

Abstract
Translation of mRNA to protein is a fundamental and highly regulated biological process. Polysome profiling is considered as a gold standard for the analysis of translational regulation. The method described here is an easy and economical way for fractionating polysomes from various plant tissues. A sucrose gradient is made without the need for a gradient maker by sequentially freezing each layer. Cytosolic extracts are then prepared in a buffer containing cycloheximide and chloramphenicol to immobilize the cytosolic and chloroplastic ribosomes to mRNA and are loaded onto the sucrose gradient. After centrifugation, six fractions are directly collected from the bottom to the top of the gradient, without piercing the ultracentrifugation tube. During collection, the absorbance at 260 nm is read continuously to generate a polysome profile that gives a snapshot of global translational activity. Fractions are then pooled to prepare three different mRNA populations: the polysomes, mRNAs bound to several ribosomes; the monosomes, mRNAs bound to one ribosome; and mRNAs that are not bound to ribosomes. mRNAs are then extracted. This protocol has been validated for different plants and tissues including Arabidopsis thaliana seedlings and adult plants, Nicotiana benthamiana, Solanum lycopersicum, and Oryza sativa leaves.
Keywords: Plant Biology, Issue 114, Translation, polysomes, RNA, sucrose gradient, fractionation, plants, Arabidopsis thaliana, Oryza sativa, Nicotiana benthamiana, Solanum lycopersicum
Introduction
Protein synthesis is an essential and energetically costly process in all cells 1. First of all, cells must invest energy in the production of the translation machinery, the ribosomes. For example an actively dividing yeast cell produces as much as 2,000 ribosomes per minute. Such a production requires up to 60% of the total transcriptional activity and up to 90% of the total splicing activity of the cell 2. In addition, energy is required for the synthesis of amino acids, aminoacyl-tRNA and peptide bonds. In plants, adding one amino acid to a peptide chain costs from 4.5 to 5.9 molecules of ATP 3. Therefore, it is not surprising that the translation of mRNA to protein is a major site of regulation, particularly when it comes to dealing with changing environmental conditions.
The initiation step of translation, that is the association of a mRNA with the ribosome, is the main target of the regulation of translation4. As a consequence of the regulation of translation as well as other post-transcriptional regulatory steps, only 40% of the variations in protein concentration can be explained by mRNA abundance 5,6. Thus, the study of total mRNA gives relatively poor information about protein abundance. On the other hand, the association of mRNA with ribosomes gives better insight into protein abundance by giving access to those mRNAs involved in translation. Actively translated mRNAs are associated with several ribosomes in structures called polysomes. Conversely, poorly translated mRNAs will be associated with only one ribosome (monosome). Consequently, the translational status of an mRNA can be evaluated by monitoring its association with ribosomes7.
This protocol describes the isolation of polysomes from six days old Arabidopsis thaliana seedlings, the subsequent isolation of RNA, and the analysis of the results. Polysomes and monosomes are separated through a sucrose density gradient. Gradients are collected into six fractions. Some of the fractions are pooled to obtain three well separated fractions: polysomes, monosomes and the light fraction (hereafter called supernatant), which contains the free 60S and 40S ribosomal subunits and mRNAs that are not associated with ribosomes. Global translation activity can be estimated by generating a polysome/monosome ratio, which is determined by integration of the area under the curve, and by comparing the polysomes profiles. mRNAs and proteins are then extracted from the different fractions and used for analysis by RT-PCR, qRT-PCR, Northern blot, microarray, western blot or proteomics. This protocol has been validated for other plants and tissues.
The equipment required to perform this protocol are commonly found in most laboratories: There is no need for a gradient maker. Freezing each layer before adding the next one prevents from any mix or disturbance of the layers. No tube piercer is used for gradient collection which can be achieved by immersion of a glass capillary tube in the gradient. Therefore, the costly ultracentrifuge tubes remain undamaged and can be re-used many times. Collectively, this makes the present protocol an easy and cheap method for polysome profiling.
Protocol
1. Preparation of 20 to 50% (w/v) Sucrose Gradients
Note: The gradients are made of 4 layers of sucrose (50%, 35% and 2 layers of 20%) in a 13.2 ml ultracentrifuge tube. In our experience, pouring the 20% sucrose in two separate layers greatly improves the quality of polysome preparations.
