

Abstract
We compared apparent origins, cellular diversity and regulation of initial axon growth for differentiating cranial sensory neurons. We assessed the molecular and cellular composition of the developing olfactory and otic placodes, and cranial sensory ganglia to evaluate contributions of ectodermal placode versus neural crest at each site. Special sensory neuron populations—the olfactory and otic placodes—as well as those in vestibulo-acoustic ganglion are entirely populated with cells expressing cranial placode-associated, rather than neural crest-associated markers. The remaining cranial sensory ganglia are a mosaic of cells that express placode-associated as well as neural crest-associated markers. We found two distinct populations of neural crest in the cranial ganglia: the first, as expected, is labeled by Wnt1:Cre mediated recombination. The second is not labeled by Wnt1:Cre recombination, and expresses both Sox10 and FoxD3. These populations—Wnt1:Cre recombined, and Sox10/Foxd3-expressing— are proliferatively distinct from one another. Together, the two neural crest-associated populations are substantially more proliferative than their placode-associated counterparts. Nevertheless, the apparently placodal and crest-associated populations are similarly sensitive to altered signaling that compromises cranial morphogenesis and differentiation. Acute disruption of either Fibroblast growth factor (Fgf) or Retinoic acid (RA) signaling alters axon growth and cell death, but does not preferentially target any of the three distinct populations. Apparently, mosaic derivation and diversity of precursors and early differentiating neurons, modulated uniformly by local signals, supports early cranial sensory neuron differentiation and growth.
Keywords: cranial sensory neurons, placodes, neural crest, Six1, Wnt1:Cre, Retinoic acid, Fgf, olfactory, otic, cranial sensory ganglia
INTRODUCTION
There is substantial variation of vertebrate craniofacial morphologies (1, 2); however, cranial sensory neuron position and identity is remarkably constant across most species: the olfactory placode/epithelium (OE) is most anterior, followed by the trigeminal (ganglion of Cranial Nerve V), geniculate (gCN VII) and vestibulo-acoustic (gCN VIII) ganglia, otic placode/vesicle (OV), the petrosal/superior (gCN IX) and nodose/jugular complex (gCN X). Despite this morphological stability it is still not known whether the anlagen of these cranial sensory populations—all derived from either the ectodermal placodes or the neural crest (3, 4)—initially share uniform molecular and cellular identities or have variable identities based upon their anterior-posterior (A-P) cranial positions. Thus, we asked whether there is uniformity or diversity among sensory neurons or their precursors as cranial sensory structures first become identifiable in the developing mouse head at mid-gestation.
Fate mapping studies over the last several decades have established that both the cranial placodes and neural crest contribute to the cranial sensory ganglia (3-5). Nevertheless, the quantitative contribution of each population to each sensory site remains unknown. Indeed, there is evidence that cranial placode and neural crest cells share a common progenitor field at early neural plate stages (5), making ultimate distinctions of derivation less certain. Moreover, the OE and OV have been considered to be exclusively placodal in origin; however, recent reports indicate that neural crest cells also contribute to varying degrees (6-8). Quantitatively distinct contributions of placode and crest may prefigure distinct sensory neuron differentiation along the A-P axis. Alternately, the early cellular constituents may be fairly uniform, and subsequent differentiation may rely upon local signals that yield divergent fates. Accordingly, we used established molecular markers and genetic approaches (9, 10) to classify and quantify placode-associated and neural crest-associated cells at each site of initial cranial sensory neuron differentiation.
A key aspect of early sensory neuron differentiation is the stereotyped trajectories of the earliest axons that extend from post-mitotic neurons at each cranial sensory site. These early axons define the primary branches of what will become a much more complicated plexus for each cranial nerve. It is well established that local signals, often produced by either placodal or cranial mesenchymal cells adjacent to each sensory site influence early cranial nerve development. In particular, Fgfs, often from placodal sites (11), and RA, primarily associated with neural crest-derived mesenchyme (12), modulate multiple aspects of sensory neuron differentiation including cranial sensory neuron survival, neurogenesis and cranial nerve differentiation. Most of the studies of the effects of these signals have focused on phenotypes in constitutively mutated mice where ligands, receptors or co-factors are non-functional from conception onward (13-17). In contrast, there are no observations, to our knowledge, that characterize the effects of these signals for sensory neuron differentiation once cranial sensory sites are established and axon outgrowth begins. Accordingly, we used acute pharmacological disruption of Fgf signal transduction or RA synthesis to assess whether these signals preferentially target distinct cell classes and whether they selectively alter axon growth.
Our data show that the OP, gCN VIII, and OV have molecular signatures consistent with primarily placodal origins, while the remaining ganglia have remarkably consistent proportions of cells with placodal and neural crest-associated molecular signatures. There are two distinct populations of neural crest-associated cells in all of the ganglia: those identified via Wnt1:Cre recombination, and those that express additional neural crest markers Sox10 and Foxd3, but are not Wnt1:Cre recombined. Disrupting Fgf or RA signaling has substantial and distinct effects on gene expression, axon outgrowth, and cell survival after the initial coalescence of sensory structures. These effects, however, are not selective for placode- versus crest-associated populations of sensory precursors or neurons. Thus, we found a molecular and cellular mosaic that defines cranial sensory precursors and neurons at midgestation—there is quantitative uniformity of placodal and neural crest-associated cells, with diversity in the responses of these cells to local signals that influence further differentiation.
MATERIALS AND METHODS
Animals
The George Washington (GW) University Animal Research Facility maintained breeding colonies of C57/BL6 (Charles River) mice carrying Wnt1:Cre and nuclear eGFP reporter alleles (Jackson). We used CD1 mice (Charles River) for microdissection for quantitative PCR (qPCR) and whole mount immuno-labeling in WT and pharmacological treatments. For proliferation analysis, BrdU (50mg/kg body weight) was injected into timed pregnant mice at E10.5. Embryos were harvested 2 hours after labeling. Standard BrdU immunolabeling techniques were used after acid treatment for antigen retrieval. Timed-pregnant females (vaginal plug =E0.5) were sacrificed at E9.0 or E10.5 by rapid cervical dislocation and embryos collected for analysis. The GW Institutional Animal Care and Use Committee approved all procedures.
Pharmacological Treatments
Timed pregnant dams (Wnt1:Cre::eGFP) were injected intraperitoneally at E9.5 with 50 mg/kg PD173074 (FgfR-I; Selleckchem), an Fgf receptor 1 and related Fgf receptors inhibitor or 100 mg/kg Di-ethyl amino benzadine (DEAB; Sigma), an RA synthesis inhibitor as described previously (18). 24 hours later, embryos were harvested for histology, qPCR whole mount immuno-labeling or acute BrdU labeling (injection 2 hours prior to collection).
Microdissection
Embryos were harvested at E10.5, and stage confirmed based upon olfactory placode differentiation, which at E10.5 is laterally placed on the head and not deeply invaginated. Embryos were collected in ice-cold RNAase-free Dulbecco's phosphate buffered saline (DPBS). Some cranial sensory ganglia (gCN VII/VIII and gCN IX/X) are not easily separable at this age, thus we dissected “ganglion complexes”. Embryos were bisected and each half trans-illuminated to visualize ganglia beneath the translucent cranial epidermis. Each structure was isolated using a micro-scalpel, and when possible the epidermis was removed. These dissected ganglia were transferred to fresh DPBS and pooled (four to six ganglia from at least 3 embryos/litter) for RNA extraction and qPCR analysis. The OE and OV were dissected from separate litters. The olfactory placodes were cut from the side of the head, and the cranial region that includes the OV was isolated. Samples were transferred to cold 1.25% pancreatin (Sigma) and 1.25 % trypsin (GIBCO) in high Ca++ medium (L15) for 30 minutes as described previously (12). Enzymatic digestion was stopped with 10% heat inactivated horse serum in L15. The OE and OV were separated from adjacent mesenchyme and epidermis using fine tungsten needles, and samples were pooled as above.
RNA isolation, cDNA synthesis and qPCR
RNA was prepared as described previously (19). qPCR was performed using a Bio-Rad CFX384 Real-Time PCR detection system. Gene-specific primers, when possible, spanned genomic intron-exon boundaries, generated amplicons between 250 and 350 bp, and were validated by melt-curve analysis (qPCR primers, Table S1). Each qPCR sample for analysis of cranial sensory neuron transcription factor expression levels consists of RNA made from a pool of 4-6 individual ganglia or ectodermal samples (OE and OV). For each transcription factor, 3-8 such pools were analyzed for each condition (WT, FgfR-I or DEAB-treated).
Quantitative and statistical analysis of expression levels
To assess relative levels of gene expression, we determined the average expression (deltaCT) across all structures and then subtracted that average from expression values in each. For comparisons of gCN V, gCN VII/VIII and gCN IX/X from WT, FgfR-I or DEAB-treated embryos, the average of WT gCN V, gCN VII/VIII and gCN IX/X mean values are subtracted from mean values of FGFRi- and DEAB-treated gCN V, gCN VII/VIII and gCN IX/X. For WT and FGFRi- or DEAB-treated OE and OV, the mean expression values of the 22 genes from E10.5 whole embryo cDNA was subtracted from each mean value of WT, FGFRi- or DEAB-treated OE or OV samples. These values were analyzed based upon highest to lowest relative expression. Student's t-tests were used to assess differences between dCT expression values in WT and treated samples.
