

Abstract
The study of neuroplasticity and molecular alterations in learned behaviors is switching from the study of whole brain regions to the study of specific sets of sparsely distributed activated neurons called neuronal ensembles that mediate learned associations. Fluorescence Activated Cell Sorting (FACS) has recently been optimized for adult rat brain tissue and allowed isolation of activated neurons using antibodies against the neuronal marker NeuN and Fos protein, a marker of strongly activated neurons. Until now, Fos-expressing neurons and other cell types were isolated from fresh tissue, which entailed long processing days and allowed very limited numbers of brain samples to be assessed after lengthy and complex behavioral procedures. Here we found that yields of Fos-expressing neurons and Fos mRNA from dorsal striatum were similar between freshly dissected tissue and tissue frozen at -80 ºC for 3 - 21 days. In addition, we confirmed the phenotype of the NeuN-positive and NeuN-negative sorted cells by assessing gene expression of neuronal (NeuN), astrocytic (GFAP), oligodendrocytic (Oligo2) and microgial (Iba1) markers, which indicates that frozen tissue can also be used for FACS isolation of glial cell types. Overall, it is possible to collect, dissect and freeze brain tissue for multiple FACS sessions. This maximizes the amount of data obtained from valuable animal subjects that have often undergone long and complex behavioral procedures.
Keywords: Neuroscience, Issue 114, Cell types, neurons, glia, antibody, mRNA, FACS
Introduction
During learning, animals form associations between complex sets of highly specific stimuli. This high-resolution information is thought to be encoded by alterations within specific patterns of sparsely distributed neurons called neuronal ensembles. Neuronal ensembles have recently been identified by the induction of immediately early genes (IEGs) such as Fos, Arc, and Zif268 and their protein products in neurons that were strongly activated during behavior or cue exposure. Fos-expressing neurons in particular have been shown to play causal roles in context and cue-specific learned behaviors 1-4. Thus, unique molecular neuroadaptations within these activated Fos-expressing neurons are top candidates for the neural mechanisms that encode learned associations formed during normal learning and abnormal learning disorders, such as addiction and post-traumatic stress disorder (PTSD) 5.
Fluorescence Activated Cell Sorting (FACS) has recently allowed analysis of unique molecular neuroadaptations within Fos-expressing neurons. Flow cytometry and cell sorting were developed in the 1960s 6,7 to characterize and isolate cells according to their light-scattering and immunofluorescent characteristics, and have long been used in immunology and cancer research. However flow cytometry and FACS requires dissociated single cells that are difficult to obtain from adult brain tissue. FACS was first used to isolate and analyze Green Fluorescent Protein (GFP)-expressing striatal neurons from transgenic mice that did not require antibody labeling 8,9. We developed an antibody-based FACS method 10 to isolate and assess molecular alterations in Fos-expressing neurons activated by drug and/or cues in wild type animals 11-15. In this method, neurons are labeled with an antibody against the general neuronal marker NeuN, while strongly activated neurons are labeled with an antibody against Fos. Although our initial method required pooling of up to 10 rats per sample for fresh tissue, subsequent modifications of the protocol allowed FACS isolation of Fos-expressing neurons and quantitative Polymerase Chain Reaction (qPCR) analysis of discrete brain areas from a single rat 13-15. Overall, unique molecular alterations were found in Fos-expressing neurons activated during a variety of context- and cue-activated behaviors in addiction research 12,14,15.
A major logistical problem with performing FACS on fresh tissue is that it takes one whole day to dissociate the tissue and process by FACS. In addition, only about four samples can be processed per day. This usually means that only one brain area can be assessed from each brain and the remaining brain areas have to be discarded. This is a major problem for low throughput behavioral procedures such as self-administration and extinction training that requires surgery and many weeks of intensive training. Furthermore, long and complicated behavioral procedures on test day makes it difficult to perform FACS on the same day. It would be a significant advantage to be able to freeze the brains from the animals immediately after behavioral testing, and then isolate Fos-expressing neurons from one or more brain areas at different times of the investigators' choosing.
Here we demonstrate that our FACS protocol can be used to isolate Fos-expressing neurons (and other cell types) from both fresh and frozen brain tissue. As an example, we isolated Fos-expressing neurons from rat striatum after acute methamphetamine injections and from naïve rats without injections (control condition). However, this FACS protocol can be used following any behavioral or pharmacological treatment. Subsequent qPCR analysis of our samples indicated that gene expression from these cell types could be assessed with similar efficiency from both fresh and frozen tissue.
Protocol
All experiments were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) of the National Institutes of Health Guide for the Care and Use of Laboratory animals 16.
Note: All the steps below use low-binding centrifuge tubes that were kept on ice unless otherwise specified.
1. Preparation Before Tissue Collection
Set the centrifuge to 4 oC.
Fire polish a set of three glass Pasteur pipettes with decreasing diameters of approximately 1.3, 0.8, and 0.4 mm for each sample.
Prepare labeled 1.7 ml-tubes containing 1 ml cold Buffer A and keep the tubes on ice.
Prepare an ice tray containing the brain slicing matrix, spatulas and glass plates (or inverted glass Petri dish) on which to perform the dissection. Pre-chill two or more razor blades on the glass plates and use tissues paper to dry all instruments that touch the tissue.
Thaw the Enzyme solution at room temperature (RT) for 30 min before tissue collection.
2. Tissue Collection and Dissection
Initiate the behavioral or pharmacological treatment 90 min before tissue collection. NOTE: For the results shown here, acute intraperitoneal injections of 5 mg/kg methamphetamine on rats in a novel environment (test condition) and naïve rats kept in their home cages (control condition) were performed.
