

Abstract
Although various therapies are available to treat cancers, including surgery, chemotherapy, and radiotherapy, cancer has been the leading cause of death in Japan for the last 30 years, and new therapeutic modalities are urgently needed. As a new modality, there has recently been great interest in oncolytic virotherapy, with measles virus being a candidate virus expected to show strong antitumor effects. The efficacy of virotherapy, however, was strongly limited by the host immune response in previous clinical trials. To enhance and prolong the antitumor activity of virotherapy, we combined the use of two newly developed tools: the genetically engineered measles virus (MV-NPL) and the multilayer virus-coating method of layer-by-layer deposition of ionic polymers. We compared the oncolytic effects of this polymer-coated MV-NPL with the naked MV-NPL, both in vitro and in vivo. In the presence of anti-MV neutralizing antibodies, the polymer-coated virus showed more enhanced oncolytic activity than did the naked MV-NPL in vitro. We also examined antitumor activities in virus-treated mice. Complement-dependent cytotoxicity and antitumor activities were higher in mice treated with polymer-coated MV-NPL than in mice treated with the naked virus. This novel, polymer-coated MV-NPL is promising for clinical cancer therapy in the future.
Introduction
There has been increasing interest in anticancer virotherapy in recent years because of its unique ability to kill cancer cells, avoidance of cross-resistance to standard anticancer therapies, and possible induction of relatively enhanced antitumor effects.1–3 Numerous viruses have been studied preclinically as virotherapeutic agents and some viruses, including measles virus (MV), have been under clinical investigation.4–6 Several reports show that the vaccine strain of MV is safe and has not reverted or converted to a pathogenic virus over the past 50 years of clinical use.7 The MV vaccine strain shows tropism toward many different types of cancer cells through the highly expressed CD46 receptors.8–19
To enhance viral genome replication in cancer cells, we generated a newly engineered MV strain, MV-NPL, which was based on the MV Edmonston vaccine strain, but had the N, P, and L genes of the wild-type MV strain. The MV-NPL strain was found to replicate more efficiently and induce greater oncolytic activity against human renal cell cancer cells in vitro and in vivo, compared to the MV vaccine strain, which occurred via faster replication and increased resistance to interferon-α.14
Several reports have shown that pre-existing or virotherapy-induced neutralizing antibodies can decrease the number of infectious viral particles in the bloodstream, resulting in reduced therapeutic efficacy.20 Recently, Yoshihara et al. coated adenoviruses by performing layer-by-layer deposition of ionic polymers (polyethyleneimine (PEI) and hyaluronic acid) to produce multilayer-coated virus particles. They reported that the infectivity of the virus in the presence of an anti-adenovirus antibody increased after multilayer coating in vitro and in vivo.21
In this study, we combined the newly engineered MV and multilayer virus-coating method with layer-by-layer deposition of ionic polymers. We demonstrated that polymer-coated MV-NPL particles had enhanced oncolytic activity even in the presence of anti-MV neutralizing antibodies compared with naked MV-NPLs, both in vitro and in vivo.
Results
High sensitivity of human cancer cells to MV-NPL-mediated oncolysis
Human CD46 expression in HEp2, A549, WiDr, and MDA-MB-231 cells was analyzed by flow cytometry. The MV receptor CD46 was highly expressed on the all human cancer cell lines studied (Figure 1a and Supplementary Table S1). These cell types were infected using multiplicities of infection (MOIs) of 0.01, 0.1, or 1, after which they were stained with crystal violet. MV-NPL infection caused dramatic cytopathic effects (CPEs) in a MOI-dependent manner in all of the human cancer cell lines examined (Figure 1b).
Figure 1.

Expression profile of CD46 and MV-NPL mediated cytotoxicity in various cancer cell lines. CD46 expression on the human cancer cell line HEp2, A549, WiDr, and MDA-MB-231 (a) and the mouse lung cancer cell lines LL/2 and LL/2-CD46 (c). The cell-surface expression level of the MV receptor CD46 on various cell lines was quantified by flow cytometry. The histograms shown represent the measured fluorescence levels of cells incubated with an isotype-control antibody (unshaded) or an anti-CD46 antibody (shaded). MV-NPL-mediated cytotoxicity against human cancer cell lines (b) and mouse lung cancer cells (d). The indicated cell lines were infected with MV-NPL at multiplicities of infection of 0.01, 0.1, or 1. Cell viability was assessed by crystal violet staining.
Mouse cancer cell lines expressing human CD46 are sensitive to MV-NPL-mediated oncolysis
To examine the in vivo antitumor effects of MVs in a preimmunized mouse model, we first established mouse cancer cells expressing human CD46. We generated LL/2-CD46 cells by lentiviral transduction with the human CD46 gene into the mouse LL/2 lung cancer cell line. LL/2 cells were negative for CD46 expression, whereas high CD46 expression was detected in LL/2-CD46 (Figure 1c). MV-NPL infection caused no CPE in LL/2 cells. In contrast, MV-NPL caused CPEs in LL/2-CD46 cells in a MOI-dependent manner (Figure 1d).
Polymer coating enhanced MV-NPL-mediated oncolysis
To characterize each MV-NPL/polymer complex, the surface charges and sizes of the complexes were assessed. The surface charge of virus particles increased proportionally to the amount of PEI added to the virus suspensions. Positively charged complexes were obtained with final PEI concentrations of 0.5 μg PEI/2.5 × 104 TCID50 of MV-NPL particles. Positively charged MV-NPL/PEI complexes were then mixed with chondroitin sulfate (CS) solution. The addition of CS effectively changed the overall complex charge to a negative charge (Figure 2a). The average sizes of MV-NPL/PEI particles and MV/PEI/CS were 1,023 and 311 nm, respectively (Supplementary Figure S1). The average size was essential for drug delivery, therefore in subsequent experiments, we used MV-NPL as the naked virus and MV-NPL/PEI/CS as the polymer-coated virus.