Immunohistochemistry and microscopy
E10.5 embryos were prepared for cryomicrotmy and antibody labeling as described previously (30). Primary antibodies include mouse anti-βIII Tubulin (TuJ1; Covance, 1:1000), rabbit anti-Six1 (Proteintech, 1:1500), chicken anti-GFP (Abcam1:1000), mouse anti-NeuN (Merck Millipore, 1:1000), mouse anti-HuC/D (16A11, Life Technologies, 1:1000), Mouse anti-BrdU (BD Biosciences, 1:100),goat anti-Sox10 (Santa Cruz Biotechnology, 1:50), mouse anti-Foxd3 (Thermo Scientific, 1:400), rabbit anti-Six4 (Proteintech Group, 1:100), mouse anti-Six1 (Atlas Antibodies, 1:200), mouse anti-Brn3a (Millipore, 1:100), mouse anti-p75 (Chemicon, 1:1000), rabbit anti-TrkA (Santa Cruz Biotechnology, 1:100), rabbit anti-TrkB (Santa Cruz Biotechnology, 1:100) and rabbit anti-cleaved Caspase 3 (Cell Signalling, 1:200). Primary antibody labeling was visualized with Alexafluor 488-, 546-, or 647- conjugated secondary antibodies (Molecular Probes, 1:2000 for 546 and 647, and 1:4000 for 488). Images were collected using a Leica Tiling microscope, or a Zeiss 710 confocal microscope.
Cell counting and analysis
Images of each ganglion or ganglion complex were collected at 20X objective magnification. Higher magnification images of dual labeled cells were collected at 40X objective magnification. The red (Six1), green (Wnt1:Cre::eGFP), blue (DAPI) channels were visualized separately and superimposed as composite images. Labeled cells were counted using video-overlay and point counting (Adobe Photoshop). Double-labeled cells were counted based on clear overlap when dual channels were analyzed. Single labeled DAPI cells were counted by visualizing all channels. We determined Six1 and eGFP “only” numbers by subtracting the number of double-labeled cells from single labeled cells of the related cell classes. Mean values between WT and treated ganglia were compared statistically using Mann-Whitney tests, and multiple comparison errors were adjusted using the Bonferoni correction. To determine proportions of proliferative or differentiated cells of each category, total number of cells positive for BrdU or NeuN were counted, then checked for overlap with Six1, GFP, Six1+GFP or DAPI and the percentages of each were determined.
Whole-mount immunohistochemistry
E10.5 embryos were prepared for whole-mount immunolabeling as described previously (20). They were incubated in anti-βIII Tubulin antibody at 4°C for 3 days, then washed in PBST repeatedly and incubated overnight in HRP-conjugated goat anti-mouse antibody (GE Healthcare Life Sciences; 1:500). Following DAB/NiCl2 visualization, embryos were dehydrated, cleared with benzyl alcohol: benzyl benzoate (BABB) and imaged using a Leica Wild M420 photomacroscope.
Quantitative assessment of altered gangliogenesis and axon growth
Pairs of images of whole-mount labeled CNs and gCNs were evaluated, blind to treatment, by five independent observers. The identity of these pairs was randomized so that no observer compared the same pairing of WT and treated structures. Images were randomized in three ways: treated images and WT images were paired randomly; left or right position was randomized, and the order of the pairs was randomized for each observer. Scoring criteria were as follows: for CN V, a score of “0” was assigned if the ganglion appeared to match WT examples, and “1” if the ganglion appeared changed from the WT state. The scores from all 5 independent observers were then collated and mean phenotypic scores determined. A mean score of 0.8 or above identified an individual nerve/ganglion as altered. Fisher's exact test was used to identify statistically significant changes in treated and untreated groups.
RESULTS
Diverse molecular identities of cranial sensory neurons and precursors
Sensory neurons in the OE, gCN V, gCN VII, gCN VIII, OV, gCN IX, and gCN X emerge at sites where cranial placodal ectoderm is directly adjacent (OE, OV) or intermixed (cranial sensory ganglia) with neural crest derived cells (3, 4, 12, 21, 22). In the mouse, the sensory epithelia or ganglia have coalesced, and substantial initial neuronal differentiation and axon outgrowth is seen at all sites by E10.5 (Figure 1, left). Labeling of cells and axons in each cranial sensory structure by the early neuronal marker βIII Tubulin suggests extensive initial differentiation of neurons at this stage. The processes of these neurons extend into the periphery as well as into the hindbrain (Figure 1, left). We used immunolabeling for the transcription factor Six1 to identify putative placode–associated cells (23, 24), and Wnt1:Cre dependent recombination (9) of a floxed nuclear eGFP reporter to identify putative neural crest associated cells (Figure 1, middle). At E10.5, Six1-expressing, presumed placode-associated cells fully populate the OE, OV and gCN VIII, whereas a combination of placode- and neural crest-associated cells constitutes the remaining cranial sensory ganglia. Differentiating neurons, recognized by expression of HuC/D (labels at all sites) and/or NeuN (only labels gCN V and VII), are found at this time (Figure 1, right). We identified neurons in both the Six1 and Wnt1:Cre labeled populations in all cranial sensory structures, although Six1-expressing cells appeared to be labeled more frequently for these neuronal markers (see below).
Figure 1.

Identity and differentiation in placode and neural crest associated cranial sensory structures. Left: Sites of cranial sensory neurons along the anterior-posterior (A-P) axis of the E10.5 mouse embryo head. This section, transverse to the A-P distribution of cranial sensory neuron sites shows that each region: the olfactory epithelium (OE), trigeminal ganglion (gCN V), geniculate ganglion (gCN VII), acoustic ganglion and otic vesicle (gCN VIII and OV), and petrosal and nodose ganglia (gCN IX and X) is a site of early neuronal differentiation and process outgrowth, based upon labeling with βIII Tubulin (cyan). Center: Immunolabeling with antisera against Six1 and for eGFP expressed based upon Wnt1:Cre recombination. The OE is completely labeled by Six1 (red), with no eGFP labeled cells (green) detected. There are many recombined eGFP labeled cells in the frontonasal mesenchyme (fnm). The medial fnm has both Six1 and eGFP labeled cells, while the lateral fnm has primarily eGFP labeled cells. gCN V, gCN VII, gCN IX and gCN X are all composed of randomly intermixed Six1 and eGFP labeled cells. In contrast, g CN VIII and the otic vesicle (OV), which gives rise to auditory/vestibular hair cells, consist primarily of Six1 labeled cells. Right: Labeling for diagnostic markers of further neuronal differentiation, HuC-D and NeuN. identity shows that both Six1 (red) and Wnt1:Cre recombined eGFP (green) expressing cells differentiate as neurons. We used the neuronal marker Hu-C/D (blue)(an RNA binding protein; REF) to label neuronal nuclei in the OE and gCN IX/X, and NeuN (blue) to label neuronal nuclei in gCN V, VII, and VIII. There are double- or triple-labeled Hu-C/D or NeuN cells for Six1 and in each structure (panels at far right, adjacent to lower magnification images of g CN V, g CN VII/VIII, g CN IX/X). In each example we present the multiple labeled cell or cells at the top, and then color separations for Hu C-D or NeuN labeling (blue), Wnt1:Cre recombination (green), or Six1 expression (red).
We next asked whether there was a consistent quantitative relationship between the contribution of putative placode- and neural crest-associated cells to early differentiating cranial sensory neuron populations. With few exceptions—single cells in a few sections in a minority of embryos—we saw only Six1-labeled cells in the OE and OV as well as gCN VIII at E10.5 (Figure 1, middle, Figure 2, left); therefore, we did not quantify proportions of placode- versus neural crest-associated cells in these structures. In the remaining cranial sensory ganglia, there are robust proportions of Six1-expressing and Wnt1:Cre recombined cells: 23 ± 2.8% (SEM; n=8) of gCN V cells express Six1, 37 ± 1.6% are Wnt1:Cre recombined, and 7 ± 0.8% are labeled for both markers (Figure 2, left and middle). Surprisingly, 33% ± 2.8% of gCN V cells are labeled neither by Six1 or Wnt1:Cre recombination. These cells, recognized using the nuclear DNA marker DAPI (Figure 2, right), are distributed throughout each ganglion, intermixed with both Six1 and Wnt1:Cre labeled populations. In gCN VII, similar proportions of Six1, Wnt1:Cre, and “DAPI-only” cells are seen: 43 ± 2.1% (SEM, n=7) are Six1-labeled, 34 ± 2.1% are Wnt1:Cre recombined, 1 ± 0.2% of the Six1 and Wnt1:Cre populations are dual-labeled, and 22 ± 2.2% of gCN VII cells are not labeled by either marker. The proportions of the distinct cell classes in the gCN IX/X complex are similar to gCN V: 18 ± 2.8% (SEM; n=8) Six1 labeled, 43 ± 3.8% Wnt1:Cre labeled, 4 ± 1.2% dual-labeled, and 35 ± 5.1% not labeled by either marker. Statistical analysis (p> 0.05; Mann Whitney; Bonferoni correction) confirms that there are no significant differences in the proportion of these cells in each of the cranial ganglia. Thus, A-P position does not influence the placode versus neural crest constituency of cranial ganglia with dual origins.
Figure 2.

Stable proportions of placodal, neural crest-derived and non-labeled cells in the cranial sensory ganglia. Left: Six1 labeling in all structures at higher magnification. The OE is populated entirely with Six1 labeled cells. There is a slight tendency for Six1 cells in gCN V and VII to be seen in more lateral (left) regions of the ganglia, but this bias is not absolute. Percentages of Six1 labeled cells (red bars), and double labeled cells (yellow bars, and insets; yellow nuclei) as well as SEMs are given in the bar histogram beneath each panel. Center: Wnt1:Cre recombined cells, labeled with a nuclear-localized eGFP reporter are largely excluded from the OE, but are quite frequent in g CN V, VII, and IX/X. Wnt1:Cre recombined cells are largely excluded from g CN VIII and the OV. The green bars in the histograms beneath each panel indicate the percentages of these cells (±SEM). Right: A substantial population of cells in the cranial ganglia, but not the OE, gCN VIII, or OV, are labeled neither by Six1 or Wnt1:Cre. These cells, recognized by nuclear labeling with the DNA dye DAPI (blue), can be easily distinguished from violet cells (Six1 labeled), turquoise cells (eGFP labeled), and cells whose nuclei appear white (Six1/eGFP double labeled). They appear randomly distributed in each ganglion. The blue bars in the histograms indicate the percentages of these cells (±SEM).