Anesthetize the rat by placing it in a glass desiccator jar with saturated isoflurane and decapitate the rat 30 - 60 sec later using a guillotine. Use scissors to remove the skin and muscle from the head and expose the skull. Use rongeurs to cut the skull and open up the foramen magnum and remove the back part of the skull. Use rongeurs to cut along the top edges of the skull to expose the brain. Be careful not to damage the brain. Use a small spatula to gently scoop under and elevate the brain. Raise the brain and cut the nerves until the brain is free 17.
- Dissect tissue using razor blades. NOTE: Obtain tissue samples using either of the following methods: (2.3.1) freshly dissected tissue; (2.3.2) frozen tissue after dissection; or (2.3.3) frozen tissue dissected from frozen whole brain. Keep the brain slicing matrix, razor blades and glass plate dry throughout the dissection and mincing processes as condensed water can lead to hypotonic lysing of cells. Use magnifying glasses to ensure accurate dissection.
- For freshly dissected tissue, place the freshly extracted brain into a brain slicing matrix (a rat brain-shaped metal mold with slots for the razor blades at 1 mm intervals) that has been chilled on ice. Insert two or more pre-chilled razor blades into the slots to cut coronal slices that contain the brain region(s) of interest. Place the cut slice on the chilled glass plate. Note: Dissect the brain regions of interest on a chilled glass plate.
- For frozen tissue after dissection, dissect the brain region(s) of interest from slices of freshly extracted brain, similar to that described above in 2.3.1. Place the dissected tissue into a microtube and rapidly freeze the tissue by submerging the microtube in -40 ºC isopentane for 20 sec. Keep dissected tissue frozen at all times in a -80 ºC freezer until further processing.
- For frozen tissue, immediately freeze the freshly extracted brain in -40 ºC isopentane and store in a sealed bag at -80 ºC for up to 6 months.
- On the day of dissection, place the frozen brain in a cryostat set at approximately -20 ºC (no warmer than -18 ºC) for approximately 2 hr to equilibrate the temperature. NOTE: Brains can be cut easily at this temperature, while much cooler temperatures make the brain too brittle to be cut safely.
- Use razor blades to cut 1 - 2 mm coronal slices of the frozen brains in a cryostat. Use a blunted 12- to 16-gauge needle to obtain tissue punches from these slices under freezing conditions. Keep dissected tissue frozen at all times until further processing.
Add 1 - 2 drops of Buffer A to cover the dissected tissue before mincing. Remove most of the white matter from the tissue using two razor blades to prevent loss of neurons during the rest of the trituration process. If starting with frozen tissue, then allow the tissue to thaw on the cold glass plate for no more than 1 min before applying Buffer A solution.
Mince the tissue 100 times in each orthogonal direction with a razor blade on a chilled glass plate. Hold the razor blade vertical to the glass plate when mincing. NOTE: Thorough mincing is critical for all protocol steps.
Use razor blades to transfer the minced tissue into microcentrifuge tubes containing 1 ml of cold Buffer A. Invert the tube 3 - 5 times to keep all minced tissue in the solution.
3. Cell Dissociation
NOTE: Use gentle end over end mixing for all of the following steps. Do not use a vortex mixer and avoid air bubbles that cause cell damage.
Centrifuge the sample tubes at 110 x g for 2 min at 4 ºC. Discard supernatant. Slowly add 1 ml of cold freshly thawed Enzyme solution (containing a mix of proteolytic enzymes) down the inner wall of the microtube. Immediately suck up the entire pellet and gently pipette up and down only 4 times with a moderately large tip diameter pipette to disperse the minced tissue pellet.
Invert the microtubes immediately to prevent the pellet from sticking to the bottom and incubate the samples with end-over-end mixing for 30 min at 4 oC.
After enzymatic tissue digestion, centrifuge the tubes at 960 x g for 2 min at 4 ºC. Remove the supernatant and add 0.6 ml of cold Buffer A. Immediately after adding the Buffer A, use the same pipette tip to disperse the pellet by sucking up the entire pellet and gently pipette up and down 5 times.
- Mechanically triturate the digested tissue and collect in 15 ml tubes using the following steps. For each sample, use a separate set of 3 fire-polished glass Pasteur pipettes with descending diameters of approximately 1.3, 0.8 and 0.4 mm attached to latex bulbs.
- Gently triturate each sample 10 times with the 1.3 mm glass pipette. Let the sample settle for 2 min on ice (the debris and undissociated cells will settle to the bottom). Collect the supernatant (~ 0.6 ml) and transfer to a 15 ml conical tube (tube #1).
- Add 0.6 ml Buffer A solution to the remaining pellet. Triturate the sample 10 times with the 0.8 mm glass pipette. Let the sample settle for 2 min on ice. Collect the supernatant (~ 0.6 ml) and transfer to the same 15 ml conical tube #1.
- Add 0.6 ml Buffer A solution to the remaining pellet. Triturate the sample 10 times with the 0.4 mm glass pipette. Let the sample settle for 2 min on ice. Collect the supernatant (~ 0.6 ml; without touching the pellet with non-dissociated cells) and transfer to the same 15 ml conical tube #1. NOTE: Keep the 0.4 mm diameter pipette in the tube #1 for the following trituration steps.
- Repeat step 3.4.3 three more times using the smallest glass pipette (~ 0.4 mm diameter), and collect the supernatant into a separate 15 ml conical tube (tube #2).
- Triturate the collected cell suspensions in tubes #1 and #2 for 10 more times using the 0.4 mm glass pipette and keep on ice.
4. Cell Fixation and Permeabilization
Prepare 4 microtubes per sample (two microtubes for cells from tube #1 and two for cells from tube #2) by adding 800 µl of 100% cold ethanol (stored at -20 oC before use) into each tube and keep them on ice. Note: The final ethanol concentration will be 50%.
Pipette ~ 800 µl of the cell suspensions (from tube#1 and tube#2) into each of the 4 tubes containing cold ethanol and invert the tubes to mix the samples.
