

Abstract
During cellular stresses, phosphorylation of eukaryotic initiation factor-2 (eIF2) elicits gene expression designed to ameliorate the underlying cellular disturbance. Central to this stress response is the transcriptional regulator activating transcription factor, ATF4. Here we describe the mechanism regulating ATF4 expression involving the differential contribution of two upstream ORFs (uORFs) in the 5′ leader of the mouse ATF4 mRNA. The 5′ proximal uORF1 is a positive-acting element that facilitates ribosome scanning and reinitiation at downstream coding regions in the ATF4 mRNA. When eIF2-GTP is abundant in nonstressed cells, ribosomes scanning downstream of uORF1 reinitiate at the next coding region, uORF2, an inhibitory element that blocks ATF4 expression. During stress conditions, phosphorylation of eIF2 and the accompanying reduction in the levels of eIF2-GTP increase the time required for the scanning ribosomes to become competent to reinitiate translation. This delayed reinitiation allows for ribosomes to scan through the inhibitory uORF2 and instead reinitiate at the ATF4-coding region. Increased expression of ATF4 would contribute to the expression of genes involved in remediation of cellular stress damage. These results suggest that the mechanism of translation reinitiation involving uORFs is conserved from yeast to mammals.
During environmental stress conditions, cells induce a program of gene expression designed to remedy cellular damage or, alternatively, to elicit apoptosis. Central to the early events in stress response pathways is a family of protein kinases that phosphorylate the α subunit of eukaryotic initiation factor-2 (eIF2) (1–10). In mammals, four eIF2 kinases have been identified, and each recognizes distinct stress signals and modulate downstream response pathways via translational control. These eIF2 kinases include general control nonderepressible-2 (GCN2) that is activated by nutritional stresses, dsRNA induced protein kinase (PKR), important for an antiviral defense pathway mediated by IFN, heme regulated inhibitor (HRI) that couples protein synthesis to the availability of heme in erythroid cells, and pancreatic eIF2 kinase, PEK (also known as Perk), important for remedying protein misfolding in the endoplasmic reticulum (ER). Phosphorylation of the α subunit of eIF2 reduces the levels of eIF2-GTP available for translation initiation, contributing to lowered global protein synthesis concurrent with induced translational expression of genes that function to alleviate stress damage in cells.
We have been interested in the molecular mechanisms by which selected mRNAs are translated in response to eIF2 phosphorylation. A classic example of such a stress remedy pathway involves the transcriptional activator GCN4 in Saccharomyces cerevisiae (1, 2). In yeast starving for nutrients, GCN2 phosphorylation of eIF2 induces translation of GCN4 mRNA. Translational expression of GCN4 occurs by a mechanism that involves four upstream ORFs (uORFs) in the 5′ noncoding portion of the GCN4 mRNA. Hinnebusch and coworkers (1, 2, 11–15) have described three hallmark features in the molecular mechanisms controlling GCN4 translation. First, the 40S ribosome bound to 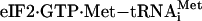 processively scans from the 5′ end of the GCN4 mRNA, eliciting translation at the 5′-proximal uORF1. Second, after synthesis of the uORF1-encoded polypeptide, ribosomes are proposed to retain association with the GCN4 mRNA and reinitiate translation at a downstream coding region. The third feature involves the timing of translation reinitiation depending on the availability of eIF2-GTP. When eIF2-GTP is readily available in nonstressed cells, ribosomes scanning downstream from uORF1 will reinitiate translation at inhibitory uORF 2, 3, or 4. However, when eIF2-GTP levels are reduced due to elevated levels of eIF2 phosphorylation in starved cells, there is a delay in reinitiation that allows the scanning ribosomes to bypass the inhibitory uORFs 2–4 and instead translate the GCN4-coding region. Elevated levels of GCN4 would then contribute to a program of gene expression that coordinates amino acid and purine metabolism, and salvaging of nutrients important for renewal (16, 17).
processively scans from the 5′ end of the GCN4 mRNA, eliciting translation at the 5′-proximal uORF1. Second, after synthesis of the uORF1-encoded polypeptide, ribosomes are proposed to retain association with the GCN4 mRNA and reinitiate translation at a downstream coding region. The third feature involves the timing of translation reinitiation depending on the availability of eIF2-GTP. When eIF2-GTP is readily available in nonstressed cells, ribosomes scanning downstream from uORF1 will reinitiate translation at inhibitory uORF 2, 3, or 4. However, when eIF2-GTP levels are reduced due to elevated levels of eIF2 phosphorylation in starved cells, there is a delay in reinitiation that allows the scanning ribosomes to bypass the inhibitory uORFs 2–4 and instead translate the GCN4-coding region. Elevated levels of GCN4 would then contribute to a program of gene expression that coordinates amino acid and purine metabolism, and salvaging of nutrients important for renewal (16, 17).
