

Abstract
The glycoprotein family of collagens represents the main structural proteins in the human body, and are key components of biomaterials used in modern tissue engineering. A technical bottleneck is the deposition of collagen in vitro, as it is notoriously slow, resulting in sub-optimal formation of connective tissue and subsequent tissue cohesion, particularly in skin models. Here, we describe a method which involves the addition of differentially-sized sucrose co-polymers to skin cultures to generate macromolecular crowding (MMC), which results in a dramatic enhancement of collagen deposition. Particularly, dermal fibroblasts deposited a significant amount of collagen I/IV/VII and fibronectin under MMC in comparison to controls.
The protocol also describes a method to decellularize crowded cell layers, exposing significant amounts of extracellular matrix (ECM) which were retained on the culture surface as evidenced by immunocytochemistry. Total matrix mass and distribution pattern was studied using interference reflection microscopy. Interestingly, fibroblasts, keratinocytes and co-cultures produced cell-derived matrices (CDM) of varying composition and morphology. CDM could be used as "bio-scaffolds" for secondary cell seeding, where the current use of coatings or scaffolds, typically from xenogenic animal sources, can be avoided, thus moving towards more clinically relevant applications.
In addition, this protocol describes the application of MMC during the submerged phase of a 3D-organotypic skin co-culture model which was sufficient to enhance ECM deposition in the dermo-epidermal junction (DEJ), in particular, collagen VII, the major component of anchoring fibrils. Electron microscopy confirmed the presence of anchoring fibrils in cultures developed with MMC, as compared to controls. This is significant as anchoring fibrils tether the dermis to the epidermis, hence, having a pre-formed mature DEJ may benefit skin graft recipients in terms of graft stability and overall wound healing. Furthermore, culture time was condensed from 5 weeks to 3 weeks to obtain a mature construct, when using MMC, reducing costs.
Keywords: Bioengineering, Issue 114, keratinocytes, fibroblasts, macromolecular crowding, 2D cell culture, 3D cell culture, skin organotypic co-cultures, extracellular matrix, collagen type I, collagen type IV, collagen type VII, in vitro skin models, dermo-epidermal junction
Introduction
The skin forms a protective barrier by preventing water loss and pathogen entry. It is made up of three major components; the stromal rich dermis1, a stratified epidermis2,3,4 on top of it, and the dermo-epidermal junction in between5,6. The dermis is composed largely of collagen and elastic fibers and is sparsely populated with fibroblasts7. In contrast, the cell-rich epidermis is composed of multiple layers of keratinocytes. The keratinocytes of the inner-most layer are proliferative and provide new basal cells which renew and replace terminally differentiating keratinocytes that constantly move to the outer-most layer of the skin and have lost their nuclei and cytoplasmic material, resulting in a cornified layer which undergoes desquamation.
The dermal-epidermal junction, a specific type of basement membrane, is a complex structure composed of interconnecting matrix molecules, which tethers the epidermis to the dermis. Collagen I fibers of the dermis interlace with collagen VII anchoring fibrils which are anchored to the collagen IV rich lamina densa. Anchoring filaments (laminin 5, collagen XVII and integrins) in turn connect the lamina densa with hemidesmosomes of basal keratinocytes. Basal keratinocytes (stratum basale) have the capacity to proliferate and renew, as well as differentiate and stratify to form the suprabasal layers; stratum spinosum, stratum granulosum, and finally the stratum corneum, which represents the contact surface of the skin with the environment. En route from the basal layer to the cornified layer, keratinocytes switch expression patterns of cytokeratins and finally undergo apoptosis and encase themselves in cornified envelopes, hulls of specific proteins that are covalently crosslinked by transglutaminase activity.
Recreating skin and its layers in vitro, including the complex structures of the dermal-epidermal junction and dermal extracellular matrix, and to emulate the cornification process, has long intrigued scientists and bioengineers as a challenging task. There have been significant advances in skin tissue engineering, for example, the successful extraction of skin cells from patient biopsies and the generation of skin organotypic cultures using patient-derived skin cells8. However, unsolved problems remain pertaining to poor secretion of extracellular matrix proteins by the skin cells themselves and resulting in sub-optimal skin models. Furthermore, the time required to generate 3D organotypic skin co-culture using current protocols varies between four to eight weeks, a timeframe which could potentially be shortened with the incorporation of macromolecular crowders. Reducing culture time saves reagent cost, reduces the incidence of cell senescence and reduces the waiting time of the patient should the product be used in the clinic.
Macromolecular crowding (MMC) involves introducing specific macromolecules to the culture medium to generate excluded volume effects. These affect enzymatic reaction rates including the proteolytic cleavage of procollagen which is tardy under standard aqueous culture conditions9-13. Under MMC, enzymatic reactions are hastened without increasing the amount of reagents14,15 resulting in, in the case of procollagen cleavage, an increased amount of collagen I molecules in crowded cultures as compared to uncrowded controls10. As the conversion of procollagen to collagen allows the formation of collagen assemblies, fibroblasts cultured with MMC for 48 hours yielded significantly more collagen I as compared to uncrowded fibroblast cultures monitored for up to four weeks11,16,17. Besides effects on enzymatic activities that affect the formation, stabilization and remodeling of ECM, MMC also has been shown to directly enhance and modulate collagen fiber formation18,19.
We present here a method to enhance the extracellular matrix (ECM) production by skin cells, in particular, dermal fibroblasts and epidermal keratinocytes. In addition, we show that the enriched ECM produced under MMC in monolayer cultures can be decellularized and used as pure cell-derived matrix (CDM).
We use a non-conventional approach to visualize and fully appreciate the ECM deposited by skin cells cultured with MMC. Interference reflection microscopy is typically used for studying cell-matrix interactions or cell-to-glass contact points. This technique was utilized in our system to view the total amount of matrix deposited on the glass surface. Interference reflection microscopy was coupled with fluorescent immunostaining to obtain the most amount of information in terms of extracellular matrix composition and pattern, in the presence and absence of MMC.
