

Abstract
The application of structure-based in silico methods to drug discovery is still considered a major challenge, especially when the x-ray structure of the target protein is unknown. Such is the case with human G protein-coupled receptors (GPCRs), one of the most important families of drug targets, where in the absence of x-ray structures, one has to rely on in silico 3D models. We report repeated success in using ab initio in silico GPCR models, generated by the predict method, for blind in silico screening when applied to a set of five different GPCR drug targets. More than 100,000 compounds were typically screened in silico for each target, leading to a selection of <100 “virtual hit” compounds to be tested in the lab. In vitro binding assays of the selected compounds confirm high hit rates, of 12–21% (full dose–response curves, Ki < 5 μM). In most cases, the best hit was a novel compound (New Chemical Entity) in the 1- to 100-nM range, with very promising pharmacological properties, as measured by a variety of in vitro and in vivo assays. These assays validated the quality of the hits as lead compounds for drug discovery. The results demonstrate the usefulness and robustness of ab initio in silico 3D models and of in silico screening for GPCR drug discovery.
Keywords: modeling, in silico screening, structure-based
G protein-coupled receptors (GPCRs) are membrane-embedded proteins, responsible for communication between the cell and its environment (1). As a consequence, many major diseases, such as hypertension, cardiac dysfunction, depression, anxiety, obesity, inflammation, and pain, involve malfunction of these receptors (2), making them among the most important drug targets for pharmacological intervention (3–5). Thus, whereas GPCRs are only a small subset of the human genome, they are the targets for ≈50% of all recently launched drugs (6). As targets of paramount importance, it is expected that drug discovery for GPCRs would benefit from the introduction of computational methodologies (7), especially as these methods can be used in conjunction with such experimental methods as high-throughput screening (8, 9), NMR, and crystallography (10).
Unfortunately, GPCRs, like other membrane-embedded proteins, have characteristics that make their 3D structure extremely difficult to determine experimentally. To date, the only GPCR for which a 3D structure was determined by x-ray crystallography is bovine rhodopsin (11), which is unique among GPCRs in that its ligand, retinal, is covalently bound and that it responds to light rather than to ligand binding. Hence, in the case of GPCRs, the limited availability of structural data has forced the computational design of ligands to heavily rely on ligand-based techniques. Indeed, for many GPCRs, the natural ligand can provide a good starting point, leading to useful pharmacophore models that can be used for identifying lead structures with novel scaffolds (6). These methods have been successfully applied for the discovery of peptide agonists to the somatostatin receptor (12) and for the discovery of nonpeptidic antagonists to the urotensin II receptor (13).
Nonetheless, structure-based drug discovery remains highly desirable for GPCRs. It is known that all GPCRs structurally consist of seven transmembrane (TM) helices joined together by three extracellular and three intracellular loops. Of particular interest to small-molecule drug discovery is the TM region of the protein. Site-directed mutagenesis studies have shown that small organic compounds (i.e., most drug compounds) bind primarily in a cleft formed by the portions of the TM domains of the protein facing the extracellular milieu. The loops and N-terminal domains are involved in binding of physiological peptide and protein ligands, but play only a minor role in the binding of drugs (14). As a consequence, modeling of the 3D structure of GPCRs has focused on the TM domain of these receptors, employing in most cases homology modeling based on 2.8-Å resolution structure of bovine rhodopsin (11) or on the 1.55- to 2.5-Å resolution x-ray structures of bacteriorhodopsin, a non-GPCR 7TM membrane protein (15, 16). These modeling efforts have been described in a recent review (14). Some of these models have been successful in screening for known ligands embedded in random libraries (17), as well as for discovery of novel dopamine D3 ligands when used in conjunction with a pharmacophore-based method (18).
Recently, we have reported a de novo GPCR modeling approach called predict, which does not rely on the rhodopsin structure and can be applied to any GPCR (14, 19–21). In the present paper, we report the successful application of predict GPCR models in blind high-throughput in silico screening for the discovery of new chemical entities that bind to five different GPCRs. These protein targets include three biogenic amine receptors [5-hydroxytryptamine (5-HT)1A, 5-HT4, and dopamine D2], a peptide receptor (NK1), and a chemokine receptor (CCR3). We show here that this approach has led to confirmed high hit rates and to the discovery of several very promising lead compounds.
Methods
In Silico Modeling. The predict algorithm and methodology were recently reported elsewhere (14, 21). Here, we shall give only a brief overview of the method. predict is a de novo GPCR modeling methodology that combines the properties of the protein sequence with those of its membrane environment, without relying on the rhodopsin (or bacteriorhodopsin) x-ray structure. The predict algorithm searches through the receptors' conformation space for the most stable 3D structure(s) of the TM domain of the GPCR protein within the membrane environment. To ensure that the final model represents the most stable conformation, the method simultaneously optimizes several thousand alternative conformations of the receptor (denoted as “decoys”). The final model is accepted only if it is significantly more stable than the majority of the decoys.