To evaluate the possibility that the quantitatively consistent “DAPI-only” population represents cells that at an earlier time express Six1 and then down-regulate the protein—suggesting a placode-association for the unlabeled cells at E10.5—we analyzed proportions of the three populations in the coalescing gCN V at E9.0, the earliest stage in mouse when gCN V is identifiable as a coherent ganglion (Figure 2, bottom row). We found approximately similar proportions of the three cell classes at E9.0 and E10.5: At E9.0, 34%± 1% (SEM, n=6) are Six1-labeled, 30%±1% are Wnt1:Cre recombined, and 31%±1% are DAPI-only. The proportions of Six1, Wnt1:Cre recombined and DAPI only cells at E9.0 are statistically indistinguishable (p> 0.05; Mann-Whitney) from those in gCN V at E10.5. Our results, therefore, define a consistent, quantitatively robust population of cells labeled by neither Six1 nor Wnt1:Cre recombination that constitute a substantial fraction of cranial sensory ganglion cells.
Placode versus neural crest signatures of distinct cranial sensory cell classes
We next asked whether the three quantitatively stable populations in the cranial ganglia: Six1-labeled cells, Wnt1:Cre-recombined cells, and the previously undetected “DAPI-only” population, have additional distinct molecular signatures that distinguish their likely origin and identity. We assessed expression of an additional placode-associated marker, Six4 (25), and found nearly complete overlap between Six1 and Six4-labeled populations (Figure 3, top left). Previous work suggests that placode-derived cranial sensory neurons express Brn3a earlier than neural crest-derived cells (26, 27). We found that nearly all Six1 labeled cells in gCN V express Brn3A at E10.5 (Figure 3, middle left). Of these cells, a large proportion (48%± 2%) also expresses NeuN (Figure 3, lower left). In contrast, very few Wnt1:Cre recombined or DAPI-only cells in gCN V express Brn3a at E10.5, and the proportion expressing NeuN is similarly low (Figure 3, graph at left). These distinctions were also seen in gCN VII and gCN IX/X (not shown) indicating that the patterns are not unique to specific ganglia. Thus, based upon multiple molecular and cellular characteristics, the vast majority of Six1 expressing cells appear to be placode-associated, and are distinct from the Wnt1:Cre recombined and “DAPI-only” populations.
Figure 3.
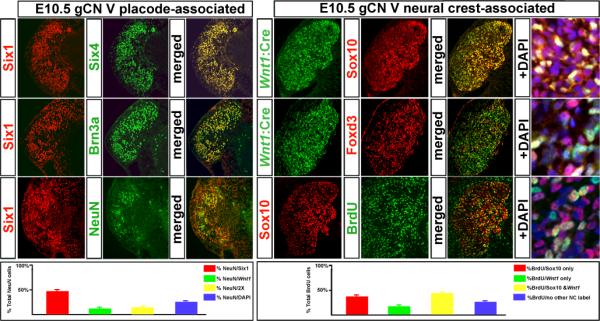
Placode versus neural crest signatures of distinct cranial sensory cell classes. The data presented here focuses on the g CN V; however, similar observations were made in the additional cranial ganglia. Left: The identity and early neuronal differentiation of cells expressing placode-associated markers. At left, cells immunolabeled with Six1 antisera (left panel, left column) to identify placode-associated cells were double-labeled with Six4 (middle column, top) or Brn3a (middle column, center), or the neuronal marker NeuN (middle column, bottom). Both Six4 and Brn3a show nearly complete identity with the Six1-lableled cells (right column, merged). Many of the NeuN labeled cells are co-labeled with Six1 (right column, bottom). At bottom left, a quantitative summary of placode-associated versus neural crest-associated labeling of g CN V cells. Right: distinct, neural crest-associated molecular signatures of Wnt1:Cre recombined and “DAPI-only” cells in g CN V. There is some overlap in the Wnt1:Cre recombined and Sox10-expressing cells (top row); however it is far from complete. In addition to the Wnt1:Cre/Sox10 labeled cells (yellow) there are several Sox10/DAPI-only cells (violet) as well as some cells that are labeled for neither neural crest-associated marker (far right, top). There is even less overlap between the Wnt1:Cre population and the Foxd3 labeled cells (middle row). There are relatively few Wnt1:Cre recombined cells that are labeled by Foxd3 (yellow; far right); however, the DAPI-only cells labeled by Foxd3 (are fairly frequent (violet; far right). BrdU is incorporated in Sox10-expressing cells (bottom panels). The overlap is extensive (yellow cells at far right); however, there also are non-labeled cells that are labeled by BrdU. At bottom, a quantitative summary of BrdU labeling seen in cells that express neural crest versus non-neural crest markers.
We next asked whether the Wnt1:Cre recombined and DAPI-only populations had molecular and cellular identities that distinguish them as related or separate cell classes associated with the neural crest. We found that the DAPI-only cells also express Sox10 and Foxd3 (Figure 3, right), both of which are selectively associated with the neural crest (28-30). As expected (28-31), a substantial proportion of Wnt1:Cre recombined cells were also labeled for Sox10 (60%±3%, SEM, n=5). A slightly lower proportion of the DAPI-only cells also express Sox10 (47%±2%), while a very small subset of Six1 expressing cells express Sox10 (8%±1%). Each of these values is statistically distinct from the others (p<0.5; Mann-Whitney). Finally, we determined whether the neural crest-associated populations are more proliferative, based upon comparison with their Six1 counterparts that generate neurons by this time. Cells expressing all neural crest-associated markers in gCN V are significantly more frequently labeled acutely by BrdU (74%±2% of the total BrdU population; SEM, n=5) than those expressing neither marker (26%%±2% SEM; Figure 3, graph at right). Within the neural crest-associated populations, however, there are distinctions in proliferative activity. The frequency of BrdU/Sox10-only expressing cells (38% ± 3%; SEM, n=5) is nearly twice that of BrdU/Wnt1:Cre recombined-only cells (17%± 2%; Figure 3, graph at right), a statistically significant difference (p < 0.009; Mann Whitney). Finally, the highest frequency of BrdU labeling is seen in Wnt1:Cre recombined cells that also express Sox10 (47% ± 4%), which is both significantly greater than the Wnt1:Cre-only/BrdU population (p<0.01) and the Sox10-only/BrdU population (p< 0.05). Thus, based upon our proliferation analysis, there are at least four classes of cells in the ganglion at E10.5: modestly proliferating placode-associated cells, and three subsets of highly, but differentially, proliferative neural crest-associated cells.
Signaling pathways that influence cranial sensory neuron differentiation
Cranial placode-and neural crest-associated cells—including sensory neurons and their progenitors as well as non-neural craniofacial structures—are well known targets of morphogenetic signals in the developing head (32-40). Two of these signals, Fgf and RA, can modify cranial placode (41) and neural crest (42, 43) cells. Most studies addressing cranial Fgf or RA signaling in the mouse have relied upon use of constitutive genetic mutants, which have concatenated phenotypes from initial early embryogenesis onward. These include extreme craniofacial disruptions that make clear interpretation of sensory neuron changes nearly impossible (44). To avoid this complication, we asked whether acute pharmacological disruption of signaling via Fgfs (45-48) or RA (32, 49-51) between E9.5 and E10.5, have differential influences on the Six1-presumed placode-associated, Wnt1:Cre recombined or “DAPI-only”/Sox10/Foxd3 neural crest-associated populations.
In response to acute Fgf inhibition (FgfR-I) at E9.5, all five cranial sensory neuron sites are noticeably smaller by E10.5 (Figure 4, left; compare with Figures 1, 3). The OE and OV are dysmorphic; nevertheless, both are still composed nearly entirely of Six1-labeled cells (Figure 4). The consequences of acute RA inhibition (DEAB) are less dramatic. The OE is slightly less invaginated and the OV slightly smaller; however, both still express Six1 exclusively (Figure 4). To determine whether disrupted Fgf or RA signaling had selective effects, we assessed proportions of Six1, Wnt1:Cre recombined and DAPI–only cells that remain in the apparently smaller cranial sensory ganglia after FgfR-I (Figure 4, left) or DEAB treatment (Figure 4, right). We found no statistically significant differences in the proportions of any of these cell classes in response to diminished Fgf or RA signaling, compared to WT values (Mann-Whitney; Bonferoni correction for multiple comparisons). Apparently, although FgfR-I and DEAB disrupts Fgf and RA signaling at each site where sensory neuron precursors and neurons are found, this disruption does not selectively compromise the proportions of any of the three distinct sub-populations that we have defined in the cranial sensory ganglia.
Figure 4.

Consequences of FgfR-I or DEAB treatment at E9.5 for cranial sensory neuron differentiation and frequency of placode (Six1; red) and neural crest (Wnt1:Cre recombined; green) cells. The overall size of the ganglia is diminished by the treatments. The proportions of each cell class, including unlabeled (DAPI cells) are shown in the histograms below each panel. We find no disproportionate statistically significant changes for any cell class compared to the WT distribution (see Figure 2) in response to either FgfR-I or DEAB.
