

Abstract
Immune system activation occurs in multiple kidney diseases and pathophysiological processes. The immune system consists of both adaptive and innate components and multiple cell types. Sometimes, the cell type of interest is present in very low numbers among the large numbers of total cells isolated from the kidney. Hence, reliable and efficient isolation of kidney mononuclear cell populations is important in order to study the immunological problems associated with kidney diseases. Traditionally, tissue isolation of kidney mononuclear cells have been performed via enzymatic digestions using different varieties and strengths of collagenases/DNAses yielding varying numbers of viable immune cells. Recently, with the development of the mechanical tissue disruptors for single cell isolation, the collagenase digestion step is avoided and replaced by a simple mechanical disruption of the kidneys after extraction from the mouse. Herein, we demonstrate a simple yet efficient method for the isolation of kidney mononuclear cells for every day immune cell extractions. We further demonstrate an example of subset analysis of immune cells in the kidney. Importantly, this technique can be adapted to other soft and non-fibrous tissues such as the liver and brain.
Keywords: Immunology, Issue 114, Kidney, immune cells, macrophages, lymphocytes, isolation, perfusion, efficient, reliable
Introduction
Immune system activation occurs in multiple kidney diseases and pathophysiological processes 6,10,11,13. Potential areas of active research encompass the various triggers for immune system activation, various cell types involved, the cytokine/chemokine pattern in a particular disease setting, modulation of all of the aforementioned processes by a particular drug etc. To exemplify, in ischemia-reperfusion injury (a model for acute kidney injury), there is an increase in immune cells or bone-marrow derived hematopoietic cells or CD45+ cells within a few hours, which is sustained through the period of repair or fibrosis (6 weeks later) 5,12. These immune cells secrete both pro-inflammatory and anti-inflammatory cytokines and chemokines to orchestrate the process of repair 5,12. Currently, the ability to use multiple fluorophores simultaneously to label cell populations in a single cell suspension has increased with the advent of modern flow cytometry machines with four to five lasers. This has substantially added to the capability to discriminate the cell populations based on their functional status 3,7. For example, to accurately label a macrophage as F4/80lowCD11bhighLy6bhighCD206low, at least 3 more fluorophores would be needed in the same sample volume to gate for live cells, CD45+ (leukocytes) and Ly6G- (neutrophils) and this is very much possible with the newer Flow Cytometers 3. However, the downstream assays for cytokine secretion, cell proliferation, cytotoxicity, macrophage activation and the quantification of numbers of various subsets of lymphocytes and monocytes not only needs good quality (live cells, singlets) but adequate numbers of cells.
The immune system in the kidney is made up of both adaptive and innate components and multiple cell types 1,7,13. For example, in mice the two kidneys together are reported to contain 2-17% (28,000-266,000) CD45+ cells of the total kidney immune cells isolated (1.4 x 106 cells) and about 5-15% (1,400-4,200) of these are CD4+ cells 1,5,12. A small percentage (5-15%, 70-630) of these CD4+ cells are FoxP3+ cells (Figure 1)1. Due to these step wise reductions in percentages of cells, sometimes the cell population of interest (in this case CD45+CD4+FoxP3+ cells) is represented by a mere ~100 cells. The small number of CD45+CD4+FoxP3+ cells makes it imperative that a large number of total cells are isolated and the cells are of good quality for downstream studies such as cytokine secretion assays. Moreover, it may be necessary to combine kidneys from 2-3 mice since the subpopulations are not represented in high enough numbers to perform quantifiable assays. Hence, reliable and efficient isolation of kidney mononuclear cell populations is desirable in order to study the immunological spectrum associated with kidney diseases.
Traditionally, for isolation of kidney mononuclear cells, investigators have used a variety of enzymatic digestions such as collagenase 1A or II including DNase 1 1,5,12. It is well known that collagenases have enzymatic activity that varies with lot numbers and by company of manufacture, necessitating titration for the optimum concentration and duration of incubation 4,14,15. In addition, digestion with collagenase adds time for mincing the kidney into small pieces, necessitates incubation of the kidney pieces in a heated (37 °C) bath and additional time for incubation in EDTA for stopping the reaction. In addition, less sterility may be achieved for some downstream procedures needing cell culture. More importantly, depending on the investigator and all the variables involved, it leads to variability in the data and interpretation across laboratories. Recently, with the development of mechanical tissue disruptors/homogenizers 16, the collagenase digestion step is completely avoided and replaced by a simple mechanical disruption of the kidneys 2. Herein, we demonstrate a simple yet efficient method for the isolation of kidney immune cells for everyday immune cell extractions. Importantly, this technique can be adapted to other soft and non-fibrous tissues such as the liver and brain 16.
