: application and engineering for industrial purposes. Crc. Cr. Rev. Biotechn. 2014;36(2):246–258. doi: 10.3109/07388551.2014.950550. [DOI] [PubMed] [Google Scholar]
- Lorenz P, Eck J. Metagenomics and industrial applications. Nat. Rev. Microbiol. 2005;3(6):510–516. doi: 10.1038/nrmicro1161. [DOI] [PubMed] [Google Scholar]
- Uchiyama T, Miyazaki K. Product-induced gene expression, a product responsive reporter assay used to screen metagenomic libraries for enzyme encoding genes. Applied and environmental microbiology. 2010;76(21):7029–7035. doi: 10.1128/AEM.00464-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn WW, Garmendia J, Galvao TC, De Lorenzo V. Surveying bio-transformations with à la carte genetic traps: translating dehydrochlorination of lindane (gamma-hexachlorocyclohexane) into lacZ-based phenotypes. Environmental microbiology. 2006;8(3):546–555. doi: 10.1111/j.1462-2920.2006.00983.x. [DOI] [PubMed] [Google Scholar]
- Tang SY, Cirino PC. Design and application of a mevalonate-responsive regulatory protein. Angew Chem. Int. Ed. 2011;50(5):1084–1086. doi: 10.1002/anie.201006083. [DOI] [PubMed] [Google Scholar]
- Jha RK, Kern TL, Fox DT, Strauss CEM. Engineering an Acinetobacter regulon for biosensing and high-throughput enzyme screening in E. coli via flow cytometry. Nucleic Acids Res. 2014;42(12):8150–8160. doi: 10.1093/nar/gku444. [DOI] [PMC free article] [PubMed] [Google Scholar]