Although there is no GCN4 ortholog in mammalian cells, the levels of a related basic zipper transcriptional regulator ATF4 are increased in response to eIF2 phosphorylation during amino acid starvation or ER stress (8, 10, 18, 19). Enhanced levels of ATF4 induce a cascade of transcriptional regulators including CHOP/GADD153 and ATF3, contributing to a program of stress gene expression important for cellular metabolism, the redox status of the cell, and apoptosis (8, 10, 18–20). Induced ATF4 expression occurs predominantly via translation control as evidenced by the preferential ATF4 mRNA association with polysomes that occurs during stress-induced eIF2 phosphorylation (18). Like the GCN4 transcripts, there are uORFs in the 5′ noncoding region of the ATF4 mRNA. In humans and mice, the first ATF4 uORF encodes a polypeptide only 3 amino acid residues in length, whereas the second uORF is 59 amino acid residues in length and overlaps the first 83 nt of the ATF4-coding region. Previously it was suggested that these uORFs contribute jointly to the inhibition of ATF4 translation (18).
In this report we address the molecular mechanisms by which elevated eIF2 phosphorylation controls mouse ATF4 expression. We find that the two uORFs contribute differentially to ATF4 translation. uORF1 is a positive-acting element that facilitates translation of the ATF4-coding region in response to stress-induced eIF2 phosphorylation. By contrast uORF2 is inhibitory, blocking ATF4 expression in nonstressed cells. These results suggest that higher eukaryotes have mechanisms of gene-specific translation control that share the hallmark features described for yeast GCN4.
Materials and Methods
Transcriptional Start Site of ATF4 mRNA. The cDNAs corresponding to the 5′ ends of the ATF4 mRNAs expressed in S/S mouse embryo fibroblast (MEF) cells treated with 0.1 μM thapsigargin (Tg) for 6 h, or no stress, were amplified by using a RNA ligase-mediated RACE kit (RLM-RACE; Ambion, Austin, TX). Total RNA was first isolated by using an RNAesy mini kit (Qiagen, Chatsworth, CA). In brief, 10 μg of total RNA was treated with calf intestinal phosphatase that removes free 5′ phosphates from RNAs other than mRNAs containing intact 5′ cap structures. The RNA was then treated with tobacco acid pyrophosphatase to remove the cap structure, leaving a 5′ monophosphate that was ligated by using T4 RNA ligase to a 45-base RNA adapter oligonucleotide that was supplied in the kit. A random-primed reverse transcription reaction and nested PCR was then performed to amplify the 5′ end of endogenous ATF4 mRNA, as well as transfected thymidine kinase (TK) minimal promoter driven ATF4-Luc transcript. The primers corresponding to the 5′ RACE adapter sequence were provided by the manufacturer. The two nested antisense primers specific to endogenous ATF4 mRNA were as follows: outer primer 5′-CCTTTCTTTATGTTTTTGGCGTC-3′ and inner primer 5′-CTCGAAGGTATCTTTGTCCGTTAC-3′. The outer primer used for amplifying the 5′ ends of ATF4-Luc transcript was 5′-TTGCCGCTGCAGAGCCTGGTGCT-3′, which was combined with the same inner primer listed above. At the end of the PCR, 3 μl of the amplified DNA products were analyzed by electrophoresis using a 2% agarose gel. The prominent DNA band was excised from the gel and sequenced (Fig. 1). The transcriptional start site was determined as the first nucleotide that is 3′ to the adapter sequence ligated to the 5′ of the mRNA transcripts.
Fig. 1.

The two uORFs present in the noncoding portion of the ATF4 mRNA are conserved among vertebrates. (A Upper) DNA was derived by 5′ RACE by using RNA prepared from S/S MEF cells treated with Tg or no stress. DNA samples were separated by electrophoresis in a 2% agarose gel. ATF4 indicates 5′ RACE products prepared by using endogenous ATF4 mRNA, and ATF4-Luc indicates products derived from ATF4-Luc transcripts. Size markers in base pairs are indicated to the right. (A Lower) Sequence of the leader of the ATF4 mRNA. A HindIII restriction site was engineered into the ATF4 DNA. The major transcription start site of the ATF4 gene was determined by sequencing of 5′ RACE products and is indicated by an arrow. ATF4 sequences upstream of this transcription initiation site may contribute to ATF4 transcription, as illustrated by a potential TATAAA box (underlined). Boxes represent uORFs 1 and 2 sequences located upstream of the ATF4-coding region. uORF1 encodes a 3-amino acid-residue polypeptide and is 87 nt upstream of uORF2. uORF2, 180 nt in length, overlaps 83 nt of the ATF4-coding region, which matches the overlap between uORF2 and the ATF4-Luc reporter. Mutations in the uORF1 and uORF2 initiation codons rendering each nonfunctional for ATF4 translation control are indicated below the sequences. Three different stem–loop structures and a 120-bp insertion were individually introduced at PstI restriction site located between uORFs 1 and 2. It is noted that an additional small ORF is present 5′ of the major transcription start site. ATF4-luciferase activity of a reporter construct containing a mutation in the ATG of this uORF was induced in response to stress similar to that measured for WT ATF4-Luc (data not shown), supporting the idea that this region is not present in the ATF4 mRNA. (B) Representative cDNAs encoding ATF4-related sequences in GenBank (accession no.), including human (BC008090), mouse (AK003001), rat (CK601272), cow (CK960046), chicken (AB013138), and zebrafish (CA470055), reveal mRNAs with a similar two-uORF configuration as described for mouse ATF4. Each panel is drawn to scale. Dark-colored boxes represent the two uORFs. The open white-colored box overlapping the uORF2 is the ATF4-coding region. The number of nucleotides between uORF1 and 2 and between start of the uORF2 and the start of the overlapping ATF4-coding region are indicated on top of each panel. The numbers mentioned below each panel represent the number of amino acids encoded by each of the uORFs.