Transcriptional changes in response to altered Fgf and RA signaling
Fgf and RA signaling are key regulators of transcription factor expression in neural progenitors, as well as in differentiating and mature neurons. Accordingly, we asked whether expression of key transcription factors in cranial sensory populations along the anterior-posterior (A-P) axis was altered systematically at cranial sensory sites in response to FgfR-I or DEAB. We selected 22 transcription factors whose activity has been associated with cranial sensory neuron genesis or differentiation based on expression as well as consequences of loss or gain of function (27, 52-65).
We first asked whether any of these transcription factors are expressed in statistically verifiable A-P gradients in non-treated OE, OV, and cranial sensory ganglia using micro-dissected samples and qPCR. Since both Fgf and RA gradients are known to influence A-P patterning in the hindbrain and neural crest (66, 67), it was surprising that none of the 22 transcription factor genes were expressed so that their A-P ranks were either in ascending or descending order across all 5 structures, nor were any expressed at constant levels in all five structures (Supplemental Figure 1). To insure that we had not overlooked subtle systematic relationships, we performed regression analyses to assess statistically whether any of the expression levels were distributed in an approximate A-P linear gradient across all five sites; we found none. We also performed this analysis excluding the OV, because of low expression levels of most of the 22 transcription factor genes we selected; nevertheless, we found no linear gradients. The initial formation of placode and neural crest precursors of the cranial sensory neurons are influenced by A-P morphogenetic gradients; nevertheless, transcription factor expression patterns no longer reflect or predict A-P position by the time that these cells have started to differentiate into neurons.
We next asked whether FgfR-I and DEAB treatment alters gene expression levels at any of the five cranial sensory neuron sites. We found that inhibiting Fgf signaling caused extensive, statistically significant changes of transcription factor expression levels (Figure 5). Only one gene, however, Dll1, changes with a consistent magnitude across all five sites. There is substantial diversity in magnitude and direction of changes for the remaining genes, and no systematic relationships between these changes and A-P position were found. Thus, although acute pharmacological inhibition of Fgf signaling has substantial effects on transcription factor expression at sites of cranial sensory neuron differentiation, there are few consistent changes across all five cranial sensory sites. DEAB treatment has more modest effects on expression levels for these 22 transcription factor genes at these sites (Figure 5). There are five genes for which acute RA inhibition has an opposite effect on expression compared to acute Fgf inhibition, consistent with the Fgf/RA antagonism reported for many non-neural regions of the developing embryo (68-72). Thus, for 22 cranial sensory precursor or neuron-associated transcription factors, in response to acute inhibition of RA synthesis and signaling, there is no consistent, gradient-based change in expression of RA-sensitive genes in developing cranial sensory neurons at E10.5.
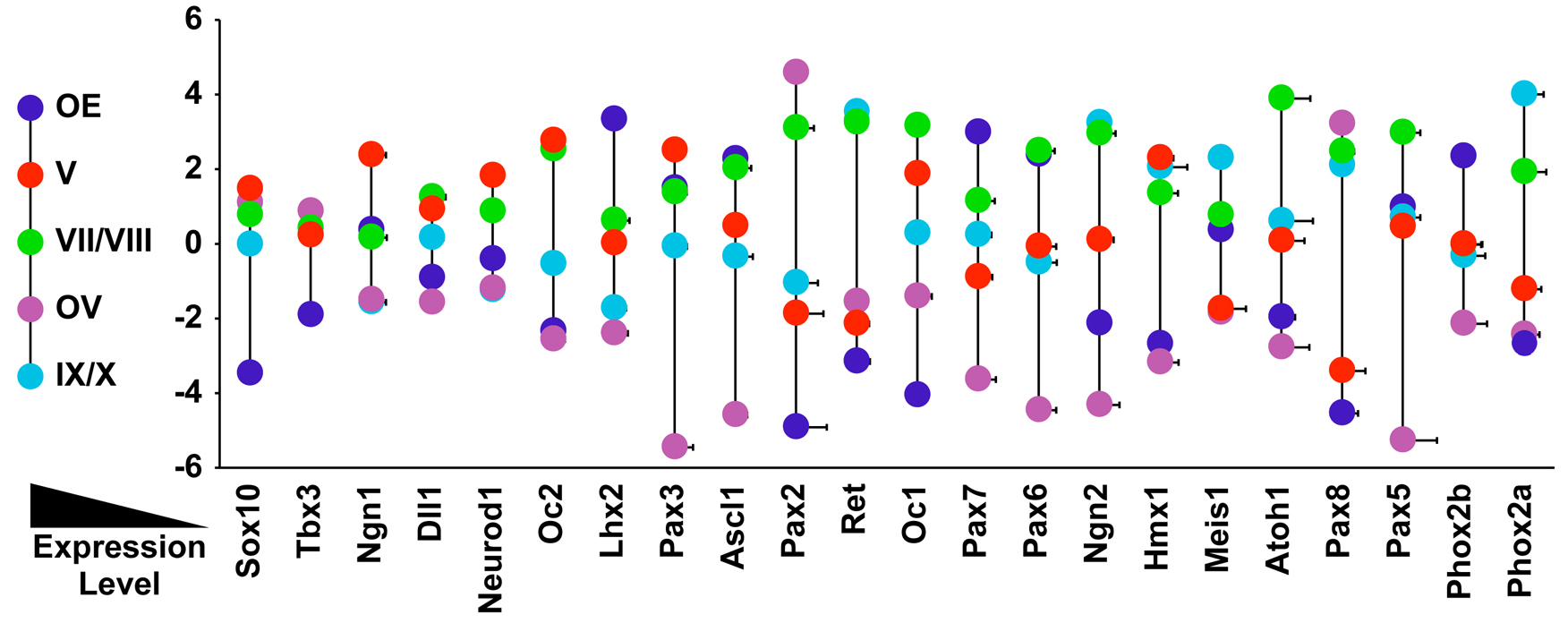
Figure 5.

Change in transcription factor expression in response to acutely diminished Fgf or RA signaling. Quantitative changes of 22 cranial sensory neuron-associated transcription factors at each of the enzymatically isolated/micro-dissected sensory neuron sites in response to FgfR-I (left side of each gray box) or DEAB (right side of each gray box) treatment. Arrows indicate direction of qPCR changes, and the sizes of the arrows are proportionate to the fold change of each altered gene. Genes are presented in order of highest to lowest expression in gCN V. The gray boxes without any arrows indicate the subset of genes whose expression did not change in response to acutely diminished Fgf or RA signaling.
Fgf and RA Signaling Influences Cranial Sensory Axon Outgrowth
Previous reports show that craniofacial morphogenesis as well as cranial nerve differentiation can be compromised by disruption of Fgf or RA signaling (71, 72). The morphological and transcriptional changes we have seen could result in altered outgrowth and targeting of cranial nerve axons, due to dysmorphic peripheral targets or direct effects on differentiating neurons. Alternately, they could be confined to diminished cell proliferation or enhanced cell death—making structures smaller, with little change in the basic trajectories or targets of cranial nerve axons. To distinguish these possibilities, we visualized cranial nerves in whole E10.5 WT, FgfR-I and DEAB treated embryos.
E9.5 FgfR-I treatment results in in craniofacial dysmorphogenesis at E10.5. These changes are consistent with, but less severe than, those caused by constitutive mutations in Fgf signaling genes (14, 15). The medial and lateral frontonasal processes are not identifiable, and the branchial arches are substantially smaller (Figure 6). In parallel, the position, growth, and targeting of cranial nerves is disrupted. The most dramatic disruptions in response to FgfR-I are seen in CNs I and V. CN VII/VIII are less compromised, and there are only modest changes in CN IX/X (Figure 6, 7). CN I axons do not make a characteristic medial turn as they approach the forebrain, nor do they enter (Figure 6, lower middle). gCN V is small and dysmorphic; it lacks an identifiable maxillary division and the ophthalmic and mandibular branches are diminished. The distance between gCN V and gCN VII, which are immediately adjacent in WT embryos (Figure 6, left), is substantially increased. gCN VII and gCN VIII are smaller; however, CN VII retains a rudimentary branching pattern similar to WT (Figure 6, middle). gCN IX and X are in approximately normal positions, and their axons are oriented appropriately. Modest changes in gCN IX/X primarily reflect anomalous axon fascicles between the two ganglia or diminished size. Thus, acute disruption of Fgf signaling modifies the trajectories of cranial nerve axons particularly in the most anterior cranial nerves (CN I, CN V).
Figure 6.

A-P distinctions in disrupted sensory ganglion differentiation and axon growth in response to acutely diminished Fgf and RA signaling. Whole E10.5 normal (wild type “WT”), FgfR-I and DEAB treated embryos labeled for βIII tubulin (see also Figure 1) to show cranial ganglia and axons. Top panels: Lateral views of cranial nerves III, V, VII, VIII, IX and X are shown for WT (left), FgfR-I treated (middle) and DEAB treated (right) E10.5 embryos. The ophthalmic (op), maxillary (mx) and mandibular (md) branches of CN V are indicated. The forebrain (fb) is labeled at the anterior of the embryo. The dual headed arrow in the middle panel indicates increased distance between gCN V and gCN VII. Arrows at gCN V in the FgfR-I treated embryo, versus those at gCN IX/X in the DEAB treated embryo indicate the anterior bias of ganglion/axon disruption in response to FgfRI versus the posterior bias in response to DEAB. Bottom panels: Anterior views of CN I (OE) in dissected specimens. The lateral and medial frontonasal processes are indicated (lnp, mnp). The arrows summarize WT and treated olfactory nerve trajectories.
Figure 7.

