

Abstract
A fundamental question in cell biology is how cell and organelle sizes are regulated. It has long been recognized that the size of the nucleus generally scales with the size of the cell, notably during embryogenesis when dramatic reductions in both cell and nuclear sizes occur. Mechanisms of nuclear size regulation are largely unknown and may be relevant to cancer where altered nuclear size is a key diagnostic and prognostic parameter. In vivo approaches to identifying nuclear size regulators are complicated by the essential and complex nature of nuclear function. The in vitro approach described here to study nuclear size control takes advantage of the normal reductions in nuclear size that occur during Xenopus laevis development. First, nuclei are assembled in X. laevis egg extract. Then, these nuclei are isolated and resuspended in cytoplasm from late stage embryos. After a 30 - 90 min incubation period, nuclear surface area decreases by 20 - 60%, providing a useful assay to identify cytoplasmic components present in late stage embryos that contribute to developmental nuclear size scaling. A major advantage of this approach is the relative facility with which the egg and embryo extracts can be biochemically manipulated, allowing for the identification of novel proteins and activities that regulate nuclear size. As with any in vitro approach, validation of results in an in vivo system is important, and microinjection of X. laevis embryos is particularly appropriate for these studies.
Keywords: Cellular Biology, Issue 114, Xenopus laevis, embryo extract, nuclear size regulation, development, nuclear shrinking assay, in vitro, organelle size scaling
Introduction
The sizes of cellular organelles typically scale with the size of the cell, and this has been perhaps best documented for the scaling of nuclear size with cell size1-10. This is particularly true during embryogenesis and cell differentiation, when dramatic reductions in both cell and nuclear size are often observed11,12. Furthermore, altered nuclear size is a key parameter in cancer diagnosis and prognosis13-17. Mechanisms that contribute to nuclear size regulation are largely unknown, in part due to the complexity and essential nature of nuclear structure and function. The method described here was developed as an in vitro assay for nuclear size scaling that is amenable to biochemical manipulation and elucidation of mechanisms of nuclear size regulation.
Xenopus laevis egg extract is a well-established system to recapitulate and study complex cellular processes in an in vitro context. These extracts have revealed new fundamental information about several cellular processes including the assembly and function of the mitotic spindle, endoplasmic reticulum, and nucleus18-22. One key advantage to the extract system is that X. laevis egg extracts represent a nearly undiluted cytoplasm whose composition can be easily altered, for instance through addition of recombinant proteins or immunodepletion. Furthermore, one is able to manipulate essential processes by employing treatments that might otherwise be lethal in an in vivo context. Modifications of the egg extract procedure allow for isolation of extracts from X. laevis embryos rather than eggs, and these embryo extracts are equally amenable to biochemical manipulation23. During X. laevis development, the single-cell fertilized embryo (~1 mm diameter) undergoes a series of twelve rapid cell divisions (stages 1 - 8) to generate several thousand 50 µm diameter and smaller cells, reaching a developmental stage termed the midblastula transition (MBT) or stage 824-26. The MBT is characterized by the onset of zygotic transcription, cell migration, asynchronous cell divisions, acquisition of gap phases, and establishment of nuclear steady-state sizes rather than continual nuclear expansion as in the pre-MBT embryo. From stage 4 to gastrulation (stages 10.5 - 12), the volume of individual nuclei decreases by more than 10-fold11.
Here, the goal is to identify mechanisms responsible for these reductions in nuclear size during developmental progression. The approach is to first assemble nuclei in X. laevis egg extract and to isolate those nuclei from the egg cytoplasm/extract. These nuclei are then resuspended in cytoplasm from late gastrula stage embryos. After an incubation period, the nuclei from egg extract become smaller in late stage embryo extract. We reasoned that this would be a useful assay for identifying cytoplasmic components present in late stage embryos that contribute to developmental nuclear size scaling. Using this assay, coupled with in vivo validation, we demonstrated that protein kinase C (PKC) contributes to developmental reductions in nuclear size in X. laevis23.
Protocol
All Xenopus procedures and studies were conducted in compliance with the NRC Guide for the Care and Use of Laboratory Animals 8th edition. Protocols were approved by the University of Wyoming Institutional Animal Care and Use Committee (Assurance # A-3216-01).
1. Preparation of X. laevis Egg Extract (adapted from27,28)
Prime female X. laevis frogs a minimum of three days and a maximum of two weeks before egg collection with a single 100 IU injection of pregnant mare serum gonadotropin (PMSG).
One day before the experiment, inject primed frogs with 800 IU of human chorionic gonadotropin (HCG) and place at 16 °C in 1 L per frog of 1/3x Marc's modified ringers (MMR). See Table 1 for all buffer compositions. Note: Some frogs will not lay eggs or will lay poor quality eggs. Typically 2 - 3 frogs are injected to ensure at least one frog will lay sufficient numbers of good quality eggs. The average frog will lay enough eggs to produce at least 1 ml of egg extract.
Prepare 1 L 1/3x MMR with cold distilled deionized water (ddH2O), 500 ml buffer B, 1 L buffer C, 500 ml buffer D, and 100 ml buffer E.
Transfer laid eggs to a glass crystallizing dish (150 x 75 mm). Using a plastic Pasteur pipet with the tip cut off, farm out white puffy activated eggs or lysed eggs. Only retain eggs with distinct and equal dark animal poles and white vegetal poles.
Prepare 13 x 51 mm centrifuge tubes with 1 ml of Buffer E supplemented with 355 µM cytochalasin D added just prior to use in step 1.7.
- Dejelly and wash the eggs.
- Wash eggs briefly with cold 1/3x MMR in an appropriately sized beaker, changing the buffer 3 - 4 times.
- Remove the jelly coats from the eggs by incubation with Buffer B. This will take anywhere from 3 - 10 min. As the cloudy jelly coats are released from the eggs, change the buffer. Note: Dejellying is complete when the eggs appear tightly packed in the corner of the dish when tilted and vegetal poles are oriented downwards. Eggs are much more fragile after dejellying, so do not incubate eggs with Buffer B for longer than necessary and subsequent washes need to be performed very carefully.