Organotypic skin co-cultures is a classical method to model the skin in vitro in a three-dimensional context. While two-dimensional co-cultures may provide significant information, it is limited when translating this data and applying it back to an in vivo environment, which is inherently a three-dimensional structure. Skin keratinocytes, in particular, are polarized and contain apical and basal segments which are essential for homeostasis and cell adherence. In addition, the expression of typical suprabasal proteins in keratinocytes above the basal layer, such as keratin 1, keratin 10 and filaggrin is only present in the course of stratification and terminal differentiation of keratinocytes. As terminal differentiation is hardly present in typical monolayer cultures, suprabasal protein expression is normally not achieved in this culture system. Therefore, organotypic cultures start submerged in culture medium, but are then lifted to an air-liquid interface to drive keratinocyte differentiation. This results in the expression of stratification markers, even cornification and a generally better reflection of epidermal physiology. While other groups have previously generated organotypic skin co-cultures successfully, the establishment of a functional dermo-epidermal junction zone has been an issue. Here, we present a new method for culturing organotypic skin co-cultures with an enhanced basement membrane, in a condensed time frame and without compromising the maturity of these constructs. This would provide skin mimetics for in vitro modeling, the study of skin biology and an assortment of screening assays.
Protocol
1. Macromolecular Crowding in 2D Skin Cell Cultures
Seed 50,000 cells (primary fibroblasts or primary keratinocytes or co-culture of primary fibroblasts and keratinocytes) per well of a 24-well plate. Seed cells in 1 ml of the corresponding cell type growth media.
Allow cells to adhere overnight, in a 37 °C incubator at 5% CO2.
Discard old media and replace with 1 ml fresh media containing macromolecular crowders and 100 µM ascorbic acid. Use a crowder cocktail consisting of 37.5 mg/ml Ficoll 70 and 25 mg/ml Ficoll 400. Use fibroblast media (FM) consisting of DMEM supplemented with 10% FBS and 1% penicillin-streptomycin antibiotics. Grow keratinocytes in a serum-free media, KSFM or CnT-57. Maintain co-cultures in a mixture of 50% FM and 50% serum-free media.
Incubate cultures for 6 days in a 37 °C incubator at 5% CO2, with a media change every 2 days.
2. Fluorescent Immunostaining on Skin Cell Cultures
Wash cell cultures twice with 1x PBS.
- Fix cell cultures with either methanol or paraformaldehyde (depending on the antibody specifications).
- For Methanol Fixation
- Aliquot 500 µl of cold methanol into each well and incubate at -20 °C for 10 min. Aspirate fixative and discard methanol waste. Air dry in a laminar fume hood.
- For Paraformaldehyde Fixation
- Aliquot 500 µl of 2% paraformaldehyde solution into each well and incubate at room temperature for 15 min. Aspirate fixative and discard paraformaldehyde waste.
- Wash with 1x PBS, for three washes, 5 min per wash.
- Aliquot 1 ml of 1x PBS into each well.
- Fluorescent Immunostaining Using Antibodies
- Aliquot 500 µl of 3% bovine serum albumin (diluted in 1x PBS) into each well. Incubate at room temperature for 30 min. Aspirate the blocking solution and discard.
- Primary Antibody
- For imaging directly from plate: Aliquot 200 µl of primary antibody (1:100 dilution in 10% goat serum) solution into each well and incubate for 90 min at room temperature.
- For coverslips: Aliquot 50 µl of primary antibody (1:100 dilution in 10% goat serum) solution onto a flat piece of Parafilm. Place coverslip onto the Parafilm, with the cell-side (part of coverslip with cells) facing the solution. Cover the entire cell-side, with no bubbles. Incubate for 90 min at room temperature.
- Aspirate primary antibody solution and discard.
- Wash with 1x PBS, for three washes, 5 min per wash.
- Secondary Antibody
- For imaging directly from plate: Aliquot 200 µl of secondary antibody (1:400 dilution in 10% goat serum) solution into each well and incubate for 30 min at room temperature, covering the plate with aluminum foil.
- For coverslips: Aliquot 50 µl of secondary antibody (1:400 dilution in 10% goat serum) solution onto a flat piece of Parafilm. Place coverslip onto the Parafilm, with the cell-side (part of coverslip with cells) facing the solution. Cover the entire cell-side, with no bubbles. Incubate for 30 min at room temperature, covering the Parafilm with aluminum foil.
- Aspirate secondary antibody solution and discard.
- Wash with 1x PBS, for three washes, 5 min per wash. Aliquot 1 ml of 1x PBS into each well.
- Mounting
- For imaging directly from plate: do not mount. Proceed to microscope.
- For imaging of coverslips: Mount coverslips, in mounting media, onto a glass slide. Dry overnight in a container covered with aluminum foil. Proceed to microscope.
Acquire Images Using a Confocal Laser Scanning Microscope with a 40X/1.30 Oil Objective.
3. Western Blotting of Skin Cell Cultures
Wash cell layers twice with 1x PBS.
Aliquot 100 µl of lysis buffer (see Materials List) into each well (6-well plate format). Scrape the cell layer with a cell scraper and transfer solution into a microcentrifuge tube.
Centrifuge solution at 15,330 x g for 30 min at 4 °C. Transfer the supernatant to another microcentrifuge tube and discard the pellet.
Mix 13 µl of each sample with 5 µl of sample buffer and 2 µl of reducing agent (see Materials List). Heat samples at 95 °C for 10 min.
Centrifuge samples at 15,330 x g for 1 min to collect condensation water. Load samples into a pre-cast PAGE gel and run at 100 V for approximately 1 hr.
To transfer separated proteins to a nitrocellulose membrane, sandwich the gel and membrane between pieces of paper and sponges (of the same size). Ensure there are no bubbles between the gel, membrane, papers and sponges. Close the sandwich cassette and run the transfer at 70 V for at 2 hr.
To check if the transfer was complete, incubate the membrane with 10 ml Ponceau S. Wash membrane with 0.1% PBS-Tween-20, three times, 10 min per wash.