Plasmid Constructions. A full-length mouse ATF4 mRNA leader and ATF4 initiation codon were fused to the luciferase-reporter gene downstream of a minimal TK promoter in plasmid p290 (Fig. 1). To generate this plasmid construct, RT-PCR was performed by using total RNA isolated from S/S MEF cells. Primer sequences based on the ATF4-leader sequence obtained from the RIKEN (The Institute of Physical and Chemical Research, Japan) database are as follows: sense 5′-GCTCAAGCTTGGCTAGGTGTCCCAC-3′ and antisense 5′-GTCATGTTGTGGGGCTTTGC-3′ (GenBank accession no. AK003001). The PCR product obtained from the above reaction was cloned between HindIII and PstI restriction sites in the plasmid construct encoding a TK-ATF4-Luc that was obtained from David Ron (New York University, New York) (18). The initiation codons of each of the uORFs in the ATF4 mRNA were mutated by using the site-directed mutagenesis kit (Stratagene) (Fig. 1). All mutations were sequenced to ensure that there were only the desired changes. Stem–loop structures between uORFs 1 and 2 were derived from previously published reports (13, 21). The insert 5′-CTGCAGCCAAGATGGCTGCAG-3′ has ΔG value of –15 kcal/mol; 5′-CTGCAGTGGTGGAGCTTCCACCACTGCAG-3′ has ΔG value of –24 kcal/mol, and 5′-CTGCAGCCACCACGGCCCCCAAGCTTGGGCCGTGGTGGCTGCAG-3′ has a ΔG value of –41 kcal/mol. The underlined portion of the sequences indicates those regions contributing to the stem structures, and the free energy values were calculated by using a web-based Vienna RNA secondary structure prediction program (http://rna.tbi.univie.ac.at/cgi-bin/RNAfold.cgi). These DNA segments were inserted into the PstI restriction site located between uORFs 1 and 2, generating various plasmids as indicated in Fig. 5. Extension of intercistronic distances between uORFs 1 and 2 was achieved by inserting a heterologous 120-bp sequence that is devoid of any start and stop codons and predicted secondary structure into the PstI site.
Fig. 5.

Model for ATF4 translational control by its leader sequences. The ATF4 mRNA is illustrated as a straight line that has uORFs 1 and 2 that are presented as boxes. The shading of the small ribosomal subunit indicates its association with eIF2-GTP bound 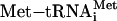 . After translation of the positive-acting uORF1, ribosomes retain the capacity to reinitiate translation at a downstream ORF. The basis for this reinitiation capacity is currently not clear. In the related GCN4 translation mechanism, the termination context of the analogous uORF1 is thought to facilitate the retention of the small ribosomal subunit with the GCN4 mRNA (1, 12, 28). After translation of the ATF4 uORF1, the 40S ribosomal subunits are proposed to resume scanning in a 5′ to 3′ direction along the ATF4 transcript. When eIF2-GTP bound
. After translation of the positive-acting uORF1, ribosomes retain the capacity to reinitiate translation at a downstream ORF. The basis for this reinitiation capacity is currently not clear. In the related GCN4 translation mechanism, the termination context of the analogous uORF1 is thought to facilitate the retention of the small ribosomal subunit with the GCN4 mRNA (1, 12, 28). After translation of the ATF4 uORF1, the 40S ribosomal subunits are proposed to resume scanning in a 5′ to 3′ direction along the ATF4 transcript. When eIF2-GTP bound 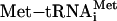 is plentiful during nonstressed conditions, the small ribosomal subunits quickly acquire the eIF2 ternary complex and, coupled with the 60S ribosome, reinitiate translation at uORF2. After translation of this inhibitory uORF2, ribosomes dissociate from the ATF4 mRNA, thereby reducing expression of the ATF4-coding region. When cells are subjected to ER stress or to nutrient deprivation, the levels of eIF2 phosphorylation are enhanced leading to reduced eIF2-GTP levels. After translation of uORF1, there is an increased time required for reacquisition of eIF2-GTP coupled
is plentiful during nonstressed conditions, the small ribosomal subunits quickly acquire the eIF2 ternary complex and, coupled with the 60S ribosome, reinitiate translation at uORF2. After translation of this inhibitory uORF2, ribosomes dissociate from the ATF4 mRNA, thereby reducing expression of the ATF4-coding region. When cells are subjected to ER stress or to nutrient deprivation, the levels of eIF2 phosphorylation are enhanced leading to reduced eIF2-GTP levels. After translation of uORF1, there is an increased time required for reacquisition of eIF2-GTP coupled 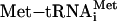 that allows a portion of the scanning 40S ribosomal subunits to scan through the negative-acting uORF2. While scanning the mRNA-leader region from beginning of uORF2 to the initiation codon of the ATF4-coding region, ribosomes reacquire the eIF2 ternary complex, facilitating translational expression of ATF4. When uORF1 is mutated, ribosomes scanning from the 5′ end of the ATF4 mRNA will initiate translation at uORF2. After translation of the inhibitory uORF2, ribosomes dissociate from the ATF4 mRNA, thus lowering translation of the ATF4-coding region even when eIF2-GTP levels are reduced in response to cellular stress. When the distance between uORF1 and uORF2 is increased compared to WT, most ribosomes are competent for reinitiation before reaching uORF2, thereby reducing ATF4 translation independent of eIF2-GTP availability.