- Wash eggs briefly with 3 - 4 changes of Buffer C.
- Wash eggs briefly with 2 changes of Buffer D.
- Wash eggs briefly with 2 changes of Buffer E.
Fill centrifuge tubes with eggs using a wide bore glass pipette or plastic dropper. Aspirate off excess buffer. To pack the eggs and remove as much buffer as possible, centrifuge in swinging bucket rotor at 212 x g for 60 sec and then at 478 x g for 30 sec. Aspirate off excess buffer. Note: It is important to remove as much excess buffer as possible at this step to ensure the egg extract is minimally diluted.
In a swinging bucket rotor, ultracentrifuge under vacuum for 15 min at 12,000 x g and 4 °C to crush eggs open and separate into different layers, with the cytoplasm being the amber-colored layer in the middle. Remove centrifuge tubes and place immediately on ice.
- Collect the extract.
- Using an 18 gauge needle and syringe, puncture the centrifuge tube just above the dark pigmented layer at the bottom of the tube and remove the middle amber-colored cytoplasmic layer. Do not collect any of the top lipid layer. Collect the cytoplasm in an appropriately sized tube. Note: Generally, 1 frog produces at least 1 ml of extract.
- Supplement cytoplasm with 1/1,000 the volume of 19.7 mM cytochalasin D, 1/1,000 the volume of LPC stock, and 1/50 the volume of 50x energy mix. Gently invert the tube to mix. Do not excessively pipet the extract.
- Keep the extract on ice and use it within 3 hr of preparation for best results.
2. Preparation of Demembranated X. laevis Sperm (adapted from29)
Note: The procedure volumes presented here are for up to 8 testes.
Remove 0.05% benzocaine (10% benzocaine stock prepared in ethanol and diluted to 0.05% in frog tank water) from 4 °C and warm to room temperature. Anesthetize and sacrifice male X. laevis frogs in benzocaine solution at room temperature for 20 - 30 min. Ensure death by absence of heartbeat by examining and feeling the chest area, by lack of response to mechanical stimulation, and/or by decapitation.
Make a U-shaped incision along the abdomen. Remove the tubulated mass of yellow fat bodies. At the top of the kidneys, locate the testes, which are oval-shaped pale pink masses about 1 cm in length29. Excise the testes and roll them on blotting paper to remove blood and other tissue.
Wash testes in Buffer T. Use a tight fitted pestle to mince testes with 1 ml of Buffer T in a 1.5 ml tube until homogenous (10 strokes or more). Centrifuge 5 - 10 sec in a mini centrifuge and collect the supernatant, removing large pieces of tissue. Repeat the centrifugation once more and collect the supernatant.
Transfer the sperm-containing solution to a 15 ml plastic tube. Increase the volume to a total of 2 ml with Buffer T. Centrifuge at 1,411 x g for 10 min at 16 °C to pellet the sperm.
Remove the supernatant and resuspend the pellet in 500 µl of Buffer T. Centrifuge at 1,411 x g for 10 min at 16 °C. Repeat this step once or twice as needed to clean the pellet of somatic and red blood cells.
Resuspend the pellet in 100 µl Buffer T and 300 µl Buffer S. Incubate at room temperature for 5 min. Note: Buffer S contains lysolecithin, which is responsible for demembranation.
Add three volumes of Buffer R (prepared fresh before use). Invert tube exactly once. Centrifuge at 1,411 x g for 15 min at 16 °C.
Resuspend the pellet in 400 µl Buffer R and then add an additional 2 ml of Buffer R. Centrifuge at 1,411 x g for 15 min at 16 °C.
Resuspend the pellet in 50 µl Buffer T.
Dilute 1 µl demembranated sperm nuclei with 9 µl Buffer T. Apply this dilution to a hemocytometer. Count the number of thin S-shaped sperm nuclei in one large 4 x 4 grid, and multiply that number by 100 to obtain the concentration of the stock in sperm nuclei per µl.
Dilute the stock to 100,000 sperm nuclei per µl with Buffer T, resulting in a 200x stock. Prepare 3 - 5 µl aliquots. Flash freeze in liquid nitrogen and store at -80 °C.
3. Nuclear Assembly
Supplement 100 µl of egg extract with 1.5 µl 35.5 mM cycloheximide (final concentration ~ 0.5 mM), 6 µl 20x calcium stock solution (final concentration ~ 0.6 mM), and 0.7 µl 200x sperm (final concentration ~ 700 sperm nuclei/µl). Scale the reaction volume up or down as necessary. Invert or gently tap to mix. Note: Extract cycled from metaphase into interphase provides a more robust and reliable platform for nuclear assembly.
Incubate in a water bath at 16 - 20 °C for 90 min. Mix by gently tapping every 15 min to ensure nuclei remain suspended in the extract.
- Monitor progress of nuclear assembly at 45 min by preparing a squash as follows.
- Pipet 2 µl of extract onto a glass slide and add 2 µl nucleus fix.
- Overlay with a 22 mm2 coverslip. Image on an epifluorescence microscope using water, oil, or air 20 - 100X objectives and a DAPI filter. Note: Nuclei can be visualized by bright-field imaging, but are much easier to visualize by fluorescence.
- Use nuclei immediately or flash freeze aliquots in liquid nitrogen and store at -80 °C. Note: Adding 4% glycerol to the nuclei prior to freezing can help to maintain their integrity. Frozen nuclei are generally still viable and capable of nuclear import, however the freezing process can lead to disrupted or more structurally fragile nuclei. Nuclei up to a year after freezing can be used.
4. Preparation of X. laevis Embryo Extract
Induce female X. laevis frogs to lay eggs as described in 1.1 and 1.2.
Isolate testes from male X. laevis frogs as described in 2.2. Submerge testes in L15 media supplemented with 50 IU/ml of penicillin and streptomycin in a glass 35 x 10 mm Petri dish. Store at 4 °C for up to two weeks.
- Collect and fertilize eggs.
- Collect freshly laid eggs by holding frogs over a glass reusable Petri dish and promote egg laying by applying gentle pressure to the lower back near the cloaca. Squeezing the frog too hard can result in injury, leading to a bloated abdomen and requiring euthanasia29.