The algorithm solves this complex search and optimization task efficiently by using a reduced representation of the protein-membrane system, which balances computational efficiency and accuracy. In this representation, each side chain is represented by two to four virtual atoms (22), allowing for an efficient search through rotamer space (23) while retaining a low dimensional representation of the system. The reduced representation is expanded to an all-atom model toward the end of the modeling process. The algorithm also takes into account, in a simplified way, the presence of the membrane environment and the different character of the membrane lipophilic core and the polar head group region. These components, as well as various protein–protein interactions, are introduced into the modeling procedure by means of the energy function (see below).
Following are the main steps in the predict algorithm (details in refs. 14 and 21). First, an extended sequence around the TM domain (but longer than it) is identified by using known methods, such as a combination of hydrophobicity and sequence conservation patterns (24). A 2D grid is used to construct thousands of alternative packing geometries that cover the protein's conformation space. Hydrophobic moments are used to rotate the helices so that the bundle presents a hydrophobic surface toward the membrane. Each decoy then undergoes a series of optimization steps, including optimization of helix orientation, helix vertical alignment (relative to the other helices and relative to the membrane/water boundary), helix position, and helical tilt angles. Each change in any of these factors is followed by stochastic simulated annealing optimization of the side-chain rotamers in the vicinity of the change. Optimization of the tilt angles is attempted along all possible three- and four-helical arches. Finally, the optimized models are ranked according to their predict energy score. Models with energy scores significantly lower than other decoys are considered solutions. Similarity clustering is then used to reduce the number of solutions. The lowest energy representative of the largest cluster is the final model. This model, still in a reduced representation, is then expanded to an all-atom model maintaining the specific side-chain rotamers that were optimized by predict.
The energy function used for optimizing the 3D conformations and for scoring the models includes two terms, an intraprotein residue–residue interaction term, and a single-residue term, reflecting its interaction with the membrane,
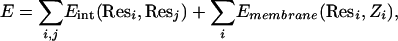 |
[1] |
where Resi and Resj are the two interacting residues, and their interaction energy Eint(Resi, Resj) is defined as
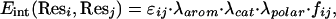 |
[2] |
where εij are the Miyazawa and Jernigan (25) contact energies between residue i and j, fij is a distance function with the general shape of a “soft” Lennard–Jones potential (a 6–4 potential in agreement with the multiatomic nature of the virtual “atoms” used in this representation, unlike the atomistic 12–6 Lennard–Jones function), λarom is an aromatic-clustering factor highlighting aromatic–aromatic interactions, λcat is a cation–π interaction factor reflecting what is recognized as an important noncovalent binding interaction in α-helical peptides (26), and λpolar is a polar–polar interaction factor that emphasizes their contribution in agreement with studies that point to specific polar interactions implicated in driving TM helix association (27, 28), especially in the hydrophilic core of GPCRs. The protein–membrane interaction term Emembrane(Resi, Zi) is a function of the chemical character of Resi and its position Zi in the direction normal to the membrane plane. The value of Zi determines whether the residue is interacting with the lipid core or with the polar head groups, adjusting the interaction accordingly.
The predict optimization algorithm is used as part of a four-step modeling process: (i) coarse modeling. predict searches through the entire protein conformation space, evenly covered by ≈1,500 decoys, to identify regions of stability; (ii) fine modeling. predict is used a second time to comb the neighborhood of the most stable “coarse” models, optimizing ≈5,000 decoy structures in the vicinity of each model. This step allows the algorithm to rapidly focus on regions of stability in the protein's energy landscape and to efficiently identify the most stable “fine” model; (iii) molecular dynamics refinement. The resulting all-atom model is minimized and then subjected to up to 300-ps molecular dynamics simulations with charmm (29) and the charmm22 force field (30). Multiple constraints are applied during the simulations to ensure that the model does not deviate significantly from the predict model. These refinement dynamics introduce helical kinks and relax the side-chain conformations; and (iv) virtual protein–ligand complex. A protein–ligand complex is carefully constructed through molecular dynamics, mimicking the experimental cocrystallization process, which locks the target in a ligand-bound conformation.
Model Validation. The absence of x-ray structures of GPCRs makes direct evaluation of the predict models difficult. The structure prediction algorithm was first validated against the bovine rhodopsin structure, which is currently the only GPCR for which the crystal structure is available (at a resolution of 2.8 Å; ref. 11). As reported elsewhere (21), the resulting predict rhodopsin model was in good agreement with the x-ray structure. The Cα rms distance (rmsd) between the 11-cis-retinal-bound predict rhodopsin model and the TM region of rhodopsin crystal structure (PDB ID code 1F88) was 2.9 Å. The heavy-atom rmsd between 11-cis retinal in the model and its conformation in the crystal structure is 0.9 Å. The location and side-chain orientation of most key residues known to be involved in retinal binding, including Glu-113, Leu-125, Met-207, Phe-208, His-211, Phe-212, and Lys-296 (31), are nicely reproduced in the model, adopting conformations similar to those in the x-ray structure. This Cα rms value is comparable with the rms obtained by Vaidehi et al. (32) when modeling rhodopsin based on the rhodopsin x-ray structure. predict also modeled correctly the unusual helical kinks observed in the x-ray structure of rhodopsin (21). Both the degree of the helical kinks and the twist angles were successfully reproduced in the predict rhodopsin model, including the inwardbent Pro kink in TM1 and the Thr-Gly kink in TM2 (33). This agreement is not trivial because the degree of twist associated with helical kinks is highly variable (34).