that allows a portion of the scanning 40S ribosomal subunits to scan through the negative-acting uORF2. While scanning the mRNA-leader region from beginning of uORF2 to the initiation codon of the ATF4-coding region, ribosomes reacquire the eIF2 ternary complex, facilitating translational expression of ATF4. When uORF1 is mutated, ribosomes scanning from the 5′ end of the ATF4 mRNA will initiate translation at uORF2. After translation of the inhibitory uORF2, ribosomes dissociate from the ATF4 mRNA, thus lowering translation of the ATF4-coding region even when eIF2-GTP levels are reduced in response to cellular stress. When the distance between uORF1 and uORF2 is increased compared to WT, most ribosomes are competent for reinitiation before reaching uORF2, thereby reducing ATF4 translation independent of eIF2-GTP availability.
Cell Culture and Dual Luciferase Assay. S/S and A/A MEF cells obtained from Randall Kaufman (University of Michigan, Ann Arbor) were immortalized by infection of a recombinant retrovirus expressing simian virus 40 large T antigen (22, 23). MEF cells were grown in 24-well plates in DMEM (BioWhittaker) supplemented with 10% FBS, 2 mM glutamine, 1 mM nonessential amino acids, 100 units of penicillin per ml, and 100 μg of streptomycin per ml. Plasmid transfections were performed by using the MEF cells at 40% confluency and the Effectene transfection reagent (Qiagen). Cotransfections were carried out in triplicate by using WT or mutant versions of the TK-ATF4-Luc fusion plasmids and a Renilla luciferase plasmid serving as an internal control (Promega). After transfection (40 h), MEF cells were treated with Tg at 0.1 μM, for 6 h, unless otherwise indicated, or with no ER stress. Dual luciferase assays were carried out as described by the Promega instruction manual. Values are a measure of a ratio of firefly vs. Renilla luciferase units (relative light units, RLU) and represent the mean values of three independent transfections. Immunoblot analysis of phosphorylated and total levels of eIF2α were carried out as described (24).
Northern Blot. MEF cells transfected with WT or various mutant ATF4-luciferase fusion constructs were treated with 0.1 μM Tg for 6 h, or no stress. Total RNA was isolated by using RNAesy kit (Qiagen), and 4 μg of RNA was separated by electrophoresis in a 1.2% agarose gel containing formaldehyde and transferred onto charged Nylon membrane. A 32P-labeled probe complementary to the luciferase reporter or actin genes was used in a Northern blot analysis to measure ATF4-Luc and actin transcripts, respectively. 18S and 28S rRNA was measured by using ethidium bromide staining and UV light.
Results and Discussion
Translational Expression of ATF4 Depends on eIF2 Phosphorylation. Translational expression of ATF4 is significantly induced in response to phosphorylation of eIF2 that occurs in response to cellular stress. To discern the contribution of the 5′ leader sequence in ATF4 translation, a segment including the mouse ATF4 mRNA-leader sequence and initiation codon were inserted into a luciferase reporter gene (Fig. 1) (18). The ATF4-luciferase fusion was expressed downstream of a minimal TK promoter in WT MEF cells. The major transcriptional start site of the ATF4-Luc reporter gene was analyzed by 5′ RACE and DNA sequencing and found to be identical to that of the endogenous ATF4 (Fig. 1). The 5′ leader sequence is 278 nt in length and contains two uORFs preceding the ATF4-coding region. The uORF1 and uORF2 have a Kozak match of A/G at –3 and G at +4 of A(+1)UG, predicting a strong preference for ribosome initiation of translation (25).