- Prepare the stock solutions. Ensure that all solutions are RNAse and DNAse free.
- Prepare 10X Salt Solution: 400 mM Tris-HCl pH 8.4, 200 mM KCl and 100 mM MgCl2.
- Prepare 2 M Sucrose Solution: For 200 ml, dissolve 137 g of sucrose in 1X Salt Solution.
Dilute the Sucrose Solution in Salt Solution, as described in Table 1, to prepare the gradients. The volumes given are for six gradients.
| Final sucrose concentration | Sucrose 2M (ml) | Salt solution 1X (ml) | Final Vol. (ml) |
| 50% | 8.8 | 3.2 | 12 |
| 35% | 12.9 | 12.1 | 25 |
| 20% | 7.4 | 17.6 | 25 |
| 20% | 5.8 | 14.2 | 20 |
Table 1. Dilutions of the Sucrose Solution to prepare six gradients.
Pour the layers as per Table 2.
| Sucrose layer | 50% | 35% | 20% | 20% |
| Vol. (ml) | 1.85 | 3.65 | 3.65 | 1.35 |
Table 2. Volume of sucrose solution per layer.
After pouring each layer, keep the tubes in a -40 °C or -80 °C freezer until complete freezing before adding the next sucrose layer. Freezing is usually achieved after 2 hr at -40 °C, but waiting about 6 hr before pouring the next layer is recommended. The last 20% layer can be frozen or added freshly on the day of the experiment. Note: Freezing each layer before adding the next one prevents from any mixing or disturbance of the layers. Gradients can be kept in a -40 °C or -80 °C freezer for at least six months.
If it has not already been done, add the last 20% layer to the gradients on the day of the experiment. Then, let the gradients thaw out in a cold room or a fridge.
2. Preparation of Cytosolic Extracts
Note: We recommend using two gradients per biological sample. 300 mg is the optimum amount of plant material to prepare two gradients when working with 6 days old Arabidopsis thaliana seedlings. When working with less translationally active tissues, the amount of plant material can be increased up to 600 mg.
Harvest six day old seedlings grown on ½ Murashige and Skoog8 medium supplemented with 1% sucrose or equivalent media by quick-freezing in liquid nitrogen
Grind plant material in a precooled mortar and pestle with liquid nitrogen.
Weigh 300 mg of powdered material in a precooled weighing dish. Perform this step quickly to avoid thawing of the sample. Note: Keep the frozen samples for no more than one week in a -80 °C freezer.
Add 2.4 ml of precooled polysome buffer (Salt Solution 4X, 5.26 mM EGTA, 0.5% (v/v) Octylphenoxy poly(ethyleneoxy)ethanol, branched, 50 µg.ml-1 cycloheximide, 50 µg.ml1 chloramphenicol) and homogenize by mixing with the pipette tip. Transfer into two 1.5 ml tubes. Work quickly to prevent the samples from warming.
Centrifuge at 16,000 x g for 15 min at 4 °C in a microcentrifuge to pellet debris. Note: To prevent RNA degradation, always keep the samples at 4°C, use RNase/DNase free solutions and work in RNase/DNase free conditions. Heparin can be added to the polysome buffer, to a final concentration of 300 µg.ml-1, to enhance RNA protection. However, since heparin can interfere with downstream analysis, it will have to be removed during the RNA precipitation step by performing lithium chloride precipitation instead of ethanol precipitation (cf. note 4.7).
3. Polysome Profiling
Carefully pipette the supernatant without disturbing the pellet. If plant fragments have been pipetted, repeat step 2.5. Load the supernatant on top of the gradient by gently pipetting onto the sidewall of the tube in a constant stream. Use one gradient for each 1.5 ml tube.
Transfer to precooled buckets and centrifuge at 175,000 x g for 2 hr 45 min at 4 °C, in an ultracentrifuge (e.g. 32,000 rpm when using a SW41 rotor).
Set up the gradient collection system as described in Figure 1. The UV cuvette has a 1 mm pathlength.