Additional validation for predict models comes from the success of these models in virtual screening, as evaluated by “enrichment factors.” The rate at which an in silico screening procedure identifies known binders from a background of random compounds, relative to simple random picking (no enrichment), is denoted as the enrichment factor. In this case, enrichment factors were obtained by virtually screening a random 10,000 drug-like compound library to which a small number of known ligands were added. The results consistently show, for a range of GPCRs, including biogenic amine, peptide, and chemokine receptors, that the predict 3D models yield enrichment factors ranging from 10- to 350-fold better than random (14, 21). These enrichment factors are similar to, and sometimes even better than, enrichment factors reported for in silico screening by using high-resolution crystal structures for non-GPCR targets (17, 35–37).
Screening Library. The screening library consists of virtual 2D representations of ≈1,600,000 drug-like compounds, obtained from electronic catalogs of >20 vendors worldwide. The library is updated quarterly, so that at any given time, it represents compounds that can be purchased on short notice. The multiple sources of the library ensure its diversity, allowing us to explore broad chemical spaces.
Library Preparation. The virtual compound library is processed before docking. This step includes 2D to 3D conversion by using concord (Tripos, St. Louis), Version 4.04, assignment of Gasteiger atom types [sybyl (Tripos), Version 6.8], assignment of atomic charges, identification of multiple anchors for docking (by using hyperion software from Predix), generating multiple conformations for these anchors by using confort (Tripos), Version 4.1, and more. Approximately 10% of the full library (on the order of 150,000 compounds) is selected for screening, according to the character of the binding site (charges, polar, or hydrophobic), and to the desired range of molecular weights, compound diversity, drug-like properties, etc.
Docking. The screening library is docked into the binding site by using the dock4.0 (Molecular Design Institute, University of California, San Francisco and ref. 38) anchor-and-grow procedure. Docking for each compound (often represented by five or more different anchor conformations) is repeated 10 times. The specific docking parameters are fine tuned for each target separately, by optimizing the docking of a small set of known binders (if available) relative to a large drug-like random compound library. Fig. 1 shows a sample enrichment graph (for the NK1 receptor).
Fig. 1.

Enrichment graph for in silico screening of 26 known NK1 antagonists embedded in the 6,200-compound random drug-like library with similar physiochemical properties after docking into the predict 3D model of the NK1 receptor. Library compounds are ranked along the x axis, according to their docking score (best scorers on the left, worst scorers near the 100% mark). The curve shows the relative ranking of the known antagonists. Enrichment at 50% is 20-fold better than with random screening (dashed line).
Scoring and Selection. A sequential application of several scoring tools and selection criteria is used, until a list of fewer than 100 virtual hits is reached (Y.M., O.M.B., S.S., B.I., A.H., M.F., O.K., S.B.-H., D.W., and S.N., unpublished work). First, an automated binding-mode analysis (by using the Predix program bma) is performed on all docked conformations to ensure proper docking. Ten additional 3D scores are calculated for the top 10% of the library with the best dock scores that pass bma [by using dock4.0, cscore (Tripos), and charmm (29)]. Specific score cutoff values or combination cutoff values are used to further narrow the list of virtual hits. The remaining compounds are further filtered by using a 3D-based principle component analysis (PCA) procedure, which is based on 3D properties of the docked compounds. The coordinates for the covariance matrix, which is diagonalized in this procedure, include the above 10 3D docking-scores, several 3D descriptors characterizing the compounds' docked conformations, and a few 2D descriptors (a total of 5–50 descriptors, depending on the target protein). The 3D-based PCA projection is first generated for a set of known compounds, binders, and nonbinders to the specific target alike. The remaining virtual hits are then projected onto this 3D-based PCA map, and the hit list is further narrowed to include only those compounds that fall within the same region of the PCA map as the known active compounds. This step typically reduces the size of the hit list by ≈50%. Finally, the remaining compounds in the hit list are clustered, with only the best-scored representative of the similarity cluster maintained.
