

Abstract
Mitotic spindle assembly, positioning and orientation depend on the combined forces generated by microtubule dynamics, microtubule motor proteins and cross-linkers. Growing microtubules can generate pushing forces, while depolymerizing microtubules can convert the energy from microtubule shrinkage into pulling forces, when attached, for example, to cortical dynein or chromosomes. In addition, motor proteins and diffusible cross-linkers within the spindle contribute to spindle architecture by connecting and sliding anti-parallel microtubules. In vivo, it has proven difficult to unravel the relative contribution of individual players to the overall balance of forces. Here we present the methods that we recently developed in our efforts to reconstitute basic mitotic spindles bottom-up in vitro. Using microfluidic techniques, centrosomes and tubulin are encapsulated in water-in-oil emulsion droplets, leading to the formation of geometrically confined (double) microtubule asters. By additionally introducing cortically anchored dynein, plus-end directed microtubule motors and diffusible cross-linkers, this system is used to reconstitute spindle-like structures. The methods presented here provide a starting point for reconstitution of more complete mitotic spindles, allowing for a detailed study of the contribution of each individual component, and for obtaining an integrated quantitative view of the force-balance within the mitotic spindle.
Keywords: Cellular Biology, Issue 114, mitotic spindle formation, spindle positioning, microfluidics, centrosomes, microtubules, dynein, kinesin-5, Ase1
Introduction
During mitosis, the chromosomes of the replicated genome are organized at the cell equator to ensure equal distribution of sister chromatids over the newly formed daughter cells. Chromosome positioning and segregation is mediated by their attachment to spindle microtubules originating from two opposing centrosomes. In addition to ensuring faithful chromosome distribution, the orientation of the mitotic spindle also dictates the cell division plane1. Coordinated orientation of cell division is essential during many stages of development and for tissue homeostasis2. Specifically, regulated mitotic spindle positioning can result in the asymmetric distribution of cellular contents or even produce daughter cells of different sizes2. Mitotic spindle assembly and orientation starts in prophase and is orchestrated by a complex interplay between cooperating and antagonizing force-generators3,4. The generated forces consist of both pulling and pushing forces. These forces originate both from spindle contacts with the cell cortex as well as from within the spindle, and can be generated by microtubule dynamics as well as by molecular motors and cross-linkers (Figure 1).
Pushing forces can be generated when microtubules grow against rigid structures such as the cell cortex. Depending on the total resisting force generated within the system, this can result in repositioning of the mitotic spindle (low forces) or trigger microtubule buckling or catastrophe (high forces)5-8. The amount of force that can be generated by pushing depends on microtubule length, since length is a strong determinant of microtubule-buckling9. In addition, microtubules that originate from one centrosome can push against the other centrosome, a mechanism that has been suggested to drive centrosome separation in early prophase3,10.
In addition to pushing forces generated by growing microtubules, microtubule ends contacting the cell cortex can also mediate pulling forces11. Studies from multiple laboratories have shown that the controlled activity of cortical force generators is required for the asymmetric positioning of mitotic spindles in vivo1. Essential to these pulling forces is cortex-localized cytoplasmic dynein (hereafter referred to as 'dynein'), a minus-end directed microtubule motor protein8,12,13. For example, in budding yeast, lateral interactions of cortical dynein with the microtubule lattice result in motor-dependent microtubule sliding along the cortex14. However, cortical pulling forces can also be generated by the ability of dynein to form end-on attachments to depolymerizing microtubules8. The energy generated by microtubule shrinkage may lead to forces that are generally an order of magnitude higher (~50 pN (reference15)) than the forces generated by individual motor proteins (~7-8 pN (ref. 16)). End-on attachment of cortical dynein to depolymerizing microtubules promotes spindle positioning in budding yeast17 and C. elegans13. Whether both motor-dependent sliding and microtubule depolymerization driven cortical pulling forces can cooperate or are mutually exclusive in vivo is at present unknown.
In addition to microtubule pushing forces and dynein-mediated cortical pulling forces, centrosome positioning is controlled from within the mitotic spindle by numerous other proteins. Kinesin-5 motors, for instance, exist as tetramers that can cross-link antiparallel interpolar microtubules, resulting in the generation of outward sliding forces18-20. Members of the kinesin-5 motor family are required for centrosome separation and bipolar spindle assembly in all eukaryotes studied, with the exception of C. elegans (reviewed in reference21).
Furthermore, diffusible cross-linkers of the Ase1/PRC1 family are also known to localize to overlapping microtubule regions of interpolar microtubules in vivo and in vitro22-27. The forces generated by Ase1-driven expansion of the overlapping microtubule-region are large enough to counterbalance kinesin-14 mediated sliding forces in vitro28,29. In cells, Ase1/PRC1 is required for stable formation of overlapping microtubule regions in the spindle midzone25-27,30, suggesting that the forces generated by diffusible cross-linkers can significantly contribute to spindle organization. Finally, forces from the mitotic chromatin and kinetochores influence mitotic spindle assembly and positioning through various pathways3. Since these components are not part of the reconstitution assays described here, they will not be discussed in detail.
Different theories exist on how the different molecular components and forces described above cooperate in spindle assembly and positioning, but we are still far from a quantitative understanding. In addition to experiments in living cells, in vitro experiments with purified components provide a powerful route to help reach this goal. Here we present a visual guide to an adapted and extended version of the recently published methods (Roth et al.31), in which basic spindles are reconstituted in water-in-oil emulsion droplets, starting with a minimal number of components. Using microfluidic techniques, spherical droplets are generated with sizes that are comparable to mitotic cells. Within these droplets, purified centrosomes and tubulin can be combined in order to study microtubule aster dynamics, force generation and aster-aster interactions. By introducing cortical (such as dynein) and inter-polar (such as kinesin-5/Ase1) force-generators, we reconstitute increasingly complex spindle-like structures that start to resemble the in vivo situation.
Protocol
1. Preparation of Microfluidics Chips
NOTE: For bottom-up study of spindle assembly, spherical water-in-oil emulsion droplets are used. These are aqueous microdroplets separated from the surrounding oil-phase by a monolayer of phospholipids and surfactant. These emulsion-droplets are produced within microfluidic polydimethylsiloxane (PDMS) chips. Changes in channel geometries and flow rates can be used to tune droplet size. In the microfluidic design described here, droplets are generated with a diameter of about 15 µm to mimic the geometrical confinement of a mammalian mitotic cell.