- Prop the dish at an approximate 45° angle, allow eggs to collect in the edge of the dish, remove all excess buffer, and add enough 1/3x MMR to barely cover the eggs. Fertilize eggs within 15 min of collection.
- Use a tightly fitted pestle to macerate and homogenize 1/4 of a testis in a 1.5 ml tube with 500 µl of high salt Modified Barth's Saline (High Salt MBS). Add macerated testes to the eggs and allow for fertilization to occur at room temperature for 15 - 20 min, then flood eggs with 1/3x MMR. Note: Effectiveness of testes decreases with storage time, therefore more testes tissue may be required for successful fertilization when using testes that have been stored for more than one week.
- Confirm fertilization by checking for contraction of the dark pigmented animal pole and by rotation of the embryos with their animal poles facing up, usually about 20 - 30 min after addition of crushed testes. Expect the first cell cleavage to occur within 90 min.
- Using a wide bore glass pipette diligently remove dead (white and puffy or uneven distribution of yolk and pigment), lysed, or unfertilized eggs (i.e., not cleaved), as they will rapidly induce death in neighboring embryos.
- Anywhere from 1 - 2.5 hr post-fertilization, dejelly embryos in 2 - 3% cysteine dissolved in 1/3 x MMR and adjusted to pH 7.9 with 10 N KOH. Perform two buffer changes that are each 3 - 4 min. Thoroughly wash the embryos with 6 - 10 brief changes of 1/3x MMR to remove all traces of cysteine. Note: Dejellied embryos have a vitelline membrane and are not as fragile as dejellied eggs.
Using a wide bore glass pipette, transfer healthy fertilized (i.e., cleaved) embryos to a Petri dish containing fresh 1/3x MMR and allow them to develop to the desired stage at RT or, if slower development is preferred, 16 °C. Continue to remove dead or lysed embryos indicated by lack of first division or white puffy appearance.
Verify the stage of the embryos by checking for nearly complete closure of the blastopore and by comparing to drawings of staged embryos available in reference materials24. Note: Embryos cultured at 16 °C will reach stage 11.5 - 12 in approximately 12 hr.
Incubate stage 11.5 - 12 embryos in fresh 1/3x MMR supplemented with 0.5 mM cycloheximide for 1 hr at RT, to arrest embryos in late interphase.
Transfer with wide bore glass or plastic pipette a minimum of 15 (preferably 30 or more) interphase-arrested embryos to a microcentrifuge tube. Note: 15 embryos are sufficient to produce approximately 20 µl of extract. Scale up as needed.
Add 1 ml egg lysis buffer (ELB) supplemented with 1 µl of LPC stock. Gently invert to wash embryos, allow embryos to fall to the bottom of tube, and remove the buffer. Repeat this wash step two more times.
Resuspend embryos in 500 µl of ELB plus 0.5 µl of LPC stock, 5 µl of 19.7 mM cycloheximide, and 5 µl of 35.5 mM cytochalasin D (to inhibit actin polymerization). Centrifuge at 200 x g for 1 min in a tabletop microcentrifuge.
Remove excess buffer and use a pestle to thoroughly crush the embryos. Centrifuge in a swinging bucket rotor for 10 min at 10,000 x g and 16 °C.
Puncture the lipid layer from the top with a 200 µl pipet tip. With a clean 200 µl pipet tip, remove the middle cytoplasmic amber-colored layer to an appropriate sized tube.
Supplement embryo extract with 1/50 the volume of 50x energy mix (to provide an ATP regeneration system), 1/100 the volume of 35.5 mM cyclohexamide, 1/500 the volume of 19.7 mM cytochalasin D, and 1/1,000 the volume of LPC stock.
Prepare a squash as described in 3.3 to visualize endogenous embryonic nuclei in the extract. Note: Embryo extract can be frozen with the endogenous nuclei at this point. Aliquot to reduce freeze/thaw cycles and store at -80 °C.
To remove nuclei from the embryo extract, dilute the extract with an equal volume of ELB containing 1/50 the volume of 50x energy mix, 1/100 the volume of 35.5 mM cyclohexamide, 1/500 the volume of 19.7 mM cytochalasin D, and 1/1,000 the volume of LPC stock. Centrifuge in a swinging bucket rotor at 17,000 x g for 15 min at 16 °C.
Collect the supernatant being careful to avoid any remaining lipid at the top. Do not disturb the pelleted nuclei at the bottom of the tube. Prepare a squash as described in 3.3 to ensure that most nuclei have been removed. Use the extract fresh or aliquot and store at -80 °C.
To heat inactivate embryo extract for control experiments, heat 30 - 100 µl aliquots of extract at 56 °C for 30 min using a thermal cycler. Allow heat inactivated extract to return to room temperature prior to use.
5. Nuclear Shrinking Assay and Immunofluorescence
Isolate nuclei assembled in egg extract by diluting 25 - 150 µl of pre-assembled nuclei in 1 ml ELB in a 1.5 ml tube. Centrifuge at 1,600 x g for 3 min at 4 °C in a tabletop microcentrifuge. Remove buffer, being careful not to disturb the fragile pellet of nuclei at the bottom of the tube.
Resuspend nuclei in a volume of embryo extract equal to the original volume of egg extract used in 5.1. Use ELB or heat inactivated extract for control reactions, as appropriate.
Gently tap the tube to break up the pellet and resuspend the nuclei. Incubate at room temperature for 90 min, gently tapping the tube to mix every 15 - 30 min. Note: Most of the nuclear shrinking occurs within the first 30 min.
- Monitor progress of nuclear shrinking by preparing a squash.
- Pipet 2 µl of extract onto a glass slide and add 2 µl nucleus fix.
- Overlay with a 22 mm2 coverslip. Image on an epifluorescence microscope.
Tap the tube to resuspend the nuclei just prior to adding 500 µl of fixative consisting of ELB, 15% glycerol, and 2.6% paraformaldehyde. Invert immediately, and place the tube on a rotator for 15 min at room temperature.