Protocol
All protocol steps performed were reviewed and approved by the University of Missouri Animal Care and Use Committee (ACUC). For this protocol, male C57Bl/6 mice aged 15 weeks were utilized although theoretically any rodent at any age can be used for experiments. Since, this is a non-survival surgery, euthanasia is achieved by exsanguination and bilateral pneumothorax.
1. Perfusion of the Kidneys
Note: Perfusion of organs such as heart, liver and kidney removes the blood which may interfere with the interpretation of data. Hence, if possible we always perfuse the organs.
Place the mouse in an isoflurane (3 ml/min) induction chamber till it is immobile. Next, remove the mouse out of the chamber, lay it dorsally on the dissecting table and place the isoflurane cone on the mouse nose and push isoflurane at the rate of 1.5 ml/min through the regulator.
Check the toe pinch response with tweezers to assess if the animal has achieved a surgical plane of anesthesia. Clean the ventral abdomen with saline and cut open the abdomen with scissors and push the abdominal organs (out of the surgical field) to the left side of the mouse.
Collect blood from the inferior vena cava with a 25 gauge needle-fixed to a 1 cc syringe and add it to a 1.5 ml microcentrifuge tube containing 10 μl of 0.5M EDTA. Note: Collection of blood is optional. Plasma may be used for biochemical measurements. Leukocytes may be used for flow cytometry assays.
Using scissors, make a small cut in the right atria to let out the mixture of blood and PBS from the next step.
Take a 50 cc syringe and fill with ice-cold PBS (20-30 cc). Next, attach a 21-25 G needle to the syringe, puncture the left ventricle at its apex (while holding the heart gently between forceps) and slowly perfuse the left ventricle with phosphate buffered saline pH 7.4 (PBS) over the course of 1-2 min. Note: The heart immediately blanches upon the first few milliliters of perfusion. Observe the liver for blanching, as this indicates that the perfusion is draining blood out of the liver via venous return through the vena cava. In addition, make sure that the lungs are not expanding during the infusion as this indicates puncture of the right ventricle. If the lungs were to expand, withdraw the needle slightly back into the left ventricle and continue the perfusion. The time taken from the first incision till euthanasia is achieved is less than 2 min
2. Dissection of Kidneys
After perfusion is complete, use scissors and forceps to dissect the right kidney from its attachments (blood vessels, fat and adrenal glands) and excise.
Gently remove the capsule covering the kidney by cutting and dissecting with a forceps. Weigh the kidney. Note: Weighing the kidney is optional. However, some experiments may result in change in kidney size/weight which may be used to normalize cell yield per gram of tissue.
After weighing the kidney, place it in 1 ml of RPMI-1640 containing 0.5% FBS or flow cytometry staining buffer solution (50 ml conical screw cap tube) and place the tube on ice till further use. Note: For ease of description, we will describe the rest of the isolation of immune cells in the flow cytometry staining buffer solution. In addition, we will describe isolation of immune cells from a single kidney, although both kidneys or 1+1/3rd of the kidneys can be used depending on the downstream experiments (optional). 1/3rd of the kidney could be frozen for protein/RNA experiments and 1/3rd can be placed in buffers for immunohistochemistry or transmission electron microscopy.
3. Homogenization of Kidneys
Label a 5-80 ml capacity homogenization bag with sample identification information, remove the inside strainer (STR) and discard it. Fill the bag with 5 ml of ice cold Flow Cytometry Staining Buffer solution and transfer kidney to the buffer. Repeat this process for a second sample or more samples that need processing.
Fold the homogenization bag in half, such that one half has the kidney submerged in the Flow Cytometry Staining Buffer solution and the other half is simply folded over it.
- Turn on the tissue homogenizer and set it to fast (setting) rpm. Place the folded homogenization bag with the kidney facing the paddle, close to the bottom edge but make sure the lower end of the bags are not sliding below the lower border of the paddles.