- Photomask Design and Mold Fabrication
- Design a photomask containing three inlet channels31. Inlet channel 1 connects to the water-phase, which contains the droplet contents (see section 3 "Reconstituting basic microtubule asters"). Inlet channel 2 connects to the oil-phase (see section 2.1 "Lipid/oil-phase preparation"). Use inlet channel 3 to dilute emulsion-droplets with additional lipid/oil-phase before observation (optional) (Figure 2a-b). NOTE: All inlet channels are followed by a dust-filter (a maze of 2 µm channels) to trap dust, PDMS particles and (protein-) aggregates and to prevent them from blocking the microfluidic channels (Figure 2d). Channels are 75 µm wide and the lipid/oil-phase and water-phase meet at a 12.5 µm wide junction where emulsion-droplets will form (Figure 2c).
- Order and obtain printed photomasks on a film substrate, with a negative polarity (the photomask will be dark with transparent designed structures).
- Mold Fabrication
- Fabricate molds by spin-coating SU-8 3025 photoresist onto a 4 inch silicon wafer using a spin-coater (500 rpm, 10 sec, followed by 1,800 rpm, 45 sec) in a clean-room environment.
- Expose the SU-8 coated silicon wafer through the photomask. Develop wafers according to the manufacturer's instructions to create channels of ~40 µm thickness.
- Making a Microfluidic chip
- Mix 10 parts PDMS pre-polymer with 1 part curing agent (total ~40 gr) in a boat-like vessel using a plastic spatula. Place PDMS containing boat-like vessel in a vacuum chamber for ~30 min. to remove air bubbles. Wrap aluminum foil around the mold to form a cup with ~1 cm upstanding edges.
- Pour the PDMS (~75% of total volume) into the mold, leave in a vacuum chamber for another ~30 min. (to remove air bubbles) and cure for 1 hr at 100 °C in an oven.
- Spin-coat the remaining PDMS onto glass slides (5 sec at 200 rpm followed by 30 sec at 4,000 rpm) followed by curing for 1 hr at 100 °C. Remove dust from glass slides using compressed air/N2-flow, pre-cleaning is not necessary. NOTE: The PDMS coated glass slides can be used both for microfluidic chip and flow-cell production (see also 2.2).
- Gently strip the PDMS off of the mold using a razor blade and punch holes (0.5 mm for inlets, 0.75 mm for outlet).Corona-treat the microfluidic chip and a PDMS-coated glass slide using a laboratory corona surface treater for a few seconds.
- Place the microfluidic chip onto the glass slide (channels facing down) and bake the chips o/n at 100 °C (Figure 3a). Store microfluidic chips for several months in a dust-free environment.
2. Microfluidic Setup
NOTE: Water-in-oil emulsion droplets are generated using a microfluidics setup and the microfluidic chips described above. The composition of the lipid/oil-phase can be varied depending on the experimental requirements. Oil-solubilized lipids will form a monolayer at the water-oil interface with their polar head-groups facing the water inside. In order to target proteins (e.g. dynein) to the droplet boundary, low amounts of biotinyl-phosphatidylethanolamine (biotinyl-PE) lipids can be added. This allows the recruitment of biotinylated dynein (see section 4.1 "Dynein targeting to the droplet cortex") via streptavidin-mediated multimerization. Droplets are stabilized by addition of a surfactant and stored in PDMS coated flow cells.
- Lipid/oil-phase Preparation
- Mix chloroform-dissolved 1,2-dioleoyl-sn-glycero-3-phospho-L-serine (DOPS) and biotinyl-PE lipids in a 4:1 molar ratio in a glass tube using glass pipets (extensively rinse pipets with chloroform before use). Prepare a total of ~250 µg lipids, resulting in a lipid concentration of ~0.5 mg/ml.
- Carefully dry the lipid mixture with N2-flow and subsequently in a vacuum chamber for ~1 hr. Dissolve the dried lipids in mineral oil and 2.5% surfactant to 0.5 mg/ml (make ~500 µl).
- Place the lipid/oil sample in an ultrasonic bath and sonicate for 30 min. at 40 kHz to completely dissolve the lipids.
- Flow-cell Preparation
- Spin-coat PDMS (see also section 1.3) onto cover glasses (5 sec at 200 rpm followed by 30 sec at 4,000 rpm) and glass slides (5 sec at 100 rpm followed by 30 sec 1,500 rpm) in order to deposit the homogeneous layer of PDMS. NOTE: For optimal imaging quality, make sure to use cover glasses with a thickness that matches the microscope objective. In this case: 1.5 (~0.17 mm).
- Cure the PDMS-coated cover glasses and glass slides for 1 hr at 100 °C in an oven. Make flow-cells by closely spacing (~2 mm) thin slices of laboratory sealing film (~3 mm in width) onto the PDMS-coated glass-slides. When stored in a dust-free environment, channels that have not been used can be used at other times.
- Cover the flow-cells with a PDMS-coated coverslip and seal by melting the laboratory sealing film ~1 min. at 100 °C, press gently (Figure 3c). Close the laboratory sealing film -glass-boundaries with Valap (Figure 3c). Store flow-cells for several months in a dust-free environment.
- PDMS Cup Preparation
- For long-term imaging, make a PDMS cup, by punching a 4 mm diameter hole in a slice of PDMS (~3 mm thick)
- Corona-treat the PDMS slice and a PDMS coated cover glass and place them on top of each other. Bake the PDMS cup o/n (at 100 °C) (Figure 5a).
- Emulsion-droplet Formation
- Monitor droplet formation on an inverted bright-field microscope. Connect the pressure controller to the microfluidic chip using PEEK tubes (diameters: 510 µm (outer) and 125 µm (inner)) (Figure 3a). Fill microfluidic chips completely with lipid/oil-phase from inlet 2.
- Introduce MRB80-based water-phase (see section 3 "Reconstituting basic microtubule asters") from inlet 1. Control droplet-size by changing lipid/oil-phase and water-phase pressures to create droplets of ~15 µm in diameter (Figure 3b). NOTE: Using this setup, use ~800 mbar for the lipid/oil-phase and ~200 mbar for the water-phase to create droplets of the desired size.
- After obtaining the desired droplet-size and required amount (~10 µl per sample), collect droplets from outlet channel and load into flow-cell (fill the flow-cell completely).