In Vitro Binding Assay. Compounds selected as virtual hits from in silico screening against GPCR targets were sent to the appropriate experimental binding assays (radioligand displacement). Compounds were initially tested at a 10-μM concentration in duplicate. Hits showing >50% inhibition at 10 μM are validated by a full-concentration dose–response curve, measured between 10–10 and 10–4 M. Compounds with experimentally validated binding affinities <5 μM are defined as actual hits (for CCR3, better than 20 μM). Specifically, for human NK1 receptor (U-373MG cells), the radioligand was [Sar-9,Met(O2)11]-SP (0.15 nM, Kd = 0.12 nM). For human recombinant CCR3 receptor (K562 cells), the radioligand was [125I]eotaxin (0.1 nM, Kd = 0.7 nM). For human recombinant 5-HT1A receptor (human embryonic kidney-293 cells), the radioligand was [3H]8-hydroxy-N,N-dipropylaminotetralin (0.5 nM, Kd = 0.5 nM). For human recombinant 5-HT4e receptor [Chinese hamster ovary (CHO) cells], the radioligand was [3H]GR-13808 (0.2 nM, Kd = 0.15 nM). For human recombinant D2 receptor (CHO cells) the radioligand was [3H]spiperone (0.3 nM, Kd = 0.17 nM).
Other in Vitro and in Vivo Assays. Lead compounds, chosen from the preliminary in vitro binding assays, were evaluated in additional in vitro and in vivo assays. These studies were carried out at external contract research organizations, and include cell-based functional assays for agonist or antagonist activity, a complete selectivity profile (in vitro binding assays for up to 60 targets, including GPCRs, ion channels, transporters, and enzymes), and human liver microsome stability assay. In some cases, the pharmacokinetic properties of the compound were measured in vivo in rats, focusing on oral bioavailability (%F) and serum half-life.
Results and Discussions
Using in silico methods for drug discovery requires the careful integration of several computational tools into a single streamlined process. In this case, the first step was predict modeling of the target GPCR. The model was then used for 3D screening of large virtual compound libraries, followed by a scoring and selection process that led to a small number of virtual hits. The virtual hits were subsequently purchased and tested in experimental binding assays to verify their activity. In this section, we present the results of this process as applied to five human GPCR drug targets: 5-HT1A, 5-HT4, Dopamine D2, NK1, and CCR3, representing biogenic amine, peptide, and chemokine receptors.
Serotonin 5-HT1A Receptor. 5-HT1A is one of 14 serotonin (5-HT) receptor subtypes. Agonist binding to 5-HT1A receptors leads to inhibition of adenylyl cyclase activity, the reduction of cAMP levels, increase in potassium (K+) conductance by regulating K+ channels, and decreasing the opening of voltage-gated calcium (Ca2+) channels (39). The 5-HT1A receptors are expressed in the CNS and have been implicated in anxiety and depression disorders. In vivo electrophysiological experiments have postulated that 5-HT1A partial agonists mediate antidepressant effects through a net increase in serotonergic neurotransmission. Although the exact mechanism of action is not fully understood, there is evidence that the physiological and behavioral responses are achieved after desensitization of 5-HT1A receptor-mediated response (40). Only one 5-HT1A agonist, buspirone (Buspar), is approved for generalized anxiety disorder, with a few other agonists of a class called azapirones, in late-stage development (e.g., gepirone ER, Variza, developed for depression, or sunepitron).
A predict 3D model of the 5-HT1A receptor was generated from its amino acid sequence, from which a receptor–ligand complex was simulated (using serotonin as the ligand). A binding pocket was easily identified on the extracellular side of the structure, allowing the 5-HT amine moiety to interact with Asp-116 in TM3 and its hydroxyl moiety to interact with Ser-199 in TM5; both in good agreement with experimental data (41, 42). Fig. 2 shows the binding mode of buspirone and of sunepitron in the 5-HT1A-binding pocket. In both cases, Asp-116 is interacting with the piperazine amine and Ser-199 interacts with the azapirones' imide moiety. In addition, the model shows that residue Phe-362 from TM6 stacks against the compounds' pyrimidine rings and that residue Asn-386 from TM7 also contributes to this interaction, in agreement with experimental data (31). Docking of a set of 24 known 5-HT1A ligands embedded in a random 10,000 drug-like compound library, with average properties similar to those of the 5-HT1A ligands (21), yielded an enrichment factor 20-fold better than random when ranked by the initial dock score (at the point where 50% of the compounds were identified). Furthermore, 88% of these known ligands were ranked among the top 10% of the dock-score ranked library (14). This study was used both for validating the model and for calibrating the docking protocol.
Fig. 2.

Buspirone (a) and sunepitron (b), two 5-HT1A partial-agonist drugs, docked in the 5-HT1A receptor-binding pocket (transparent surface). The compounds are shown in space-fill-form to highlight their binding mode. Key protein residues responsible for ligand binding are shown. Also shown are the chemical structures of the two drugs.
Docking and scoring of a 40,000-compound screening library led to the selection of 78 virtual hits (Table 1). Experimental in vitro binding assays confirmed 16 hits with a Ki < 5 μM, reflecting a 21% hit rate, with the best hit being a novel 1.0-nM compound (PRX-93009). Furthermore, the 16 hits represented five distinct chemical scaffolds and 87% of them (14 of 16 compounds) were found to be novel chemical entities, not covered by any patent or publication (other than the suppliers' catalog).