Block with 10 ml of 5% milk for 1 hr at room temperature. Wash membrane with 0.1% PBS-Tween-20, three times, 10 min per wash.
Incubate with 10 ml primary antibody for 90 min (mouse anti-collagen VII, LH 7.2, 1:1,000 dilution in 5% milk). Wash membrane with 0.1% PBS-Tween-20, three times, 10 min per wash.
Incubate with 10 ml secondary antibody for 30 min (goat anti-mouse-HRP, 1:2,000 dilution in 5% milk). Wash membrane with 0.1% PBS-Tween-20, three times, 10 min per wash.
Transfer the membrane to a cassette and add 1 ml of ECL Detection Reagent. Place a clear, thin plastic sheet over the membrane.
To detect chemiluminescence, place a light-sensitive film over the plastic sheet and close the cassette. After 2-5 min, remove the film from the cassette and place it into a developer.
4. Making and Using a Cell-derived Matrix
Seed 50,000 cells (primary fibroblasts or primary keratinocytes or co-culture of primary fibroblasts and keratinocytes) per well of a 24-well plate.
Allow cells to adhere overnight, in a 37 °C incubator at 5% CO2.
Discard old media and replace with fresh media containing macromolecular crowders and 100 µM ascorbic acid. The crowder cocktail consists of 37.5 mg/ml Ficoll 70 and 25 mg/ml Ficoll 400. Fibroblast media (FM) consists of DMEM supplemented with 10% FBS and 1% pen-strep. Keratinocytes are grown in a serum-free media, KSFM or CnT-57. Maintain co-cultures in a mixture of 50% FM and 50% KSFM or CnT-57.
Incubate cultures for 6 days in a 37 °C incubator at 5% CO2, changing media every 2 days.
- Decellularize cell layers to obtain a cell-derived matrix (f-mat = fibroblast-derived matrix, k-mat = keratinocyte-derived matrix, co-mat = matrix derived from a co-culture of fibroblasts and keratinocytes).
- Wash cell layers twice in 1x PBS.
- Add 250 µl of 0.5% sodium deoxycholate. Incubate, on ice, for 10 min. Aspirate cell lysate.
- Repeat Step 4.5.2 thrice.
- Wash cell-derived matrix twice in distilled H2O. Keep cell-derived matrix in 1x PBS. Matrices can be stored for up to 1 week at 4 °C.
- Prepare Secondary Cell Suspension (For Example, Seeding Keratinocytes on Top of f-mat).
- Aspirate 1x PBS.
- Seed 50,000 keratinocytes on top of the cell-derived matrix (f-mat). Allow cells to adhere overnight, in a 37 °C incubator at 5% CO2. Note: Assays may be performed directly on the adherent cells.
5. Characterization of the Cell-derived Matrix
Fluorescent immunostaining: Refer to Protocol 2.
- Interference Reflection Microscopy
- Seed cells on to glass coverslips, following Protocol 4.1-4.5.
- After obtaining the cell-derived matrix, fix samples following Protocol 2.2. Note: Antibody staining is optional. If required, refer to Protocol 2.3.
- Perform image acquisition by using a confocal laser scanning microscope with a 40X/1.30 oil objective. Set the pinhole at 74 mm.
- Use LP 505 for the interference reflection microscopy (IRM) channel and BP 575-615 IR for the fluorescence red channel for filters. Use NT 80/20 for the IRM channel and HFT 405/488/561, NFT 565, plate for the fluorescence red channel for the beam splitters. Use the following lasers: DPSS 561-10 (wavelength 561 nm) at 1.1% power for the fluorescence red channel and HeNe633 (wavelength 633 nm) at 5.0% power for the IRM channel.
6. 3D Organotypic Skin Co-culture
- Cast a Collagen Gel, with Fibroblasts Encapsulated, in a Cell-culture Insert (6-well plate format).
- Aliquot 10 ml of rat tail collagen type I to a cold beaker.
- Add 1 ml of cold DMEM. Add 0.5 ml of 1 M NaOH to neutralize the acidic collagen. Add 0.5 ml of fibroblast suspension (containing 1,200,000 fibroblasts).
- Transfer the solution, in a cold pipette, to the cell-culture inserts within a 6-well plate. 12 ml of total solution is evenly divided into 6 cell-culture inserts.
- Incubate the gel in a 37 °C incubator at 5% CO2, for 1 hr.
- After the gel has solidified, submerge the gel in FM. After 24 hr, discard the old media and replace with fresh media, containing macromolecular crowders. Add a volume of 2 ml to the interior of the cell-culture insert and 2 ml to the outside of the cell-culture insert.
- After 24 hr, Seed Keratinocytes on top of the Collagen Gel.
- Trypsinize keratinocytes using 0.125% trypsin/chelating agent for 5 min at 37 °C. Tap sides of flask gently to dislodge cells and neutralize trypsin with serum containing media. Count cells and prepare keratinocyte suspension (300,000 keratinocytes per gel and suspended in 200 µl media).
- Aspirate old media from cell-culture insert. Add keratinocytes on top of the gel.
- Incubate the gel in a 37 °C incubator at 5% CO2, for 1 hr.
- After keratinocytes have attached to the gel surface, submerge the gel with 2 ml KSFM or CnT-57 to the inside of the cell-culture insert and 2 ml of FM to the outside of the cell-culture insert.
After 24 hr, discard the old media and replace with fresh media, containing macromolecular crowders.
- After 7 days of submerged culture, raise the culture to an air-liquid interface.
- Transfer the cell-culture insert to a deep-well plate. Add 10 ml stratification media to the outside of the cell-culture insert. Keep the interior of the cell-culture insert dry.
Incubate cultures in a 37 °C incubator at 5% CO2 and change media every 3 days, for the next 14 days. Note: After 2 weeks at the air-liquid interface, the skin equivalent is mature and can be harvested (either snap frozen or formalin fixed).
7. Harvest and Characterization of Organotypic Skin Equivalents
Aspirate stratification media.