Incubate tubes on ice for 15 min while inverting the tubes every 5 min.
After fixation/permeabilization, centrifuge the tubes at 1,700 x g for 4 min at 4 ºC and discard the supernatant. Leave 50 µl of solution to prevent cell loss. NOTE: The pellets at this stage are white and stick less to the wall of the tubes than before permeabilization. Therefore, remove the supernatant carefully by touching the wall of the microtube where there is no pellet and draw up the supernatant slowly using a micropipette.
5. Cell Filtration
- Re-suspend the pellets from step 4.4 with cold PBS as follows:
- Add 550 μl of cold PBS to one of the two microtubes coming from tube #1, and use a moderately large diameter pipette tip to gently pipette the cell suspension up and down 5 times. Repeat the same for one microtube coming from tube #2.
- For cells from tube #1, using the same pipette, transfer the first cell suspension to the second pellet. Resuspend the second pellet by gently pipetting the combined cell suspension up and down 5 times.
- For cells from tube #2, resuspend and combine the pellets (i.e., third and fourth microtubes) as described in 5.1.2.
- Filter the two cell suspensions from steps 5.1.2 and 5.1.3 separately using two different pairs of cell strainers (100 µm and 40 µm pore sizes) as follows:
- Pre-wet each cell strainer by adding PBS to the inside of the strainer. Use a pipette to remove the excess PBS from the outside of the strainer.
- Pipette the cell suspension from step 5.1.2 evenly onto the first cell strainer with 100 µm pore size. Collect the flow-through in a 50 ml tube on ice. Use the pipette to collect the flow- through attached on the outer side bottom of the cell strainer.
- Use the same pipette to transfer the flow-through from the 100 μm cell strainer to the 40 μm cell strainer. Collect this flow-through in another 50 ml tube on ice. When transferring the flow through from 40 μm cell strainer, change to new pipette tips to avoid contamination from 100 μm cell strainer.
- Repeat these steps for the cell suspension from step 5.1.3 using a new set of filters.
Combine the two flow-through from each sample for a final volume of approximately 1 ml per sample.
6. Incubation with Antibodies
NOTE: Use small aliquots of the brain cell suspensions to prepare multiple control samples for setting appropriate flow cytometer settings prior to running the main sample. For each antibody labeling, prepare control samples that include no antibodies, only the secondary antibody (or other fluorescent label), and then both primary and secondary antibodies (or other fluorescent label). Use these controls to set the gates that separate specific versus non-specific labeling in the flow cytometer. When possible, use fluorochromes that do not require compensation. Increase the final volume of the gating control samples to a final volume of 700 µl by adding PBS prior to adding the primary antibodies.
Prepare light-scattering and DAPI control sample by transferring a 100 µl sample of the filtered cells to a 1.7 ml microtube. Add 600 µl of cold PBS with 1 µg/ml DAPI for nuclei staining. Incubate for 10 min with end-over-end mixing, and wash (as indicated in steps 6.4.2 to 6.4.5) before passing through the flow cytometer. NOTE: This sample is used only to set the cell/nuclei gate prior to sorting the remaining cells in the FACS machine.
Prepare single immunolabeled control samples by transferring 100 µl of the filtered cells to a 1.7 ml microtube. Add 600 µl of cold PBS with one antibody (e.g., NeuN antibody). Add 600 µl of cold PBS to a microtube with another antibody (e.g., Fos antibody). Add the corresponding secondary antibody if it is not a direct-conjugated primary antibody. NOTE: These samples are used to determine the appropriate photomultiplier tube (PMT) voltage, and to assess the overlap of the two fluorescent signals if necessary. Antibody concentrations should be titrated to achieve the best signal-to-noise ratios in all channels. Overlapping fluorescent signals can also be compensated by internal calibration of different fluorescent channels within the flow cytometer.
Prepare double fluorescent-labeled negative control sample by transferring 100 µl of the filtered cells to a 1.7 ml microtube. Add 600 µl of cold PBS with the secondary antibodies alone or IgG direct-conjugated primary antibodies. NOTE: This control sample can be used every time before sorting to determine the background fluorescence for each fluorophore.
- Prepare main sample to be sorted.
- Transfer 700 μl of the filtered cells into a 1.7 ml microtube. Add 7 µl (1:100) of primary Fos antibody conjugated directly to Alexa Fluor 647 (anti-Fos-AF647) and 1.4 µl (1:500) of primary NeuN antibody conjugated directly to Phycoerythrin (anti-NeuN-PE). Incubate tubes for 30 min at 4 ºC with end-over-end mixing.
- At the end of the incubation, add 800 µl of cold PBS to each microtube, including the main sample and the control samples, and mix by inversion.
- Centrifuge microtubes at 1,300 x g for 3 min at 4 ºC. Discard the supernatant, leaving 50 µl supernatant to prevent cell loss.
- Add 1 ml of cold PBS to the pellet and resuspend cells using a pipette with a moderately large tip diameter.
- Centrifuge at 1,300 x g for 3 min at 4 ºC. Discard the supernatant.
- Re-suspend the pellet in 500 µl cold PBS (adjust final volume depending the size of the pellet).
- Transfer and filter the suspension into a round-bottom FACS sample tube with a cell strainer cap (40 µm). Keep the immunolabeled cells on ice and immediately perform FACS. Note: For filtering, touch the pipette tip vertically on the strainer cap and push the cell suspension through it.
- Set flow cytometer gates using control samples.
- Use the light-scattering and DAPI control sample to identify the neuronal cell population from the total events and cellular debris. NOTE: Cell bodies in this preparation are only 1 - 2% of the total events (the rest are debris and broken cellular processes). Based on DAPI fluorescent signal (indicating nuclei) and Forward scatter (size of the cell) it is possible to locate cell bodies and gate backwards based on the Forward and Side light-scattering properties of the cells (as shown in Figure 1 A,B).