Table 1. Summary of 3D structure-based in silico screening for GPCR targets.
| Target | Compounds screened in silico | Compounds tested in vitro | Hits <5 μM | Hit rate, % | Best hit, nM | Percent novel | Best novel hit, nM |
|---|---|---|---|---|---|---|---|
| 5-HT1A | 40,000 | 78 | 16 | 21 | 1.0 | 87 | 1.0 (PRX-93009) |
| NK1 | 150,000 | 53 | 8 | 15 | 56 | 100 | 56 (PRX-96026) |
| 5-HT4 | 150,000 | 93 | 19 | 21 | 1.6 | 42 | 21 (PRX-93046) |
| D2 | 120,000 | 42 | 7 | 17 | 58 | 30 | 58 (PRX-92026) |
| CCR3 | 120,000 | 43 | 5* | 12 | 12,000 | 60 | 12,000 (PRX-94042) |
Hit is defined as Ki < 20 μM.
To address whether these hits represent real leads for drug discovery, the best 5-HT1A hit, compound PRX-93009, was subjected to additional in vitro and in vivo assays (Table 2). It was found that in addition to its 1-nM binding affinity, PRX-93009 also tested as a partial agonist in a cell-based assay, showing 65% activity relative to 5-HT with EC50 = 21 nM. The compound also showed good pharmacokinetic properties in rats, with a 2-h serum half-life p.o. and 5% oral bioavailability. These properties compare favorably with buspirone, the only 5-HT1A agonist approved as a drug, which has Ki = 20 nM, is a partial agonist with 50% activity relative to 5-HT with EC50 = 80 nM in a cell-based assay, has a 1-h serum half-life p.o. in rats, and as little as 1% oral bioavailability. The main downside of PRX-93009 was its selectivity profile, showing high affinity to the α1 adrenergic receptor (Ki = 6.6 nM) as well as to several other receptors. As such, this selectivity issue was the focus of a lead optimization process that started from this lead compound and was able to quickly convert it into a very selective compound.
Table 2. In vitro and in vivo assay results for lead compounds discovered by in silico screening on the predict 3D models.
| Target GPCR | Lead compound | Ki, nM | Agonist/antagonist | Selectivity | Other assays |
|---|---|---|---|---|---|
| 5-HT1A | PRX-93009 | 1 | EC50 = 21 nM | 60-target panel: α1 = 7 nM, | Rat PK: 2-h t1/2 p.o. |
| 65% partial agonist | 10-100 nM: four targets | 5% oral bioavailability | |||
| 100-500 nM: two targets | |||||
| NK1 | PRX-96026 | 56 | EC50 = 950 nM | NK2 = 1,700 nM | |
| antagonist | NK3 > 10,000 nM | ||||
| 27-target panel: | |||||
| 500-1,000 nM: two targets | |||||
| 5-HT4 | PRX-93046 | 21 | EC50 = 200 nM | 50-receptor panel: | 46 min in human liver microsomes |
| 18% partial agonist | 500-2,000 nM: one target |
Tachykinin NK1 Receptor. The NK1 receptor is one of three tachykinin receptor subtypes, whose endogenous ligand is the Substance P peptide. The NK1 receptor is distributed in the central and peripheral nervous system and is implicated in depression, asthma, emesis, anxiety, and pain (43). There is only one marketed drug that targets this receptor, aprepitant (Emend), which is approved for emesis.
The predict 3D model of the NK1 receptor was generated from sequence and a receptor–ligand complex was simulated with aprepitant (Ki = 0.9 nM). This model was discussed in detail in ref. 21. A binding pocket was easily identified on the extracellular side of the model, located between helices 4, 5, and 6. Docking a set of 26 known NK1 small-molecule ligands embedded in a 6,200 random compound library with similar physicochemical properties yielded an enrichment factor 20-fold better than random (for 50% of the known identified, all of which were ranked within the top 2.5% of the ranked library). Eighty percent of the ligands were identified within the top 10% of the ranked library (Fig. 1).
Docking of a 150,000-compound library led to the selection of 53 virtual hits. An in vitro binding assay confirmed eight hits with Ki < 5 μM, reflecting a 15% hit rate, with the best hit being a novel 56-nM compound (PRX-96026). These eight hits fell into five distinct chemical scaffolds and all (100%) of them were found to be novel (Table 1).
Additional studies confirmed the drug-like quality of this lead compound (Table 2). PRX-96026 tested as an antagonist in a cell-based functionality assay, with EC50 = 950 nM. The compound was selective relative to the other two neurokinin receptors, with a Ki of 1,700 nM to NK2 and >10,000 nM to NK3, reflecting a selectivity ratio of 1:30:180. In a selectivity panel of 27 targets (mostly GPCRs), PRX-96026 showed an excellent selectivity profile, with affinities in the 500–1,000 nM range to only two other targets.