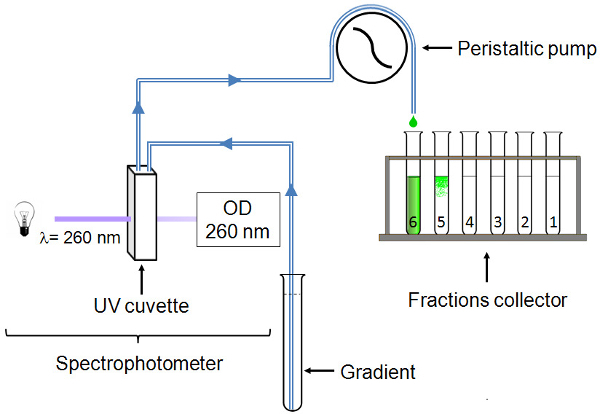 Figure 1. Gradient collection system. The UV cuvette is connected by polyvinyl chloride tubing to a glass capillary tube that descends to the bottom of the gradient. The gradient progresses through the system thanks to a peristaltic pump. OD260 is continuously read and 2 ml fractions are collected. Please click here to view a larger version of this figure.
Figure 1. Gradient collection system. The UV cuvette is connected by polyvinyl chloride tubing to a glass capillary tube that descends to the bottom of the gradient. The gradient progresses through the system thanks to a peristaltic pump. OD260 is continuously read and 2 ml fractions are collected. Please click here to view a larger version of this figure.
Adjust the fraction collector to RT, and set the carousel at a speed that will allow the collection of 2 ml fractions. Use pre-cooled collection tubes.
Collect the fractions from bottom to top using a glass capillary tube connected by polyvinyl chloride tubing to the gradient collection system. Use a plastic paraffin film to secure the connection between the polyvinyl chloride tubing and the glass capillary tube.
Read the absorbance at 260 nm from the bottom to the top of the gradient. When the entire gradient is collected, place the collection tubes on ice before RNA extraction.
4. RNA Extraction
Pool the fractions collected from two gradients, in 50 ml capped centrifuge tubes, as follows: Polysomes: fractions 1 to 3 (12 ml) Monosomes: fraction 4 (4 ml) Supernatant: fractions 5 and 6 (8 ml)
To each fraction, add 1 vol. 8M guanidine hydrochloride, 50 µg acryl carrier and 1.5 vol. isopropanol. Note: Acryl carrier is linear acrylamide, used as a coprecipitant to improve the recovery of nucleic acids during alcohol precipitation.
Mix by inverting the tubes and precipitate O/N at -20 °C.
Centrifuge at 175,000 x g for 1 hr at 4 °C, in an ultracentrifuge (32,000 rpm in a SW32 rotor). If the precipitation step is performed in a tube which is not compatible with ultracentrifugation, transfer to a suitable tube.
Discard supernatant and dissolve the pellet in 200 µl RNAse-free TE buffer (10 mM Tris-HCl pH 7.5 - 1 mM EDTA). Transfer to a fresh 1.5 ml tube.
Extract RNA by adding 1 vol. water saturated phenol (pH 6.6) and 1 vol. chloroform:isoamyl alcohol (24:1). Mix vigorously. Centrifuge at 15,000 x g for 20 min at 4 °C. Transfer the aqueous phase to a new 1.5 ml tube. Note: Acid phenol (pH 4.5) can be used in order to minimize DNA contamination.
Precipitate RNA by adding 1/10 vol. 3 M sodium acetate and 3 vol. 100% ethanol. Allow precipitation at -80 °C for 20 min or O/N at -20 °C. Centrifuge at 15,000 x g for 15 min at 4 °C. Note: If using heparin in the polysome buffer (cf. note 2.5), perform a lithium chloride (LiCl) precipitation instead of ethanol precipitation to properly remove any trace of heparin that could interfere with downstream analysis9. After extraction, add LiCl to a final concentration of 2.5 M. Allow precipitation for 30 min at -20 °C or O/N at 4 °C. Centrifuge at 15,000 x g for 15 min at 4 °C.
Wash the pellet with 500 µl 75% ethanol. Air dry the pellet and dissolve in 30 µl TE buffer. Assess RNA quality by electrophoresis on an RNAse free 1.2% agarose gel (100V, 15 min) or by capillary electrophoresis methods.