Quantitative consistency of cranial nerve axon growth/ganglion morphogenesis changes in response to acutely diminished Fgf and RA signaling. Left: Higher magnification views of each cranial nerve/ganglion shown in Figure 7. Arrows indicate key changes scored by the 5 observers as described in Materials and Methods. Right: Histograms summarizing the numbers and percentages of CNs scored as altered by FgfR-I (left) or DEAB (right) treatment. The red bars at right in each histogram indicate the frequency of identifiable changes for each nerve for each set of treated embryos. The green bars indicate the small number of normal nerves scored as altered in a way consistent with DEAB treatment.
Craniofacial dysmorphogenesis in response to disrupted RA signaling is less severe, but still discernible, consistent with previous reports of mutations in RA signaling genes as well as teratogenic manipulations (73). The E10.5 embryos are slightly smaller, the eye is reduced in size, and the maxillary process is not as prominent as in WT embryos (Figure 6, right). The medial and lateral frontonasal processes are distinct; however, the degree of OE invagination is not as advanced as in the WT (Figures 4, 6, 7). In DEAB treated embryos, CN I axons make a medial turn as they exit into the mesenchyme; however, they continue medially and ventrally, rather than turning dorsally to enter the forebrain as in WT. Although smaller, gCN V has all three divisions, and its position immediately adjacent to gCN VII is not different from that in the WT. CN V axons seem to be sparse and less tightly fasiculated. gCN VII and gCN VIII are smaller than WT, and CN VII axons appear diminished, nevertheless, their branching pattern is not disrupted (Figure 6). The most substantial changes are seen for CN IX/X. gCN IX is displaced, hypotrophic, either fused with gCN X or completely separated, and it does not extend axons into the peripheral or central nervous system. Thus, acute disruption of RA signaling substantially changes cranial nerve axon growth at several sites, with the most dramatic changes seen posteriorly (CN IX and X).
To quantify the consistency and robustness of these changes across multiple embryos and litters, we imaged each cranial nerve in at least 10 and as many as 35 embryos from multiple litters of WT, Fgfr-I and DEAB treated embryos. Thus, we assessed between 20 and 70 individual cranial nerves at each site for each treatment, using a multi-observer, randomized “forced choice” approach (see Methods). We found remarkably consistent, statistically significant changes (Fisher's Exact, p < 3 ×10−10) across all of the CNs and ganglia in FgfR-I and DEAB treated embryos compared to WT (Figure 7). These changes do not approximate normal variation in WT embryos at this stage (20). Indeed, only one WT CN I and two WT CNs VII/VIII were scored as altered, similar to changes seen in DEAB-treated embryos. In contrast, after FgfR-I treatment, 95% of CN I, 95% of CN V, 98% of CN VII/VIII, and 83% of CN IX/X, are altered as described above. In response to acute RA inhibition, 90% of CN I, 57% of CN V, 72% of CN VII/VIII and 84% of CN IX/X are altered. Thus the “penetrance” of morphogenetic changes for each cranial nerve was robust and consistent, with FgfR-I most severely compromising anterior CNs, and DEAB altering posterior CNs.
Cellular correlates of altered cranial nerve differentiation
Changes in the sizes of cranial sensory neuron structures, levels of gene expression and extent of axon outgrowth in response to FgfR-I or DEAB treatments could all be the result of having fewer neurons differentiate at these sites. Both decreased cell proliferation, by diminishing cell and axon numbers, and increased cell death, by eliminating cells and thereby axons, could account for CN axon changes. Accordingly, we assessed the integrity of both of these processes at each cranial sensory site after acute inhibition of either Fgf or RA signaling.
We assessed proliferative activity in WT, FgfR-I treated and DEAB treated gCN V following BrdU labeling on E10.5, 2 hours prior to collecting the embryos for analysis. We found that this metric of cell proliferation was unchanged by either acute reduction of Fgf or RA signaling. Thus, For WT embryos, the proportion of BrdU labeled cells was 40% ± 2% (SEM, n=6), for FgfR-I, 40% ± 3% (n=5), and for DEAB, 43% ± 1% (n=6). These values are statistically indistinguishable (p> 0.05; Mann-Whitney). Based upon the proportion of S-phase labeled cells in gCN V, which is nevertheless compromised by both FgfR-I and DEAB treatment, it seems unlikely that there are major alterations in cell proliferation at sites of sensory neuron differentiation in response to acutely altered Fgf or RA signaling.
We next examined the extent of cell death based upon activated Caspase 3 labeling. In response to acutely diminished Fgf signaling, there is substantial enhancement of cell death (Figure 8, left). Enhanced frequency of activated caspase-labeled cells is seen in the frontonasal mesenchyme adjacent to the OE, in the lateral portion of gCN V, in the mesenchyme adjacent to the OV, but not in gCN IX/X. In contrast, acute disruption of RA signaling elicits a distinctly different pattern of cell death (Figure 8, left). No Caspase 3 labeled cells can be seen in the OE or frontonasal mesenchyme. Their presence, compared to controls, also appears diminished in gCN V as well as gCN VII/VIII, and adjacent mesenchyme. In contrast, there is a substantial enhancement of Caspase 3 labeled cells in gCN IX/X. These data indicate that acute disruption of Fgf and RA signaling modulates apoptotic cell death at sites of cranial sensory neuron differentiation. Diminished Fgf signaling enhances cell death more noticeably in anterior structures, whereas disrupted RA signaling enhances cell death more noticeably in posterior ganglia.
Figure 8.

A-P distinctions in apoptotic cellular responses, but not neurotrophin receptor expression changes in response to acutely diminished Fgf and RA signaling. Left: Activated Caspase 3 immunolabeled cells at WT, FgfR-I and DEAB treated cranial sensory neuron sites. There is an apparently consistent low frequency of caspase labeled cells, some of which overlap with Wnt1:Cre recombined cells and some that do not, in each of the structures. In response to FgfR-I, Caspase 3-labelled cells increase in frequency in the mesenchyme adjacent to the OE, in gCN V, and in the mesenchyme adjacent to the OV. There is little caspase 3 labeling in gCN IX/X in response to FgfR-I. In contrast, the frequency of caspase labeled cells in response to DEAB is little changed from the normal in the OE, gCN V, gCN VII/VIII and OV. There is noticeably increased frequency, however, in gCN IX/X. Center: qPCR analysis of neurotrophin receptor expression, associated with distinct classes of cranial sensory neurons in all five structures analyzed, in response to FgfR-I and DEAB. Both treatments cause increased expression of subsets of Trk receptors and p75 across all of the cranial sensory ganglia. Right: Immunolabeling for Trk receptor and p75 proteins in WT (left), FgfRI-treated (center), and DEAB-treated (right) in gCN V. The increased protein expression in treated samples compared to untreated samples is consistent with increased expression seen in mRNA levels by qPCR.
Finally, we asked whether expression levels of a key set of regulators of cranial sensory neuron differentiation, growth, and survival, the neurotrophin receptors p75, TrkA, B, and C, were altered by acute disruption of Fgf or RA signaling. We first assessed expression at the mRNA level in dissected OE, OV, and ganglia at E10.5. We found that levels of most of the Trks are increased in the three cranial sensory ganglion complexes in response to both FgfR-I and DEAB (Figure 8, middle). The magnitude of these increases varies between 1 and 3 fold. Only TrkA declines, and these change are limited to the OE and OV. To further assess these apparent changes, we evaluated p75 and Trk protein expression immunohistochemically. Comparison of protein expression in gCN V is consistent with the qPCR data showing increased expression of p75, TrkA, B and C, are increased in expression. The location of increased expression appears to differ for each protein: p75 is enriched at the margins of the ganglion, TrkA in a posterior region, and TrkB and C in the outer part of the ganglion, where Six1 and NeuN cells are also concentrated (see Figures 2 and 3). Thus, acute disruption of Fgf and RA signaling elicits substantial changes in cell death, and compensatory changes in neurotrophin signaling. The predominantly up-regulation of neurotrophin receptors may be a compensatory mechanism to maintain survival or growth in the context of diminished trophic support caused by disrupted Fgf or RA signaling.
DISCUSSION
The initial establishment of cranial sensory sites depends upon relatively stable contributions from placode-associated and distinct classes of neural crest-associated cells early in mid-gestation, accompanied by local signaling that likely modifies gene expression and differentiation in each distinct population. Our analysis of placode- and neural crest-associated populations in the OE, OV, and coalescing cranial ganglia identified a novel population of neural crest-associated cells. Neural crest-associated cells in g CN V, g CN VII and g CN IX/X can be divided into those that are Wnt1:Cre recombined, and those that are not, but express other neural crest markers including Sox10 and Foxd3, and are significantly more proliferative. The proportions of these cells that constitute g CN V, g CN VII and g CN IX/X are consistent and do not seem to be preferentially altered by acute manipulation of Fgf or RA signaling. Nevertheless, disrupting Fgf and RA signaling acutely at midgestation results in diverse changes in gene expression, axon outgrowth, cell survival and death at cranial sensory sites. Together, our results show that the initial derivations of cranial sensory neurons are fairly uniform based upon contributions of placode-associated and neural crest-associated cells. Once cranial sensory neurons have begun differentiating, local signaling modifies axon growth and cell survival at each site.
Analyzing initial cranial sensory neuron development
We combined several approaches to evaluate likely derivation, transcriptional state, and signaling in cranial sensory structures at midgestation in the mouse. To quantify likely placode derivation, we used established molecular markers, Six1 and Six4, which are selective for placode derivatives (23-25). To assess neural crest derivation we used an established genetic fate mapping method, via Wnt1:Cre recombination (9). Nevertheless, Six1 and Wnt1:Cre alone do not completely account for all of the cells in several nascent ganglia including gCN V, gCN VII, and g CN IX/X. Moreover, we found differences in Wnt1:Cre labeling based upon the reporter allele used—the nuclear GFP reporter shows little recombination in the OE and OV, while a cytoplasmic reporter detects some cells descended from Wnt1 expressing precursors in the OE and OV (7, 8). To define the likely identity of the non-Wnt1:Cre recombined cells, we used additional neural crest associated markers, Sox10 and Foxd3. The non-Six1/Six4 and non-Wnt1:Cre recombined express both markers at high frequency. Finally, we used assessment of proliferation based upon BrdU labeling to determine whether the Wnt1:Cre and non-Wnt1:Cre populations were distinct. We found that there are substantial, significant differences between each population, thus reinforcing the impression that each subset is a distinct contributor to the cranial ganglia. It is also clear that single markers of cranial sensory neuron or precursor identity are insufficient to definitively identify cell classes during midgestation in the mouse.