Serotonin 5-HT4 Receptor. One of the 14 serotonin receptor subtypes, 5-HT4 is expressed in many organs, including the gastrointestinal tract and CNS (44). These receptors are positively coupled to adenylate cyclase and are known to exert such neurochemical responses as serotonin, acetylcholine, and dopamine release. Potential indications for this target include irritable bowel syndrome (IBS) and Alzheimer's disease (agonist). The 5-HT4 agonist tegaserod (Zelnorm) is a marketed drug for IBS. Another 5-HT4 agonist, prucalopride (Resolor), may undergo phase III clinical trials. A well known 5-HT4 agonist drug that was withdrawn from the market due to the QT interval prolongation is cisapride (Propulsid).
The predict model of the 5-HT4 receptor was virtually complexed with GR-113808 (Kd = 0.15 nM). A binding pocket was easily identified on the extracellular side of the model, located between helices 3, 5, 6, and 7, in agreement with mutation data (45). Fig. 3 shows the binding mode of GR-113808 in the receptor's binding pocket. Docking a set of 19 known 5-HT4 ligands embedded in a 10,000-random compound library with similar physicochemical properties gave an enrichment factor 50-fold better than random (at the point where 50% of the compounds were identified), identifying 95% of these known ligands within the top 10% of the dock score-ranked library.
Fig. 3.

GR-113808, a potent 5-HT4 ligand, docked in the receptor 3D model. The compound's amine group is 4.1 Å from Asp-100 (TM3), and the ester interacts with Ser-197 (TM5, 3.9 Å); Phe-297 (TM7) and Asn-279 (TM6) interact with the indole group of the ligand, and Tyr-302 (TM7) is within 5 Å from the sulfonamide. Also shown is the chemical structure of the compound.
A 150,000-compound library was docked and 93 virtual hits selected. In vitro binding assay confirmed the binding of 19 compounds with Ki < 5 μM (21% hit rate). These hits reflect four distinct chemical scaffolds and 42% of them were found to be novel (Table 1). The best hit was a known 1.6-nM binder; however, the best novel hit was not far behind with Ki = 21 nM (PRX-93046).
Additional studies confirmed the drug-like quality of PRX-93046. It tested as a partial agonist in a cell-based assay, showing 18% of 5-HT activity with EC50 = 200 nM. In a 50-target selectivity assay, the compound showed affinity in the 500- to 2,000-nM range to only one additional target. The compound also has excellent metabolic stability, with a 46-min half-life in human liver microsomes (Table 2).
Dopamine D2 Receptor. Dopamine is the predominant catecholamine neurotransmitter in mammalian brains, where it affects locomotor activity, cognition, emotion, and other functions (46). The predict 3D model of the 5-HT4 receptor was generated from sequence and a receptor–ligand complex was simulated with D2 antagonist fluphenazine (Ki = 0.32–4 nM, details reported in ref. 21). Docking a set of 43 known D2 agonists and antagonists embedded in a 10,000-random compound library yielded an enrichment factor 17- and 9-fold better than random, respectively (at the point where 50% of the compounds were identified), identifying 85% of the antagonists and 70% of the agonists within the top 10% of the dock score-ranked library.
Docking a 120,000-compound library into the 3D model resulted in 42 virtual hits. In vitro binding assays confirmed the binding of seven compounds with Ki < 5 μM, reflecting a 17% hit rate (Table 1). The best hit was a novel 58-nM compound (PRX-92026). No additional studies were performed on this compound.
Chemokine CCR3 Receptor. The chemokine CCR3 receptor is involved in the inflammatory response. Although the binding of chemokines, which are small proteins, involves the N terminus and extracellular loops, studies have shown that small-molecule antagonists bind within the TM domain of the receptor (47). As discussed elsewhere (21), the predict model of the CCR3 receptor shows a binding pocket between TMs 1, 2, 3, and 7, in agreement with experimental data. The receptor–ligand complex of CCR3 was generated by using the potent CCR3 small-molecule antagonist (Ki = 5 nM, compound d36 in ref. 48).
Docking a set of 22 known compounds embedded in a 10,000-random compound library yielded an enrichment factor 45-fold better than random (at the 50% mark), identifying 86% of these known ligands (19 of 22 compounds) within the top 15% of the dock score-ranked library. Subsequently, a 120,000-compound library was docked into the 3D model, leading to a selection of 43 virtual hits. In vitro binding assays confirmed five hits with Ki < 20 μM, reflecting a 12% hit rate. The best hit was a novel 12-μM compound (PRX-94042). While less potent than in previous studies, this hit is acceptable for chemokine receptor screening. No additional studies were performed on this compound.
Summary
We have reported herein the repeated successful use of predict 3D GPCR models for actual blinded structure-based in silico screening. For five GPCR drug targets; biogenic amine, peptide, and chemokine receptor, this methodology was successful in identifying high-quality hits, including promising lead compounds for multiple drug discovery programs. As will be reported elsewhere, some of these lead compounds were later successfully optimized by using the 3D structure of the target GPCR as a guideline. This work paves the way to a broader application of 3D models in GPCR drug discovery, compensating for the limited number of x-ray structures available in this important field.