Prepare spin down tubes by outfitting 15 ml round bottom glass tubes with round bottom plastic inserts (design and schematics available upon request). Add 5 ml of nuclear cushion buffer. Drop a 12 mm circular coverslip into the tube, being sure it lays flat on top of the plastic insert.
Gently layer the solution containing fixed nuclei on top of the nuclear cushion using a wide-bore pipet tip. Centrifuge in a swinging bucket rotor for 15 min at 1,000 x g and 16 °C.
Use an aspirator to remove all of the buffer and pull the plastic insert to the top of the tube. Remove the coverslip with a pair of fine forceps, being careful to note the side of the coverslip onto which the nuclei were spun. Post-fix in cold methanol (stored at -20° C) for 5 min at RT.
Transfer the coverslip nucleus side up onto a sheet of plastic paraffin film lining a large plastic petri dish. Place wet disposable wipes along the side of the dish to prevent dehydration.
Rehydrate nuclei on the coverslip with 500 µl of PBS-NP40 (1x PBS plus 0.1% NP40) and aspirate after 5 - 10 sec.
Carefully layer 75 µl of PBS-3% Bovine Serum Albumin (BSA) onto the coverslip. Allow to block 1 hr at room temperature or overnight at 4 °C.
Remove blocking solution, and incubate with primary antibody (e.g., mAb414 against the nuclear pore complex, 1:1,000) diluted in PBS-3% BSA for 1 hr at room temperature or overnight at 4 °C. Wash with five immediate changes of PBS-NP40.
Incubate with secondary antibody diluted in PBS-3% BSA for 1 hr at RT. Wash with five immediate changes of PBS-NP40.
Incubate with 10 µg/ml Hoechst diluted in PBS-3% BSA for 5 min at RT. Wash five times with PBS-NP40. Remove all excess buffer.
Mount the coverslip onto a slide with 5 µl antifade mounting medium. Seal with clear nail polish. Image immediately using an epifluorescence microscope with 20 - 100x objectives or store at 4 °C for later imaging. Note: Perform quantification as previously described23.
Representative Results
Assembly of Nuclei in Egg Extract
The first steps of this protocol are to prepare X. laevis egg extract (Protocol 1) and demembranated sperm nuclei (Protocol 2). These reagents are then used to assemble nuclei de novo (Protocol 3). Figure 1 shows some representative data. Addition of calcium drives the meiotically arrested egg extract into interphase, and the cycloheximide keeps the extract arrested in interphase. By 30 - 45 min after initiation of the reaction, the initially thin S-shaped sperm nuclei should begin to thicken into slug-shaped chromatin masses. Successful formation of an intact nuclear envelope can be assessed by import and retention of a fluorescently labeled import cargo, such as GST-GFP-NLS (data not shown), or by rim staining for the nuclear pore complex using mAb414 that recognizes FG-repeat containing nucleoporins (Figure 1A). After nuclear assembly occurs, nuclear shape should become more rounded and the nuclear volume should increase as the nuclear envelope expands and nuclear proteins are imported (Figure 1B-C). Failure to observe formation of an intact nuclear envelope or nuclear growth indicates a poor quality egg extract. In this case, it is best to start again with a new batch of eggs.
Removal of Endogenous Nuclei from Embryo Extract The next step is to prepare a late stage embryo extract (Protocol 4). After initial preparation of the extract and prior to removal of nuclei, it is best to prepare a Hoechst-stained squash of a small aliquot of the extract. Visualization of many small embryonic nuclei is a good indication of success in preparing the extract (Figure 2A). As endogenous embryonic nuclei can interfere with the downstream analysis, it is also important to check an aliquot of the extract after removing nuclei by dilution and centrifugation. Compared to the first squash, very few nuclei should be left in the extract (Figure 2B). If there are still many nuclei present, a second round of centrifugation is required.
Nuclear Shrinking AssayFigure 3 shows representative results for the nuclear shrinking assay (Protocol 5). Egg extract nuclei resuspended in late stage embryo extract become smaller over time (Figure 3B). A good negative control for the assay is to incubate nuclei with embryo extract that has been heat inactivated (Figure 3A). As nuclei fail to shrink in heat inactivated extract, this provides some confidence that observed nuclear shrinking with untreated embryo extract is not a consequence of osmotic effects. The extent of nuclear size reductions observed varies depending on the particular batch of egg extract nuclei as well as the quality of the embryo extract. In our experience, nuclear shrinking is always observed (e.g., in more than 40 different embryo extracts), however it is important to repeat these experiments multiple times with different nuclei and extracts to address the inherent variability of the assay. Figure 4 shows a schematic of the entire protocol.
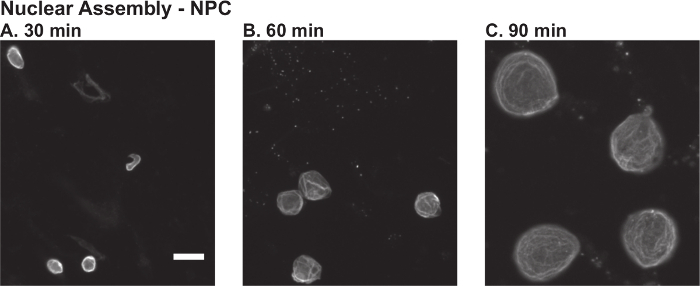 Figure 1.Nuclear Assembly Time Course inX. laevis Egg Extract. Nuclei were assembled de novo in X. laevis egg extract as described in Protocol 3. Aliquots from the same reaction were fixed and visualized by immunofluorescence using an antibody against the nuclear pore complex (NPC) (mAb414) at: A) 30 min, B) 60 min, and C) 90 min after the initiation of nuclear assembly. The immunofluorescence protocol is described in Protocol 5. The scale bar is 25 µm. Please click here to view a larger version of this figure.
Figure 1.Nuclear Assembly Time Course inX. laevis Egg Extract. Nuclei were assembled de novo in X. laevis egg extract as described in Protocol 3. Aliquots from the same reaction were fixed and visualized by immunofluorescence using an antibody against the nuclear pore complex (NPC) (mAb414) at: A) 30 min, B) 60 min, and C) 90 min after the initiation of nuclear assembly. The immunofluorescence protocol is described in Protocol 5. The scale bar is 25 µm. Please click here to view a larger version of this figure.