Expression of ATF4 protein was previously shown to be increased in response to ER stress (8, 10, 18, 26). Supporting these observations, expression of the ATF4-luciferase was increased 6-fold in MEF cells containing WT eIF2α (S/S) treated with Tg, a known ER stress agent that rapidly induces eIF2α phosphorylation (Fig. 2). By contrast, there was no increase in ATF4-luciferase levels in response to ER stress of MEF cells containing a mutant version of eIF2α with Ala substituted for the phosphorylated residue Ser-51 (A/A). Levels of the ATF4-Luc mRNA were comparable in the stressed and nonstressed S/S MEF cells, supporting a translational mechanism for induced ATF4 expression (Fig. 3). These results are consistent with an earlier report that ATF4 expression is induced in response to eIF2 phosphorylation, and the 5′ noncoding portion of the ATF4 mRNA can facilitate this translation control when fused to a heterologous reporter gene (18).
Fig. 2.

Phosphorylation of eIF2 is required for ATF4 translational expression. (Upper)WT(S/S) or mutant MEF cells containing eIF2α with Ala substituted for Ser-51 (A/A) were treated with 1.0 μM Tg for the indicated number of hours, or no stress (0), and phosphorylation of eIF2α was measured by immunoblot by using Ab that specifically recognizes eIF2α phosphorylated at Ser-51. Total eIF2α was similarly analyzed by using Ab that recognizes both phosphorylated and nonphosphorylated forms of eIF2α. (Lower) The uORFs in the ATF4-Luc reporter construct are indicated as boxes labeled 1 or 2 (not to scale). The S/S and A/A MEF cells were cotransfected with the ATF4-Luc plasmid and a Renilla luciferase plasmid that served as an internal control for normalization. The transfected cells were subsequently treated with the indicated concentrations of Tg for 6 h or no ER stress agent (0 μM). Relative light units (RLU) is a ratio of firefly luciferse units normalized for Renilla luciferase units, and each value was derived from three independent transfections. White-colored bars represent values obtained from nonstressed MEF cells, and gray-colored bars represent values from cells subjected to ER stress.
Fig. 3.

Levels of ATF4-Luc mRNAs are similar between nonstressed and ER stressed MEF cells. Total RNA was prepared from S/S and A/A MEF cells expressing WT ATF4-Luc or mutant versions defective in uORF1 or uORF2 as indicated. The same amount of each total RNA was separated by gel electrophoresis, and 18S and 28S rRNA was visualized by staining with ethidium bromide (Middle). RNA was then transferred to membrane filters, and ATF-Luc transcripts were measured by using a radiolabeled probe complementary to the luciferase reporter gene and autoradiography (Top). Northern blot analyses were also carried out by using a radiolabeled actin probe to ensure characterization of similar amounts of RNA (Bottom). Either a stem–loop with a ΔG = –41 kcal/mol or a 120-bp insert was included in the leader of the ATF4-Luc reporter construct as indicated.
uORF1 and uORF2 Contribute Differentially to ATF4 Translational Control. To determine the roles of uORF1 and uORF2 in the mechanisms regulating ATF4 expression, the initiation codons for each uORF were mutated to AGG, rendering them nonfunctional for translation initiation. The ATF4-Luc reporter plasmid containing the uORF1 and uORF2 mutants individually or in combination were introduced into the S/S or A/A MEF cells and assessed for expression during ER stress. Mutation of uORF1 resulted in a severe reduction in ATF4-Luc expression in S/S MEF cells, independent of stress that was comparable to luciferase levels measured in A/A cells (Fig. 4A). By comparison, when uORF2 was mutated there was a ≈35-fold increase in ATF4-Luc compared to nonstressed WT ATF4-Luc. The increased expression associated with the uORF2 mutation was found in either the S/S or A/A MEF cells irrespective of ER stress (Fig. 4A). Levels of the mutant versions of the ATF4-Luc mRNA were similar between stressed and nonstressed conditions (Fig. 3). When the uORF1 and uORF2 mutations were combined into a single ATF4-Luc reporter construct there were elevated levels of ATF4-Luc in either the S/SorA/A MEF cells, independent of treatment with Tg (Fig. 4A). These results support the idea that uORF1 is a positive element in ATF4 translational control whose function is required to enhance ATF4 expression in response to eIF2 phosphorylation induced during ER stress. By contrast, uORF2 is an inhibitory element that when deleted allows for constitutively high levels of ATF4 translation that is independent of eIF2 phosphorylation. Only after uORF2 is eliminated is uORF1 dispensable for ATF4 expression (Fig. 4A). Therefore uORF1 and uORF2 have opposing roles in the regulation of ATF4 translation, with uORF1 enabling ribosomes to overcome the repressing affects of uORF2.
Fig. 4.