Figure 2.

Polymer-coated virus production and coated-virus-mediated cytotoxicity. Zeta-potential of virus particles (2.5 × 104 TCID50) changed after the addition of either PEI or CS (a i,ii). Naked- or polymer-coated virus-mediated cytotoxicity in human HEp2, A549, WiDr, and MDA-MB-231 cancer cells in the presence or absence of an anti-MV neutralizing antibody. The indicated cells were infected with MV-NPL at multiplicities of infection (MOIs) of 0.01, 0.1, or 1. Cell viability was assessed by crystal violet staining (b). Cell viability of the HEp2, A549, WiDr, and MDA-MB-231 cancer cell lines after viral infection. To assess cell viabilities, the OD at 570 nm was measured after solubilization by adding 1% SDS to the remaining cells (c). Naked- or polymer-coated virus-mediated cytotoxicity in parental mouse LL/2 lung cancer cells and LL/2 cells expressing CD46 (LL/2-CD46) (d). The values shown in panel c are the mean ± SD for three replicates. NS, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. NV, naked virus; CV, coated virus; w, with; w/o, without; NAb, anti-MV neutralizing antibody; EC50, effective concentration 50.
To evaluate the effect of polymer coating on CPE, HEp2 cells were treated with naked virus or polymer-coated virus, in the presence or absence of an anti-MV neutralizing antibody. Syncytia formation were observed In the absence of the neutralizing anti-MV antibody, greater and MOI-dependent CPEs were observed by the polymer-coated virus, compared with those observed using the naked virus (Figure 2b). The polymer-coated virus showed significantly higher CPE than did the naked virus (Figure 2c) even in the presence of the antibodies (P < 0.0001). The polymer-coated virus showed significantly higher cytotoxicity also in A549 cells, WiDr cells, and MDA-MB-231 cells than did the naked virus (Figure 2c). We next tested the CPEs of the naked or polymer-coated viruses in LL/2 and LL/2-CD46 cells. The polymer-coated virus showed higher cytotoxicity than did the naked virus (Figure 2d). Cell lines of human HEp2 (Supplementary Figure S2a) and mouse LL/2-CD46 (Supplementary Figure S2b) were infected with the polymer-coated virus and showed syncytia formation under light microscope.
Polymer-coated virus infection inhibited tumor growth in immunodeficient mice bearing human immortalized cancer cells
To evaluate the antitumor activity of the polymer-coated virus in vivo, nude mice bearing subcutaneous human cancer cells were treated intratumorally once per week with Tris-HCl (vehicle control), naked virus, or polymer-coated virus. Using the HEp2 tumor xenograft model, intratumoral injection with naked virus or polymer-coated virus resulted in decreased tumor volumes, compared with those observed in control mice (P < 0.05 and P < 0.05, respectively, Figure 3a). In the subcutaneous A549 and WiDr tumor-xenograft models, treatment with naked virus or polymer-coated virus also suppressed tumor growth (Figure 3b,c).
Figure 3.
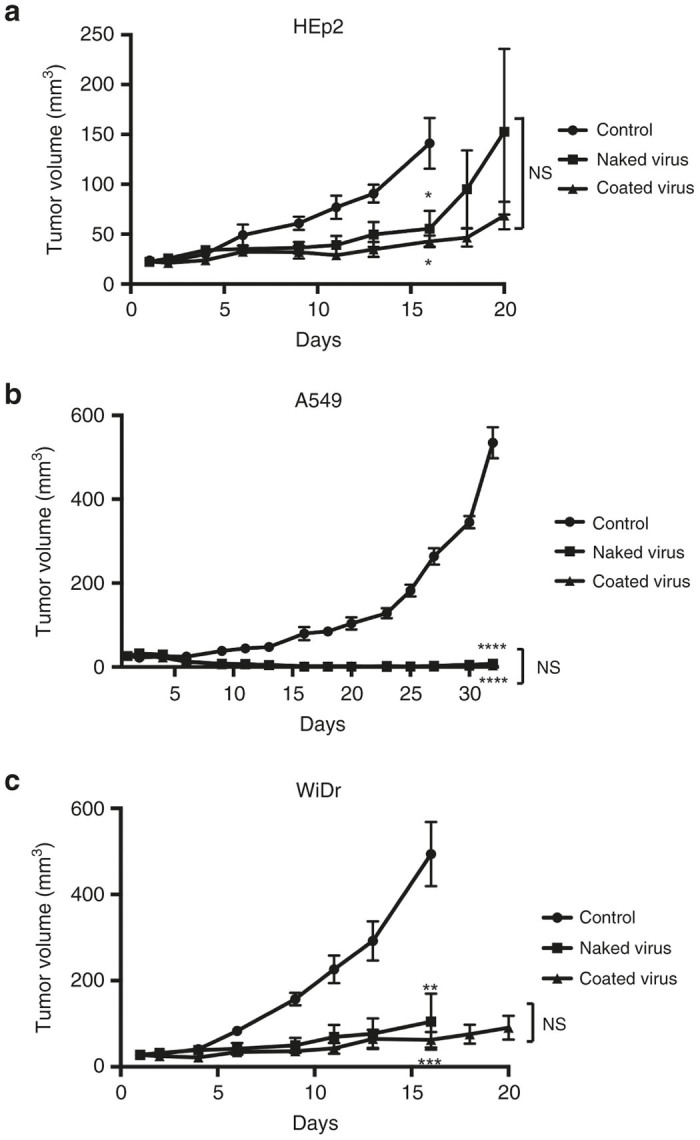
In vivo antitumor effects of MV. Nude mice with xenograft tumors forming after subcutaneous injection of human HEp2 (a), A549 (b), or WiDr (c) cells, were intratumorally injected with Tris-HCl (control), naked virus, or polymer-coated virus (coated virus) on days 1, 8, and 15. Values shown in a, b, and c are the mean ± SEM for 6 (b, c) or 7 (a) female nude mice. NS, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
The polymer-coated virus suppressed tumor growth, induced antitumor immunity, and prolonged the survival of immunocompetent, preimmunized mice bearing LL/2-CD46 cells