Using tweezers, transfer the culture inserts onto a paper towel and dab away remaining media from the base of the insert.
Using a scalpel, cut along the inner circumference of the culture insert.
- Fix the organotypic skin co-culture (together with the PET membrane).
- Snap Freeze
- Transfer the gel construct to a cryomold. Cover the interior of the cryomold with OCT compound. Place the cryomold into liquid nitrogen for 10 sec.
- Using tweezers, remove the frozen cryomold from the liquid nitrogen, wrap in aluminum foil and store the frozen samples in a -80 °C freezer.
- Section frozen samples with a cryostat, with a section thickness of 7 µm.
- Formalin Fixation
- Place a 'biopsy sponge pad' into the 'biopsy cassette'. Transfer the gel construct onto the sponge pad. Place another sponge pad gently on top of the gel construct. Close the cassette.
- Fully submerge the cassette in 10% neutral buffered formalin for 48 hr, at room temperature. Remove the cassette and discard formalin waste.
- Embed the fixed sample into a wax block by first dehydrating with an ethanol series and then gradually infiltrating the sample with wax. Section the wax block with a microtome.
- Characterization of the Skin Equivalent
- Immunostaining
- For frozen Sections:
- Incubate sections in 10% goat serum (diluted in 1x PBS) for 1 hr at room temperature. Wash sections in 1x PBS to remove the goat serum.
- Incubate sections with primary antibody (1:100 dilution in 10% goat serum) for 90 min at room temperature. Wash sections in 1x PBS, three times, 5 min per wash.
- Incubate sections with secondary antibody (1:400 dilution in 10% goat serum) for 30 min at room temperature, in a container covered with aluminum foil. Wash sections in 1x PBS, three times, 5 min per wash.
- Mount slides, with mounting media, onto a coverglass. Cover samples with aluminum foil and allow to dry overnight.
- Image using a confocal microscope with a 40X/1.30 oil objective.
- For Formalin Fixed Paraffin Embedded Sections:
- Dewax sections (and rehydrate) in descending percentages of ethanol to water (Xylene - 100% ethanol - 90% ethanol - 80% ethanol - 70% ethanol - water). Fully submerge samples in solutions. Each dewaxing step should be 3 min.
- Incubate sections in 1% hydrogen peroxide for 30 min. Wash sections in 1x PBS for 5 min.
- Incubate sections in citrate solution (10 ml citrate solution is dissolved in 90 ml water) at 120 °C for 20 min. Allow slides to cool overnight, at room temperature. Wash sections in 1x PBS, three times, 5 min per wash.
- Incubate sections in 10% goat serum (diluted in 1x PBS) for 1 hr at room temperature. Wash sections in 1x PBS to remove the goat serum.
- Incubate sections with primary antibody (1:100 dilution in 10% goat serum) for 90 min at room temperature. Wash sections in 1x PBS, three times, 10 min per wash.
- Incubate sections with HRP-labeled polymer (undiluted) for 30 min, at room temperature. Wash sections with 1x PBS, three times, 10 min per wash.
- To a microcentrifuge tube, combine 1 ml DAB Substrate (3,3'-diaminobenzidine) with 1 drop of chromogen. Incubate the sections with this solution for 15 sec - 5 min (brown color = positive signal).
- To stop the reaction, wash the sections in running water.
- Counterstain nuclei with hematoxylin, for 5 min. Rinse sections in running water.
- Incubate sections in acid alcohol solution, for 1 min. Rinse sections in running water.
- Incubate sections in Scott's tap water solution, for 2 min. Rinse sections in running water.
- Optional: Incubate sections in eosin for 1 min and wash in running water.
- Dehydrate sections in ascending percentages of ethanol to xylene (70% ethanol - 80% ethanol - 90% ethanol - 100% ethanol - xylene). Ensure that samples are fully submerged for 3 min per stage.
- Mount slides, using mounting media, to a coverglass. Air dry overnight.
- Image using a confocal microscope with a 40X/1.30 oil objective.
- Transmission Electron Microscopy (To Visualize Anchoring Fibrils)
- Fix samples in 2.5% glutaraldehyde for 72 hr.
- Wash samples in 1x PBS, three times, 10 min per wash.
- Cut into small pieces (1 mm3). Post-fix samples in 1% osmium tetroxide, pH 7.4, for 1 hr at room temperature. Wash samples in distilled water. Caution: Osmium tetroxide is poisonous and toxic and should be handled carefully in a fume hood.
- Dehydrate samples through an ascending ethanol series (25% ethanol - 50% ethanol - 75% ethanol - 95% ethanol - 100% ethanol - acetone) for 20 min per step.
- Infiltrate by soaking samples in 100% acetone: araldite resin (1:1) for 30 min at room temperature and then at 1:3 for 1 hr, followed by 1:6 overnight. Soak sample in fresh araldite resin for 2 hr at room temperature, followed by a second change of fresh araldite resin for 1 hr at 40 °C. Leave the third fresh araldite resin change for 1 hr at 45 °C and leave the fourth fresh araldite resin change for 1 hr at 50 °C.
- Incubate samples at 60 °C for 24 hr to allow polymerization.
- Section samples using a glass knife, to obtain semi-thin sections with a thickness of 1 µm. Stain section with methylene blue to observe histology. Section the sample to obtain ultra-thin sections of 70 nm and collect each section onto a copper grid.
- Stain sections with 5% uranyl acetate solution for 15 min followed by 3% lead citrate solution for 10 min.
- Acquire images with a transmission electron microscope (100 kV), at a magnification of 30,000X.
Representative Results
Macromolecular crowding was able to enhance ECM deposition, in particular, fibroblasts deposited more collagen I, IV and fibronectin as compared to control cultures (Figure 1, Cell layer; 1A, collagen I; 1B, collagen IV; 1C, fibronectin). Upon decellularization, it was evident that fibroblasts were the main depositors of collagen I, IV and fibronectin as compared to keratinocytes (Figure 1, Matrix).