- Use the single immunolabeled control samples to determine the PMT voltage settings, and to analyze spillover of emission from one specific fluorochrome into another filter/channel (e.g., Alexa Fluor 488/FITC or Phycoerythrine). If fluorescent signals overlap, correct the overlap by adjusting compensation levels manually.
- Use the double fluorescent-labeled negative control to set up the threshold for the positive population. NOTE: The signal coming from this sample is thought to be due to auto-fluorescence and the cells of the main sample located above this threshold can be considered as positive. Typically, the threshold parameters for sorting are more stringent and approximately the top 2/3 of the labeled cells above the negative control are actually sorted.
Sort the neurons from the main sample by FACS into 1.7 ml microtubes. Collect the cells into 50 µl of RNA extraction buffer. After sorting, vortex and centrifuge the tube and mix the suspension by pipetting up and down 10 times to recover all sorted cells from the walls of the tube.
Incubate the sorted cell suspension at 42 ºC for 30 min to ensure that all cells are lysed prior to RNA extraction, and then centrifuge at 2,800 x g for 2 min at 4 ºC.
Transfer the supernatant to an RNase-free tube for long-term storage at -80 ºC or immediately process the sample for RNA isolation, cDNA synthesis, target gene pre-amplification and qPCR, as described previously 13-15.
Representative Results
Sorting Fos-positive and Fos-negative neurons from fresh and frozen dorsal striatum tissue from single rats after acute methamphetamine injections.
The protocol described above was used to sort Fos-positive and Fos-negative neurons from a single rat dorsal striatum 90 min after an intraperitoneal injection of methamphetamine (5 mg/kg). Naïve rats in their home cages were used as controls. Dorsal striatum tissue was processed either immediately after its collection (fresh tissue) or processed after being frozen and stored in -80 ºC for 1, 7 or 21 days. The results from these freezing time points were not significantly different from each other, so the data were pooled.
To set up the sorting conditions on the instrument, control samples (described above) from either the home cage control or methamphetamine-injected groups were used. When each particle or event in these samples passes through the flow cell in the cytometer, they are given an identification number that is associated with the event's light scattering and fluorescence characteristics and recorded in a spreadsheet. The events in this sheet can then be organized in scattergrams or density plots for these characteristics (Figure 1). Events with similar characteristics can then be grouped or 'gated' by different pairs of characteristics to determine the gates such as those containing cells, neurons, or Fos-positive neurons. Once the gates are determined using the control samples described above, the main sample is injected into the flow cytometer and sorted according to these gates. The cell population was gated from all events (cells and debris) based on their forward light scatter (FSC; an indication of the particle's size) and side light scatter (SSC; an indication of the particle's granularity). As shown in the FSC versus SSC dot plot (Figure 1A and 1B), the density plot of all the events reveals a small homogenous population with similar size and granularity, in addition to a large heterogeneous population (Figure 1A and 1B). A small homogeneous population that contains cell bodies was identified and gated as "Cells", based on FSC and SSC characteristics from previous studies 11,13-15 and labeling with DAPI. A slightly higher percentage of cells were obtained from frozen tissue (2.6%) than from fresh tissue (1.9%). The large heterogeneous population is mostly debris, presumably from dendrites and axonal processes.
Next, single cells from the "Cells" gate were identified based on their size, which was indicated by the width of the FSC signal for each event on the x-axis. Events that were larger than single cells were considered cell aggregates and excluded from the "Single cell gate. All of the subsequent analysis steps were conducted within this "Single cell" gate (Figure 1C and 1D). Majority of this single cell population (> 92%) was positive for DAPI staining (data not shown).
First, neurons were identified based on their PE fluorescence intensity that indicated NeuN-immunoreactivity (Figure 1E and 1F). Neurons comprised approximately 24% of all events from the "Single cell" gate for fresh tissue and 38% for frozen tissue. Almost all of the events in this "Neuron" gate (98% in fresh tissue and 99% in frozen tissue) were positive for DAPI staining of DNA in the cell nuclei (Figure 1G and 1H). The remaining DAPI-labeled events in the Single cell gate are NeuN-negative and include glia, oligodendrocytes and microglia, as confirmed by qPCR in Figure 3.
Then neurons were classified as Fos-positive or Fos-negative neurons based on their Alexa Fluor 647 fluorescence signal (Fos-immunoreactivity). Unlike cell-type markers, Fos expression levels in neurons increases gradually due to differing levels of neural activity and time course. Hence, there will not be a clear cut threshold between Fos-positive versus Fos-negative neurons. In the first step (used only for off-line analyses to calculate percentages of Fos-positive neurons), we defined the threshold for Fos-positive neurons based on the maximum Alexa-647 fluorescence (for Fos) from the NeuN-negative population in the naïve home cage control rats. Fos-positive neurons are indicated in the blue squares from different experimental conditions in Figure 2. In both fresh and frozen tissues, the percentage Fos-positive neurons from methamphetamine-injected rats (1.9 - 2.2%, Figure 2C and 2D) was twice as compared to home cage rats (0.7 - 0.8%, Figure 2A and 2B).
Confirming cell-type specific genes from FACS-sorted cells using target gene pre-amplification and RT-PCR.