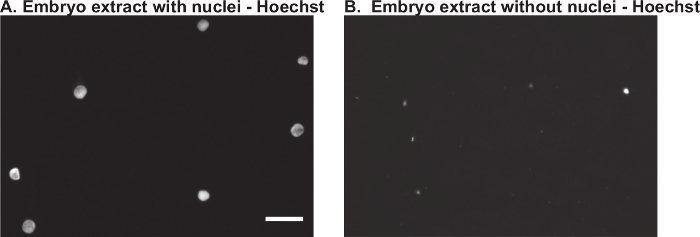 Figure 2. Removal of Nuclei from X. laevis Embryo Extract.X. laevis embryo extract was prepared from stage 11.5 - 12 embryos as described in Protocol 4. A small aliquot of extract was fixed and stained with Hoechst: A) prior to removal of nuclei, and B) after removal of nuclei by centrifugation. The scale bar is 25 µm. Please click here to view a larger version of this figure.
Figure 2. Removal of Nuclei from X. laevis Embryo Extract.X. laevis embryo extract was prepared from stage 11.5 - 12 embryos as described in Protocol 4. A small aliquot of extract was fixed and stained with Hoechst: A) prior to removal of nuclei, and B) after removal of nuclei by centrifugation. The scale bar is 25 µm. Please click here to view a larger version of this figure.
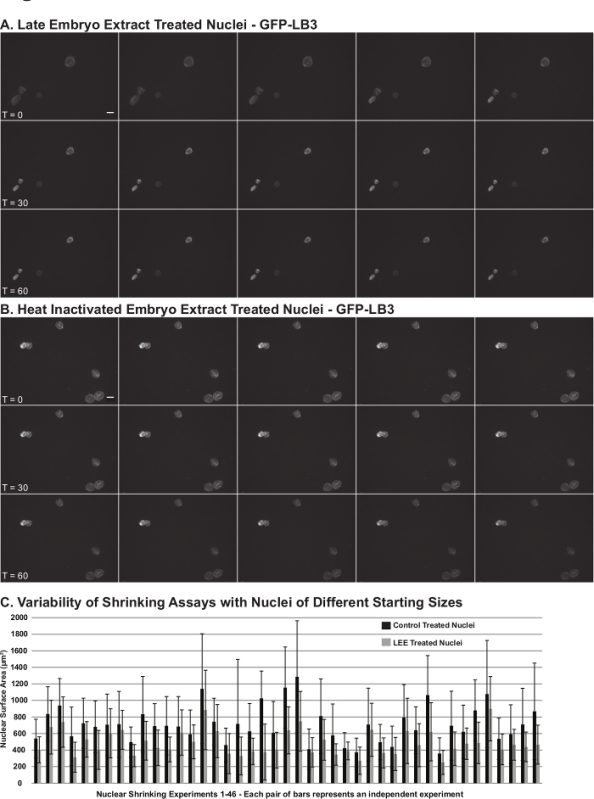 Figure 3. Representative Data from the Nuclear Shrinking Assay. Nuclei were assembled de novo in X. laevis egg extract supplemented with recombinant GFP-LB3 (to visualize the nuclear lamina). As described in Protocol 5, egg extract nuclei were isolated and resuspended in stage 11.5 - 12 embryo extract from which endogenous embryonic nuclei had been removed. Live time-lapse imaging was performed at 30 sec intervals for 90 min. Figure panels show images acquired at 6 min intervals for nuclei incubated in: A) late stage embryo extract (LEE), and B) heat inactivated (HI) embryo extract. Images from the time courses proceed from left-to-right. The scale bar is 25 µm. C) Nuclear shrinking data from 46 different egg and embryo extracts. Bars show the means for > 240 nuclei. Error bars are standard deviation. Please click here to view a larger version of this figure.
Figure 3. Representative Data from the Nuclear Shrinking Assay. Nuclei were assembled de novo in X. laevis egg extract supplemented with recombinant GFP-LB3 (to visualize the nuclear lamina). As described in Protocol 5, egg extract nuclei were isolated and resuspended in stage 11.5 - 12 embryo extract from which endogenous embryonic nuclei had been removed. Live time-lapse imaging was performed at 30 sec intervals for 90 min. Figure panels show images acquired at 6 min intervals for nuclei incubated in: A) late stage embryo extract (LEE), and B) heat inactivated (HI) embryo extract. Images from the time courses proceed from left-to-right. The scale bar is 25 µm. C) Nuclear shrinking data from 46 different egg and embryo extracts. Bars show the means for > 240 nuclei. Error bars are standard deviation. Please click here to view a larger version of this figure.
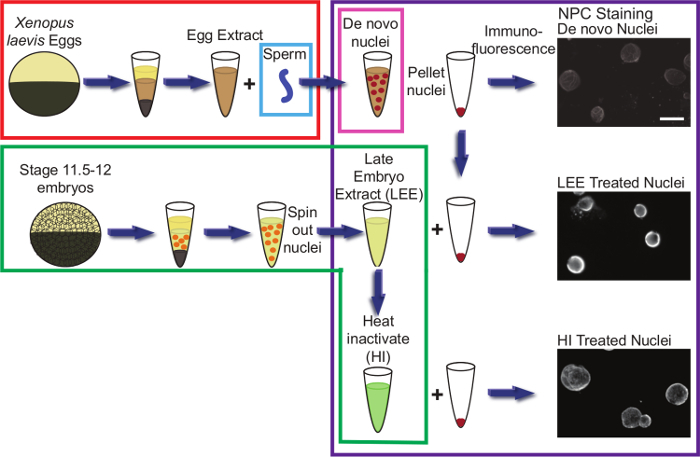 Figure 4.Schematic of the Nuclear Shrinking Assay. Each colored box represents one protocol. Red indicates preparation of X. laevis egg extract (Protocol 1). Blue indicates preparation of demembranated X. laevis sperm (Protocol 2). Pink indicates de novo nuclear assembly in egg extract (Protocol 3). Green indicates preparation of X. laevis embryo extract (Protocol 4). Purple indicates the nuclear shrinking assay protocol and immunofluorescence (Protocol 5). Portions of this figure have been reused from23. Please click here to view a larger version of this figure.