In Figure 2, it was observed that fibroblasts rather than keratinocytes were the main producers of collagen VII. This is the first report of collagen VII deposited successfully in vitro (Benny et al, 2015).
The cell-derived matrix was characterized through immunofluorescence using specific antibodies. While this is a classic approach, it was important to visualize the ECM in its totality and fully appreciate the effect of the macromolecular crowders. Using interference reflection microscopy (IRM), the full extent of the matrix was captured (Figure 3). An overlay of the antibody staining with the IRM image shows the relative quantity of that ECM component in relation to the total amount of ECM.
In a 3D in vitro skin model, MMC condensed the culture time from 5 weeks to 3 weeks to obtain a mature organotypic skin co-culture (Figure 4). A hematoxylin and eosin staining showed that at 3 weeks, the culture with macromolecular crowders consisted of a pluri-stratified epidermis and stromal rich dermis, as compared to uncrowded control cultures which lacked a completely differentiated epidermis. In addition, intense and continuous collagen VII immunostaining was detected at the dermal-epidermal junction of crowded cell cultures, as compared to a weak and spotty collagen VII immunostaining in control cultures. Transmission electron microscopy (Figure5) showed the presence of anchoring fibrils in organotypic cultures, showing functional collagen VII.
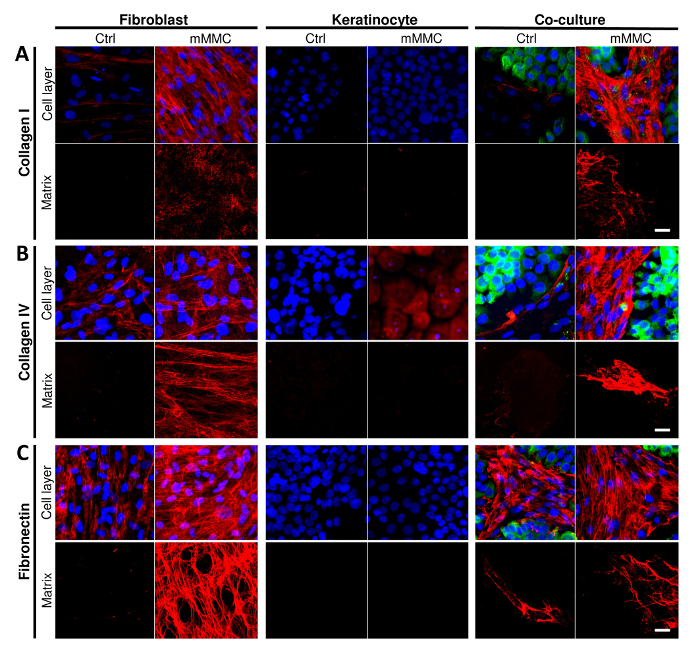 Figure 1: Mixed macromolecular crowding (mMMC) enhances the deposition of dermal-epidermal junction components in vitro. (A) Collagen I deposition is enhanced by mMMC (cell layer and matrix) in fibroblasts only. Crowding of co-cultures produce the most collagen I and show that keratinocytes stimulated collagen I production by fibroblasts. (B) Collagen IV deposition by fibroblasts is enhanced by crowding. This is seen even more clearly in crowded co-cultures. Of note, keratinocytes stained for collagen IV show mostly cell-associated or intracellular collagen IV, but not a pericellular matrix. In co-cultures, both cell types segregate with collagen IV being predominantly associated with fibroblasts sparing keratinocyte islands. (C) Fibronectin deposition was only seen with fibroblasts, and therein strongly enhanced by crowding (cell layer and matrix). In co-cultures, a reticular mesh of fibronectin was associated with fibroblasts only, sparing islands of keratinocytes. Scale bars = 20 µm. This figure has been modified from Benny et al., 2015. Please click here to view a larger version of this figure.
Figure 1: Mixed macromolecular crowding (mMMC) enhances the deposition of dermal-epidermal junction components in vitro. (A) Collagen I deposition is enhanced by mMMC (cell layer and matrix) in fibroblasts only. Crowding of co-cultures produce the most collagen I and show that keratinocytes stimulated collagen I production by fibroblasts. (B) Collagen IV deposition by fibroblasts is enhanced by crowding. This is seen even more clearly in crowded co-cultures. Of note, keratinocytes stained for collagen IV show mostly cell-associated or intracellular collagen IV, but not a pericellular matrix. In co-cultures, both cell types segregate with collagen IV being predominantly associated with fibroblasts sparing keratinocyte islands. (C) Fibronectin deposition was only seen with fibroblasts, and therein strongly enhanced by crowding (cell layer and matrix). In co-cultures, a reticular mesh of fibronectin was associated with fibroblasts only, sparing islands of keratinocytes. Scale bars = 20 µm. This figure has been modified from Benny et al., 2015. Please click here to view a larger version of this figure.
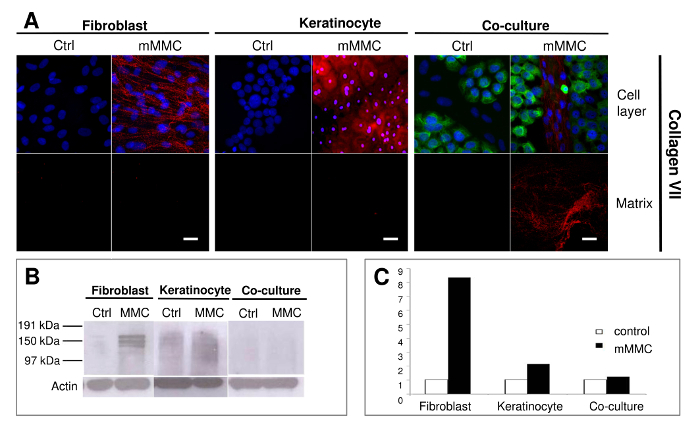 Figure 2: Mixed macromolecular crowding (mMMC) facilitates the deposition of anchoring fibril building collagen VII. (A) A reticular deposition pattern of collagen VII deposition is evident with fibroblasts only under mMMC. In co-cultures, extracellular collagen VII is strongly associated with fibroblast colonies in between keratinocyte islands. Keratinocytes show pericellular and intracellular collagen VII more strongly expressed in the presence of mMMC. After cell lysis, collagen VII footprints are seen in a fine granular layer in mMMC-treated fibroblast cultures, but a discernible fibrillar deposition is retrieved from co-cultures. (B) Immunoblot analysis of lysed cell layers shows that both crowded fibroblasts and keratinocyte cultures contain significantly more collagen VII compared with uncrowded controls. The retrieved collagen VII is mainly pericellular derived. (C) Densitometric analysis of B shows that mMMC increases the amount of cell-associated collagen VII by a factor of 8 in fibroblasts and a factor of 2 in keratinocytes. Scale bars = 20 µm. This figure has been modified from Benny et al., 2015. Please click here to view a larger version of this figure.