The therapeutic effects of the polymer-coated virus in preimmunized mice bearing LL/2-CD46 xenograft tumors were then investigated. Subcutaneous administration of MV-NPL led to anti-MV humoral immune responses in immunocompetent mouse (Supplementary Figure S3a, b). Mice bearing LL/2-CD46 tumors were administered intratumorally once per week with Tris-HCl (vehicle control), naked virus, or polymer-coated virus (Figure 4a). Intratumoral administration of the polymer-coated virus exhibited a greater antitumor effect than did the naked virus (Figure 4b). Mice injected with naked or polymer-coated virus survived longer than control mice (P = 0.063 or P < 0.001, respectively). Mice injected with the polymer-coated virus showed a tendency for longer survival compared with mice injected with the naked virus, though the difference was not statistically significant (P = 0.079) because of the small sample sizes (Supplementary Figure S4). We next examined the serum-mediated cancer cell cytotoxicity of antibody-mediated complement-dependent cytotoxicity (CDC) in virus-treated mice. The CDC activities in the sera of mice injected with polymer-coated virus were significantly higher than those of mice injected with Tris-HCl (P < 0.0001) or the naked virus (P < 0.01; Figure 4c).
Figure 4.

In vivo antitumor effects of polymer-coated virus on subcutaneous LL/2-CD46 tumors in preimmunized C57BL/6N mice. Immune-competent C57BL/6N mice were preimmunized with 1 × 106 TCID50 of MV-NPL. The mice were subsequently injected intratumorally with Tris-HCl (control), naked virus, or polymer-coated virus (coated virus) on days 1, 8, and 15 (a). Tumor volumes (mean ± SEM) were monitored for 9 female C57BL/6N mice, according to protocol A (b). CDC activities in the sera of mice injected with Tris-HCl (control), naked virus, or polymer-coated virus (coated virus) on days 1, 8, and 15 using protocol A are shown as the cell viability (c). Preimmunized mice were intratumorally injected with 1 × 105 TCID50, 2.5 × 105 TCID50, or 5 × 105 TCID50 of polymer-coated virus on days 1, 8, and 15 (d). Tumor volumes (mean ± SEM) were monitored in 5 female C57BL/6N mice, according to protocol B (e). CDC activities in the sera of mice treated according to protocol B were shown as the cell viability (f). NS, not significant; **P < 0.01; ***P < 0.001; ****P < 0.0001.
We next examined the in vivo antitumor effects of three doses of polymer-coated virus in preimmunized mice bearing LL/2-CD46 tumors (Figure 4d). Antitumor effects were observed in a dose-dependent manner, although these differences were not statistically significant (Figure 4e). We also tested CDC activities with sera obtained from mice injected with three doses of polymer-coated virus at each different dose level. Significantly higher CDC activities were observed in mice treated with higher doses of polymer-coated virus (Figure 4f).
Mechanism of enhanced antitumor activity mediated by polymer-coated viruses
To assess the mechanism of enhanced anti-tumor activity mediated by polymer-coated viruses, we investigated how the polymer-coated virus infected the tumor cells. First, we observed the entry of polymer-coated virus into WiDr cells by transmission electron microcopy (TEM) examination. Numerous viral particles bound to the plasma membrane (Figure 5b), but only a few virus particles (black arrowhead) were observed after infection with the naked virus (Figure 5a). We also investigated entry of the polymer-coated virus into A549 cells, and similar results were observed (Supplementary Figure S5). Second, we tested whether anti-CD46 antibodies could block CPEs mediated by the naked virus or the polymer-coated virus. HEp2 cell monolayers were inoculated with naked virus or polymer-coated virus in the presence or absence of anti-CD46 Abs. Anti-CD46 antibodies blocked oncolysis mediated by the naked virus, though the difference was not statistically significant. Anti-CD46 antibodies also blocked polymer-coated virus-mediated oncolysis modestly (Figure 6).
Figure 5.