Figure 4.Schematic of the Nuclear Shrinking Assay. Each colored box represents one protocol. Red indicates preparation of X. laevis egg extract (Protocol 1). Blue indicates preparation of demembranated X. laevis sperm (Protocol 2). Pink indicates de novo nuclear assembly in egg extract (Protocol 3). Green indicates preparation of X. laevis embryo extract (Protocol 4). Purple indicates the nuclear shrinking assay protocol and immunofluorescence (Protocol 5). Portions of this figure have been reused from23. Please click here to view a larger version of this figure.
| Buffer name | Buffer composition |
| Marc's Modified Ringers (MMR), 1/3x | 33 mM NaCl, 0.7 mM KCl, 0.3 mM MgSO4, 0.7 mM CaCl2, 1.7 mM HEPES, pH 7.4 |
| Extract buffer (XB), 10x | 1 M KCl, 1 mM CaCl2, 10 mM MgCl2, 500 mM sucrose, 100 mM HEPES, pH 7.8 (pH adjusted with 10 N KOH, stored at 4 °C) |
| Buffer B | 4% Cysteine dissolved in 0.8x XB (prepared with cold ddH2O, pH adjusted to 7.8 with 10 N NaOH) |
| Buffer C | Mix 100 m of 10x XB with 900 ml of cold ddH2O |
| Buffer D | Buffer C plus 5 mM EGTA and 800 µM MgCl2 |
| LPC stock, 1,000x | 10 mg/ml each of leupeptin, pepstatin, and chymostatin dissolved in DMSO (stored in aliquots at -20 °C) |
| Buffer E | 100 ml Buffer D plus 100 µl LPC stock |
| Energy mix, 50x | 190 mM creatine phosphate disodium, 25 mM ATP disodium salt, 25 mM magnesium chloride |
| Buffer T | 15 mM PIPES, 15 mM NaCl, 5 mM EDTA, 7 mM MgCl2, 80 mM KCl, 0.2 M sucrose, pH 7.4 (filter sterilize and stored at 4 °C) |
| Buffer S | 20 mM maltose and 0.05% lysolecithin prepared in Buffer T |
| Buffer R | Buffer T plus 3% bovine serum albumin |
| Calcium stock solution, 20x | 10 mM CaCl2, 0.1 M KCl, 1 mM MgCl2 (stored at -20 °C) |
| Nucleus fix | 125 µl 2 M sucrose, 12.5 µl 1 M HEPES pH 7.9, 250 µl 37% formaldehyde, 112 µl ddH2O, 0.5 µl 10 mg/ml Hoechst (stored at room temperature for up to two weeks) |
| Modified Barth's Saline (MBS), 10x | 880 mM NaCl, 10 mM KCl, 50 mM HEPES, 25 mM NaHCO3, pH 7.8 |
| High Salt MBS | 1x MBS plus 0.7 mM CaCl2 and 20 mM NaCl |
| Egg lysis buffer, ELB | 250 mM sucrose, 50 mM KCl, 2.5 mM MgCl2, 10 mM HEPES, pH 7.8 |
| Nuclear cushion buffer | 1x XB, 0.2 M sucrose, 25% glycerol (filter sterilized and stored at 4 °C) |
Table 1. Buffers. Compositions of all buffers mentioned in this protocol are described in this table.
Discussion
Here is presented a novel method to study mechanisms of nuclear size regulation during X. laevis development. Developmental progression is associated with dramatic changes in cell physiology, metabolism, division rates, and migration, as well as alterations in the sizes of cells and intracellular structures. These varied processes are complex and essential, so it is difficult to study just one of these aspects of development in an in vivo setting. The X. laevis embryo extract and nuclear shrinking assay described here allow for the study of size regulatory mechanisms in an in vitro context, circumventing the complex signals and physical constraints of the in vivo system. Furthermore, the open nature of the extract allows for its facile biochemical manipulation.
Troubleshooting some steps of the protocol may be necessary. Problems with nuclear assembly and growth can result from poor quality eggs, incomplete demembranation of sperm nuclei, use of sperm nuclei at too high of a concentration, or incubating nuclear assembly reactions at too low of a temperature. Removal of endogenous embryonic nuclei from late embryo extract can be difficult due to the small size of these nuclei. This step in the protocol may require more than one round of centrifugation or the use of alternate rotors to completely remove all nuclei. Failure of nuclei to become smaller in the shrinking assay may be due to poor quality embryos, starting with nuclei that are too small, or too low of an incubation temperature. Poor quality eggs and embryos are indicated by a large proportion of white puffy activated eggs, unequal distribution of yolk and pigment granules, and eggs laid in strings. Spinning down fixed nuclei onto coverslips occasionally causes fragmentation of the nuclei, which can be avoided by decreasing the centrifugation speed or increasing the glycerol and sucrose concentrations in the nucleus cushion buffer. Troubleshooting antibody selection and dilution for immunofluorescence of nuclei is also often necessary. Critical steps within these protocols include: using high quality eggs and embryos, removing activated and lysed eggs and embryos, ensuring sperm nuclei are fully demembranated and appropriately diluted, monitoring nuclear assembly and growth, correctly staging embryos, completely removing endogenous embryonic nuclei from late stage embryo extracts, centrifuging fixed nuclei onto glass coverslips, and immunofluorescent staining of nuclei for visualization and quantification.
Nuclear size differences have long been noted between different species, cell types, disease states, and stages of embryonic development, indicating that nuclear size is tightly regulated1-10. Cancer of the prostate, breast, and ovary, to name a few, are all diagnosed and staged based on graded increases in nuclear size14,30-32. It is unclear to what extent changes in nuclear size are a cause or consequence of disease, and whether normal scaling of nuclear size contributes to proper development. Answering these functional questions necessitates an understanding of the mechanisms that control nuclear size. The assay presented here provides one approach to identifying novel pathways and proteins that regulate nuclear size, with potential relevance to cancer and development.