Acknowledgments
We thank Adam Muzikant for assistance.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: GPCR, G protein-coupled receptor; TM, transmembrane; 5-HT, 5-hydroxytryptamine.
References
- 1.Horn, F., Weare, J., Beukers, M. W., Horsch, S., Bairoch, A., Chen, W., Edvardsen, O., Campagne, F. & Vriend, G. (1998) Nucleic Acids Res. 26, 275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilson, S. & Bergsma, D. (2000) Drug Des. Discovery 17, 105–114. [PubMed] [Google Scholar]
- 3.Sautel, M. & Milligan, G. (2000) Curr. Med. Chem. 7, 889–896. [DOI] [PubMed] [Google Scholar]
- 4.Schoneberg, T., Schulz, A. & Gudermann, T. (2002) Rev. Physiol. Biochem. Pharmacol. 144, 143–227. [PubMed] [Google Scholar]
- 5.Rattner, A., Sun, H. & Nathans, J. (1999) Annu. Rev. Genet. 33, 89–131. [DOI] [PubMed] [Google Scholar]
- 6.Klabunde, T. & Hessler, G. (2002) ChemBioChem 3, 928–944. [DOI] [PubMed] [Google Scholar]
- 7.Rodrigues, A. D. & Lin, J. H. (2001) Curr. Opin. Chem. Biol. 5, 396–401. [DOI] [PubMed] [Google Scholar]
- 8.Bajorath, J. (2002) Nat. Rev. Drug Discov. 1, 882–894. [DOI] [PubMed] [Google Scholar]
- 9.Jenkins, J. L., Kao, R. Y. & Shapiro, R. (2003) Proteins 50, 81–93. [DOI] [PubMed] [Google Scholar]
- 10.van Dongen, M. J., Uppenberg, J., Svensson, S., Lundback, T., Akerud, T., Wikstrom, M. & Schultz, J. (2002) J. Am. Chem. Soc. 124, 11874–11880. [DOI] [PubMed] [Google Scholar]
- 11.Palczewski, K., Kumasaka, T., Hori, T., Behnke, C. A., Motoshima, H., Fox, B. A., Trong, I. L., Teller, D. C., Okada, T., Stenkamp, R. E., et al. (2000) Science 289, 739–745. [DOI] [PubMed] [Google Scholar]
- 12.Veber, D. F. (1992) in Peptides, Chemistry, and Biology: Proceedings of the 12th American Peptide Symposium, eds. Smith, J. A. & Rivier, J. E. (ESCOM, Leiden, The Netherlands), pp. 3–14.
- 13.Flohr, S., Kurz, M., Kostenis, E., Brkovich, A., Fournier, A. & Klabunde, T. (2002) J. Med. Chem. 45, 1799–1805. [DOI] [PubMed] [Google Scholar]
- 14.Becker, O. M., Shacham, S., Marantz, Y. & Noiman, S. (2003) Curr. Opin. Drug Discov. Devel. 6, 353–361. [PubMed] [Google Scholar]
- 15.Pebay-Peyroula, E., Rummel, G., Rosenbusch, J. P. & Landau, E. M. (1997) Science 277, 1676–1681. [DOI] [PubMed] [Google Scholar]
- 16.Luecke, H., Schobert, B., Richter, H. T., Cartailler, J. P. & Lanyi, J. K. (1999) J. Mol. Biol. 291, 899–911. [DOI] [PubMed] [Google Scholar]
- 17.Bissantz, C., Bernard, P., Hibert, M. & Rognan, D. (2003) Proteins 50, 5–25. [DOI] [PubMed] [Google Scholar]
- 18.Varady, J., Wu, X., Fang, X., Min, J., Hu, Z., Levant, B. & Wang, S. (2003) J. Med. Chem. 46, 4377–4392. [DOI] [PubMed] [Google Scholar]
- 19.Becker, O. M., Shacham, S., Topf, M. & Naor, Z. (2000) World Patent WO-02/15106-A2.