uORF1 functions as a positive regulator, and uORF2 is inhibitory in a scanning mechanism that regulates the translation of ATF4 mRNA. Schematic representation of the WT and different mutant versions of the ATF4-leader sequences fused to luciferase are shown to the left of each luciferase measurement. (A) A box represents the WT version of uORF 1 and uORF2, and an X indicates a nonfunctional uORF due to a mutation in the initiation codon. S/S and A/A MEF cells were cotransfected with the indicated ATF4-Luc plasmid and a control Renilla luciferase plasmid. The transfected cells were treated with Tg for 6 h (gray and black bars) or no ER stress agent (white and stippled bars). Relative light units (RLU) is a ratio of firefly luciferse units normalized for Renilla luciferase units, and each value was derived from three independent transfections. For clarity the histogram is represented in two different scales. (B) Three stem–loop structures with the indicated ΔG values in kcal/mol were inserted between uORF1 and uORF2 in the WT ATF4-Luc construct. Alternatively, the stem–loop structures were inserted in ATF4-leader regions containing an uORF1 mutation (C) or an uORF2 mutation (D). (E) A 120-bp sequence was inserted in the ATF4-leader region between uORF1 and uORF2. The transfected S/S MEF cells were treated with Tg for 6 h (gray bar) or no ER stress agent (white bar), and the RLU was measured as described for A.
Presence of Stem–Loop Structures or Insert Sequences in the ATF4 Leader Decrease Its Expression. One of the key features of GCN4 translational control is ribosome scanning 5′ to 3′ along the GCN4 mRNA. Such directional scanning is instrumental to the sequence of events beginning with uORF1 translation, followed by a reinitiation event that can be delayed with reduced eIF2-GTP levels resulting from eIF2 phosphorylation. To address the importance of ribosome scanning in the regulation of ATF4 translation, DNA segments encoding stem–loop structures with increasing stabilities were engineered between uORF1 and uORF2 in the ATF4-Luc reporter gene and introduced into the S/S MEF cells (Fig. 4B). The presence of a stem–loop with ΔG = –15 kcal/mol in the leader sequence reduced the ATF4-Luc expression to ≈40% of the WT levels measured during ER stress. Furthermore, insertion of stem–loops with progressively greater stability (ΔG = –24 and –41 kcal/mol) inhibited luciferase expression to levels measured for ATF4-Luc containing an uORF1 mutation (Fig. 4 B and C). Insertion of these stem–loop structures in an ATF4-Luc reporter construct containing an uORF2 mutation also led to progressively lower expression that was correlated with the increased stability of the stem–loop (Fig. 4D). Levels of ATF4-Luc transcripts containing the most stable stem–loop structure were similar, with the exception of the reporter construct containing the uORF2 mutant which appeared to have elevated levels in the nonstressed condition (Fig. 3). These results support the model whereby translation of the ATF4-coding region requires ribosome scanning after synthesis of the uORF1-encoded polypeptide.
A central tenet of the GCN4 translational model is that reduced eIF2-GTP levels delay translation reinitiation, allowing scanning ribosomes to bypass inhibitory uORFs and instead translate the GCN4-coding region. We wanted to determine whether the timing of reinitiation is also fundamental to translation of ATF4 mRNA. Such timing of translation reinitiation infers that there is an optimal distance between uORF1 and uORF2 that would facilitate reinitiation at inhibitory uORF2 during nonstressed conditions, but allow for a bypass of the uORF2 in response to eIF2 phosphorylation. To test this premise, we introduced a 120-bp segment devoid of significant secondary structures between uORF1 and uORF2 in the ATF4-Luc reporter construct. Insertion of the 120-bp segment would give reinitiating ribosomes increased time to acquire eIF2-GTP before encountering the initiation codon at uORF2. Therefore, ribosomes would reinitiate at uORF2 even when eIF2-GTP levels are reduced in response to eIF2 phosphorylation. Introduction of the 120-bp sequence was found to significantly reduce ATF4-Luc expression in the S/S MEF cells subjected to ER stress compared to the WT version of ATF4-Luc reporter (Fig. 4E). Similar levels of the ATF4-Luc mRNA with the insertion were found between the nonstressed and ER stressed conditions (Fig. 3). The 120-bp segment was also inserted in the ATF4-Luc constructs containing the uORF1 or uORF2 mutations, resulting in minimal changes in ATF4-Luc expression compared to the ATF4-Luc containing only the corresponding uORF mutation. The ATF4-Luc reporter with the combined uORF1 mutation and 120-bp insert displayed low levels of luciferase expression independent of ER stress, whereas the ATF4-Luc with the insertion and uORF2 mutation gave constitutively high luciferase levels (Fig. 4E). This latter observation demonstrates that the 120-bp insertion does not inherently contribute to reduced translation. Overall, these results support the model that there is an optimum distance between the uORF1 and uORF2 in the ATF4 mRNA leader that allows scanning ribosomes to differentially reinitiate depending on the levels of available eIF2-GTP.