- Close the ends of the flow-cell carefully using Valap (incompletely closed flow-cells can result in extensive movement of the droplets, making it difficult to image them). In addition, prevent the formation of air bubbles which results in droplet movement.
- Rinse the PEEK tubes extensively with isopropanol before and after use to prevent sample cross-contamination and clogging.
3. Reconstituting Basic Microtubule Asters
NOTE: Droplet contents can be varied depending on the experimental requirements. All buffers are MRB80-based (80 mM PIPES, 1 mM EGTA, 4 mM MgCl2 (pH 6.8)), and all samples are prepared on ice. In general, droplets always contain centrosomes, components required for microtubule polymerization, an oxygen scavenger system and blocking agent (see section 3.1). Microtubule force-generators and required cofactors and targeting-factors can be included if desired (see sections 4.1 and 4.2).
- General setup for aster formation in emulsion droplets (Figure 3d)
- Prepare glucose oxidase (20 mg/ml glucose oxidase in 200 mM DTT and 10 mg/ml catalase) mix in advance and store in small aliquots at -80 °C.
- Prepare a 'blocking mix' containing κ-casein, bovine serum albumin (BSA), Tween-20 and fluorescent dextran (as a neutral marker) (see Table 1) in advance and store in small aliquots at -80 °C. Prevent repeated freeze-thaw cycles. Ensure all components are freshly prepared.
- Purify centrosomes from human lymphoblastic KE37 cell lines as described by Moudjou et al.32 and store at -150 °C in small aliquots.
- Thaw centrosomes at room temperature and incubate at 37 °C for ~20 min. before use to ensure proper microtubule nucleation. NOTE: This promotes microtubule nucleation during the experiment.
- In the mean time, prepare 'assay mix', containing tubulin (fluorescent and 'dark'), guanosine triphosphate (GTP), an oxygen scavenger system (glucose, glucose-oxidase, catalase and 1,4-dithiothreitol (DTT)), molecular force generators (e.g. microtubule motors/cross-linkers), adenosine triphosphate (ATP) and an ATP-regenerating system (phosphoenolpyruvate (PEP), pyruvate kinase (PK) and lactate dehydrogenase (LDH)) on ice (see Table 2).
- Pre-cool the airfuge rotor on ice. Spin down the sample in the cooled airfuge rotor (30 psi for 3 min).Add pre-heated centrosomes (optimize the amount of centrosomes to aim for droplets containing 1-2 centrosomes).
- Use the combined mix to produce emulsion droplets using the methods described in section 2.3.
| Component | Stock concentration | Final Concentration (in assay) | Volume | Comment |
| Dextran-647nm | 75 μM | 3.75 uM (0.6 μM) | 5 μl | |
| κ-casein | 20 mg/ml | 12.5 mg/ml (2 mg/ml) | 62.5 μl | |
| Tween-20 | 5% | 0.375% (0.06%) | 7.5 μl | |
| BSA | 200 mg/ml | 12 mg/ml (1.9 mg/ml) | 6 μl | |
| MRB80 | 19 μl | |||
| 100 μl final | Store in 2 μl aliquots |
Table 1: Constituents of 'Blocking Mix'. Constituents of blocking mix, required for the reconstitution of spindle-like structures in water-in-oil emulsion droplets. For description, see main text section 3 "Reconstituting basic microtubule asters". For details on materials, see "Materials Table.
| Component | Stock concentration | Final Concentration | Volume | Comment |
| Blocking mix | 1.6 μl | |||
| Tubulin | 500 μM | 30 μM | 0.6 μl | |
| Tubulin-488/561 | 50 μM | 3 μM | 0.6 μl | |
| GTP | 50 mM | 5 mM | 1.0 μl | |
| Dynein-TMR | 178 nM | 30 nM | 1.7 μl | |
| Streptavidin | 5 mg/ml | 200 nM | 0.4 μl | For cortical Dynein only |
| Kinesin-5 | 1.6 mM | 80 nM | 0.5 μl | |
| ATP | 25 mM | 1 mM | 0.4 μl | For motors only |
| PEP | 0.5 M | 25.6 mM | 0.5 μl | For motors only |
| PK/LDH | 800 U/1,100 U | 23 U/32 U | 0.3 μl | For motors only |
| Ase1-GFP | 400 nM | 80 nM | 2.0 μl | |
| Glucose | 2.5 M | 50 mM | 0.3 μl | Last step |
| Glucose-oxidase | 50x | 1.0x | 0.3 μl | Last step |
| MRB80 | x | |||
| 9 μl final |
Table 2: Constituents of 'Assay Mix'. Constituents of assay mix, required for the reconstitution of spindle-like structures in water-in-oil emulsion droplets. For description, see main text section 3 "Reconstituting basic microtubule asters". For details on materials, see Materials Table.
4. Introducing Spindle Assembly-factors
NOTE: In the assays described here, a green fluorescent protein (GFP)-tagged truncated version of S. cerevisiae dynein was used that is artificially dimerized by means of an amino-terminal Glutathione S-transferase (GST)-tag33. This protein is purified through an affinity tag that contains two copies of the protein A IgG-binding domain. The variant used here is labeled with a tetramethylrhodamine (TMR) label onto the carboxy-terminal and the N-terminal SNAP-tag is used to biotinylate the purified proteins. For construct details, see Roth et al.31; for purification details, see Reck-Peterson et al.33. GFP-biotin-dynein-TMR can be targeted to the droplet-cortex through the formation of biotin-streptavidin complexes that link dynein to biotinyl-PE lipids (Figure 3e).
- Dynein Targeting to the Droplet Cortex
- Include GFP-biotin-dynein-TMR into the 'assay mix' (see Table 2) at a final droplet-concentration of 30 nM. Include Streptavidin into 'assay mix' (see Table 2) at a final droplet-concentration of 200 nM. NOTE: Dynein depends on ATP-hydrolysis for step-wise movement on microtubules. Therefore, include ATP and an ATP regenerating system (containing PEP and PK/LDH) into the 'assay mix' (see Table 2). NOTE: Recombinant full-length S. pombe GFP-Cut7 (kinesin-5) was kindly provided by the Diez lab (Center for Molecular Bioengineering, Technische Universitat Dresden, Dresden, Germany), see . Recombinant full-length histidine-tagged S. pombe GFP-Ase1 was kindly provided from the Jansons lab (Wageningen University, Wageningen, The Netherlands) and added to the 'assay mix' at concentrations at 80 nM.