- Process the second sample simultaneously as there are 2 paddles. Select time as 120 sec. Observe the paddles move back and forth to homogenize the kidneys. Note: Meanwhile, this downtime can be used to prepare 2 more kidney (or other tissue) samples while the first two are processing.
After completing with first set of homogenization, transfer the bags to ice and reload the tissue homogenizer with the next set of samples and so on. Visually ensure that the kidneys are adequately homogenized and no large chunks are visible. Note: Generally, 2 min in the tissue homogenizer at fast rpm is adequate for the kidney.
Place a 100 µm cell strainer into the 50 ml conical tube on ice. Next, pipette the homogenate completely on to the cell strainer. Set a floor centrifuge at 50 x g for 1 min at 4 °C and load the samples into the centrifuge. Note: Centrifugation will ensure that all homogenate (containing cells) has passed through the cell strainer.
Remove the tubes from the centrifuge and place them back on ice. Discard the cell strainers and keep the homogenate. Add equal volume (~ 6 ml) of 1x RBC lysis buffer to the cell pellet/supernatant and pipette up and down several times to make a uniform cell suspension. Visually, observe some of the red color in the suspension decrease immediately. Leave the tubes on ice for 5 min to achieve maximum RBC lysis. Note: the RBC lysis step can be skipped if the investigator is confident of the kidney perfusion and less cell manipulation is desired. We have observed a significant increase in kidney cell yield by not subjecting the cell suspension to RBC lysis.
- Spin the tubes in the centrifuge that has been set to 500 x g, 4 °C for 5 min to form a cell pellet. Note: During steps 3.6 and 3.7, prepare for colloidal silica (Percoll) based density gradient centrifugation.
- Take 10 ml round bottom polypropylene tubes and label them with corresponding sample numbers and place them in a tube rack. Note: The following describes how to make enough density gradient reagents for 4 kidney samples.
- Take 7 ml of Flow Cytometry Staining Buffer solution and pipette it into a 15 ml conical tube. Add 1 ml of 2 M sucrose to prepare 0.25 M sucrose.
- Next, take 7.2 ml of density gradient reagent and pipette it into another 15 ml conical tube. Add 1.55 ml of Flow Cytometry Staining Buffer solution and 1.25 ml of 2 M sucrose to prepare 72% density gradient reagent and 0.25 M sucrose.
- Pipette 1 ml of 72% density gradient reagent into one round bottom polypropylene tube for each sample.
4. Gradient Centrifugation
After Step 3.7, take the tubes out and gently decant the supernatant in one smooth motion and also decant the next drop of liquid that forms while the tube is held upside down. Note: This step is important since leaving behind excess liquid can change the density gradient enough to create variation in yields. When the tube is set back upright, ~ 200-300 µl of liquid remains with the cell pellet.
Pipette 1 ml of 0.25 M sucrose onto the cell pellet and gently but vigorously pipette up and down to bring the cells to a uniform suspension. Next, add the cell suspension to 1 ml 72% density gradient reagent (3.7.4) and once again pipette vigorously to mix the cells to form a 36% density gradient reagent.
Take 1 ml of the 72% density gradient reagent into a polypropylene Pasteur (transfer) pipette. With even pressure on the bulb (keeps the liquid column in the tip and prevents bubble formation), insert the Pasteur pipette to the bottom of the cell suspension (36% density gradient reagent) gently and unload the 72% density gradient reagent to form a distinct heavier layer.
Pick the sample tubes gently and place into the centrifuge, which is at room temperature for this step. Spin for 20-30 min at 500 x g.
Meanwhile, prepare another two sets of polypropylene round bottom centrifuge tubes with the corresponding sample identifiers.
After Step 4.4 is complete, remove the sample containing polypropylene tubes gently, with minimal shaking and set it down on the tube rack.
- Pick up the first sample tube and its corresponding empty tube. With a polypropylene Pasteur pipette, gently but thoroughly with measured suction, remove the upper "junk layer" or "gooey material" and put it into the empty round bottom centrifuge tube.
- Proceed removing the upper part of the gradient till the "buffy coat" is reached (may appear off-white with a reddish tinge [red blood cells]). Stop suctioning if a height of ~3 mm is reached from the "buffy coat". Note: Ensure that this stuff is completely gone from the upper part of the gradient before continuing to suction.
Take a fresh Pasteur pipette and a fresh round bottom centrifuge tube and gently remove the buffy coat. Continue to suction 3 - 5 mm below the buffy coat, to ensure that most of the cells are collected and added to the 2nd new round bottom tube with appropriate sample identifier.