Regulation of ATF4 Translation Follows the Hallmark Features Described for Yeast GCN4. The molecular mechanism inducing ATF4 translation in response to eIF2 phosphorylation shares hallmark features described by Hinnebusch (1, 2) for yeast GCN4. The sequence of events in ATF4 translation follow the processive scanning of ribosomes from the 5′ end of the ATF4 mRNA as illustrated in Fig. 5. These events begin with the translation of the 5′ proximal uORF1. The uORF1 is a positive element in ATF4 translation, and a mutation that blocked translation of this coding region prevented expression of ATF4 even during stress conditions when eIF2 was phosphorylated (Figs. 4 and 5). After translation of the positive-acting uORF1, ribosomes resume scanning along the mRNA and reinitiate translation at a downstream coding region. The importance of ribosome scanning after translation of uORF1 is highlighted by our observation that insertion of stem–loop sequences downstream of uORF1 significantly reduces ATF4 expression even during stress conditions that enhance eIF2 phosphorylation (Fig. 5). It is noteworthy that a previous study describing mouse ATF4 translation in response to cellular stress suggested that uORF1 and uORF2 functioned as tandem inhibitory elements of ATF4-luciferase expression (18). We are not yet certain of the basis for the apparent difference between the previous and present studies. One possible explanation is that the ATF4-luciferase construct used in our studies contained an extended ATF4 segment upstream of the ATF4-luciferase reporter. This configuration resulted in the major transcriptional start site and 5′ mRNA leader being identical to that chararacterized for endogenous mouse ATF4. The transcription initiation site did not appear to be analyzed in the earlier study.
An important feature of the ATF4 regulatory mechanism concerns the timing of the translation reinitiation event. When eIF2-GTP is abundant in nonstressed cells, ribosomes scanning downstream of uORF1 will reinitiate at the next coding region, uORF2 (Fig. 5). After translation of uORF2, ribosomes are thought to dissociate from the ATF4 mRNA. Emphasizing the inhibitory role of uORF2 in ATF4 translation is the observation that a mutation blocking uORF2 translation contributed to elevated expression of ATF4 independent of phosphorylation of eIF2 (Fig. 4A). By comparison, during stress conditions there is enhanced phosphorylation of eIF2 that reduces the levels of eIF2-GTP. Lowered levels of eIF2·GTP·Met-tRNAiMet complex increases the time required for the scanning ribosome to become competent to reinitiate translation. This delayed reinitiation would allow for a portion of the ribosomes to bypass the inhibitory uORF2. During the interval between the uORF2 initiation codon and the ATF4-coding region, scanning ribosomes would have sufficient time to reacquire eIF2·GTP·Met-tRNAiMet and initiate ATF4 translation (Fig. 5). Increased expression of ATF4 would contribute to activation of genes contributing to remediation of cellular stress damage.
In this model, the primary role of uORF1 is to overcome the uORF2 repression of ATF4 translation. This premise is supported by the observation that the uORF2 mutation overcame the requirement of uORF1 for induced ATF4 translation in response to cellular stress (Fig. 4). The 87-nt spacing between uORF1 and uORF2 appears to be optimized to delineate between the changes in eIF2-GTP levels in cells subjected to stressed and nonstressed conditions. When an additional 120-nt sequence was inserted between uORF1 and uORF2, there was significantly reduced expression of ATF4 even during stressed conditions (Fig. 4E). This extended sequence between uORF1 and uORF2 would allow the scanning ribosome additional time to reacquire the eIF2·GTP·Met-tRNAMeti complex, even during stressed conditions, facilitating reinitiation of translation at the inhibitory uORF2 (Fig. 5). This reduction in ATF4 expression was not evident when uORF2 was mutated, emphasizing the role of the uORF2 in diminishing ATF4 translation and demonstrating that the inclusion of insertion sequence did not impede ribosome scanning before translation initiation. Elevated ATF4 synthesis in response to cellular stress contributes in a dosage-dependent fashion to a program of induced gene expression. ATF4 activation of transcription contributes to amino acid synthesis and import, glutathionine biosynthesis, regulation of apoptosis, and cellular differentiation (8, 10, 18–20, 27). Together, expression of this combination of genes is thought to provide cellular protection against a variety of environmental insults. This model is also supported by the high degree of conservation of the disposition of short uORFs in ATF4-related mRNAs in different vertebrates (Fig. 1B).
Acknowledgments
We thank Drs. David Ron and Randal Kaufman for providing plasmids and cell lines, Sheree Wek, Mandar Joshi, and Jill Sergesketter for technical assistance, and the Biochemistry Biotechnology Facility at Indiana University for technical support. This study was supported in part by Grants GM49164 and GM643540 from the National Institutes of Health.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: uORF, upstream ORF; MEF, mouse embryo fibroblast; ER, endoplasmic reticulum; eIF2, eukaryotic initiation factor-2; TK, thymidine kinase; Tg, thapsigargin; GCN, general control nonderepressible; ATF, activating transcription factor.