5. Data Analysis
Export raw data to a graphic and data analysis software. Note: As shown in Figure 2A and B, the raw profiles give qualitative information: the number of polysome peaks that can be seen, the height of the monosome peak, and the presence of a shoulder corresponding to the free ribosome subunits.
Merge profiles to allow comparison of the profiles between samples. Use the screen reader tool to determine the X value of the monosome peak for each sample and align the monosome peaks by removing extra data points at the beginning of the curves. Identify the lowest point of the curve and determine its Y value (using the screen reader tool). Set the lowest point Y value to 0 by using the "set columns value" function.
Determine the polysomes/monosomes ratio by integrating the area under the curve from raw profiles (Figure 3C). Use the data selector tool to define the border of the area to be integrated. Then use the integrate function (Analysis-Mathematics).
Normalize the curves to the monosome peak by dividing all data points of a curve by the Y value of the summit of the monosome peak (determined using the screen reader tool). Select the column, then under Analysis-Mathematics, select Normalize and choose the "divided by a specified value" methods. This step allows comparison of the relative level of polysomes (Figure 3D).
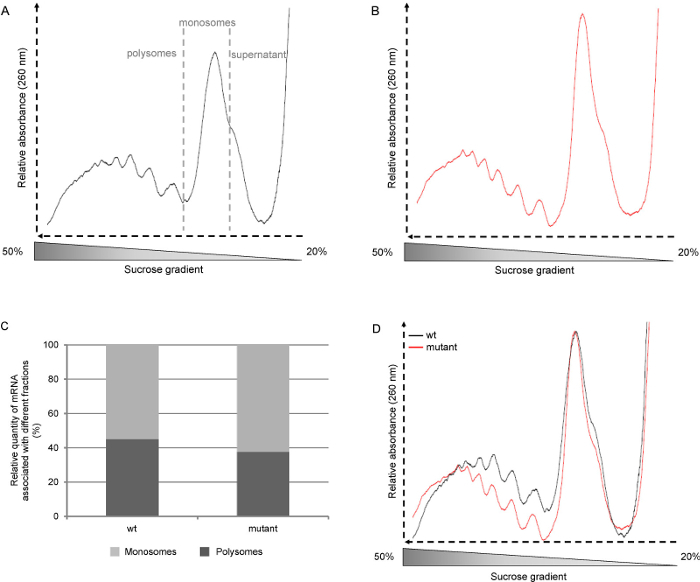 Figure 2. Representative polysome profiles.Arabidopsis thaliana wild-type (wt, ecotype Col-0) and mutant seedlings were grown for six days on ½ Murashige and Skoog medium under long day photoperiods (16 hr light, 8 hr dark). A: raw polysome profiles from wt seedlings. B: raw polysome profiles from mutant seedlings. C: Determination of the percentage of polysomes and monosomes by integration of the area under the curve D:Polysome profiles normalized to the monosome peak. Please click here to view a larger version of this figure.
Figure 2. Representative polysome profiles.Arabidopsis thaliana wild-type (wt, ecotype Col-0) and mutant seedlings were grown for six days on ½ Murashige and Skoog medium under long day photoperiods (16 hr light, 8 hr dark). A: raw polysome profiles from wt seedlings. B: raw polysome profiles from mutant seedlings. C: Determination of the percentage of polysomes and monosomes by integration of the area under the curve D:Polysome profiles normalized to the monosome peak. Please click here to view a larger version of this figure.
Representative Results
In the literature, polysome profiles are often shown from the light fraction to the heavy fraction as a result of the way the gradients are collected, i.e. from the top to the bottom. Since in the protocol described here the gradients are collected from the bottom to the top, the profiles we show start with the heavy fraction (the polysomes) and go to the light fraction (free ribosome subunits and RNAs) (Figure 2A). We then collect each gradient in six 2 ml fractions, but smaller fractions can be collected if a more detailed analysis of polysome content has to be carried out.