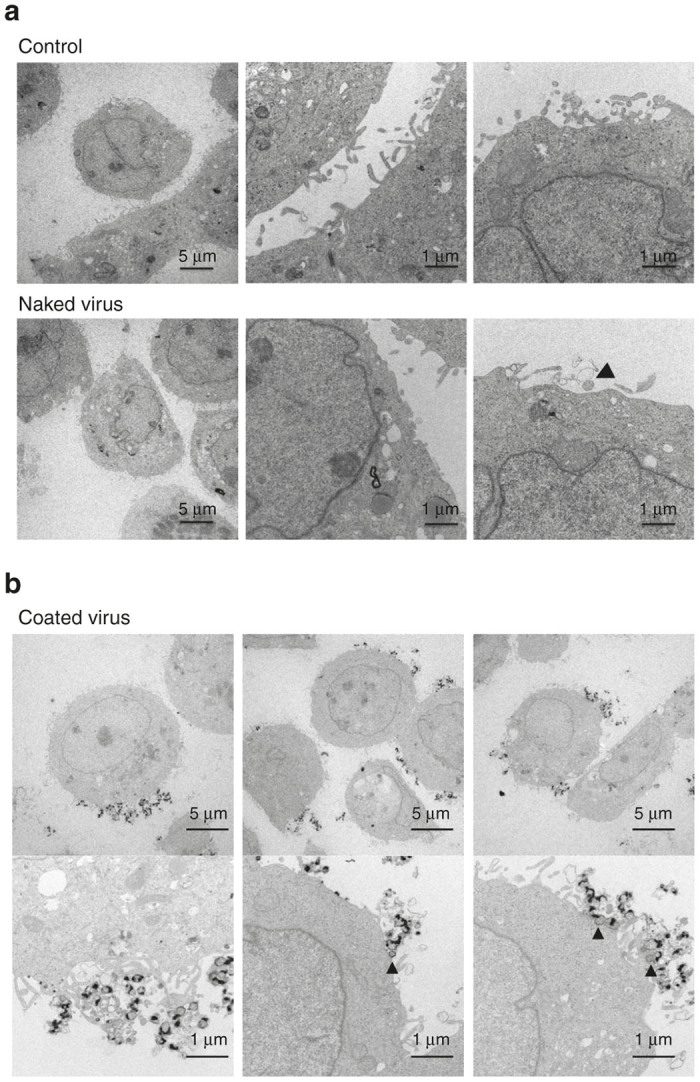
Electron microscopic images of human cancer cells after viral infection. WiDr cells or A549 cells were administered with Tris-HCl (control), naked virus, or polymer-coated virus (coated virus) at an MOI of 1 for 1 hour and incubated for another 12 hours. Black arrow heads and white arrow heads show virus particles. (a) WiDr cells after infection with control or the naked virus. (b) WiDr cells infected with the coated virus.
Figure 6.
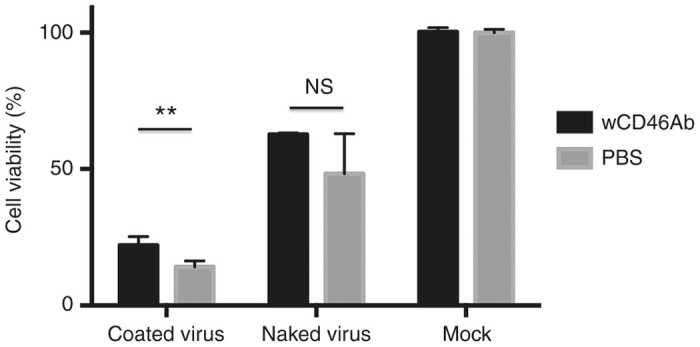
Blocking the effect of anti-CD46 antibodies on virus treatment. HEp2 cells were infected with naked virus or polymer-coated virus (coated virus) in the presence or absence of anti-CD46 antibodies. At 2 days postinfection, cell viabilities were measured by performing crystal violet assays. *P < 0.05; **P < 0.01.
Discussion
Several studies using measles virus infected carrier cells to evade or overcome the inhibitory effects of antiviral antibodies have already been reported.22–26 A clinical trial testing mesenchymal stem cell delivery of measles virus is ongoing (NCT02068794). The other strategy to evade antiviral immunity is surface modification of viruses. A variety of polymers have been proposed to shield the surface of therapeutic virus vectors and enabled the virus to remain longer period in the blood circulation even in the presence of the antivirus antibody.27–29 “Shielding” with the other virus protein is also one of the strategies to escape from anti-MV neutralization.30 Nevertheless, although polymer-coated viruses could successfully evade host’s immune systems in vitro and in vivo and be located inside the tumors by enhanced permeability and retention (EPR) effects, their ability to infect tumor cells is often decreased.31 Our results demonstrated that the polymer-coated MV-NPL had higher oncolytic activity in the presence of anti-MV neutralizing antibodies compared with naked MV-NPL, both in vitro and in vivo. Here, we report for the first time, to our knowledge, several novel findings regarding the combined use of oncolytic MV and the drug-delivery system of polymer-coated viruses. First, we demonstrated that polymer coating itself enhanced MV-NPL-mediated oncolysis. Although we cannot clearly explain, TEM examination suggested that polymer-coated viruses easily and strongly bound to the membrane of cancer cells than noncoated viruses. This binding might have helped polymer-coated virus to have enhanced antitumor efficacy. Second, the polymer-coated virus induced effectively antitumor immunity. Previous preclinical and clinical data suggested that in some cases virotherapy induced anticancer immunity.2,3,32–35 CDC activities were studied in archival blood samples from patients who received Pexa-Vec therapy in clinical trials.36,37 In our experiments, the sera from mice injected with polymer-coated virus showed higher CDC activity than those injected with the naked virus. The mechanism of higher CDC activity after coated virus treatment remains unclear. Not only tumor-specific antibodies but also virus-specific antibodies or nonspecific antibodies might be related to induction of CDC activities. Previous reports showed that the released danger signals and dead tumor cells after oncolytic virus infection induced immune responses, such as induction of antibody-mediated cytotoxicity via tumor-specific cytotoxic T lymphocytes (CTLs).32 In our experiment to assess CTL activities, splenocytes in mice injected with the naked virus or the polymer-coated virus showed no cytotoxic effects on LL/2-CD46 cells (data not shown). Lack of MHC Class I expression on LL/2-CD46 (data not shown), namely their non-immunogenic phenotype, possibly resulted in the lack of CTL-activity induction. Third, anti-CD46 antibodies modestly blocked polymer-coated virus-mediated oncolysis. On the other hand, anti-CD44 antibodies did not block polymer-coated virus-mediated oncolysis (data not shown). TEM examination showed engrafted virus particles (white arrow head) after polymer-coated virus infection (Supplementary Figure S5). The result suggested another mechanism (potential use of alternative MV receptors (nectin-4 or SLAM) or nonspecific transduction) may be involved in the cellular uptake of polymer-coated virus, other than CD46 receptor-mediated cell entry.