In the second step, to ensure sorting of only Fos-positive neurons from the main sample for subsequent mRNA analyses and reducing the inclusion of Fos-negative cells, the Alexa-647 fluorescence threshold was raised so that only the upper two-thirds of Fos-positive events in the blue squares were sorted and collected. This threshold has at least 10-fold higher fluorescence (higher Fos expression per cell) than the threshold used above to calculate percentage of Fos-positive neurons in the samples. Four populations of cells were sorted from a single tissue sample, including NeuN-negative+Fos-negative (~ 5,000 events), NeuN-negative+Fos-positive (ranged 25 - 83 events), NeuN-positive+Fos-negative (~ 5,000 events) and NeuN-positive+Fos-positive (ranged 44 - 133 events for the home group, 185 - 450 events for the Methamphetamine group). First, the expression of cell-type specific genes in NeuN-positive (including both Fos-positive and Fos-negative) and NeuN-negative population (including both Fos-positive and Fos-negative) was confirmed (Figure 3). Gapdh was used as reference/housekeeping gene, based on results from our previous study 13. To control for different levels of RNA in the sample and cDNA in the PCR reactions, duplex qPCR reactions were performed using primers for both the target gene and Gapdh cDNA. Ct values for the Gapdh housekeeping gene were kept between 15 and 31 for accurate measurements. For both fresh and frozen tissue, NeuN mRNA levels were significantly greater (approximately 8-fold) in the NeuN-positive population than in the NeuN-negative population (p < 0.05; Figure 3A; data combined from home and methamphetamine group). In contrast, for both fresh and frozen tissue, Gfap (glial cell marker; Figure 3B) and Oligo2 (oligodendrocyte cell marker; Figure 3C) mRNA levels were greater in the NeuN-negative population than in the NeuN-positive population (p < 0.05). A similar trend of higher Iba1 (microglial cell marker; Figure 3D) mRNA levels was observed in the NeuN-negative population, but this was not statistically significant. Some samples contained extremely low levels of particular mRNAs that could not be accurately measured in a particular cell type (e.g., Gfap mRNA in NeuN-labeled neurons). Maximum Ct values were defined as 35 to eliminate values that were too low to be accurately detected. Higher Ct values exceeded the confidence limit in our qPCR assays.
Fos mRNA expression was examined in the NeuN-positive neuronal population from rats that received a single injection of methamphetamine (Figure 4). For both fresh and frozen tissue, Fos mRNA levels were greater in the Fos-positive neurons than in the Fos-negative neurons (p < 0.05). Moreover, while there was a 3-fold increase of Fos mRNA in Fos-positive neurons from the fresh tissue, a 11-fold increase of Fos mRNA in Fos-positive neurons from the frozen tissue was detected. Taken together, these mRNA results confirm the identity of the Fos-positive neurons, Fos-negative neurons and non-neuronal population from both fresh and frozen tissue. Furthermore, cell type-specific genes and other genes of interest can also be analyzed from sorted cells under both fresh and frozen conditions.
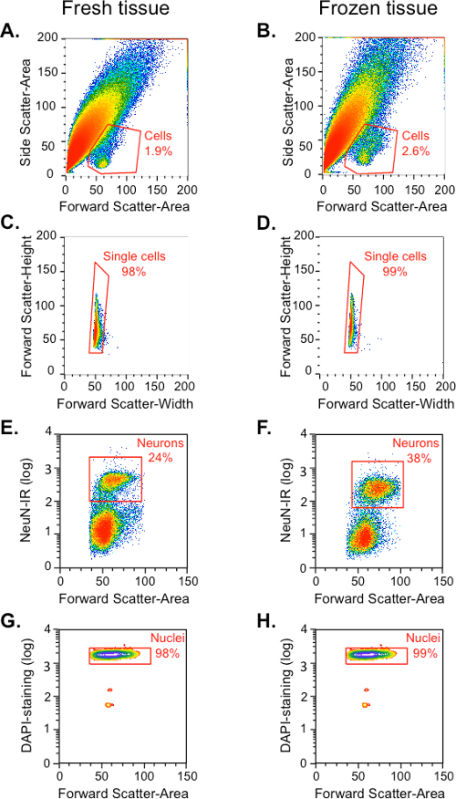 Figure 1.Gating of Dissociated and Labeled Cells from Fresh and Frozen Rat Dorsal Striatum. The 'Cells' gate was determined by forward and side scatter properties (A-D) and confirmed by positive labeling with nuclear DAPI staining (G-H). The 'Neurons' gate within the 'Cells' population was determined by fluorescence for NeuN (PE; E-F). (A-B) Cell gate: Linear plot of all events, based on their forward scatter (X-axis, cell size) and side scatter (Y-axis, granularity). (C-D) Single cells gate: Linear plot of forward scatter height (Y-axis) and width (X-axis) within the cell gate shown in A-B. (E-F) Neuron gate: Logarithmic plot of immunofluorescence for PE-labeled NeuN (Y-axis) within the single cells gate shown in C-D reveals neurons in the upper cluster of events and non-neuronal cells in the lower cluster. (G-H) Nuclei staining: Logarithmic plot of fluorescence for DAPI-labeled nuclei (Y-axis) within the neuronal cell gate. Please click here to view a larger version of this figure.
Figure 1.Gating of Dissociated and Labeled Cells from Fresh and Frozen Rat Dorsal Striatum. The 'Cells' gate was determined by forward and side scatter properties (A-D) and confirmed by positive labeling with nuclear DAPI staining (G-H). The 'Neurons' gate within the 'Cells' population was determined by fluorescence for NeuN (PE; E-F). (A-B) Cell gate: Linear plot of all events, based on their forward scatter (X-axis, cell size) and side scatter (Y-axis, granularity). (C-D) Single cells gate: Linear plot of forward scatter height (Y-axis) and width (X-axis) within the cell gate shown in A-B. (E-F) Neuron gate: Logarithmic plot of immunofluorescence for PE-labeled NeuN (Y-axis) within the single cells gate shown in C-D reveals neurons in the upper cluster of events and non-neuronal cells in the lower cluster. (G-H) Nuclei staining: Logarithmic plot of fluorescence for DAPI-labeled nuclei (Y-axis) within the neuronal cell gate. Please click here to view a larger version of this figure.