To assess how diversity is modulated in apparently fairly uniform populations of placodal and neural crest-associated cells in the cranial ganglia, we manipulated two local signals, Fgf and RA, known to influence cranial development, including cranial ganglion and nerve development. We acutely diminished signaling via Fgfs and RA pharmacologically rather than genetically. Our data are therefore not complicated by concatenation of earlier developmental disruptions, as in constitutive mutants, or incompleteness of recombination to inactivate Fgf or RA-associated genes. It would be impossible using current murine genetic approaches to diminish Fgf or RA signaling in all cranial sensory neuron sites at mid-gestation without dramatically compromising the entire developing head (16, 74). Thus, our pharmacological manipulations provide a temporally precise, reliable means of evaluating local signaling via Fgf and RA in the developing head between E9.5 and E10.5.
Two populations of neural crest-associated cells in the cranial ganglia
Previous studies have used Wnt1:Cre recombination to genetically identify dorsal/alar cells in the neural tube as neural crest that migrates peripherally, thus enabling the mapping of these cells from their point of origin in the CNS to their targets (9, 10, 75). We used this genetic approach expecting that all, or nearly all, of the neural crest cells that contribute to cranial sensory structures would be labeled by Wnt1:Cre, as suggested in the literature, while remaining cells would likely be labeled by markers associated with the cranial placodes. Our goal was to establish a clear quantitative accounting of the proportions of placode and neural crest-associated cells in the mouse embryo as cranial ganglia form, which to our knowledge had not been done previously. We confirmed a contribution of Wnt1:Cre recombined cells in gCN V, VII, IX/X (which in the mouse forms a complex, thus making further distinctions difficult) at consistent proportions at each site. We assumed that placode-associated markers would label the additional cells, thus indicating that, as argued previously by several investigators, placode contributions are dominant in the cranial sensory ganglia. Instead, the placode contribution, based upon labeling with Six1 and Six4, to gCN V, gCN VII and gCN IX/X was statistically indistinguishable from that of the Wnt1:Cre recombined presumed neural crest, leaving unaccounted for a substantial population of cells. These cells could have been either placode-associated or neural crest-associated cells not labeled by Wnt1:Cre recombination. They are indeed neural crest-associated based upon multiple markers and distinctions in proliferative activity. Thus, we have defined a previously unreported distinction in cranial neural crest that contributes to sensory ganglia: those associated with Wnt1 expression in the dorsal neural tube and those that are not.
We do not know the ultimate origin of the second population of neural crest, beyond its likely location outside of the zone of Wnt1 expression in the dorsal neural tube. Alternately, this novel population may be generated earlier than Wnt1 is expressed in the neural tube (76), distinguishing the two populations temporally, or it may arise from more laterally placed neural tube cells, consistent with arguments that the zone of neural crest generation is broader than previously thought (77). Moreover, we do not know if this novel population of neural crest cells has distinct fates. It is possible that these cells are biased toward generating glial constituents of the ganglia, based upon some of their molecular and cellular properties (29, 55). It also remains possible that they are not distinct in their potentials and fates, but represent a transient state that is primarily developmental in its significance. Resolving these questions will require additional embryological as well as genetic fate mapping studies. The consistency of proportions of each class of neural crest in the cranial ganglia indicates that there is likely to be fairly robust regulation of the epithelial/mesenchymal transitions for each population at the neural tube, and for the migration of each population in the developing head.
Morphogenetic signals and cranial sensory differentiation
It is fairly well established that during the earliest stages of vertebrate cranial patterning and morphogenesis, Fgf and RA signaling act in a graded and antagonistic manner to establish A-P organization (5, 78). Accordingly, we assumed that there would likely be evidence of continued A-P distinctions in cellular composition, transcription factor expression and differentiation in cranial sensory populations at mid-gestation that reflects the earlier patterning, especially that via Fgf and RA. Nevertheless, we found very little evidence of robust A-P relationships in cellular composition, quantitative levels of transcription factor expression, or graded A-P sensitivity to acutely altered Fgf or RA signaling for cranial sensory sites across the A-P axis at E10.5. Instead, by E10.5 each site has a distinct signature of expression and differentiation responses to altered Fgf and RA signaling, indicating that the early gradient while clearly essential for initial establishment of the A-P axis, is not maintained. Our results show that each site at mid-gestation responds uniquely to changes in Fgf and RA signaling, and that the changes include local alterations in transcription factor expression levels, ganglion morphogenesis, axon growth and cell death.
The axon trajectory changes we identified are in some cases similar to those reported previously for RA signaling pathway mutations (71). For the most part, however, they represent novel and robust alterations in differentiation due to acutely, rather than constitutively, disrupted signaling via each pathway. These changes are not only distinct for Fgf versus RA disruption, they also vary over the A-P array of cranial sensory sites so that anterior sites are more compromised by disrupted Fgf signaling, and posterior sites are more compromised by altered RA signaling. Moreover, the preferential changes in CN I, the maxillary branch of CN V CN IX in response to acute disruption of Fgf versus RA signaling indicate that there is selectivity for altering cranial nerve developmental response to these two signals. The targets for this selectivity remain to be identified: there may be direct, differential actions on specific populations of sensory neurons that constitute distinct nerve branches or nerves, or there may be distinct influences on target structures, leading secondarily to changes in axon growth and ganglion integrity. Our evidence indicating extensive changes in expression of neurotrophin receptors, which are established as key regulators of cranial sensory neuron development and axon growth (79), is consistent with changes in axon growth we report. The causes of these changes—either direct effects on sensory neurons or axons, or indirect responses of altered neurotrophin ligand expression in target structures—are not yet known. Together, the consequences of acute disruption of morphogenetic signaling on axon growth indicate that once a fairly uniform population of placode and/or neural crest derived cells has been established at each cranial sensory site, diversity in developmental programs may be executed locally based upon local activity of distinct signaling molecules.
Craniofacial and Cranial Sensory Development: Beyond A-P Patterning
One of the major challenges for understanding craniofacial developmental disruption is the apparent selectivity for many morphogenetic anomalies (80-84). Cranial morphogenesis and related cranial sensory neuron function can be independently altered at each of the sites we characterized. The functional consequences of such disruptions include altered sense of smell, reproductive difficulties due to lack of genesis or migration of GNRH neurons in the nascent olfactory placode (85); eye movements, hearing, feeding and swallowing, cardiovascular regulation, or gastric motility. Such changes can result in long term deficits in sensory capacity, growth and nutritional status. Our results suggest that restricted craniofacial dysfunction reflects local mechanisms that act upon relatively uniform populations of placode-associated and neural crest-associated cells at each cranial sensory site. The most severe consequences for sensory neuron development due to genetic or environmental alterations of these local mechanisms may be confined to a particular region or nerve, limiting deleterious effects in other critical cranial sensory neuron populations. Thus, our data provides two novel insights into clinically significant cranial sensory dysfunction. First, it likely reflects local rather than global disruption of cranial development, resulting in anosmia in some, eye movement disorders in others, hearing disruption in an additional subset, feeding and swallowing difficulties in many, and cardiovascular, respiratory or gastro-intestinal difficulties in additional individuals. Second, there may be mechanisms that preserve most local cranial sensory neuron differentiation across the A-P axis of the head in the context of selective mutational or environmental compromise of a specific cranial sensory neuron population.
Supplementary Material
{kind=link}
Highlights.