Several important issues should be clarified. First, whereas we are planning to initiate a preclinical trial employing intratumoral delivery because of the safety and convenience, intravenous administration is strongly preferred for the treatment of advanced metastatic cancers. The efficacy of virotherapy by intravenous administration is often decreased in the presence of neutralizing antibodies, further preclinical study in vitro and in vivo are needed to assess the efficacy of intravenous administration using the polymer-coated virus. Second, using the current production method, concentrated polymer-coated viral stocks were difficult to obtain because of the formation of aggregates. For clinical use of this newly developed coated MV in cancer treatment, the development of large-scale virus production and virus purification is required. We have already developed a stable method for large-scale virus production and virus purification using liquid chromatography (http://mhlw-grants.niph.go.jp/niph/search/NIDD00.do?resrchNum=201307017A). We expect our newly developed highly purification virus method would enable us to produce high titer polymer-coated virus without formation of aggregate. Third, the safety of polymer-coated virus remains unclear. Engineered MV has also been tested in clinical trials against cutaneous T-cell lymphoma, multiple myeloma, ovarian cancer, malignant mesothelioma, and glioblastoma multiforme. Dose-limiting toxicity was not observed to date in these trials.5,6 To confirm the safety of polymer-coated virus, a preclinical dose-escalation study in nonhuman primates is now underway (http://mhlw-grants.niph.go.jp/niph/search/NIDD00.do?resrchNum=201307017A).
In summary, polymer-coated MV-NPL showed enhanced oncolytic activity even in the presence of anti-MV neutralizing antibodies in vitro and in vivo, compared with the naked MV-NPL. This novel polymer-coated MV-NPL shows promise for cancer therapy in the future.
Materials and Methods
Cell culture
The human A549 lung cancer cell line, the human MDA-MB-231 breast cancer cell line, the mouse LL/2 lung cancer cell line, and the African green monkey Vero cell line were obtained from the American Type Culture Collection (Rockville, MD). The human WiDr colon cancer cell line was obtained from the Japanese Collection of Research Bioresources (JCRB, Tokyo, Japan). The human HEp2 laryngeal cancer cell line was kindly provided by Dr. Hiroyuki Shimizu (National Institute of Infectious Diseases, Tokyo, Japan). All cells were cultured in Dulbecco’s modified Eagle’s medium, supplemented with 10% fetal bovine serum and 1% antibiotics. Cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2.
Gene transduction
Human cDNA encoding CD46 was amplified from the KPON-57 acute lymphoblastic lymphoma cell line38 by the reverse transcription-polymerase chain reaction, using a forward primer (5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTGCCGCGCATGGAGCCTCCCGGCCGCCGC-3′) and a reverse primer (5′-GGGGACCACTTTCTACAAGAAAGCTGGGTTAATAGAGTCAAAAGATGAACTGGCAAA-3′). The amplicon was inserted into the pDONR222 entry vector (Invitrogen, Carlsbad, CA) by performing a BP recombinase reaction with GatewayBP Clonase (Invitrogen). The CD46 gene was inserted into the CSII-EF-RfA plasmid vector (kindly gifted from Dr. Hiroyuki Miyoshi, Riken, Tsukuba, Japan) by performing an LR recombinase reaction with GatewayLR Clonase (Invitrogen). The plasmid vector was cotransfected into 293T cells, as previously described.39 The supernatant was collected 72 hours after transfection, filtered (0.45 µm), concentrated by high-speed centrifugation, and frozen at −80 °C until use. LL/2 cells expressing CD46 were generated by transducing these cells with a lentiviral vector expressing CD46.
Flow cytometry
Cells were harvested, washed in phosphate-buffered saline containing 2% bovine serum albumin, and incubated with a phycoerythrin-labeled mouse anti-human CD46 antibody (eBioscience, San Diego, CA). A phycoerythrin-labeled mouse IgG1 (BioLegend, San Diego, CA) was used for CD46 staining as an isotype control according to the manufacturer’s instructions. The washed cells were analyzed on a Becton Dickinson FACScan Plus flow cytometer (Becton Dickinson, Mountain View, CA). Analysis was performed using CellQuest software (BD Bioscience, Heidelberg, Germany).
Virus preparation and titering
MV-NPL was generated as described previously.14 Briefly, MV-NPL was amplified by first infecting Vero cells grown in 15-cm dishes at 95% confluence (MOI = 0.01) and incubating them for 1 hour at 37 °C. Fresh media were added and the cells were further incubated at 32 °C. After complete CPE was observed (almost 72 hours postinfection), cell lysates were harvested. Viruses were harvested by three repeated freeze-thaw cycles and separated from the cells by a 15-minute centrifugation at 3,000 rotations/minute. Virus titers were determined by performing serial-dilution assays, as follows. Serial dilutions (10-fold) of virus samples were prepared in OPTI-MEM, and 50 µl of each dilution was used to infect eight replicate wells containing 5 × 103 Vero cells in 100 µl DMEM with 10% fetal bovine serum. Five days after the infection, the wells were scored for the presence of more than 50% cell death, and the log10 TCID50 was calculated by the method of Karber.40
Polymer coating
Linear polyethyleneimine hydrochloride (PEI Max; MW 40,000) was obtained from Polyscience (Warrington, UK). CS (MW 10,000) was gifted from Seikagaku Corporation (Tokyo, Japan). The preparation of polymer-coated MV-NPL was performed as described previously.21 MV-NPL particles were suspended in aqueous 20-mmol/l Tris-HCl solution. To obtain MV-NPL/PEI complexes, the PEI solution was added to the virus suspension (2.5 × 104 TCID50) and the suspension was vortexed. Next, CS solution was added to MV-NPL/PEI (0.5 µg) complexes to obtain MV-NPL/PEI/CS complexes, and the resultant suspension was vortexed. Zeta potentials and sizes were measured with a Zetasizer Nano ZS particle analyzer (Malvern Instruments, Malvern, UK).