5. Imaging
NOTE: During the experiment, microtubule growth can be controlled by changing the temperature. In choosing the imaging conditions, trade-offs have to be made between high-quality imaging and the ability to monitor spindle assembly for long time-periods. To compare the kinetics of spindle formation under different conditions, droplets with different contents can be mixed and imaged in the same flow channel.
- Basic Imaging Settings
- Image in a temperature-controlled environment using an enclosed chamber. Visualize individual microtubules after ~30 min at 26 °C. While imaging, promote microtubule-growth by increasing the temperature up to ~30 °C. NOTE: The settings below are specific to our microscope setup and can vary with different systems and different operating software. All of the assays described here are imaged on a motorized inverted system with a spinning disk confocal head and operated using imaging software such as Andor iQ 3.1. Obtain all images with a 100X oil immersion objective and EmCCD camera.
- Set excitation lasers 488, 561 and 641 nm to roughly ~10% intensity. Go to the 'acquisition' panel, click on the 'AOTF' tab and slide laser intensities to 10%. Click on 'record' to store the settings.
- For z-projections, take stacks with 1 µm intervals (~20 images/droplet). Go to the main "camera' panel, click on 'edit z' and set 'Z step' to 1.0. Click on 'next' to store the settings.
- Set maximum linear EM-gain by clicking on the 'camera' tab in the 'acquisition' panel and slide the EM gain bar to 300. Set exposure times to 200 msec in the same tab. Click on 'record' to store the settings.
- Live Imaging
- For live imaging, using the software controls make z-projections every 2 min. for the duration of 1-2 hr by reducing the exposure times to ~100 msec. (as described in 5.1.4) and increase z-intervals to 2 µm (as described in 5.1.3). Set the time series in the main 'camera' panel, click on 'repeat t' and set to 2 min. intervals for a total time of 120 min.
- For live assays, use a PDMS cup instead of a regular flow-cell to ensure that the droplets are as immobile as possible.
- Combined Imaging of 2 Conditions
- Produce droplets using microfluidics and store on top of a PDMS coated glass slide on ice. This prevents microtubule nucleation until the second set of droplets has been formed. Before producing the second set of droplets, rinse the PEEK tubes and microfluidic chip with MRB80 and completely refill microfluidic chip with the lipid/oil-phase.
- Generate droplets with and without fluorescent dextran (or using dextran with different wavelength fluorophores) in order to discriminate between droplets with different colors. After producing the second batch of droplets, gently mix the droplets by pipetting and load into a single flow-cell/PDMS-cup.
- Image Analysis.
- Analyze images using Fiji (open source software available online)34.
- For live assays, convert stacks into hyperstacks using 'image'-'hyperstacks'-'stack to hyperstack'. Set the correct number of z-slices and time-frames.
- In order to make z-projections, choose 'image'-'stacks'-'z-project'. Choose 'max intensity' projection.
- For 3D-reconstructions, first go to 'image'-'properties' and set the proper pixel with, pixel heigth, voxel depth and frame intervals. Click 'OK'. Choose 'image'-'stacks'-'3D project', check the 'interpolate' box and click 'OK'.
Representative Results
The methods presented in the previous sections allow the reconstitution of spindle-like structures with increasing complexity using the geometrical confinement of water-in-oil emulsion droplets. This section describes representative results that qualitatively demonstrate the capability of these assays.
During mitosis, when the bipolar spindle is assembled, cells round up to form spheres with a diameter of roughly 15 µm, as measured for human cells. This characteristic mitotic cell shape provides a geometrical boundary that both restricts and directs spindle size and orientation35,36. Microfluidic techniques, provide a level of geometrical confinement that accurately resembles the situation observed in living cells and therefore permits the bottom-up reconstruction of mitotic spindles in vitro.
Microtubule asters are by themselves already capable of complex behavior when microtubule growth is restricted by geometrical boundaries. As microtubules grow, pushing forces generated by the incorporation of new tubulin dimers drive the two centrosomes to opposing sides of the emulsion droplets. At first, centrosomes freely diffuse within the confined volume (Figure 4a, left panel). After about 20-30 min, the first microtubules become visible and centrosome diffusion becomes restricted as microtubules grow against the cortex in all directions. Asters with microtubules of intermediate length (roughly 50% of the droplet diameter) can (sterically) repel each another, with microtubules pushing against each other, the other centrosomes and the cortex. This results in a typical 'bipolar' spindle-like arrangement with the two centrosomes opposing each other (Figure 4a, middle panel). When microtubules grow longer than ~50% of the droplet-diameter, the centrosomes get pushed further to opposing boundaries of the droplet, with microtubules growing along the droplet cortex (Figure 4a, right panel). It is important to note that microtubule growth rates in these assays are very sensitive to both temperature and tubulin-concentrations. These parameters will therefore strongly affect the time when nucleation is first observed and when steady-state aster positions are reached.
In cells, cortical pulling forces are generated through the formation of load-bearing attachments between microtubule plus-ends and cortex-associated dynein. In animal cells, this association depends on the Gαi/LGN/NuMA complex, which is targeted to the plasma membrane via N-terminal myristoylation of Gαi1. In these reconstitution assays, the requirement of the Gαi/LGN/NuMA complex is bypassed by directly coupling dynein to biotinylated lipids through the formation of biotin-streptavidin-biotin complexes. These bonds are relatively stable (KD ~10-14 M)37 and form rapidly (usually within 10 min after emulsion droplet formation, before microtubule nucleation becomes apparent) (Figure 4b). In the presence of cortical dynein, centrosomes typically retain a more central position, whereas in the absence of dynein, centrosomes are pushed to opposite sides of the droplet cortex (Figure 4c). We reason this is the result of two effects, which prevent and counteract microtubule pushing forces. (1) Dynein directly promotes microtubule catastrophes, thereby restricting microtubule length and preventing excessive microtubule buckling, and (2) cortical pulling forces lead to net centering forces on the individual asters8, which counteract the aster-aster repulsion forces.