Add 1-2 ml of Flow Cytometry Staining Buffer solution to the 2nd tube and mix thoroughly by pipetting up and down.
Spin down the cells at 500 x g for 5 min at 4 °C. A 2nd wash step with Flow Cytometry Staining Buffer solution is optional. Resuspend in 500 µl of Flow Cytometry Staining Buffer solution and proceed to count cells using a hemocytometer.
5. Optional (Cell Labeling)
Aliqout each sample for the same number of cells. Add the blocking antibody at a dilution of 0.5 µg per 106 cells (anti- CD16/CD32 FcR) and incubate on ice for 15 min.
Add the desired cocktail of primary antibodies (conjugated to the appropriate fluorophores) that are determined to work well with each other by the investigator, prior to the actual experiments. Incubate on ice for 30 min. Wash the cells with PBS. Note: For the demonstration of kidney immune cell labeling for this manuscript, we used a T-lymphocyte and a macrophage/dendritic cell panel. The T-lymphocyte panel consisted of Fixable Viability Stain FVS510, anti-CD45 (clone: 30-F11) BV421, Anti-Foxp3 (Clone: FJK-16s) APC, Anti-CD127 (Clone: A7R34) PE/Cy7, anti-CD44 (Clone: IM7) PerCP/Cy5.5, anti-CD4 (Clone: RM4-5) APC-Cy7, anti-CD8 (Clone: 53-6.7) BV785. The macrophage panel consisted of Fixable Viability Stain FVS510, anti-CD45 (clone: 30-F11) BV421, Anti-Ly6G (Clone: 1A8) FITC, Anti-CD11b (Clone: M1/70) PerCP-Cy5.5, Anti-F4/80 (Clone: BM8) APC, Anti-CD11c (Clone: N418) BV785.
Add the Fixable Viability Stain and leave on bench-top for 5 min. Wash the cells with Flow Cytometry Staining Buffer solution to remove excess of the Fixable Viability Stain.
Add Fixation/Permeabilization reagent and incubate overnight at 4 ºC. Wash in permeabilization solution.
For intracellular stain, block again with anti-CD16/32 FcR for 10min. Add the intracellular stains and incubate for 20-30 min. Wash 2x in permeabilization solution.
Resuspend in Flow Cytometry Staining Buffer solution and run samples through FACS machine 3,9.
Representative Results
The number of panels that can be run depends on the number of immune cells that can be reliably extracted out of the kidneys. Herein, we demonstrate the ability to run 2 panels, one for T-lymphocytes and one for macrophages/dendritic cells. On the T-lymphocyte panel, we first look at the forward scatter (FSC) and side scatter (SSC) pattern and delineate the population of interest as shown in Figure 1 (top left dot plot). Next, a viability marker, in this case a fixable viability stain (FVS510) is used to exclude the dead cells from the analysis (Figure 1, top middle dot plot). CD45, a marker for bone marrow derived hematopoietic cells is typically used to delineate the kidney immune cells and these typically range from 2-18% of the total kidney immune cells depending on the age, sex, strain, inflammatory condition of the mouse (Figure 1, top right dot plot). From the CD45+ bone marrow derived hematopoietic cells, one can either gate on CD3 epsilon as a marker for T-lymphocytes or proceed to CD4+/CD8+ to separate the effector/cytotoxic cells from the bone marrow derived hematopoietic cells (Figure 1, bottom right dot blot). Typically, more CD4+ than CD8+ cells are seen. Further characterization of the CD4+ cells revealed ~8.8% FoxP3+ cells that are likely T-regulatory cells (Figure 1, bottom middle dot plot). Further detailed characterization may include markers such as CD25 and CD127 to distinguish which subset is altered depending on the population of interest. CD127 is largely a marker for FoxP3- cells and this is further dissected out by using CD44 as marker of activation, CD44+CD127lo/- best resemble activated effector cells versus CD44+CD127+ cells that best resemble effector memory cells.