Figure 2: Mixed macromolecular crowding (mMMC) facilitates the deposition of anchoring fibril building collagen VII. (A) A reticular deposition pattern of collagen VII deposition is evident with fibroblasts only under mMMC. In co-cultures, extracellular collagen VII is strongly associated with fibroblast colonies in between keratinocyte islands. Keratinocytes show pericellular and intracellular collagen VII more strongly expressed in the presence of mMMC. After cell lysis, collagen VII footprints are seen in a fine granular layer in mMMC-treated fibroblast cultures, but a discernible fibrillar deposition is retrieved from co-cultures. (B) Immunoblot analysis of lysed cell layers shows that both crowded fibroblasts and keratinocyte cultures contain significantly more collagen VII compared with uncrowded controls. The retrieved collagen VII is mainly pericellular derived. (C) Densitometric analysis of B shows that mMMC increases the amount of cell-associated collagen VII by a factor of 8 in fibroblasts and a factor of 2 in keratinocytes. Scale bars = 20 µm. This figure has been modified from Benny et al., 2015. Please click here to view a larger version of this figure.
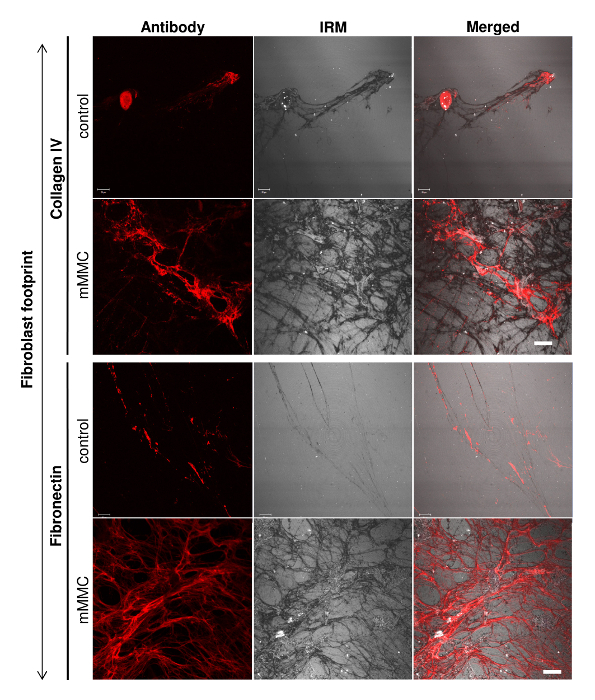 Figure 3: Fibroblast footprints contain more total ECM as visualized by interference reflection microscopy (IRM). On analysis of individual ECM components (collagen IV and fibronectin), culture under mMMC enhanced the extracellular deposition. To visualize the total ECM deposited, IRM was used to quantify all ECM as antibody staining had its limitations. IRM clearly showed the total matrix quantity and pattern under mMMC as compared with control conditions. Scale bar = 20 µm. This figure has been modified from Benny et al., 2015. Please click here to view a larger version of this figure.
Figure 3: Fibroblast footprints contain more total ECM as visualized by interference reflection microscopy (IRM). On analysis of individual ECM components (collagen IV and fibronectin), culture under mMMC enhanced the extracellular deposition. To visualize the total ECM deposited, IRM was used to quantify all ECM as antibody staining had its limitations. IRM clearly showed the total matrix quantity and pattern under mMMC as compared with control conditions. Scale bar = 20 µm. This figure has been modified from Benny et al., 2015. Please click here to view a larger version of this figure.
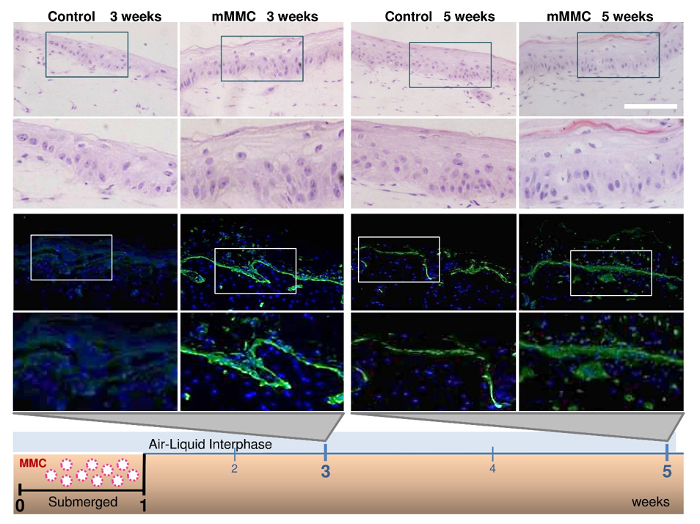 Figure 4: Mixed macromolecular crowding (mMMC) during the submerged phase enhances maturation of the dermal-epidermal junction (DEJ) in skin equivalents. Fibroblast-containing collagen gels were seeded with keratinocytes on top and kept submerged for 1 week, then lifted to an air-liquid interface. In the classical protocol, collagen VII (green) was absent after a total of 3 weeks in culture, but appeared in skin equivalents after 5 weeks. In contrast, under mMMC, collagen VII was already strongly evident after 3 weeks and even more strongly stained after 5 weeks compared with standard cultures. Hematoxylin and eosin (20X magnification) staining confirmed that with this rapid protocol, stratification and maturity of the skin equivalent were maintained and accelerated. Scale bar = 100 µm. This figure has been modified from Benny et al., 2015. Please click here to view a larger version of this figure.