The diffusible cross-linker Ase1 induces forces that tend to increase the overlapping region of anti-parallel microtubules29. Consistent with this, in the presence of Ase1 (and absence of dynein) centrosomes are found close together with Ase1 localizing to bundled interpolar microtubules (Figure 4d). Members of the kinesin-5 family drive centrosome separation by providing pushing forces from within the spindle (see introduction). In the presence of Cut7 (the S. pombe kinesin-5 ortholog), centrosomes are pushed to opposite sides of the emulsion droplets, even in the presence of Ase1 (Figure 4e). By combining cortical and inter-polar force generators, the level of complexity can be further increased in these experiments, eventually leading to a comprehensive understanding of bipolar mitotic spindle assembly. A detailed quantitative description of these results will be available in the future (Roth et al., manuscript in preparation).
In addition to observing the position of the centrosomes at fixed moments in time, and relating their behavior to the observed lengths of the microtubules, valuable information can be obtained by following the positioning process over time. By reducing exposure times and laser intensities, it is now possible to follow aster positioning at 2 min intervals for at least 1 hr with only ~30% bleaching. This allows monitoring of aster assembly and positioning from freely diffusing centrosomes (0 min.) via centralized (12 min.) to decentralized asters (36 min and further) (Figure 5c). This facilitates the study of both steady-state behavior and spindle assembly kinetics. By combining droplets with different contents in the same sample, it is, for example, possible to directly compare centrosome positioning and spindle assembly kinetics in droplets with and without cortical force generators (Figure 5b).
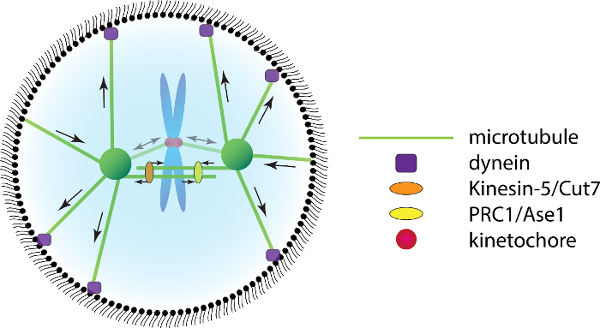 Figure 1:Forces Acting on the Mitotic Spindle. Several force-generating molecules act on microtubules of the mitotic spindle to promote spindle formation and positioning. Microtubules that grow into the cell cortex generate a pushing force on the centrosome. Cortical dynein (purple) captures depolymerizing microtubules and generates a pulling force on the centrosomes. Within the spindle, Kinesin-5/Cut7 motor proteins provide an outward sliding force on anti-parallel microtubules, whereas cross-linkers of the PRC1/Ase1 family generate an opposing outward sliding force. Please click here to view a larger version of this figure.
Figure 1:Forces Acting on the Mitotic Spindle. Several force-generating molecules act on microtubules of the mitotic spindle to promote spindle formation and positioning. Microtubules that grow into the cell cortex generate a pushing force on the centrosome. Cortical dynein (purple) captures depolymerizing microtubules and generates a pulling force on the centrosomes. Within the spindle, Kinesin-5/Cut7 motor proteins provide an outward sliding force on anti-parallel microtubules, whereas cross-linkers of the PRC1/Ase1 family generate an opposing outward sliding force. Please click here to view a larger version of this figure.
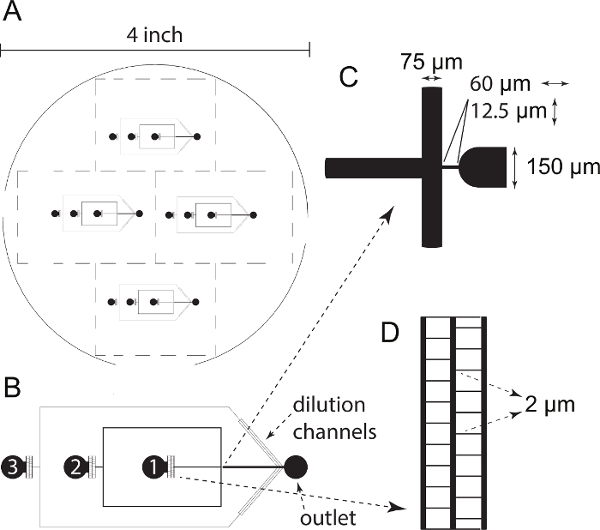 Figure 2: Microfluidic Chip Design. (A) Design of 4 inch photomask containing 4 microfluidic chips. (B) Detailed representation of a single chip. Chips contain one outlet channel and three inlet channels followed by a dust-filter. Inlet channel 1 contains the water-phase, channel 2 the lipid/oil-phase and channel 3 can be used to dilute the formed droplets with additional lipid/oil-phase. (C) Detailed representation of the junction where the lipid/oil-phase (coming from the top and bottom) meets with the water-phase (coming from the left). At the junction, droplets will form and flow towards the outlet channel on the right. (D) Detailed representation of a dust-filter with 2 µm channels. Please click here to view a larger version of this figure.
Figure 2: Microfluidic Chip Design. (A) Design of 4 inch photomask containing 4 microfluidic chips. (B) Detailed representation of a single chip. Chips contain one outlet channel and three inlet channels followed by a dust-filter. Inlet channel 1 contains the water-phase, channel 2 the lipid/oil-phase and channel 3 can be used to dilute the formed droplets with additional lipid/oil-phase. (C) Detailed representation of the junction where the lipid/oil-phase (coming from the top and bottom) meets with the water-phase (coming from the left). At the junction, droplets will form and flow towards the outlet channel on the right. (D) Detailed representation of a dust-filter with 2 µm channels. Please click here to view a larger version of this figure.