Crystal violet assay
Cells were plated at a density of 1 × 104 or 2 × 104 cells/well in 96-well plates. Cells were infected with naked virus (MV-NPL) or polymer-coated virus (MV-NPL/PEI/CS), with or without 100-fold diluted anti-MV polyclonal antibodies (Kaketsu-Ken, Tokyo, Japan), of which anti-MV neutralizing titer was twice. After 2 to 5 days, cells were fixed and stained with 0.5% crystal violet in 50% methanol at room temperature, washed with water, and air-dried. Cell viabilities were measured as previously described.41 The remaining cells were solubilized by adding 100 µl of 1% SDS, and the optical density (OD) at 570 nm was measured in multi-well plate reader.
Anti-MV neutralizing assay
Vero cells were seeded at a density of 5 × 103 cells per well in 96-well plates. Anti-MV polyclonal antibodies or heat-inactivated mouse serum were diluted in phosphate-buffered saline using twofold serial dilutions, and 100 TCID50/25 µl MV-NPL was added to each well. The cultures were maintained at 37 °C for 5 days, after which CPEs were measured. The neutralizing titer was calculated as the reciprocal of the highest dilution at which no CPE was noted in any of the replicate wells.
In vivo study
The experimental protocol was approved by the Institutional Animal Care and Use Committee at Kyushu University. In immune-deficient mouse models, 1 × 106 HEp2 cells, 3 × 106 A549 cells, or 3 × 106 WiDr cells were injected subcutaneously in the right flanks of female Balb-c/nu/nu mice. When the tumor size reached 3 to 5 mm, on average by 2 days post-tumor inoculation, the mice were assigned to the following three groups: group I, control (Tris-HCl); group II, naked virus (2.5 × 105 TCID50); and group III, polymer-coated virus (2.5 × 105 TCID50). The viruses were intratumorally injected once per week. Tumor sizes were measured using a caliper three times per week, and volumes were calculated using the following formula: (L × W × W)/2, where L equals the tumor length and W equals the tumor width. Animals with large tumors (length >10 mm) were sacrificed for ethical reasons, and this was recorded as the date of death for survival studies.
In the immune-competent mouse model, C57BL/6N mice were immunized by subcutaneous injection of MV-NPL (1 × 106 TCID50) 3 weeks before testing. LL/2-CD46 cells (2 × 105) were inoculated subcutaneously in the right flanks of female C57BL/6N mice. When the tumor sizes reached 3 to 5 mm in diameter (4 to 5 days post-tumor inoculation), the mice were randomized to three to four groups. Protocol A was performed with three groups as described above. Protocol B involved the following four groups: group I, control (Tris-HCl); group II, polymer-coated virus 1 × 105 TCID50; group III, polymer-coated virus 2.5 × 105 TCID50; and group IV, polymer-coated virus 5 × 105 TCID50. The viruses were diluted in 100 µl Tris-HCl and injected intratumorally once a week. Tumor sizes were measured as described above. Animals with large tumors (length >10 mm) were sacrificed as discussed above.
CDC assays
CDC assays were conducted as described previously.37 Briefly, mice sera were collected at day 7 (if terminated at day 14) or day 14. LL/2-CD46 cells were seeded into 96-well plates and subsequently incubated with eightfold diluted serum samples at 37 °C for 4 hours. Tumor cell viabilities were determined using an MTS Cell Proliferation Assay Kit, following the manufacturer’s protocol (Promega, Madison, WI) after 4 hours of coculture. Cells were subsequently washed in phosphate-buffered saline and incubated with 100 µl of fresh medium and 20 µl of MTS solution at 37 °C for 1 hour. Cell viabilities were measured by reading the OD at 490 nm. Cell viability following exposure to serum from MV-administrated mice was normalized to cell viability following exposure to serum from control mice (set as 100%).
Electron microscopy
Electron-microscopy examinations were performed as previously described.33 WiDr or A549 cells were infected with naked virus or polymer-coated virus at an MOI of 1 for 1 hour and cultured for 12 hours. Viral particles and target cells were fixed with 2% glutaraldehyde and scraped. Carrier and target cells were further fixed with 2% buffered-osmium tetroxide for 1 hour and embedded in Epon epoxy resin (Electron Microscopy Sciences, Ft Washington, PA). Thin sections were stained with uranyl acetate and lead citrate and examined. Electron micrographs were taken on a Tecnai 20 200-kV transmission electron microscope (Philips, Hillsboro, OR).
Antibody-blocking assay
Monolayer HEp2 cells were inoculated with naked or polymer-coated virus at a MOI of 0.5 in the presence or absence of phycoerythrin-labeled anti-human CD46 antibodies (eBioscience, San Diego, CA). Two days after beginning infection, the cells were stained with crystal violet. Cell viability was measured by performing a crystal violet assay, as described above.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA). Statistical analysis was conducted using a two-tailed unpaired Student’s t-test. Spearman’s correlation tests were used to determine the strength of relationships between CDC activities and tumor volumes or survival times. Survival curves were plotted according to the Kaplan–Meier method, and survival times of the injected groups were compared using the log-rank test. P < 0.05 was considered statistically significant.