For the macrophage/dendritic cell panel, the initial gates are the same, including the FVS510 based separation of live/dead (Figure 2, top 2nd from left dot plot) and CD45 gates to separate the bone marrow derived hematopoietic cells (top 2nd from right dot plot). Next, the CD45+ cells can be gated several ways. We gated them on Ly6G here to separate neutrophils from the rest of the monocytes i.e. macrophages/dendritic cells, (Figure 2, top right dot plot). Next, Ly6G- cells were gated on CD11b and F4/80 to identify subsets of macrophages and dendritic cells (Figure 2, bottom 2nd from left dot plot). Gating on Ly6C identifies the percentage of infiltrating/inflammatory monocytes/macrophages (bottom 2nd from right histogram) and CCR2 and CX3CR1 identifies the macrophages that were recruited (Figure 2, bottom right dot plot). Bottom left most panel identifies most cells as dendritic cells, a lower portion of which were recruited via CCR2 expression.
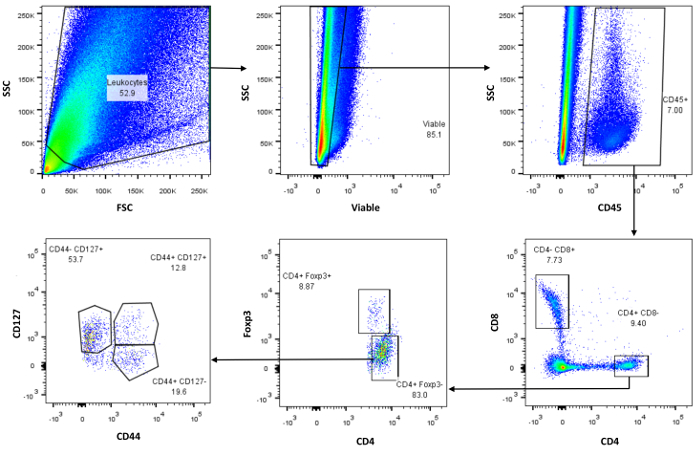 Figure 1:Representative Dot Plots Displaying T-Lymphocyte Subset Isolation. Top left is a typical forward (FSC) and side (SSC) scatter profile of kidney immune cells in the Flo Jo software. Top middle panel is gated on a Fixable Viability Stain (FVS510) and the live population on the left is then gated on CD45 for bone marrow derived immune cells. CD45+ cells are gated for CD4 (effector) and CD8 (cytotoxic) cells (Lower right panel). Middle panel shows the number of CD4+ cells that are FoxP3+ (T regulatory cells). Lower left panel attempts to tease out how many of the CD4+Foxp3- cells are activated effector CD44+CD127lo/- versus memory effector CD44+CD127+. Please click here to view a larger version of this figure.
Figure 1:Representative Dot Plots Displaying T-Lymphocyte Subset Isolation. Top left is a typical forward (FSC) and side (SSC) scatter profile of kidney immune cells in the Flo Jo software. Top middle panel is gated on a Fixable Viability Stain (FVS510) and the live population on the left is then gated on CD45 for bone marrow derived immune cells. CD45+ cells are gated for CD4 (effector) and CD8 (cytotoxic) cells (Lower right panel). Middle panel shows the number of CD4+ cells that are FoxP3+ (T regulatory cells). Lower left panel attempts to tease out how many of the CD4+Foxp3- cells are activated effector CD44+CD127lo/- versus memory effector CD44+CD127+. Please click here to view a larger version of this figure.
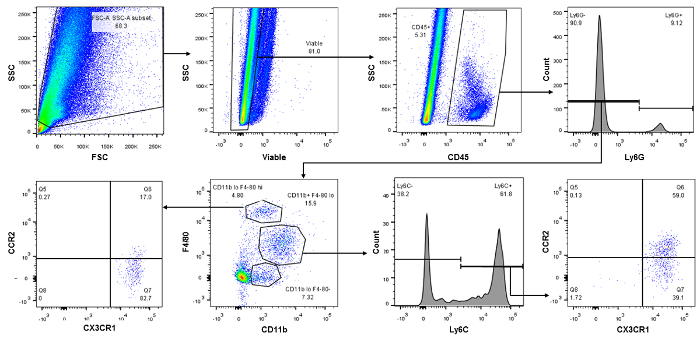 Figure 2:Representative Dot Plots Displaying Monocyte-Cell Subset Isolation. Again, top left is a typical forward (FSC) and side (SSC) scatter profile of kidney immune cells in the Flo Jo software. Top 2nd from left dot plot is gated on a Fixable Viability Stain (FVS510) and the live population on the left is then gated on CD45 for bone marrow derived immune cells (Top 2nd from right dot plot). CD45+ cells are gated for Ly6G (neutrophils, top right dot plot) in order to separate for macrophages and dendritic cells. Gating on Ly6C identifies the percentage of infiltrating/inflammatory monocytes/macrophages (bottom 2nd from right histogram) and CCR2 and CX3CR1 identifies the macrophages that were recruited (Figure 2, bottom right dot plot). Bottom left most panel identifies most cells as dendritic cells, a lower portion of which were recruited via CCR2 expression. Please click here to view a larger version of this figure.