Cranial sensory neuron lineage or transcription factors do not follow A-P gradient
Disrupting Fgfs or RA signaling does alter these in an A-P gradient
Diminished Fgf enhances anterior cell death; diminished RA enhances it posteriorly
Diminished Fgf disrupts anterior nerves; diminished RA affects posterior ones
ACKNOWLEDGEMENTS
We thank Anastas Popratiloff for assistance with imaging. HD040677 supports the GW Microscopic Imaging and analysis core. DC011534 (A-S.L.), HD083157 (A-S.L. and SAM), and DE022065 (SAM) supported this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Brugmann SA, Kim J, Helms JA. Looking different: understanding diversity in facial form. American journal of medical genetics. Part A. 2006;140(23):2521–2529. doi: 10.1002/ajmg.a.31361. [DOI] [PubMed] [Google Scholar]
- 2.Young NM, et al. Embryonic bauplans and the developmental origins of facial diversity and constraint. Development. 2014;141(5):1059–1063. doi: 10.1242/dev.099994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.D'Amico-Martel A, Noden DM. Contributions of placodal and neural crest cells to avian cranial peripheral ganglia. The American journal of anatomy. 1983;166(4):445–468. doi: 10.1002/aja.1001660406. [DOI] [PubMed] [Google Scholar]
- 4.Ayer-Le Lievre CS, Le Douarin NM. The early development of cranial sensory ganglia and the potentialities of their component cells studied in quail-chick chimeras. Developmental biology. 1982;94(2):291–310. doi: 10.1016/0012-1606(82)90349-9. [DOI] [PubMed] [Google Scholar]
- 5.Saint-Jeannet JP, Moody SA. Establishing the pre-placodal region and breaking it into placodes with distinct identities. Developmental biology. 2014;389(1):13–27. doi: 10.1016/j.ydbio.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saxena A, Peng BN, Bronner ME. Sox10-dependent neural crest origin of olfactory microvillous neurons in zebrafish. eLife. 2013;2:e00336. doi: 10.7554/eLife.00336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Forni PE, Taylor-Burds C, Melvin VS, Williams T, Wray S. Neural crest and ectodermal cells intermix in the nasal placode to give rise to GnRH-1 neurons, sensory neurons, and olfactory ensheathing cells. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31(18):6915–6927. doi: 10.1523/JNEUROSCI.6087-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Freyer L, Aggarwal V, Morrow BE. Dual embryonic origin of the mammalian otic vesicle forming the inner ear. Development. 2011;138(24):5403–5414. doi: 10.1242/dev.069849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Current biology : CB. 1998;8(24):1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- 10.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127(8):1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- 11.Pirvola U, et al. FGF/FGFR-2(IIIb) signaling is essential for inner ear morphogenesis. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2000;20(16):6125–6134. doi: 10.1523/JNEUROSCI.20-16-06125.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LaMantia AS, Bhasin N, Rhodes K, Heemskerk J. Mesenchymal/epithelial induction mediates olfactory pathway formation. Neuron. 2000;28(2):411–425. doi: 10.1016/s0896-6273(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 13.Sun X, Meyers EN, Lewandoski M, Martin GR. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes & development. 1999;13(14):1834–1846. doi: 10.1101/gad.13.14.1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deng CX, et al. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes & development. 1994;8(24):3045–3057. doi: 10.1101/gad.8.24.3045. [DOI] [PubMed] [Google Scholar]
- 15.Feldman B, Poueymirou W, Papaioannou VE, DeChiara TM, Goldfarb M. Requirement of FGF-4 for postimplantation mouse development. Science. 1995;267(5195):246–249. doi: 10.1126/science.7809630. [DOI] [PubMed] [Google Scholar]
- 16.Niederreither K, Subbarayan V, Dolle P, Chambon P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nature genetics. 1999;21(4):444–448. doi: 10.1038/7788. [DOI] [PubMed] [Google Scholar]
- 17.Frank DU, et al. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development. 2002;129(19):4591–4603. doi: 10.1242/dev.129.19.4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maynard TM, et al. 22q11 Gene dosage establishes an adaptive range for sonic hedgehog and retinoic acid signaling during early development. Human molecular genetics. 2013;22(2):300–312. doi: 10.1093/hmg/dds429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maynard TM, et al. A comprehensive analysis of 22q11 gene expression in the developing and adult brain. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(24):14433–14438. doi: 10.1073/pnas.2235651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karpinski BA, et al. Dysphagia and disrupted cranial nerve development in a mouse model of DiGeorge (22q11) deletion syndrome. Disease models & mechanisms. 2014;7(2):245–257. doi: 10.1242/dmm.012484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D'Amico-Martel A. Temporal patterns of neurogenesis in avian cranial sensory and autonomic ganglia. The American journal of anatomy. 1982;163(4):351–372. doi: 10.1002/aja.1001630407. [DOI] [PubMed] [Google Scholar]
- 22.Montcouquiol M, Kelley MW. Planar and vertical signals control cellular differentiation and patterning in the mammalian cochlea. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2003;23(28):9469–9478. doi: 10.1523/JNEUROSCI.23-28-09469.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brugmann SA, Pandur PD, Kenyon KL, Pignoni F, Moody SA. Six1 promotes a placodal fate within the lateral neurogenic ectoderm by functioning as both a transcriptional activator and repressor. Development. 2004;131(23):5871–5881. doi: 10.1242/dev.01516. [DOI] [PubMed] [Google Scholar]
- 24.Schlosser G, Ahrens K. Molecular anatomy of placode development in Xenopus laevis. Developmental biology. 2004;271(2):439–466. doi: 10.1016/j.ydbio.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 25.Konishi Y, Ikeda K, Iwakura Y, Kawakami K. Six1 and Six4 promote survival of sensory neurons during early trigeminal gangliogenesis. Brain research. 2006;1116(1):93–102. doi: 10.1016/j.brainres.2006.07.103. [DOI] [PubMed] [Google Scholar]
- 26.Artinger KB, Fedtsova N, Rhee JM, Bronner-Fraser M, Turner E. Placodal origin of Brn-3-expressing cranial sensory neurons. Journal of neurobiology. 1998;36(4):572–585. doi: 10.1002/(sici)1097-4695(19980915)36:4<572::aid-neu10>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 27.Blentic A, Chambers D, Skinner A, Begbie J, Graham A. The formation of the cranial ganglia by placodally-derived sensory neuronal precursors. Molecular and cellular neurosciences. 2011;46(2):452–459. doi: 10.1016/j.mcn.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 28.Dottori M, Gross MK, Labosky P, Goulding M. The winged-helix transcription factor Foxd3 suppresses interneuron differentiation and promotes neural crest cell fate. Development. 2001;128(21):4127–4138. doi: 10.1242/dev.128.21.4127. [DOI] [PubMed] [Google Scholar]
- 29.Britsch S, et al. The transcription factor Sox10 is a key regulator of peripheral glial development. Genes & development. 2001;15(1):66–78. doi: 10.1101/gad.186601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Honore SM, Aybar MJ, Mayor R. Sox10 is required for the early development of the prospective neural crest in Xenopus embryos. Developmental biology. 2003;260(1):79–96. doi: 10.1016/s0012-1606(03)00247-1. [DOI] [PubMed] [Google Scholar]
- 31.Hari L, et al. Temporal control of neural crest lineage generation by Wnt/beta-catenin signaling. Development. 2012;139(12):2107–2117. doi: 10.1242/dev.073064. [DOI] [PubMed] [Google Scholar]
- 32.LaMantia AS, Colbert MC, Linney E. Retinoic acid induction and regional differentiation prefigure olfactory pathway formation in the mammalian forebrain. Neuron. 1993;10(6):1035–1048. doi: 10.1016/0896-6273(93)90052-s. [DOI] [PubMed] [Google Scholar]
- 33.Tucker ES, et al. Proliferative and transcriptional identity of distinct classes of neural precursors in the mammalian olfactory epithelium. Development. 2010;137(15):2471–2481. doi: 10.1242/dev.049718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Whitesides J, Hall M, Anchan R, LaMantia AS. Retinoid signaling distinguishes a subpopulation of olfactory receptor neurons in the developing and adult mouse. The Journal of comparative neurology. 1998;394(4):445–461. [PubMed] [Google Scholar]
- 35.Creuzet S, Schuler B, Couly G, Le Douarin NM. Reciprocal relationships between Fgf8 and neural crest cells in facial and forebrain development. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(14):4843–4847. doi: 10.1073/pnas.0400869101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lassiter RN, Reynolds SB, Marin KD, Mayo TF, Stark MR. FGF signaling is essential for ophthalmic trigeminal placode cell delamination and differentiation. Developmental dynamics : an official publication of the American Association of Anatomists. 2009;238(5):1073–1082. doi: 10.1002/dvdy.21949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawauchi S, et al. Fgf8 expression defines a morphogenetic center required for olfactory neurogenesis and nasal cavity development in the mouse. Development. 2005;132(23):5211–5223. doi: 10.1242/dev.02143. [DOI] [PubMed] [Google Scholar]
- 38.Trumpp A, Depew MJ, Rubenstein JL, Bishop JM, Martin GR. Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes & development. 1999;13(23):3136–3148. doi: 10.1101/gad.13.23.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu D, Marcucio RS. A SHH-responsive signaling center in the forebrain regulates craniofacial morphogenesis via the facial ectoderm. Development. 2009;136(1):107–116. doi: 10.1242/dev.026583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneider RA, Hu D, Rubenstein JL, Maden M, Helms JA. Local retinoid signaling coordinates forebrain and facial morphogenesis by maintaining FGF8 and SHH. Development. 2001;128(14):2755–2767. doi: 10.1242/dev.128.14.2755. [DOI] [PubMed] [Google Scholar]