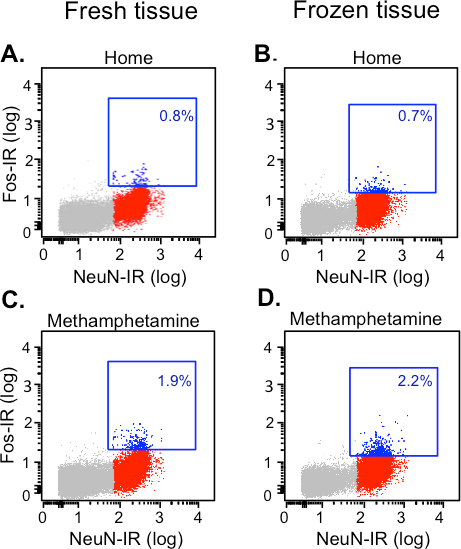 Figure 2.Gating of Fos-positive and Fos-negative Neurons from Fresh and Frozen Rat Dorsal Striatum Based on Double Labeling for NeuN and Fos. The fresh and frozen dorsal striatum from rats (taken directly from their home cage or 90 min after a single intraperitoneal injection of methamphetamine) were dissociated and labeled with the directly conjugated antibodies against NeuN and Fos and sorted using a FACS machine. Sorting was based on phycoerythrin (PE)-labeled NeuN immunofluorescence (X-axis) and Alexa 647-labeled Fos immunofluorescence (Y-axis). Fos-positive (blue dots) and Fos-negative (red dots) neurons were located in the upper and lower right quadrants, respectively. The dot plots show NeuN-positive cells (neurons) and NeuN-negative cells (grey dots); the blue squares indicate neurons with above-threshold Fos expression. (A-B) Home group: naïve rats taken directly from their home cages. (C-D) Methamphetamine group: rats that received single injections of methamphetamine. The thresholds for Fos-positive neurons were selected to be just above maximal Alexa-647 fluorescence (Fos-IR) observed for NeuN-negative cells in the home cage control group. While 0.7 - 0.8% of all neurons are Fos-positive in the home group, 1.9 - 2.2% of all neurons are Fos-positive in the methamphetamine group.
Figure 2.Gating of Fos-positive and Fos-negative Neurons from Fresh and Frozen Rat Dorsal Striatum Based on Double Labeling for NeuN and Fos. The fresh and frozen dorsal striatum from rats (taken directly from their home cage or 90 min after a single intraperitoneal injection of methamphetamine) were dissociated and labeled with the directly conjugated antibodies against NeuN and Fos and sorted using a FACS machine. Sorting was based on phycoerythrin (PE)-labeled NeuN immunofluorescence (X-axis) and Alexa 647-labeled Fos immunofluorescence (Y-axis). Fos-positive (blue dots) and Fos-negative (red dots) neurons were located in the upper and lower right quadrants, respectively. The dot plots show NeuN-positive cells (neurons) and NeuN-negative cells (grey dots); the blue squares indicate neurons with above-threshold Fos expression. (A-B) Home group: naïve rats taken directly from their home cages. (C-D) Methamphetamine group: rats that received single injections of methamphetamine. The thresholds for Fos-positive neurons were selected to be just above maximal Alexa-647 fluorescence (Fos-IR) observed for NeuN-negative cells in the home cage control group. While 0.7 - 0.8% of all neurons are Fos-positive in the home group, 1.9 - 2.2% of all neurons are Fos-positive in the methamphetamine group.
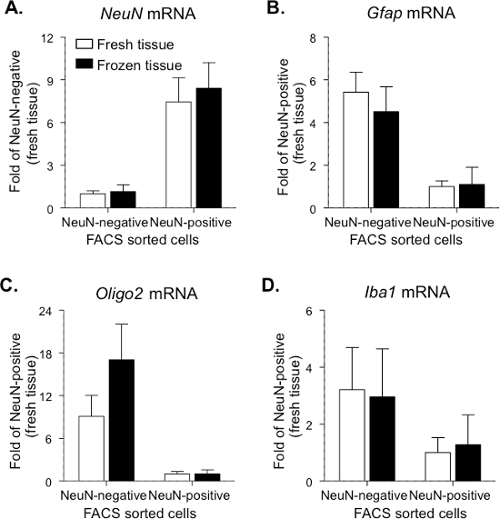 Figure 3.Cell-type Specific Gene Expression in FACS-sorted Cells from Fresh and Frozen Rat Dorsal Striatum. NeuN-positive neurons (Fos-positive and Fos-negative) and NeuN-negative cells (Fos-positive and Fos-negative) were sorted using the described protocol and mRNA expression levels of cell-type specific genes were used to confirm cell sorting. (A) NeuN is a marker for neuronal cells. (B) Gfap is a marker for glial cells. (C) Oligo2 is a marker for oligodendrocyte cells. (D) Iba1 is a marker for microglial cells. For NeuN mRNA, data are presented as mean±SEM of fold values relative to expression levels in NeuN-negative cells from the fresh tissue (n = 9 - 14). For Gfap (n = 6 - 12), Oligo2 (n = 9 - 11) and Iba1 (n = 5 - 8) mRNA, data are presented as mean ± SEM of fold values relative to expression levels in NeuN-positive cells from the fresh tissue. Please click here to view a larger version of this figure.
Figure 3.Cell-type Specific Gene Expression in FACS-sorted Cells from Fresh and Frozen Rat Dorsal Striatum. NeuN-positive neurons (Fos-positive and Fos-negative) and NeuN-negative cells (Fos-positive and Fos-negative) were sorted using the described protocol and mRNA expression levels of cell-type specific genes were used to confirm cell sorting. (A) NeuN is a marker for neuronal cells. (B) Gfap is a marker for glial cells. (C) Oligo2 is a marker for oligodendrocyte cells. (D) Iba1 is a marker for microglial cells. For NeuN mRNA, data are presented as mean±SEM of fold values relative to expression levels in NeuN-negative cells from the fresh tissue (n = 9 - 14). For Gfap (n = 6 - 12), Oligo2 (n = 9 - 11) and Iba1 (n = 5 - 8) mRNA, data are presented as mean ± SEM of fold values relative to expression levels in NeuN-positive cells from the fresh tissue. Please click here to view a larger version of this figure.