References
- 1.Hinnebusch, A. G. (1997) J. Biol. Chem. 272, 21661–21664. [DOI] [PubMed] [Google Scholar]
- 2.Hinnebusch, A. G. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Hershey, J. W. B. & Mathews, M. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 185–244.
- 3.Wek, R. C. (1994) Trends Biochem. Sci. 19, 491–496. [DOI] [PubMed] [Google Scholar]
- 4.Dever, T. E. (2002) Cell 108, 545–556. [DOI] [PubMed] [Google Scholar]
- 5.Williams, B. R. (1999) Oncogene 18, 6112–6120. [DOI] [PubMed] [Google Scholar]
- 6.Kaufman, R. J. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Hershey, J. W. B. & Mathews, M. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 503–528.
- 7.Chen, J.-J. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Hershey, J. W. B. & Mathews, M. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 529–546.
- 8.Harding, H. P., Calfon, M., Urano, F., Novoa, I. & Ron, D. (2002) Annu. Rev. Cell Dev. Biol. 18, 575–599. [DOI] [PubMed] [Google Scholar]
- 9.Ron, D. & Harding, H. P. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Hershey, J. W. B. & Mathews, M. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 547–560.
- 10.Kaufman, R. J., Scheuner, D., Schroder, M., Shen, X., Lee, K., Lin, C. Y. & Arnold, S. M. (2002) Nat. Rev. Mol. Cell Biol. 3, 411–421. [DOI] [PubMed] [Google Scholar]
- 11.Mueller, P. P. & Hinnebusch, A. G. (1986) Cell 45, 201–207. [DOI] [PubMed] [Google Scholar]
- 12.Miller, P. F. & Hinnebusch, A. G. (1988) Genes Dev. 8, 1217–1225. [DOI] [PubMed] [Google Scholar]
- 13.Abastado, J. P., Miller, P. F., Jackson, B. M. & Hinnebusch, A. G. (1991) Mol. Cell. Biol. 11, 486–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dever, T. E., Feng, L., Wek, R. C., Cigan, A. M., Donahue, T. F. & Hinnebusch, A. G. (1992) Cell 68, 585–596. [DOI] [PubMed] [Google Scholar]
- 15.Gaba, A., Wang, Z., Krishnamoorthy, T., Hinnebusch, A. G. & Sachs, M. S. (2001) EMBO J. 20, 6453–6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinnebusch, A. G. & Natarajan, K. (2002) Eukaryotic Cell 1, 22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Natarajan, K., Meyer, M. R., Jackson, B. M., Slade, D., Roberts, C., Hinnebusch, A. G. & Marton, M. J. (2001) Mol. Cell. Biol. 21, 4347–4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harding, H. P., Novoa, I., Zhang, Y., Zeng, H., Wek, R., Schapira, M. & Ron, D. (2000) Mol. Cell 6, 1099–1108. [DOI] [PubMed] [Google Scholar]
- 19.Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., Sadri, N., Yun, C., Popko, B., Paules, R., et al. (2003) Mol. Cell 11, 619–633. [DOI] [PubMed] [Google Scholar]
- 20.Jiang, H. Y., Wek, S. A., McGrath, B. C., Lu, D., Hai, T., Harding, H. P., Wang, X., Ron, D., Cavener, D. R. & Wek, R. C. (2004) Mol. Cell. Biol. 24, 1365–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kozak, M. (1989) Mol. Cell. Biol. 9, 5134–5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scheuner, D., Song, B., McEwen, E., Liu, C., Laybutt, R., Gillespie, P., Saunders, T., Bonner-Weir, S. & Kaufman, R. J. (2001) Mol. Cell 7, 1165–1176. [DOI] [PubMed] [Google Scholar]
- 23.Jiang, H. Y., Wek, S. A., McGrath, B. C., Scheuner, D., Kaufmann, R. J., Cavener, D. R. & Wek, R. C. (2003) Mol. Cell. Biol. 23, 5651–5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vattem, K., Staschke, K. A. & Wek, R. C. (2001) Eur. J. Biochem. 268, 3674–3684. [DOI] [PubMed] [Google Scholar]
- 25.Kozak, M. (1991) J. Biol. Chem. 263, 19867–19870. [PubMed] [Google Scholar]
- 26.Harding, H. P., Zhang, Y., Bertolotti, A., Zeng, H. & Ron, D. (2000) Mol. Cell 5, 897–904. [DOI] [PubMed] [Google Scholar]
- 27.Yang, X., Matsuda, K., Bialek, P., Jacquot, S., Masuoka, H. C, Schinke, T., Li L., Brancorsini, S., Sassone-Corsi, P., Townes, T. M., et al. (2004) Cell 117, 387–398. [DOI] [PubMed] [Google Scholar]
- 28.Grant, C. M. & Hinnebusch, A. G. (1994) Mol. Cell. Biol. 14, 606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]