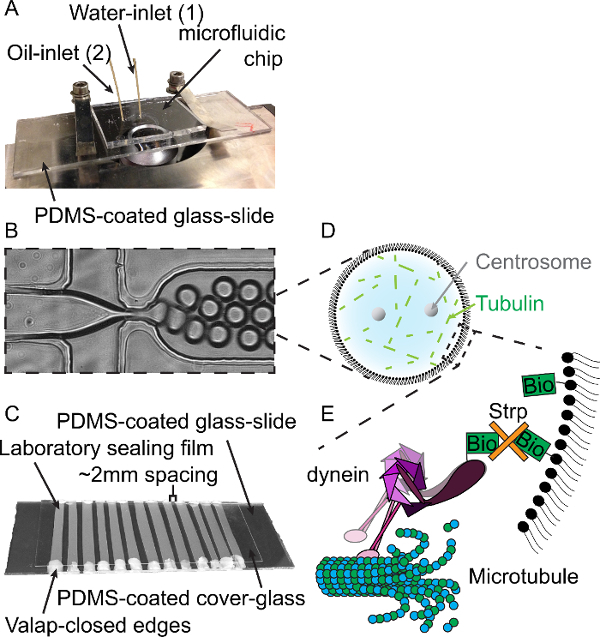 Figure 3:Methodology for Water-in-Oil Emulsion Droplet Formation. (A) Microfluidic chip and microfluidics tubing on a bright-field microscope. Both the water- and lipid/oil-phase tubes are connected to the chip inlets 1 and 2 respectively. (B) Restriction on microfluidics chip where the water- and lipid/oil-phases meet. By changing the pressures, droplets size can be controlled. (C) Design of PDMS-coated flow-cells. After loading the droplets into the flow-cell, the open ends are closed with additional Valap. (D) Schematic of droplet formation. Droplets are used to encapsulate centrosomes, tubulin and additional components required for mitotic spindle formation. (E) Schematic of cortical-dynein targeting. Biotinylated (Bio) dynein is targeted to biotinylated lipids through streptavidin (Strp). Please click here to view a larger version of this figure.
Figure 3:Methodology for Water-in-Oil Emulsion Droplet Formation. (A) Microfluidic chip and microfluidics tubing on a bright-field microscope. Both the water- and lipid/oil-phase tubes are connected to the chip inlets 1 and 2 respectively. (B) Restriction on microfluidics chip where the water- and lipid/oil-phases meet. By changing the pressures, droplets size can be controlled. (C) Design of PDMS-coated flow-cells. After loading the droplets into the flow-cell, the open ends are closed with additional Valap. (D) Schematic of droplet formation. Droplets are used to encapsulate centrosomes, tubulin and additional components required for mitotic spindle formation. (E) Schematic of cortical-dynein targeting. Biotinylated (Bio) dynein is targeted to biotinylated lipids through streptavidin (Strp). Please click here to view a larger version of this figure.
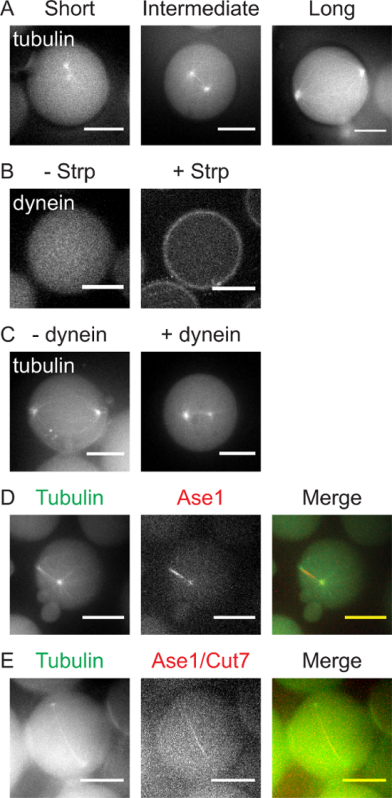 Figure 4:Representative Results of Spindle-Reconstitutions with Increasing Complexity. (A) Maximum intensity projections of droplets containing two centrosomes showing examples of short, intermediate, and long microtubules. Centrosomes and microtubules are visualized by the addition of 10% HiLyte-488 labeled Tubulin. Scale bars = 10 µm. (B) Localization of GFP-biotin-dynein-TMR in the absence (-, left) and presence (+, right) of streptavidin (Strp). (C) Microtubule aster positioning in the absence (-, left) or presence (+, right) of cortical GFP-biotin-dynein-TMR. (D) Microtubule aster (left panel, green) positioning in the presence of Ase1 (middle panel, red). (E) Microtubule aster (left panel, green) positioning in the presence of Ase1 and Cut7 (kinesin-5) (middle panel, red). Please click here to view a larger version of this figure.
Figure 4:Representative Results of Spindle-Reconstitutions with Increasing Complexity. (A) Maximum intensity projections of droplets containing two centrosomes showing examples of short, intermediate, and long microtubules. Centrosomes and microtubules are visualized by the addition of 10% HiLyte-488 labeled Tubulin. Scale bars = 10 µm. (B) Localization of GFP-biotin-dynein-TMR in the absence (-, left) and presence (+, right) of streptavidin (Strp). (C) Microtubule aster positioning in the absence (-, left) or presence (+, right) of cortical GFP-biotin-dynein-TMR. (D) Microtubule aster (left panel, green) positioning in the presence of Ase1 (middle panel, red). (E) Microtubule aster (left panel, green) positioning in the presence of Ase1 and Cut7 (kinesin-5) (middle panel, red). Please click here to view a larger version of this figure.
 Figure 5: Imaging Aster Positioning Dynamics in vitro. (A) Design of a PDMS cup that allows droplet imaging for up to several hr. (B) Generation, storage and imaging of droplets (transmitted) containing different contents, illustrated by absence (left) inclusion (right) of fluorescent dextran (Alexa 647). (C) Single plane images of a 60 min time-lapse (2 min intervals) taken from a z-stack (2 µm distances) of a droplet containing a single centrosome. Please click here to view a larger version of this figure.
Figure 5: Imaging Aster Positioning Dynamics in vitro. (A) Design of a PDMS cup that allows droplet imaging for up to several hr. (B) Generation, storage and imaging of droplets (transmitted) containing different contents, illustrated by absence (left) inclusion (right) of fluorescent dextran (Alexa 647). (C) Single plane images of a 60 min time-lapse (2 min intervals) taken from a z-stack (2 µm distances) of a droplet containing a single centrosome. Please click here to view a larger version of this figure.
Discussion
The methods described here present recent efforts to reconstitute basic mitotic spindles in water-in-oil emulsion droplets. Studying spindle assembly within geometrical boundaries provides numerous advantages. Important feedback mechanisms exist between microtubules of the mitotic spindle and the geometrical constraints in which they are assembled. Microtubules can generate pushing forces when they grow into geometrical boundaries, which can result in mitotic spindle displacement. In addition, growing into a physical barrier can induce microtubule catastrophes. Also, spindle assembly within a confined geometry can lead to a gradual depletion of individual components, such as tubulin, which in turn directly affects the microtubule growth rate36. These are all essential determinants of spindle assembly, which can be studied using the assays described here.