- 20.Shacham, S., Topf, M., Avisar, N., Glazer, F., Marantz, Y., Bar-Haim, S., Noiman, S., Naor, Z. & Becker, O. M. (2001) Med. Res. Rev. 21, 472–483. [DOI] [PubMed] [Google Scholar]
- 21.Shacham, S., Marantz, Y., Bar-Haim, S., Kalid, O., Warshaviak, D., Avisar, N., Inbal, B., Heifetz, A., Fichman, M., Topf, M., et al. (2004) Proteins, in press. [DOI] [PubMed]
- 22.Herzyk, P. & Hubbard, R. E. (1993) Proteins 17, 310–324. [DOI] [PubMed] [Google Scholar]
- 23.Ponder, J. W. & Richards, F. M. (1987) J. Mol. Biol. 193, 775–791. [DOI] [PubMed] [Google Scholar]
- 24.Mirzadegan, T., Benko, G., Filipek, S. & Palczewski, K., (2003) Biochemistry 42, 2759–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyazawa, S. & Jernigan, R. L. (1996) J. Mol. Biol. 256, 623–644. [DOI] [PubMed] [Google Scholar]
- 26.Gromiha, M. M. (2003) Biophys. Chem. 103, 251–258. [DOI] [PubMed] [Google Scholar]
- 27.Zhou, F. X., Cocco, M. J., Russ, W. P., Brunger, A. T. & Engelman, D. M. (2000) Nat. Struct. Biol. 7, 154–160. [DOI] [PubMed] [Google Scholar]
- 28.Cosson, P., Lankford, S. P., Bonifacino, J. S. & Klausner, R. D. (1991) Nature 351, 414–416. [DOI] [PubMed] [Google Scholar]
- 29.Brooks, B. R., Bruccoleri, R. E., Olafson, B. D., States, D. J., Swaminathan, S. & Karplus, M. (1983) J. Comput. Chem. 4, 187–217. [Google Scholar]
- 30.MacKerell, A. D. D., Jr., Bashford, D., Bellott, M., Dunbrack, R. L., Jr., Evanseck, J., Field, M. J., Fischer, S., Gao, J., Guo, H., Ha, S., et al. (1998) J. Phys. Chem. B. 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- 31.Shi, L. & Javitch, J. A. (2002) Annu. Rev. Pharmacol. Toxicol. 42, 437–467. [DOI] [PubMed] [Google Scholar]
- 32.Vaidehi, N., Floriano, W. B., Trabanino, R., Hall, S. E., Freddolino, P., Choi, E. J., Zamanakos, G. & Goddard, W. A., III (2002) Proc. Natl. Acad. Sci. USA 99, 12622–12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gray, T. M. & Matthews, B. W. (1984) J. Mol. Biol. 175, 75–81. [DOI] [PubMed] [Google Scholar]
- 34.Piela, L., Nemethy, G. & Scheraga, H. A. (1987) Biopolymers 26, 1587–1600. [DOI] [PubMed] [Google Scholar]
- 35.Charifson, P. S., Corkery, J. J., Murcko, M. A. & Walters, W. P. (1999) J. Med. Chem. 42, 5100–5109. [DOI] [PubMed] [Google Scholar]
- 36.Wang, R. & Wang, S. (2001) J. Chem. Inf. Comput. Sci. 41, 1422–1426. [DOI] [PubMed] [Google Scholar]
- 37.Oshiro, C., Bradley, E. K., Eksterowicz, J., Evensen, E., Lamb, M. L., Lanctot, J. K., Putta, S., Stanton, R. & Grootenhuis, P. D. (2004) J. Med. Chem. 47, 764–767. [DOI] [PubMed] [Google Scholar]
- 38.Ewing, T. J., Makino, S., Skillman, A. G. & Kuntz, I. D. (2001) J. Comput. Aided Mol. Des. 15, 411–428. [DOI] [PubMed] [Google Scholar]
- 39.Barnes, N. M. & Sharp, T. (1999) Neuropharmacology 38, 1083–1152. [DOI] [PubMed] [Google Scholar]
- 40.De Vry, J. (1995) Psychopharmacology 121, 1–26. [DOI] [PubMed] [Google Scholar]
- 41.Jacoby, E., Fauchere, J.-L., Raimbaud, E., Ollivier, S., Michel, A. & Spedding, M. (1999) Quant. Struct.–Act. Relat. 18, 561–571. [Google Scholar]
- 42.Ho, B. Y., Karschin, A., Branchek, T., Davidson, N. & Lester, H. A. (1992) FEBS Lett. 312, 259–262. [DOI] [PubMed] [Google Scholar]
- 43.Severini, C., Improta, G., Falconieri-Erspamer, G., Salvadori, S. & Erspamer, V. (2002) Pharmacol. Rev. 54, 285–322. [DOI] [PubMed] [Google Scholar]
- 44.Eglen, R. M., Wong, E. H., Dumuis, A. & Bockaert, J. (1995) Trends Pharmacol. Sci. 16, 391–398. [DOI] [PubMed] [Google Scholar]
- 45.Mialet, J., Dahmoune, Y., Lezoualc'h, F., Berque-Bestel, I., Eftekhari, P., Hoebeke, J., Sicsic, S., Langlois, M. & Fischmeister, R. (2000) Br. J. Pharmacol. 130, 527–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ballesteros, J. A., Shi, L. & Javitch, J. A. (2001) Mol. Pharmacol. 60, 1–19. [PubMed] [Google Scholar]
- 47.Paterlini, M. G. (2002) Biophys. J. 83, 3012–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Danak, D., Christmann, L. T., Darcy, M. G., Keenan, R. M., Knight, S. D., Lee, J., Ridgers, L. H., Sarau, H. M., Shah, D. H., White, J. R. & Zhang, L. (2001) Bioorg. Med. Chem Let. 11, 1445–1450. [DOI] [PubMed] [Google Scholar]