Acknowledgments
The authors thank Michiyo Okada and Michiko Ushijima (Kyushu University) for providing technical assistance. The authors also appreciate the technical support provided by members of the Research Support Center at the Graduate School of Medical Sciences (Kyushu University). This work was supported by a grant from the Ministry of Health Labour and Welfare (Grant No. 23080101) of Japan.
K.T. has minor stock option rights with Oncolys BioPhama Inc. No potential conflicts are disclosed by the other authors.
References
- Beljanski, V and Hiscott, J (2012). The use of oncolytic viruses to overcome lung cancer drug resistance. Curr Opin Virol 2: 629–635. [DOI] [PubMed] [Google Scholar]
- Gauvrit, A, Brandler, S, Sapede-Peroz, C, Boisgerault, N, Tangy, F and Gregoire, M (2008). Measles virus induces oncolysis of mesothelioma cells and allows dendritic cells to cross-prime tumor-specific CD8 response. Cancer Res 68: 4882–4892. [DOI] [PubMed] [Google Scholar]
- Greiner, S, Humrich, JY, Thuman, P, Sauter, B, Schuler, G and Jenne, L (2006). The highly attenuated vaccinia virus strain modified virus Ankara induces apoptosis in melanoma cells and allows bystander dendritic cells to generate a potent anti-tumoral immunity. Clin Exp Immunol 146: 344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, SJ, Peng, KW and Bell, JC (2012). Oncolytic virotherapy. Nat Biotechnol 30: 658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanis, E, Hartmann, LC, Cliby, WA, Long, HJ, Peethambaram, PP, Barrette, BA et al. (2010). Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res 70: 875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Msaouel, P, Opyrchal, M and Galanis, E (2011). Translational research in oncolytic measles virotherapy: early discoveries and future steps. Future Microbiol 6: 125–128. [DOI] [PubMed] [Google Scholar]
- Lievano, F, Galea, SA, Thornton, M, Wiedmann, RT, Manoff, SB, Tran, TN et al. (2012). Measles, mumps, and rubella virus vaccine (M-M-R™II): a review of 32 years of clinical and postmarketing experience. Vaccine 30: 6918–6926. [DOI] [PubMed] [Google Scholar]
- Parrula, C, Fernandez, SA, Zimmerman, B, Lairmore, M and Niewiesk, S (2011). Measles virotherapy in a mouse model of adult T-cell leukaemia/lymphoma. J Gen Virol 92(Pt 6): 1458–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, KW, Ahmann, GJ, Pham, L, Greipp, PR, Cattaneo, R and Russell, SJ (2001). Systemic therapy of myeloma xenografts by an attenuated measles virus. Blood 98: 2002–2007. [DOI] [PubMed] [Google Scholar]
- Penheiter, AR, Wegman, TR, Classic, KL, Dingli, D, Bender, CE, Russell, SJ et al. (2010). Sodium iodide symporter (NIS)-mediated radiovirotherapy for pancreatic cancer. AJR Am J Roentgenol 195: 341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuong, LK, Allen, C, Peng, KW, Giannini, C, Greiner, S, TenEyck, CJ et al. (2003). Use of a vaccine strain of measles virus genetically engineered to produce carcinoembryonic antigen as a novel therapeutic agent against glioblastoma multiforme. Cancer Res 63: 2462–2469. [PubMed] [Google Scholar]
- Iankov, ID, Msaouel, P, Allen, C, Federspiel, MJ, Bulur, PA, Dietz, AB et al. (2010). Demonstration of anti-tumor activity of oncolytic measles virus strains in a malignant pleural effusion breast cancer model. Breast Cancer Res Treat 122: 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly, OG, Errington-Mais, F, Steele, L, Hadac, E, Jennings, V, Scott, K et al. (2013). Measles virus causes immunogenic cell death in human melanoma. Gene Ther 20: 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, X, Nakamura, T, Okazaki, T, Inoue, H, Takahashi, A, Miyamoto, S et al. (2010). Enhanced antitumor effects of an engineered measles virus Edmonston strain expressing the wild-type N, P, L genes on human renal cell carcinoma. Mol Ther 18: 544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H, Peng, KW, Dingli, D, Kratzke, RA and Russell, SJ (2010). Oncolytic measles viruses encoding interferon beta and the thyroidal sodium iodide symporter gene for mesothelioma virotherapy. Cancer Gene Ther 17: 550–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutzen, B, Pierson, CR, Russell, SJ, Galanis, E, Raffel, C and Studebaker, AW (2012). Treatment of medulloblastoma using an oncolytic measles virus encoding the thyroidal sodium iodide symporter shows enhanced efficacy with radioiodine. BMC Cancer 12: 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisgerault, N, Guillerme, JB, Pouliquen, D, Mesel-Lemoine, M, Achard, C, Combredet, C et al. (2013). Natural oncolytic activity of live-attenuated measles virus against human lung and colorectal adenocarcinomas. Biomed Res Int 2013: 387362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, SC, Wang, WL, Cai, WS, Jiang, KL and Yuan, ZW (2012). Engineered measles virus Edmonston strain used as a novel oncolytic viral system against human hepatoblastoma. BMC Cancer 12: 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, KW, TenEyck, CJ, Galanis, E, Kalli, KR, Hartmann, LC and Russell, SJ (2002). Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res 62: 4656–4662. [PubMed] [Google Scholar]