The described assays are very sensitive to small concentration differences of the droplet components. This is the case for tubulin, where minor concentration differences can result in either too slow or too rapid microtubule growth. In this case, microtubule growth rates can often still be corrected by adjusting the temperature during the first ~ 30 min of the experiment. Mitotic spindle assembly and positioning is also very sensitive to variations in the concentration of (cortical) force generators. This is likely to depend on the concentration, purity and activity of the purified components and needs to be titrated carefully for every newly purified protein.
In addition, certain trade-offs have to be made in imaging settings, depending on the nature of the assay. Initially, high levels of soluble fluorescent tubulin dimers make it difficult to image individual microtubules. As microtubules grow longer and soluble tubulin is slowly depleted, the signal-to-noise ratio gradually becomes higher and allows the imaging of individual microtubules. In contrast to setups with open systems, the geometrical confinement prevents the exchange of fluorescent molecules and oxygen scavenger system components within a bulk reservoir, resulting in significant bleaching. Especially for monitoring spindle assembly and positioning over time, it is required to image with low light intensities, even in the presence of a potent oxygen scavenger system.
These methods enable the bottom-up reconstitution of simplified spindle-like structures in vitro. In addition to the components introduced here, many other factors have been described to influence bipolar spindle formation, positioning, orientation, shaping, etc.3. This system can be further expanded to study the individual and combined effects of additional force generators. Important contributors to spindle formation that are currently absent from this system are the mitotic chromosomes. Mitotic chromatin has been shown to direct spindle formation by (1) chromokinesins that can generate so-called 'polar-ejection forces'3, (2) the formation of a RanGTP gradient by the chromatin-bound RanGEF RCC138, (3) kinetochores that can interact with (depolymerizing) microtubules39 and (4) the chromatin itself, which can function as a mechanical spring in-between opposing microtubules40.
Finally, the described system currently provides limited temporal and spatial control over the activity and localization of introduced force-generators. In cells, many spindle assembly factors localize to specific compartments or are active within a restricted time-window. A striking example is the activity of dynein, which is specifically enriched at the posterior side of the C. elegans one-cell stage embryo to promote asymmetric spindle positioning and cell division. In this system, we are currently exploring the possibility of mimicking such temporal and asymmetric activity using methods borrowed from opto-genetic techniques. In addition, as is the case for the C. elegans embryo and cells with a rigid cell wall, many cells do not round up in mitosis. The shape of the geometrical confinement will have a fundamental impact on the forces acting on the spindle41. It will therefore be important to reconstitute spindle assembly and positioning in non-spherical droplets as well, potentially using more complex microfluidic systems that allow shaping and storing of emulsion droplets42,43. Finally, in tissues, cell-cell and cell-substrate signaling and attachment provide external cues that can be translated into directed spindle orientation, as is often observed in epithelial cells and stem cells. All of these specialized aspects have not been taken into account in this current study and might provide future challenges to build towards more in vivo-like reconstitutions of mitotic spindle assembly and positioning.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
We like to thank the members of the Dogterom lab for valuable discussions. We acknowledge Stefan Diez and Marcel Janson for reagents, and Samara Reck-Peterson for help with the purification of dynein. This work was supported by funding from the European Research Council (ERC Synergy grant: MODEL-CELL).
References
- Kotak S, Gonczy P. Mechanisms of spindle positioning: cortical force generators in the limelight. Curr Opin Cell Biol. 2013;25(6):741–748. doi: 10.1016/j.ceb.2013.07.008. [DOI] [PubMed] [Google Scholar]
- Williams SE, Fuchs E. Oriented divisions, fate decisions. Curr Opin Cell Biol. 2013;25(6):749–758. doi: 10.1016/j.ceb.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum ME, Medema RH. Mechanisms of centrosome separation and bipolar spindle assembly. Dev Cell. 2010;19:797–806. doi: 10.1016/j.devcel.2010.11.011. [DOI] [PubMed] [Google Scholar]
- Civelekoglu-Scholey G, Scholey JM. Mitotic force generators and chromosome segregation. Cell Mol Life Sci. 2010;67(13):2231–2250. doi: 10.1007/s00018-010-0326-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faivre-Moskalenko C, Dogterom M. Dynamics of microtubule asters in microfabricated chambers: the role of catastrophes. Proc Natl Acad Sci U S A. 2002;99(26):16788–16793. doi: 10.1073/pnas.252407099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janson ME, de Dood ME, Dogterom M. Dynamic instability of microtubules is regulated by force. J Cell Biol. 2003;161(6):1029–1034. doi: 10.1083/jcb.200301147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laan L, Husson J, Munteanu EL, Kerssemakers JW, Dogterom M. Force-generation and dynamic instability of microtubule bundles. Proc Natl Acad Sci U S A. 2008;105(26):8920–8925. doi: 10.1073/pnas.0710311105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laan L, et al. Cortical dynein controls microtubule dynamics to generate pulling forces that position microtubule asters. Cell. 2012;148(3):502–514. doi: 10.1016/j.cell.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogterom M, Yurke B. Measurement of the force-velocity relation for growing microtubules. Science. 1997;278(5339):856–860. doi: 10.1126/science.278.5339.856. [DOI] [PubMed] [Google Scholar]
- Cytrynbaum EN, Scholey JM, Mogilner A. A force balance model of early spindle pole separation in Drosophila embryos. Biophys J. 2003;84(2 Pt 1):757–769. doi: 10.1016/S0006-3495(03)74895-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski C, Srayko M, Nedelec F. Cortical microtubule contacts position the spindle in C. elegans embryos. Cell. 2007;129(3):499–510. doi: 10.1016/j.cell.2007.03.027. [DOI] [PubMed] [Google Scholar]