- 41.Monks DC, Morrow BE. Identification of putative retinoic acid target genes downstream of mesenchymal Tbx1 during inner ear development. Developmental dynamics : an official publication of the American Association of Anatomists. 2012;241(3):563–573. doi: 10.1002/dvdy.23731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song Y, Hui JN, Fu KK, Richman JM. Control of retinoic acid synthesis and FGF expression in the nasal pit is required to pattern the craniofacial skeleton. Developmental biology. 2004;276(2):313–329. doi: 10.1016/j.ydbio.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 43.Sulik KK, Cook CS, Webster WS. Teratogens and craniofacial malformations: relationships to cell death. Development. 1988;103(Suppl):213–231. doi: 10.1242/dev.103.Supplement.213. [DOI] [PubMed] [Google Scholar]
- 44.Hebert JM. FGFs: Neurodevelopment's Jack-of-all-Trades - How Do They Do it? Frontiers in neuroscience. 2011;5:133. doi: 10.3389/fnins.2011.00133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Crossley PH, Martin GR. The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development. 1995;121(2):439–451. doi: 10.1242/dev.121.2.439. [DOI] [PubMed] [Google Scholar]
- 46.Sun SK, et al. Epibranchial and otic placodes are induced by a common Fgf signal, but their subsequent development is independent. Developmental biology. 2007;303(2):675–686. doi: 10.1016/j.ydbio.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 47.Martin K, Groves AK. Competence of cranial ectoderm to respond to Fgf signaling suggests a two-step model of otic placode induction. Development. 2006;133(5):877–887. doi: 10.1242/dev.02267. [DOI] [PubMed] [Google Scholar]
- 48.Walshe J, Maroon H, McGonnell IM, Dickson C, Mason I. Establishment of hindbrain segmental identity requires signaling by FGF3 and FGF8. Current biology : CB. 2002;12(13):1117–1123. doi: 10.1016/s0960-9822(02)00899-0. [DOI] [PubMed] [Google Scholar]
- 49.Maden M. Role and distribution of retinoic acid during CNS development. International review of cytology. 2001;209:1–77. doi: 10.1016/s0074-7696(01)09010-6. [DOI] [PubMed] [Google Scholar]
- 50.Dupe V, Lumsden A. Hindbrain patterning involves graded responses to retinoic acid signalling. Development. 2001;128(12):2199–2208. doi: 10.1242/dev.128.12.2199. [DOI] [PubMed] [Google Scholar]
- 51.Durston AJ, et al. Retinoic acid causes an anteroposterior transformation in the developing central nervous system. Nature. 1989;340(6229):140–144. doi: 10.1038/340140a0. [DOI] [PubMed] [Google Scholar]
- 52.Nicolay DJ, Doucette JR, Nazarali AJ. Transcriptional regulation of neurogenesis in the olfactory epithelium. Cellular and molecular neurobiology. 2006;26(4-6):803–821. doi: 10.1007/s10571-006-9058-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tremblay P, Kessel M, Gruss P. A transgenic neuroanatomical marker identifies cranial neural crest deficiencies associated with the Pax3 mutant Splotch. Developmental biology. 1995;171(2):317–329. doi: 10.1006/dbio.1995.1284. [DOI] [PubMed] [Google Scholar]
- 54.Hodge LK, et al. Retrograde BMP signaling regulates trigeminal sensory neuron identities and the formation of precise face maps. Neuron. 2007;55(4):572–586. doi: 10.1016/j.neuron.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 55.Kim J, Lo L, Dormand E, Anderson DJ. SOX10 maintains multipotency and inhibits neuronal differentiation of neural crest stem cells. Neuron. 2003;38(1):17–31. doi: 10.1016/s0896-6273(03)00163-6. [DOI] [PubMed] [Google Scholar]
- 56.Carney TJ, et al. A direct role for Sox10 in specification of neural crest-derived sensory neurons. Development. 2006;133(23):4619–4630. doi: 10.1242/dev.02668. [DOI] [PubMed] [Google Scholar]
- 57.Tsuzuki T, et al. Spatial and temporal expression of the ret proto-oncogene product in embryonic, infant and adult rat tissues. Oncogene. 1995;10(1):191–198. [PubMed] [Google Scholar]
- 58.Koblar SA, et al. Pax-3 regulates neurogenesis in neural crest-derived precursor cells. Journal of neuroscience research. 1999;56(5):518–530. doi: 10.1002/(SICI)1097-4547(19990601)56:5<518::AID-JNR7>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 59.Tiveron MC, Hirsch MR, Brunet JF. The expression pattern of the transcription factor Phox2 delineates synaptic pathways of the autonomic nervous system. The Journal of neuroscience : the official journal of the Society for Neuroscience. 1996;16(23):7649–7660. doi: 10.1523/JNEUROSCI.16-23-07649.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kolterud A, Alenius M, Carlsson L, Bohm S. The Lim homeobox gene Lhx2 is required for olfactory sensory neuron identity. Development. 2004;131(21):5319–5326. doi: 10.1242/dev.01416. [DOI] [PubMed] [Google Scholar]
- 61.Morin X, et al. Defects in sensory and autonomic ganglia and absence of locus coeruleus in mice deficient for the homeobox gene Phox2a. Neuron. 1997;18(3):411–423. doi: 10.1016/s0896-6273(00)81242-8. [DOI] [PubMed] [Google Scholar]
- 62.Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. Expression and interactions of the two closely related homeobox genes Phox2a and Phox2b during neurogenesis. Development. 1997;124(20):4065–4075. doi: 10.1242/dev.124.20.4065. [DOI] [PubMed] [Google Scholar]
- 63.Ma Q, Chen Z, del Barco Barrantes I, de la Pompa JL, Anderson DJ. neurogenin1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron. 1998;20(3):469–482. doi: 10.1016/s0896-6273(00)80988-5. [DOI] [PubMed] [Google Scholar]
- 64.Cau E, Casarosa S, Guillemot F. Mash1 and Ngn1 control distinct steps of determination and differentiation in the olfactory sensory neuron lineage. Development. 2002;129(8):1871–1880. doi: 10.1242/dev.129.8.1871. [DOI] [PubMed] [Google Scholar]
- 65.Hogan BL, et al. Small eyes (Sey): a homozygous lethal mutation on chromosome 2 which affects the differentiation of both lens and nasal placodes in the mouse. Journal of embryology and experimental morphology. 1986;97:95–110. [PubMed] [Google Scholar]
- 66.Schilling TF. Anterior-posterior patterning and segmentation of the vertebrate head. Integrative and comparative biology. 2008;48(5):658–667. doi: 10.1093/icb/icn081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moens CB, Prince VE. Constructing the hindbrain: insights from the zebrafish. Developmental dynamics : an official publication of the American Association of Anatomists. 2002;224(1):1–17. doi: 10.1002/dvdy.10086. [DOI] [PubMed] [Google Scholar]
- 68.Diez del Corral R, et al. Opposing FGF and retinoid pathways control ventral neural pattern, neuronal differentiation, and segmentation during body axis extension. Neuron. 2003;40(1):65–79. doi: 10.1016/s0896-6273(03)00565-8. [DOI] [PubMed] [Google Scholar]
- 69.Cunningham TJ, et al. Antagonism between retinoic acid and fibroblast growth factor signaling during limb development. Cell reports. 2013;3(5):1503–1511. doi: 10.1016/j.celrep.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Diez del Corral R, Storey KG. Opposing FGF and retinoid pathways: a signalling switch that controls differentiation and patterning onset in the extending vertebrate body axis. BioEssays : news and reviews in molecular, cellular and developmental biology. 2004;26(8):857–869. doi: 10.1002/bies.20080. [DOI] [PubMed] [Google Scholar]
- 71.Niederreither K, et al. The regional pattern of retinoic acid synthesis by RALDH2 is essential for the development of posterior pharyngeal arches and the enteric nervous system. Development. 2003;130(11):2525–2534. doi: 10.1242/dev.00463. [DOI] [PubMed] [Google Scholar]
- 72.Trokovic N, Trokovic R, Partanen J. Fibroblast growth factor signalling and regional specification of the pharyngeal ectoderm. The International journal of developmental biology. 2005;49(7):797–805. doi: 10.1387/ijdb.051976nt. [DOI] [PubMed] [Google Scholar]
- 73.White JC, Highland M, Kaiser M, Clagett-Dame M. Vitamin A deficiency results in the dose-dependent acquisition of anterior character and shortening of the caudal hindbrain of the rat embryo. Developmental biology. 2000;220(2):263–284. doi: 10.1006/dbio.2000.9635. [DOI] [PubMed] [Google Scholar]
- 74.Paek H, Hwang JY, Zukin RS, Hebert JM. beta-Catenin-dependent FGF signaling sustains cell survival in the anterior embryonic head by countering Smad4. Developmental cell. 2011;20(5):689–699. doi: 10.1016/j.devcel.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chai Y, et al. Fate of the mammalian cranial neural crest during tooth and mandibular morphogenesis. Development. 2000;127(8):1671–1679. doi: 10.1242/dev.127.8.1671. [DOI] [PubMed] [Google Scholar]
- 76.Rhinn M, Dierich A, Le Meur M, Ang S. Cell autonomous and non-cell autonomous functions of Otx2 in patterning the rostral brain. Development. 1999;126(19):4295–4304. doi: 10.1242/dev.126.19.4295. [DOI] [PubMed] [Google Scholar]
- 77.Weston JA, Thiery JP. Pentimento: Neural Crest and the origin of mesectoderm. Developmental biology. 2015;401(1):37–61. doi: 10.1016/j.ydbio.2014.12.035. [DOI] [PubMed] [Google Scholar]
- 78.Moody SA, LaMantia AS. Transcriptional regulation of cranial sensory placode development. Current topics in developmental biology. 2015;111:301–350. doi: 10.1016/bs.ctdb.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annual review of neuroscience. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Oystreck DT, Engle EC, Bosley TM. Recent progress in understanding congenital cranial dysinnervation disorders. Journal of neuro-ophthalmology : the official journal of the North American Neuro-Ophthalmology Society. 2011;31(1):69–77. doi: 10.1097/WNO.0b013e31820d0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Abadie V, Couly G. Congenital feeding and swallowing disorders. Handbook of clinical neurology. 2013;113:1539–1549. doi: 10.1016/B978-0-444-59565-2.00024-1. [DOI] [PubMed] [Google Scholar]
- 82.Valdes-Socin H, et al. Reproduction, smell, and neurodevelopmental disorders: genetic defects in different hypogonadotropic hypogonadal syndromes. Frontiers in endocrinology. 2014;5:109. doi: 10.3389/fendo.2014.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Eppsteiner RW, Smith RJ. Genetic disorders of the vestibular system. Current opinion in otolaryngology & head and neck surgery. 2011;19(5):397–402. doi: 10.1097/MOO.0b013e32834a9852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fekete DM. Development of the vertebrate ear: insights from knockouts and mutants. Trends in neurosciences. 1999;22(6):263–269. doi: 10.1016/s0166-2236(98)01366-6. [DOI] [PubMed] [Google Scholar]
- 85.Forni PE, Wray S. GnRH, anosmia and hypogonadotropic hypogonadism--where are we? Frontiers in neuroendocrinology. 2015;36:165–177. doi: 10.1016/j.yfrne.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.