Figure 2:Representative Dot Plots Displaying Monocyte-Cell Subset Isolation. Again, top left is a typical forward (FSC) and side (SSC) scatter profile of kidney immune cells in the Flo Jo software. Top 2nd from left dot plot is gated on a Fixable Viability Stain (FVS510) and the live population on the left is then gated on CD45 for bone marrow derived immune cells (Top 2nd from right dot plot). CD45+ cells are gated for Ly6G (neutrophils, top right dot plot) in order to separate for macrophages and dendritic cells. Gating on Ly6C identifies the percentage of infiltrating/inflammatory monocytes/macrophages (bottom 2nd from right histogram) and CCR2 and CX3CR1 identifies the macrophages that were recruited (Figure 2, bottom right dot plot). Bottom left most panel identifies most cells as dendritic cells, a lower portion of which were recruited via CCR2 expression. Please click here to view a larger version of this figure.
Table 1: Reagents and Equipment Needed for Immune Cell Isolation From the Kidney. See Materials Table.
Table 2: Table Depicting total Cells and CD45+ Isolated in the Corresponding Prep. Please click here to download this file.
Discussion
We have presented here a methodology to obtain immune cells from the kidney in a reliable and efficient manner. The major modification to the widely used collagenase digestion step (mechanical disruption of tissue) saves about 30 min and the isolation of a large number of viable immune cells takes under two hours for 4 kidney samples. Moreover, depending on our research question, we now only use a single kidney (the other kidney can be used for protein analysis by Western blots, immunohistochemistry and mRNA analysis by PCR) for our immune cell isolation and routinely get ~2 x 106 cells per isolation. We have used this method to isolate large number of immune cells from the liver and heart (multienzyme digestion for the heart combined with mechanical disruption, data not shown) but are confident that mechanical disruption can be applied to other non-fibrous tissues such as the brain 16.
As with any protocol to isolate immune cells from tissues, adhering to detail is critical for success. If perfusion of the kidneys is critical, then practice is needed in placing the needle into the heart to prevent it from fibrillating or from penetrating too far to the right ventricle. An alternative to putting a needle into the heart is to cannulate the aorta above the renal artery and perfuse the kidneys 8. In addition, there are other suggestions from the authors to make the isolation of immune cells reliable and uniform. The kidneys should be adequately mashed and no large chunks should be visible. If this is not the case, then repetition of the mechanical disruption should solve the problem. The mechanically disrupted tissue should be passed through the right sized filter in order to include cells of all sizes. For example, some macrophage/dendritic cells populations may be excluded if using a 40 µm mesh as opposed to using a 100 µm mesh (macrophage/dendritic cell sizes vary between 10-40 µm). However, on the flip side, more kidney epithelial and other cells could be included in the final analysis changing the percentages of immune cell populations. We believe that being all inclusive increases the representation of underrepresented populations and hence improves the reliability of the prep.
When adding the cell suspension to the density gradient reagent, the cells should be resuspended in no more than 200-300 µl of the Flow Cytometry Staining Buffer solution. Formation of the 36%:72% gradient should be ensured. For this, introduction of the transfer pipette carrying the 72% density gradient reagent into the 36% density gradient reagent should be gentle, should not leave bubbles and a clear demarcation seen between the gradients. As far as possible, centrifugation of cells in density gradient reagent should be done at room temperature. Complete removal of the "cell debris" at the top surface of the gradient after the spin should be ensured. Otherwise, it will interfere with the cell yield and the quality of the prep. Take care to exclude the tiniest cells on the hemocytometer. These are left over RBCs. This is particularly important if the RBC lysis step is omitted. There is a limitation to the method. Not every cell isolated using this method is an immune cell. A large percentage of cells are epithelial cells (includes proximal tubule, distal tubule and glomerulus). However, this limitation can be used to an investigator's advantage as questions pertaining to non-immune cells can be answered.