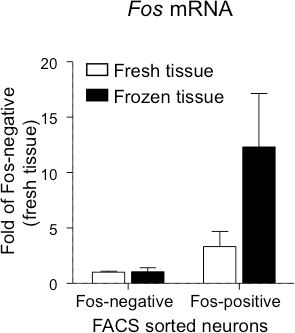 Figure 4.Fos mRNA Levels in FACS-sorted Neurons from Fresh or Frozen Rat Dorsal Striatum after Methamphetamine Injection. Fos-positive and Fos-negative neurons were sorted as described in the protocol above and mRNA levels of Fos was confirmed. Data are presented as mean ± SEM of fold values relative to expression levels in Fos-negative neurons from the fresh tissue (n = 3 - 4).
Figure 4.Fos mRNA Levels in FACS-sorted Neurons from Fresh or Frozen Rat Dorsal Striatum after Methamphetamine Injection. Fos-positive and Fos-negative neurons were sorted as described in the protocol above and mRNA levels of Fos was confirmed. Data are presented as mean ± SEM of fold values relative to expression levels in Fos-negative neurons from the fresh tissue (n = 3 - 4).
Discussion
FACS can be used to sort neurons and other cell types from either fresh or frozen adult brain tissue. As mentioned in the introduction, the ability to use frozen tissue allows optimal utilization of samples from animals that have undergone complex and protracted behavioral procedures, such as self-administration and relapse studies in addiction research. These behavioral procedures usually takes 1 - 2 hr or longer, and require all animals (10 - 20 total) be tested on the same day 13,18. It takes ~ 4 hr to process 4 freshly dissected brain tissue samples, which includes brain dissection, cell dissociation, permeabilization and immunolabeling. After processing, sorting each sample by FACS takes between 20 - 30 min. The final step of heating the sorted cells in the lysis buffer requires another 30 min. In total, ~ 7 hr are required from brain dissection to the final RNA-containing cell-lysis solution step. However, it is now possible to freeze whole brains or freshly dissected brain regions immediately after testing, and process them later. This greatly simplifies testing and permits multiple brain areas to be sorted on a different day. Freezing the tissue prior to FACS may also improve results. More NeuN-positive neurons are obtained from frozen tissue samples. This might be explained in part by increased NeuN-labeling due to disruption of cell membranes during freezing and subsequent thawing, which would increase antibody access to intracellular proteins such as NeuN and Fos. Similar effects have been observed with fibroblast, epithelial cells and HeLa cells 19.
The yield of cells and neurons can be maximized using the following steps. During dissection of the tissue, remove majority of white matter (corpus callosum and anterior commissure for the dorsal striatum and nucleus accumbens, respectively). This prevents tissue from adhering to the plastic tips and glass pipettes in the subsequent steps. Use Buffer A (see specification in Table of Materials and Reagents) to keep the dissected tissue covered. This improves the quality of cells during mincing of the tissue with the razor blade (step 2.5). The extra set of 3 trituration steps with the smallest glass pipette (step 3.9) and subsequent filtering with a second set of cell strainers (step 5) doubles the yield of cells and neurons. Reusing the cell strainers will clog the filters and lower cell yield. Be gentle with the cells when resuspending the pellets and during trituration. The use of a moderately large pipette tip during these steps will reduce cell damage due to shear stress. Carefully watch the pellet when removing supernatant after permeabilization with ethanol (step 4.4). The pellet is less compact and can be distributed along the wall of the microcentrifuge tube. Rough calculations suggest that the yield of Fos-positive neurons is approximately 10% of the starting number in the dissected striatal tissue sample.
The current protocol can be modified to suit other experimental aims. In the current protocol, tissue was collected 90 min after methamphetamine injections because Fos expression reaches maximal levels at this time, which is crucial for efficient sorting of Fos-expressing neurons. However, tissue can be collected at any time point for FACS, depending on the specific aim of the experiment. For initial tissue sample storage, we have successfully used freshly dissected tissue, frozen tissue after dissection, or frozen tissue dissected from frozen whole brain to isolate Fos-expressing neurons using FACS followed by qPCR analysis of gene expression. For fixation and permeabilization, the duration and temperature in this protocol (4 ºC for 15 min) was optimized to permeabilize both cytoplasmic and nuclear membranes, as well as to fix the tissue. Variations in duration and/or temperature can change the final results. Other fixation solutions such as paraformaldehyde and non-ionic detergents such as saponin, which have worked for embryonic and post-natal neurons 20, may also work for neurons from adult brain. Furthermore, myelin removal beads can be used as a modification of the current protocol as shown recently for FACS from brain tissue 21.
The current protocol has two important limitations to consider. First, this protocol is not suited for detecting cell phenotype markers that are expressed primarily in synapses (e.g., dopamine or glutamate receptors) because these cellular processes are removed from the cell bodies during the trituration and dissociation steps. A second possible limitation is that the RNA lengths obtained from FACS-sorted cells may not be sufficient for all RNA analysis methods. RNA integrity numbers (RIN) for RNA obtained from FACS sorted cells are between 2.5- 3.5, which corresponds to shorter average RNA lengths. Here, the shorter RNA size was compensated by using relatively short qPCR amplicons of 80 - 100 bp, as described previously13-15. We have also used these shorter length RNAs successfully for microarray analysis11. Nevertheless, short RNA lengths obtained may not be adequate for all RNA analysis methods. It should be emphasized that PCR primers that specifically targeted exon-exon junctions found only in fully spliced mRNA in the cytosol were used. These primers do not detect intron-containing RNA from the nucleus. Furthermore, we have previously used this protocol with antibodies and fresh brain tissue to detect tyrosine hydroxylase in the cytosol and D1 dopamine receptors in the cellular membrane 10. Detection of these cytosolic and cell membrane markers indicated that the dissociation procedure produces largely intact cells, and not simply nuclei.