The Xenopus egg and embryo extract systems provide particularly powerful approaches to investigate mechanisms of organelle size regulation. While Xenopus extracts have been used for decades to study various aspects of cell cycle regulation and organelle assembly and dynamics, these extracts have more recently provided insights into size regulation of the nucleus3,23,33, mitotic spindle34-36, and chromosomes37-39. One of the major advantages of working with X. laevis embryo extracts is that they can be isolated from different stages of development, representing cytoplasm from differently sized cells containing nuclei of differing sizes. The method for investigating nuclear re-sizing presented here is physiologically relevant and offers a simplified system with which to identify mechanisms of nuclear size regulation. One complication of studying size regulation in vivo is that it is difficult to distinguish effects of spatial constraints and volume-limited components on size versus developmentally regulated changes in the expression or activities of size-controlling factors40. The approach described here focuses on developmentally controlled nuclear size regulators, using a robust nuclear re-sizing assay that is amenable to biochemical dissection.
Using the methods presented here, we identified protein kinase C (PKC) as a novel regulator of nuclear size. Furthermore, we showed that PKC activity and nuclear localization increase during X. laevis development23,41. A key limitation to the nuclear shrinking assay is that it is performed entirely in vitro. While Xenopus extract systems provide a simplified platform to discover new factors that regulate size, it is important that these results are validated in vivo. There are a variety of different approaches to corroborate physiological significance. X. laevis embryos can be microinjected with mRNA or morpholinos to alter the levels of nuclear scaling factors identified in extract, and then the effects on nuclear size can be quantified. Treatment of embryos with cell-permeable small molecule inhibitors or activators is also possible. For instance, we observed nuclear shrinking in live embryos treated with a phorbol ester, and this shrinking occurred within interphase without a requirement for nuclear envelope breakdown and passage through mitosis23. Another approach is to alter the levels of nuclear scaling factors in tissue culture cells to assess conservation of nuclear size control mechanisms outside of Xenopus.
What are the functional implications of altered nuclear size? One possibility is that nuclear size influences chromosome and chromatin organization, thereby altering gene expression. This idea can be directly tested as mechanisms of nuclear size regulation are further elucidated. Interestingly, PKC signaling and nuclear size are both routinely deregulated in cancer cells, often representing more aggressive metastatic disease13,14,42,43. Whether there is a direct relationship between deregulated PKC signaling and altered nuclear size in cancer remains to be shown. There is still much to be learned about nuclear size regulation, and the assay presented here, or variants of it, may still yield new information about mechanisms of nuclear size control. Furthermore, this assay could easily be adapted for the study of scaling mechanisms of other organelles and intracellular structures such as the mitotic spindle, endoplasmic reticulum, chromosomes, and Golgi, to name a few.
Disclosures
The authors have nothing to disclose.
Acknowledgments
Members of the Levy and Gatlin labs as well as colleagues in the Department of Molecular Biology offered helpful advice and discussions. Rebecca Heald provided support in the early stages of developing this protocol. This work was supported by the NIH/NIGMS (R15GM106318) and the American Cancer Society (RSG-15-035-01-DDC).
References
- Conklin E. Cell size and nuclear size. J. Exp. Embryol. 1912;12:1–98. [Google Scholar]
- Wilson EB. The Cell in Development and Heredity. The Macmillan Company; 1925. pp. 727–733. [Google Scholar]
- Levy DL, Heald R. Nuclear size is regulated by importin alpha and Ntf2 in Xenopus. Cell. 2010;143(2):288–298. doi: 10.1016/j.cell.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YH, Marshall WF. Scaling properties of cell and organelle size. Organogenesis. 2010;6(2):88–96. doi: 10.4161/org.6.2.11464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters AD, Bommakanti A, Cohen-Fix O. Shaping the nucleus: Factors and forces. J Cell Biochem. 2012;113(9):2813–2821. doi: 10.1002/jcb.24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster M, Witkin KL, Cohen-Fix O. Sizing up the nucleus: nuclear shape, size and nuclear-envelope assembly. J Cell Sci. 2009;122(Pt 10):1477–1486. doi: 10.1242/jcs.037333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edens LJ, White KH, Jevtic P, Li X, Levy DL. Nuclear size regulation: from single cells to development and disease. Trends Cell Biol. 2013;23(4):151–159. doi: 10.1016/j.tcb.2012.11.004. [DOI] [PubMed] [Google Scholar]
- Jevtic P, Edens LJ, Vukovic LD, Levy DL. Sizing and shaping the nucleus: mechanisms and significance. Curr Opin Cell Biol. 2014;28:16–27. doi: 10.1016/j.ceb.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DL, Heald R. Mechanisms of intracellular scaling. Annu Rev Cell Dev Biol. 2012;28:113–135. doi: 10.1146/annurev-cellbio-092910-154158. [DOI] [PubMed] [Google Scholar]
- Vukovic LD, Jevtic P, Edens LJ, Levy DL. New insights into the mechanisms and functions of nuclear size regulation. Int Rev Cell Mol Biol. 2016;322:1–59. doi: 10.1016/bs.ircmb.2015.11.001. [DOI] [PubMed] [Google Scholar]
- Jevtic P, Levy DL. Nuclear size scaling during Xenopus early development contributes to midblastula transition timing. Curr Biol. 2015;25(1):45–52. doi: 10.1016/j.cub.2014.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y, Iwabuchi M, Ohsumi K, Kimura A. Intranuclear DNA density affects chromosome condensation in metazoans. Mol Biol Cell. 2013;24(15):2442–2453. doi: 10.1091/mbc.E13-01-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink D, Fischer AH, Nickerson JA. Nuclear structure in cancer cells. Nat Rev Cancer. 2004;4(9):677–687. doi: 10.1038/nrc1430. [DOI] [PubMed] [Google Scholar]