Figure 4: Mixed macromolecular crowding (mMMC) during the submerged phase enhances maturation of the dermal-epidermal junction (DEJ) in skin equivalents. Fibroblast-containing collagen gels were seeded with keratinocytes on top and kept submerged for 1 week, then lifted to an air-liquid interface. In the classical protocol, collagen VII (green) was absent after a total of 3 weeks in culture, but appeared in skin equivalents after 5 weeks. In contrast, under mMMC, collagen VII was already strongly evident after 3 weeks and even more strongly stained after 5 weeks compared with standard cultures. Hematoxylin and eosin (20X magnification) staining confirmed that with this rapid protocol, stratification and maturity of the skin equivalent were maintained and accelerated. Scale bar = 100 µm. This figure has been modified from Benny et al., 2015. Please click here to view a larger version of this figure.
 Figure 5: Evidence of de novo formation of anchoring fibrils in skin equivalents generated under mMMC. Ultrastructural studies of the nascent dermal-epidermal junction of organotypic co-cultures after a 3 week culture protocol with mMMC (A, B) suggests structures akin to anchoring fibrils (arrows) that are absent in non-crowded skin equivalents (C) after 3 weeks of culture. This figure has been modified from Benny et al., 2015. Please click here to view a larger version of this figure.
Figure 5: Evidence of de novo formation of anchoring fibrils in skin equivalents generated under mMMC. Ultrastructural studies of the nascent dermal-epidermal junction of organotypic co-cultures after a 3 week culture protocol with mMMC (A, B) suggests structures akin to anchoring fibrils (arrows) that are absent in non-crowded skin equivalents (C) after 3 weeks of culture. This figure has been modified from Benny et al., 2015. Please click here to view a larger version of this figure.
Discussion
Enhanced extracellular matrix is obtained upon introduction of macromolecular crowders to cell culture, owing to the excluded volume effect which increases the propeptide cleavage by proteinases. This results in extracellular matrix, in particular collagen, to be processed faster and deposited on the culture surface. While other groups have obtained thick fibroblast cell layers, it involved a culture time of several weeks20. In contrast, MMC described in this Protocol, dramatically shortens the culture time while increasing the amount of ECM. The increase in reaction rates in crowded cell cultures has also been reported in chondrocyte matrix deposition21, extracellular matrix organization22,23 and adipogenesis24.
After decellularization of crowded cell layers, cell-derived matrices are harvested and used for various tissue engineering applications, one of which is the seeding of secondary cell cultures on top of the cell-derived matrix25,26. As the matrices are cell-derived and natural products, degradation might occur if kept at non-ideal conditions for extended periods of time. A significant benefit of the cell-derived matrix, obtained under MMC, is that it is cell-derived and cell-specific. This could be a potential alternative to commercially available coatings which often are expensive and from animal sources. The skin equivalents must be handled with care, especially prior to fixation.
The use of interference reflection microscopy (IRM) in this Protocol shows, in much greater detail, the cell-derived matrix obtained with MMC. As compared with conventional fluorescent immunocytochemistry, only two or three proteins can be viewed at a time. With IRM, the total matrix can be visualized, with minimal manipulation to the sample. When immunocytochemistry was merged with the IRM field, it provided important information as to the ECM protein deposited in relation to the total amount of ECM. For example, it was observed that fibroblasts produced most ECM components, but a fibroblast-derived matrix was largely composed of fibronectin.
Fibroblasts were also found to be the main producers of collagen VII, a feat which could be enhanced by MMC in vitro. This Protocol could have implications in therapies pertaining to the skin disorder, recessive dystrophic epidermolysis bullosa, where there is defective collagen VII.
Finally, the application of MMC in a 3D model to generate organotypic skin co-cultures, provides a useful tool in the advancement of skin research. Since MMC allows for the generation of skin equivalents with complete stratification, a mature dermal-epidermal junction delivered in a condensed time frame, this would be largely beneficial in the modeling of skin diseases, the study of skin biology in vitro, as well as the production of skin grafts for burn and chronic wounds.
Disclosures
The authors have no conflicting interests to disclose.
Acknowledgments
This work was supported by the Biomedical Research Council, Singapore through core support to the Institute of Medical Biology and grant SPF2013/005. M.R. was supported by a NUS Faculty Research Committee Grant (Engineering in Medicine) (M.R.) R-397-000-081-112, and the NUS Tissue Engineering Program (NUSTEP). The authors would like to thank Professor Irene Leigh for providing the collagen VII antibody. Electron microscopy work was carried out at the National University of Singapore Electron Microscopy Unit.