Overall, FACS isolation of neurons from frozen samples opens up other applications. No differences in sorting or gene expression were observed (data not shown) when samples were stored 3 days or 3 weeks at -80ºC. Three weeks is a reasonable time to sort all samples from a specific experiment (20 - 40 samples) and isolate RNA for gene expression. Usable RNA has also been obtained from brain tissue stored for 6 months at -80 ºC (data not shown here). Thus, this protocol could be useful for FACS isolation of post-mortem frozen human brain samples that were stored for several months or even years.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by the NIDA Intramural Research Program (Bruce T. Hope, Yavin Shaham). F.J.R. was supported by an appointment to the NIDA Research Participation Program sponsored by the National Institutes of Health and administered by the Oak Ridge Institute for Science and Education, and received additional financial support from a Becas-Chile scholarship managed by CONICYT and the Universidad de los Andes, Santiago, Chile. The Johns Hopkins FACS Core facility was supported by Award P30AR053503 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institute of Health.
References
- Bossert JM, et al. Ventral medial prefrontal cortex neuronal ensembles mediate context-induced relapse to heroin. Nat Neurosci. 2011;14:420–422. doi: 10.1038/nn.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz FC, et al. Role of nucleus accumbens shell neuronal ensembles in context-induced reinstatement of cocaine-seeking. J Neurosci. 2014;34:7437–7446. doi: 10.1523/JNEUROSCI.0238-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanous S, et al. Role of orbitofrontal cortex neuronal ensembles in the expression of incubation of heroin craving. J Neurosci. 2012;32:11600–11609. doi: 10.1523/JNEUROSCI.1914-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koya E, et al. Targeted disruption of cocaine-activated nucleus accumbens neurons prevents context-specific sensitization. Nat Neurosci. 2009;12:1069–1073. doi: 10.1038/nn.2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz FC, Rubio FJ, Hope BT. Using c-fos to study neuronal ensembles in corticostriatal circuitry of addiction. Brain Research. 2015;1628:157–173. doi: 10.1016/j.brainres.2014.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamentsky LA, Melamed MR. Spectrophotometric cell sorter. Science. 1967;156:1364–1365. doi: 10.1126/science.156.3780.1364. [DOI] [PubMed] [Google Scholar]
- Kamentsky LA, Melamed MR, Derman H. Spectrophotometer: new instrument for ultrarapid cell analysis. Science. 1965;150:630–631. doi: 10.1126/science.150.3696.630. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Karsten SL, Gray M, Geschwind DH, Yang XW. FACS-array profiling of striatal projection neuron subtypes in juvenile and adult mouse brains. Nat Neurosci. 2006;9:443–452. doi: 10.1038/nn1654. [DOI] [PubMed] [Google Scholar]
- Lobo MK. Molecular profiling of striatonigral and striatopallidal medium spiny neurons past, present, and future. Int Rev Neurobiol. 2009;89:1–35. doi: 10.1016/S0074-7742(09)89001-6. [DOI] [PubMed] [Google Scholar]
- Guez-Barber D, et al. FACS purification of immunolabeled cell types from adult rat brain. J Neurosci Methods. 2012;203:10–18. doi: 10.1016/j.jneumeth.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guez-Barber D, et al. FACS identifies unique cocaine-induced gene regulation in selectively activated adult striatal neurons. J Neurosci. 2011;31:4251–4259. doi: 10.1523/JNEUROSCI.6195-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanous S, et al. Unique gene alterations are induced in FACS-purified Fos-positive neurons activated during cue-induced relapse to heroin seeking. J Neurochem. 2013;124:100–108. doi: 10.1111/jnc.12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu QR, et al. Detection of molecular alterations in methamphetamine-activated Fos-expressing neurons from a single rat dorsal striatum using fluorescence-activated cell sorting (FACS) J Neurochem. 2014;128:173–185. doi: 10.1111/jnc.12381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio FJ, et al. Context-induced reinstatement of methamphetamine seeking is associated with unique molecular alterations in Fos-expressing dorsolateral striatum neurons. J Neurosci. 2015;35:5625–5639. doi: 10.1523/JNEUROSCI.4997-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, et al. Incubation of methamphetamine craving is associated with selective increases in expression of Bdnf and trkb, glutamate receptors, and epigenetic enzymes in cue-activated fos-expressing dorsal striatal neurons. J Neurosci. 2015;35:8232–8244. doi: 10.1523/JNEUROSCI.1022-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- US National Research Council. Guide for the care and use of laboratory animals. 2011.
- Koya E, Margetts-Smith G, Hope BT. Daun02 inactivation of behaviourally-activated Fos-expressing neuronal ensembles. Current Protocols. 2016. [DOI] [PMC free article] [PubMed]
- Cruz FC, et al. New technologies for examining the role of neuronal ensembles in drug addiction and fear. Nat Rev Neurosci. 2013;14:743–754. doi: 10.1038/nrn3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardones G, Gonzalez A. Selective plasma membrane permeabilization by freeze-thawing and immunofluorescence epitope access to determine the topology of intracellular membrane proteins. J Immunol Methods. 2003;275:169–177. doi: 10.1016/s0022-1759(03)00015-2. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, et al. DeCoN: genome-wide analysis of in vivo transcriptional dynamics during pyramidal neuron fate selection in neocortex. Neuron. 2015;85:275–288. doi: 10.1016/j.neuron.2014.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz JM. Using fluorescence activated cell sorting to examine cell-type-specific gene expression in rat brain tissue. J Vis Exp. 2015. p. e52537. [DOI] [PMC free article] [PubMed]