- Carminati JL, Stearns T. Microtubules orient the mitotic spindle in yeast through dynein-dependent interactions with the cell cortex. J Cell Biol. 1997;138(3):629–641. doi: 10.1083/jcb.138.3.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen-Ngoc T, Afshar K, Gonczy P. Coupling of cortical dynein and G alpha proteins mediates spindle positioning in Caenorhabditis elegans. Nat Cell Biol. 2007;9(11):1294–1302. doi: 10.1038/ncb1649. [DOI] [PubMed] [Google Scholar]
- Moore JK, Cooper JA. Coordinating mitosis with cell polarity: Molecular motors at the cell cortex. Semin Cell Dev Biol. 2010;21(3):283–289. doi: 10.1016/j.semcdb.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishchuk EL, Molodtsov MI, Ataullakhanov FI, McIntosh JR. Force production by disassembling microtubules. Nature. 2005;438(7066):384–388. doi: 10.1038/nature04132. [DOI] [PubMed] [Google Scholar]
- Toba S, Watanabe TM, Yamaguchi-Okimoto L, Toyoshima YY, Higuchi H. Overlapping hand-over-hand mechanism of single molecular motility of cytoplasmic dynein. Proc Natl Acad Sci U S A. 2006;103(15):5741–5745. doi: 10.1073/pnas.0508511103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Hoopen R, et al. Mechanism for astral microtubule capture by cortical Bud6p priming spindle polarity in S. cerevisiae. Curr Biol. 2012;22(12):1075–1083. doi: 10.1016/j.cub.2012.04.059. [DOI] [PubMed] [Google Scholar]
- Kapitein LC, et al. The bipolar mitotic kinesin Eg5 moves on both microtubules that it crosslinks. Nature. 2005;435(7038):114–118. doi: 10.1038/nature03503. [DOI] [PubMed] [Google Scholar]
- van den Wildenberg SM, et al. The homotetrameric kinesin-5 KLP61F preferentially crosslinks microtubules into antiparallel orientations. Curr Biol. 2008;18(23):1860–1864. doi: 10.1016/j.cub.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein LC, et al. Microtubule cross-linking triggers the directional motility of kinesin-5. J Cell Biol. 2008;182(3):421–428. doi: 10.1083/jcb.200801145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferenz NP, Gable A, Wadsworth P. Mitotic functions of kinesin-5. Semin Cell Dev Biol. 2010;21(3):255–259. doi: 10.1016/j.semcdb.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuyler SC, Liu JY, Pellman D. The molecular function of Ase1p: evidence for a MAP-dependent midzone-specific spindle matrix. Microtubule-associated proteins. J Cell Biol. 2003;160(4):517–528. doi: 10.1083/jcb.200210021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loiodice I, et al. Ase1p organizes antiparallel microtubule arrays during interphase and mitosis in fission yeast. Mol Biol Cell. 2005;16(4):1756–1768. doi: 10.1091/mbc.E04-10-0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitein LC, et al. Microtubule-driven multimerization recruits ase1p onto overlapping microtubules. Curr Biol. 2008;18(21):1713–1717. doi: 10.1016/j.cub.2008.09.046. [DOI] [PubMed] [Google Scholar]
- Janson ME, et al. Crosslinkers and motors organize dynamic microtubules to form stable bipolar arrays in fission yeast. Cell. 2007;128(2):357–368. doi: 10.1016/j.cell.2006.12.030. [DOI] [PubMed] [Google Scholar]
- Bieling P, Telley IA, Surrey T. A minimal midzone protein module controls formation and length of antiparallel microtubule overlaps. Cell. 2010;142(3):420–432. doi: 10.1016/j.cell.2010.06.033. [DOI] [PubMed] [Google Scholar]
- Mollinari C, et al. PRC1 is a microtubule binding and bundling protein essential to maintain the mitotic spindle midzone. J Cell Biol. 2002;157(7):1175–1186. doi: 10.1083/jcb.200111052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun M, et al. Adaptive braking by Ase1 prevents overlapping microtubules from sliding completely apart. Nat Cell Biol. 2011;13(10):1259–1264. doi: 10.1038/ncb2323. [DOI] [PubMed] [Google Scholar]
- Lansky Z, et al. Diffusible crosslinkers generate directed forces in microtubule networks. Cell. 2015;160(6):1159–1168. doi: 10.1016/j.cell.2015.01.051. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Sato M, Fujita A, Yamamoto M, Toda T. The roles of fission yeast ase1 in mitotic cell division, meiotic nuclear oscillation, and cytokinesis checkpoint signaling. Mol Biol Cell. 2005;16(3):1378–1395. doi: 10.1091/mbc.E04-10-0859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S, Laan L, Dogterom M. Reconstitution of cortical Dynein function. Methods Enzymol. 2014;540:205–230. doi: 10.1016/B978-0-12-397924-7.00012-1. [DOI] [PubMed] [Google Scholar]
- Moudjou M, Bornens M. Celis JE, editor. Method of centrosome isolation from cultured animal cells. Cell Biology: A laboratory handbook. 1998. pp. 111–119.
- Reck-Peterson SL, et al. Single-molecule analysis of dynein processivity and stepping behavior. Cell. 2006;126(2):335–348. doi: 10.1016/j.cell.2006.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster OM, et al. Mitotic rounding alters cell geometry to ensure efficient bipolar spindle formation. Dev Cell. 2013;25(3):270–283. doi: 10.1016/j.devcel.2013.03.014. [DOI] [PubMed] [Google Scholar]
- Good MC, Vahey MD, Skandarajah A, Fletcher DA, Heald R. Cytoplasmic volume modulates spindle size during embryogenesis. Science. 2013;342(6160):856–860. doi: 10.1126/science.1243147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green NM. Avidin and streptavidin. Methods Enzymol. 1990;184:51–67. doi: 10.1016/0076-6879(90)84259-j. [DOI] [PubMed] [Google Scholar]
- Moore W, Zhang C, Clarke PR. Targeting of RCC1 to chromosomes is required for proper mitotic spindle assembly in human cells. Curr Biol. 2002;12(16):1442–1447. doi: 10.1016/s0960-9822(02)01076-x. [DOI] [PubMed] [Google Scholar]
- Akiyoshi B, et al. Tension directly stabilizes reconstituted kinetochore-microtubule attachments. Nature. 2010;468(7323):576–579. doi: 10.1038/nature09594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrimore J, et al. DNA loops generate intracentromere tension in mitosis. J Cell Biol. 2015;210(4):553–564. doi: 10.1083/jcb.201502046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minc N, Burgess D, Chang F. Influence of cell geometry on division-plane positioning. Cell. 2011;144(3):414–426. doi: 10.1016/j.cell.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberner N, et al. In vitro systems for the study of microtubule-based cell polarity in fission yeast. Methods Cell Biol. 2015;128:1–22. doi: 10.1016/bs.mcb.2015.02.008. [DOI] [PubMed] [Google Scholar]
- Boukellal H, Selimovic S, Jia Y, Cristobal G, Fraden S. Simple robust storage of drops and fluids in a microfluidic device. Lab Chip. 2009;9(2):331–338. doi: 10.1039/b808579j. [DOI] [PubMed] [Google Scholar]