References
- Breitkreutz D, Mirancea N, Nischt R. Basement membranes in skin: unique structures with diverse functions. Histochem Cell Biol. 2009;132(1):1–10. doi: 10.1007/s00418-009-0586-0. [DOI] [PubMed] [Google Scholar]
- Lane EB, et al. mutation in the conserved helix termination peptide of keratin 5 in hereditary skin blistering. Nature. 1992;356(6366):244–246. doi: 10.1038/356244a0. [DOI] [PubMed] [Google Scholar]
- Fuchs E, Raghavan S. Getting under the skin of epidermal morphogenesis. Nat Rev Genet. 2002;3(3):199–209. doi: 10.1038/nrg758. [DOI] [PubMed] [Google Scholar]
- Niessen CM. Tight junctions/adherens junctions: basic structure and function. J Invest Dermatol. 2007;127(11):2525–2532. doi: 10.1038/sj.jid.5700865. [DOI] [PubMed] [Google Scholar]
- Jarvelainen H, Sainio A, Koulu M, Wight TN, Penttinen R. Extracellular matrix molecules: potential targets in pharmacotherapy. Pharmacol Rev. 2009;61(2):198–223. doi: 10.1124/pr.109.001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer L, Schaefer RM. Proteoglycans: from structural compounds to signaling molecules. Cell Tissue Res. 2010;339(1):237–246. doi: 10.1007/s00441-009-0821-y. [DOI] [PubMed] [Google Scholar]
- Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123(Pt 24):4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghalbzouri A, Jonkman M, Dijkman R, Ponec M. Basement membrane reconstruction in human skin equivalents is regulated by fibroblasts and/or exogenously activated keratinocytes. J Invest Dermatol. 2005;124(1):79–86. doi: 10.1111/j.0022-202X.2004.23549.x. [DOI] [PubMed] [Google Scholar]
- Lareu RR, Arsianti I, Harve KS, Peng YX, Raghunath M. In Vitro Enhancement of Collagen Matrix Formation and Crosslinking for Applications in Tissue Engineering--a Preliminary Study. Tiss Eng. 2007;13(2):385–391. doi: 10.1089/ten.2006.0224. [DOI] [PubMed] [Google Scholar]
- Lareu RR, et al. Collagen matrix deposition is dramatically enhanced in vitro when crowded with charged macromolecules: the biological relevance of the excluded volume effect. FEBS Lett. 2007;581(14):2709–2714. doi: 10.1016/j.febslet.2007.05.020. [DOI] [PubMed] [Google Scholar]
- Lareu RR, Harve KS, Raghunath M. Emulating a crowded intracellular environment in vitro. dramatically improves RT-PCR performance. Biophys Biochem Res Comm. 2007;363(1):171–177. doi: 10.1016/j.bbrc.2007.08.156. [DOI] [PubMed] [Google Scholar]
- Harve KS, Ramakrishnan V, Rajagopalan R, Raghunath M. Macromolecular crowding in vitro as means of emulating cellular interiors: when less might be more. Proc Natl Acad USA Sci. 2008;105(51):E119. doi: 10.1073/pnas.0810077106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, et al. The Scar-in-a-Jar: Studying antifibrotic lead compounds from the epigenetic to extracellular level in a single well. Br J Pharmacol. 2009;158(5):1196–1209. doi: 10.1111/j.1476-5381.2009.00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harve KS, Lareu R, Rajagopalan R, Raghunath M. Understanding how the crowded interior of cells stabilizes DNA/DNA and DNA/RNA hybrids-in silico predictions and in vitro evidence. Nucleic Acids Res. 2009;38(1):172–181. doi: 10.1093/nar/gkp884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satyam A, et al. Macromolecular crowding meets tissue engineering by self-assembly: A paradigm shift in regenerative medicine. Adv Mat. 2014;26(19):3024–3034. doi: 10.1002/adma.201304428. [DOI] [PubMed] [Google Scholar]
- Chen CZC, Loe F, Blocki A, Peng Y, Raghunath M. Applying macromolecular crowding to enhance extracellular matrix deposition and its remodeling in vitro for tissue engineering and cell-based therapies. Adv Drug Deliv Rev. 2011;63(4-5):277–290. doi: 10.1016/j.addr.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Rashid R, et al. Novel use for Polyvinylpyrrolidone as a Macromolecular Crowder for Enhanced Extracellular Matrix Deposition and Cell Proliferation. Tiss Eng C. 2014. [DOI] [PMC free article] [PubMed]
- Dewavrin JY, Hamzavi N, Shim VPW, Raghunath M. Tuning the Architecture of 3D Collagen Hydrogels by Physiological Macromolecular Crowding. Acta Biomaterialia. 2014;10(10):4351–4359. doi: 10.1016/j.actbio.2014.06.006. [DOI] [PubMed] [Google Scholar]
- Dewavrin JY, et al. Synergistic Rate Boosting of Collagen Fibrillogenesis in Heterogeneous Mixtures of Crowding Agents. J Phys Chem B. 2015;119(12):4350–4358. doi: 10.1021/jp5077559. [DOI] [PubMed] [Google Scholar]
- Auger FA, Berthod F, Moulin V, Pouliot R, Germain L. Tissue engineered skin substitutes: from in vitro constructs to in vivo applications. Biotechnol Appl Biochem. 2004;39:263. doi: 10.1042/BA20030229. [DOI] [PubMed] [Google Scholar]
- Chen B, et al. Macromolecular crowding effect on cartilaginous matrix production: a comparison of two-dimensional and three-dimensional models. Tiss Eng Part C Methods. 2013;19(8):586–595. doi: 10.1089/ten.tec.2012.0408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeiger AS, Loe FC, Li R, Raghunath M, van Vliet KJ. Macromolecular crowding directs extracellular matrix organization and mesenchymal stem cell behavior. PLOS One. 2012;7(5):e37904. doi: 10.1371/journal.pone.0037904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, et al. Macromolecularly crowded in vitro microenvironments accelerate the production of extracellular matrix-rich supramolecular assemblies. Scientific Reports. 2015;5:8729. doi: 10.1038/srep08729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang XM, et al. Macromolecular crowding amplifies adipogenesis of human bone marrow-derived MSCs by enhancing the pro-adipogenic microenvironment. Tiss Eng Part A. 2014;20(5-6):966–981. doi: 10.1089/ten.tea.2013.0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YX, et al. Human Fibroblast Matrices Bioassembled Under Macromolecular Crowding Support Stable Propagation Of Human Embryonic Stem Cells. J Tiss Eng Regen Med. 2012;6(10):e74–e84. doi: 10.1002/term.1560. [DOI] [PubMed] [Google Scholar]
- Benny P, Badowski C, Lane EB, Raghunath M. Making More Matrix: Enhancing the deposition of dermal-epidermal junction components in vitro and accelerating organotypic skin culture development, using macromolecular crowding. Tiss Eng A. 2015;21(1-2):182–192. doi: 10.1089/ten.tea.2013.0784. [DOI] [PMC free article] [PubMed] [Google Scholar]