To conclude, the method for isolation of immune cells from the kidney presented here is widely applicable and can be adapted by most labs to their research. We believe that this technique can be used for other non-fibrous tissues such as liver and brain as well. Isolation of a large number of immune cells will also help to design more ambitious downstream experiments such as proliferation assays, cytokine secretion assays and isolation of a subset of cells based on surface antigens. Ultimately, we hope that this method will standardize immune cell isolation i.e. avoid various levels of enzymatic digestion of surface antigens.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work is supported by a Research Grant from Dialysis Clinics Inc. and from the University of Missouri Research Board Grant.
References
- Ascon DB, et al. Phenotypic and functional characterization of kidney-infiltrating lymphocytes in renal ischemia reperfusion injury. J. Immunol. 2006;177(5):3380–3387. doi: 10.4049/jimmunol.177.5.3380. [DOI] [PubMed] [Google Scholar]
- Ascon M, et al. Renal ischemia-reperfusion leads to long term infiltration of activated and effector-memory T lymphocytes. Kidney Int. 2009;75(5):526–535. doi: 10.1038/ki.2008.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belliere J, et al. Specific macrophage subtypes influence the progression of rhabdomyolysis-induced kidney injury. J. Am. Soc. Nephrol. 2015;26(6):1363–1377. doi: 10.1681/ASN.2014040320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, et al. Collagenase digestion down-regulates the density of CD27 on lymphocytes. J. Immunol. Methods. 2014;413:57–61. doi: 10.1016/j.jim.2014.06.017. [DOI] [PubMed] [Google Scholar]
- Dong X, et al. Resident dendritic cells are the predominant TNF-secreting cell in early renal ischemia-reperfusion injury. Kidney Int. 2007;71(7):619–628. doi: 10.1038/sj.ki.5002132. [DOI] [PubMed] [Google Scholar]
- Hickey FB, Martin F. Diabetic kidney disease and immune modulation. Curr. Opin. Pharmacol. 2013. [DOI] [PubMed]
- Kawakami T, et al. Resident renal mononuclear phagocytes comprise five discrete populations with distinct phenotypes and functions. J. Immunol. 2013;191(6):3358–3372. doi: 10.4049/jimmunol.1300342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, et al. Isolating glomeruli from mice: A practical approach for beginners. Exp. Ther. Med. 2013;5(5):1322–1326. doi: 10.3892/etm.2013.1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picot J, Guerin CL, Le Van KC, Boulanger CM. Flow cytometry: retrospective, fundamentals and recent instrumentation. Cytotechnology. 2012;64(2):109–130. doi: 10.1007/s10616-011-9415-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricardo SD, van GH, Eddy AA. Macrophage diversity in renal injury and repair. J. Clin. Invest. 2008;118(11):3522–3530. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Iturbe B, Pons H, Herrera-Acosta J, Johnson RJ. Role of immunocompetent cells in nonimmune renal diseases. Kidney Int. 2001;59(5):1626–1640. doi: 10.1046/j.1523-1755.2001.0590051626.x. [DOI] [PubMed] [Google Scholar]
- Vielhauer V, et al. Efficient renal recruitment of macrophages and T cells in mice lacking the duffy antigen/receptor for chemokines. Am. J. Pathol. 2009;175(1):119–131. doi: 10.2353/ajpath.2009.080590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisheit CK, Engel DR, Kurts C. Dendritic Cells and Macrophages: Sentinels in the Kidney. Clin. J. Am. Soc. Nephrol. 2015. [DOI] [PMC free article] [PubMed]
- Williams SK, McKenney S, Jarrell BE. Collagenase lot selection and purification for adipose tissue digestion. Cell Transplant. 1995;4(3):281–289. doi: 10.1177/096368979500400306. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, et al. Deterioration and variability of highly purified collagenase blends used in clinical islet isolation. Transplantation. 2007;84(8):997–1002. doi: 10.1097/01.tp.0000284979.48497.de. [DOI] [PubMed] [Google Scholar]
- Zhou J, Nagarkatti P, Zhong Y, Nagarkatti M. Immune modulation by chondroitin sulfate and its degraded disaccharide product in the development of an experimental model of multiple sclerosis. J. Neuroimmunol. 2010;223(1-2):55–64. doi: 10.1016/j.jneuroim.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]