- Kreppel, F and Kochanek, S (2008). Modification of adenovirus gene transfer vectors with synthetic polymers: a scientific review and technical guide. Mol Ther 16: 16–29. [DOI] [PubMed] [Google Scholar]
- Yoshihara, C, Hamada, K and Koyama, Y (2010). Preparation of a novel adenovirus formulation with artificial envelope of multilayer polymer-coatings: therapeutic effect on metastatic ovarian cancer. Oncol Rep 23: 733–738. [PubMed] [Google Scholar]
- Ong, HT, Hasegawa, K, Dietz, AB, Russell, SJ and Peng, KW (2007). Evaluation of T cells as carriers for systemic measles virotherapy in the presence of antiviral antibodies. Gene Ther 14: 324–333. [DOI] [PubMed] [Google Scholar]
- Mader, EK, Maeyama, Y, Lin, Y, Butler, GW, Russell, HM, Galanis, E et al. (2009). Mesenchymal stem cell carriers protect oncolytic measles viruses from antibody neutralization in an orthotopic ovarian cancer therapy model. Clin Cancer Res 15: 7246–7255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, KW, Dogan, A, Vrana, J, Liu, C, Ong, HT, Kumar, S et al. (2009). Tumor-associated macrophages infiltrate plasmacytomas and can serve as cell carriers for oncolytic measles virotherapy of disseminated myeloma. Am J Hematol 84: 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C, Russell, SJ and Peng, KW (2010). Systemic therapy of disseminated myeloma in passively immunized mice using measles virus-infected cell carriers. Mol Ther 18: 1155–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castleton, A, Dey, A, Beaton, B, Patel, B, Aucher, A, Davis, DM et al. (2014). Human mesenchymal stromal cells deliver systemic oncolytic measles virus to treat acute lymphoblastic leukemia in the presence of humoral immunity. Blood 123: 1327–1335. [DOI] [PubMed] [Google Scholar]
- Morrison, J, Briggs, SS, Green, N, Fisher, K, Subr, V, Ulbrich, K et al. (2008). Virotherapy of ovarian cancer with polymer-cloaked adenovirus retargeted to the epidermal growth factor receptor. Mol Ther 16: 244–251. [DOI] [PubMed] [Google Scholar]
- Kim, J, Kim, PH, Kim, SW and Yun, CO (2012). Enhancing the therapeutic efficacy of adenovirus in combination with biomaterials. Biomaterials 33: 1838–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesfay, MZ, Kirk, AC, Hadac, EM, Griesmann, GE, Federspiel, MJ, Barber, GN et al. (2013). PEGylation of vesicular stomatitis virus extends virus persistence in blood circulation of passively immunized mice. J Virol 87: 3752–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miest, TS, Yaiw, KC, Frenzke, M, Lampe, J, Hudacek, AW, Springfeld, C et al. (2011). Envelope-chimeric entry-targeted measles virus escapes neutralization and achieves oncolysis. Mol Ther 19: 1813–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subr, V, Kostka, L, Selby-Milic, T, Fisher, K, Ulbrich, K, Seymour, LW et al. (2009). Coating of adenovirus type 5 with polymers containing quaternary amines prevents binding to blood components. J Control Release 135: 152–158. [DOI] [PubMed] [Google Scholar]
- Inoue, H and Tani, K (2014). Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ 21: 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada, K, Desaki, J, Nakagawa, K, Zhang, T, Shirakawa, T, Gotoh, A et al. (2007). Carrier cell-mediated delivery of a replication-competent adenovirus for cancer gene therapy. Mol Ther 15: 1121–1128. [DOI] [PubMed] [Google Scholar]
- Liu, BL, Robinson, M, Han, ZQ, Branston, RH, English, C, Reay, P et al. (2003). ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther 10: 292–303. [DOI] [PubMed] [Google Scholar]
- Thorne, SH, Hwang, TH, O’Gorman, WE, Bartlett, DL, Sei, S, Kanji, F et al. (2007). Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963. J Clin Invest 117: 3350–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo, J, Reid, T, Ruo, L, Breitbach, CJ, Rose, S, Bloomston, M et al. (2013). Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med 19: 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, MK, Breitbach, CJ, Moon, A, Heo, J, Lee, YK, Cho, M et al. (2013). Oncolytic and immunotherapeutic vaccinia induces antibody-mediated complement-dependent cancer cell lysis in humans. Sci Transl Med 5: 185ra63. [DOI] [PubMed] [Google Scholar]
- Inukai, T, Sugita, K, Mitsui, K, Iijima, K, Goi, K, Tezuka, T et al. (2000). Participation of granulocyte colony-stimulating factor in the growth regulation of leukemia cells from Philadelphia chromosome-positive acute leukemia and blast crisis of chronic myeloid leukemia. Leukemia 14: 1386–1395. [DOI] [PubMed] [Google Scholar]
- Kurita, R, Sasaki, E, Yokoo, T, Hiroyama, T, Takasugi, K, Imoto, H et al. (2006). Tal1/Scl gene transduction using a lentiviral vector stimulates highly efficient hematopoietic cell differentiation from common marmoset (Callithrix jacchus) embryonic stem cells. Stem Cells 24: 2014–2022. [DOI] [PubMed] [Google Scholar]
- Hamilton, MA, Russo, RC and Thurston, RV (1977). Trimmed Spearman-Karber method for estimating median lethal concentrations in toxicity bioassays. Environ Sci Technol 11: 714–719. [Google Scholar]
- Miyazaki, T, Futaki, S, Suemori, H, Taniguchi, Y, Yamada, M, Kawasaki, M et al. (2012). Laminin E8 fragments support efficient adhesion and expansion of dissociated human pluripotent stem cells. Nat Commun 3: 1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.