- Jevtic P, Levy DL. Mechanisms of nuclear size regulation in model systems and cancer. Adv Exp Med Biol. 2014;773:537–569. doi: 10.1007/978-1-4899-8032-8_25. [DOI] [PubMed] [Google Scholar]
- Chow KH, Factor RE, Ullman KS. The nuclear envelope environment and its cancer connections. Nat Rev Cancer. 2012;12(3):196–209. doi: 10.1038/nrc3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom JH, Ten Kate FJ, Schroeder FH, van der Heul RO. Morphometrically estimated variation in nuclear size. A useful tool in grading prostatic cancer. Urol Res. 1990;18(2):93–99. doi: 10.1007/BF00302467. [DOI] [PubMed] [Google Scholar]
- Dey P. Cancer nucleus: morphology and beyond. Diagn Cytopathol. 2010;38(5):382–390. doi: 10.1002/dc.21234. [DOI] [PubMed] [Google Scholar]
- Cross MK, Powers MA. Learning about cancer from frogs: analysis of mitotic spindles in Xenopus egg extracts. Dis Model Mech. 2009;2(11-12):541–547. doi: 10.1242/dmm.002022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124(3):573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- Chan RC, Forbes DI. In vitro study of nuclear assembly and nuclear import using Xenopus egg extracts. Methods Mol Biol. 2006;322:289–300. doi: 10.1007/978-1-59745-000-3_20. [DOI] [PubMed] [Google Scholar]
- Edens LJ, Levy DL. In: Xenopus Development. Kloc M, Kubiak JZ, editors. John Wiley & Sons, Inc; 2014. pp. 325–345. [Google Scholar]
- Levy DL, Heald R. Biological Scaling Problems and Solutions in Amphibians. Cold Spring Harb Perspect Biol. 2016;8(1):a019166. doi: 10.1101/cshperspect.a019166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edens LJ, Levy DL. cPKC regulates interphase nuclear size during Xenopus development. J Cell Biol. 2014;206(4):473–483. doi: 10.1083/jcb.201406004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal Table of Xenopus laevis (Daudin) 2nd edn. North-Holland Publishing Company; 1967. [Google Scholar]
- Newport J, Kirschner M. A major developmental transition in early Xenopus embryos: II. Control of the onset of transcription. Cell. 1982;30(3):687–696. doi: 10.1016/0092-8674(82)90273-2. [DOI] [PubMed] [Google Scholar]
- Newport J, Kirschner M. A major developmental transition in early Xenopus embryos: I. characterization and timing of cellular changes at the midblastula stage. Cell. 1982;30(3):675–686. doi: 10.1016/0092-8674(82)90272-0. [DOI] [PubMed] [Google Scholar]
- Maresca TJ, Heald R. Methods for studying spindle assembly and chromosome condensation in Xenopus egg extracts. Methods Mol Biol. 2006;322:459–474. doi: 10.1007/978-1-59745-000-3_33. [DOI] [PubMed] [Google Scholar]
- Hannak E, Heald R. Investigating mitotic spindle assembly and function in vitro using Xenopus laevis egg extracts. Nat Protoc. 2006;1(5):2305–2314. doi: 10.1038/nprot.2006.396. [DOI] [PubMed] [Google Scholar]
- Murray AW. Cell cycle extracts. Methods Cell Biol. 1991;36:581–605. [PubMed] [Google Scholar]
- Khan MA, et al. Quantitative alterations in nuclear structure predict prostate carcinoma distant metastasis and death in men with biochemical recurrence after radical prostatectomy. Cancer. 2003;98(12):2583–2591. doi: 10.1002/cncr.11852. [DOI] [PubMed] [Google Scholar]
- Tan PH, Goh BB, Chiang G, Bay BH. Correlation of nuclear morphometry with pathologic parameters in ductal carcinoma in situ of the breast. Mod Pathol. 2001;14(10):937–941. doi: 10.1038/modpathol.3880415. [DOI] [PubMed] [Google Scholar]
- Zeimet AG, et al. DNA ploidy, nuclear size, proliferation index and DNA-hypomethylation in ovarian cancer. Gynecol Oncol. 2011;121(1):24–31. doi: 10.1016/j.ygyno.2010.12.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theerthagiri G, Eisenhardt N, Schwarz H, Antonin W. The nucleoporin Nup188 controls passage of membrane proteins across the nuclear pore complex. J Cell Biol. 2010;189(7):1129–1142. doi: 10.1083/jcb.200912045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuhr M, et al. Evidence for an upper limit to mitotic spindle length. Curr Biol. 2008;18(16):1256–1261. doi: 10.1016/j.cub.2008.07.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughlin R, Wilbur JD, McNally FJ, Nedelec FJ, Heald R. Katanin contributes to interspecies spindle length scaling in Xenopus. Cell. 2011;147(6):1397–1407. doi: 10.1016/j.cell.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilbur JD, Heald R. Mitotic spindle scaling during Xenopus development by kif2a and importin. eLife. 2013;2:e00290. doi: 10.7554/eLife.00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieserman EK, Heald R. Mitotic chromosome size scaling in Xenopus. Cell Cycle. 2011;10(22):3863–3870. doi: 10.4161/cc.10.22.17975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresca TJ, Freedman BS, Heald R. Histone H1 is essential for mitotic chromosome architecture and segregation in Xenopus laevis egg extracts. J Cell Biol. 2005;169(6):859–869. doi: 10.1083/jcb.200503031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCallum DE, Losada A, Kobayashi R, Hirano T. ISWI remodeling complexes in Xenopus egg extracts: identification as major chromosomal components that are regulated by INCENP-aurora B. Mol Biol Cell. 2002;13(1):25–39. doi: 10.1091/mbc.01-09-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring NW, Hyman AA. Organelle growth control through limiting pools of cytoplasmic components. Curr Biol. 2012;22(9):R330–R339. doi: 10.1016/j.cub.2012.03.046. [DOI] [PubMed] [Google Scholar]
- Du Toit A. Development: Honey, I shrunk the nucleus. Nat Rev Mol Cell Biol. 2014;15(10):633. doi: 10.1038/nrm3872. [DOI] [PubMed] [Google Scholar]
- Buchner K. The role of protein kinase C in the regulation of cell growth and in signalling to the cell nucleus. J Cancer Res Clin Oncol. 2000;126(1):1–11. doi: 10.1007/pl00008458. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target. Nat Rev Drug Discov. 2012;11(12):937–957. doi: 10.1038/nrd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]