Merging and normalizing the curves to the monosome peak (Figure 2D) allows the comparison of profiles from different lines or growth conditions. This provides information on the relative amount of polysomes independently of the level of initiation. Another way to analyze the profiles is to calculate the area under the curve, thus, one can determine the relative quantity of mRNA associated with either the monosomes or the polysomes (Figure 2C). This ratio is specific to a plant and growth conditions. However, this approach may not be relevant in the case of poorly translationally active tissues.
We have used this method for A. thaliana whole seedlings, young and old rosettes, as well as for N. benthamiana (Figure 3C), S. lycopersicum (Figure 3E) and O. sativa leaves (Figure 3D). The profile shape depends on the growth conditions, plant age and the tissues analyzed. Here we used A. thaliana six days old seedlings. At this stage, the translational activity is high, and the profile shows well shaped peaks (Figure 2A and B). This is also the case when A. thaliana flowers are used (Figure 3A). When using 4 weeks old A. thaliana rosettes (Figure 3B), the samples mostly contain fully developed adult leaves where cells do not divide. Hence, the overall amount of polysomes and monosomes is lower. The profile shows fewer, but still well shaped polysome peaks. Next to the monosome peak, the shoulder shows the large amount of free 60S ribosomal subunits. With other kind of plants or tissues, the polysome peaks may be barely visible. This is the case when using 30 days old O. sativa leaves (Figure 3D). Even when the amount of mRNAs involved in translation is very low (no polysome peak can be seen on the profile), the presence of the monosome peak indicates that the fractionation was properly done and that mRNAs can be further extracted from the fraction for further analysis. The quality of extracted RNA quality is assessed by agarose gel electrophoresis (Figure 4). The 25S and 18S cytosolic ribosomal RNA should be clearly visible on the gel. Lower bands corresponding to chloroplastic ribosomal RNA should also be visible when RNA are extracted from green tissues 10.
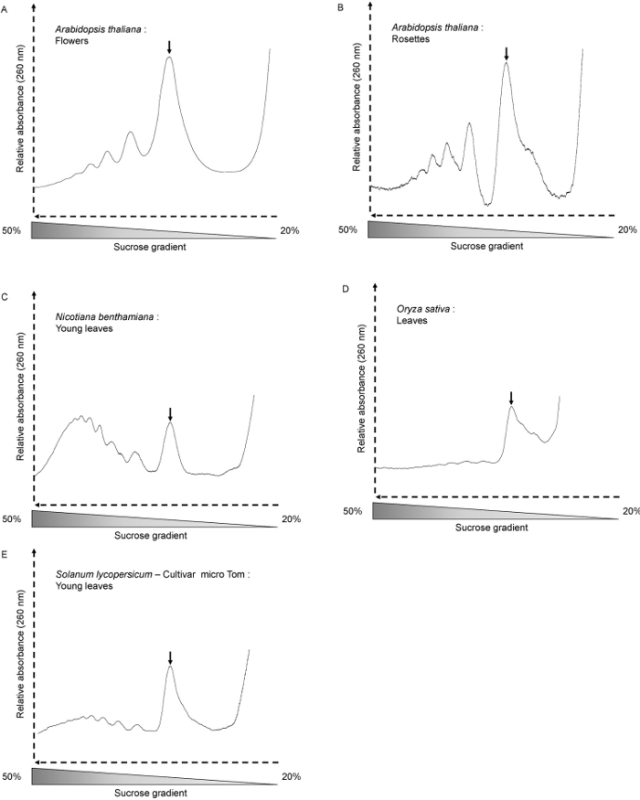 Figure 3.Polysome profiles from different plant material and species. (A)Arabidopsis thaliana flowers (300mg), (B)
Arabidopsis thaliana 4 weeks old rosettes (600mg), (C) Nicotiana benthamiana (young leaves of 40 days old short day-grown plants - 300mg), (D)
Oryza sativa (leaves of 30 days old plants - 300mg), (E) Solanum lycopersicum (young leaves of 35 days old short day-grown plants - 300mg). The monosome peaks are indicated by arrows. Please click here to view a larger version of this figure.
Figure 3.Polysome profiles from different plant material and species. (A)Arabidopsis thaliana flowers (300mg), (B)
Arabidopsis thaliana 4 weeks old rosettes (600mg), (C) Nicotiana benthamiana (young leaves of 40 days old short day-grown plants - 300mg), (D)
Oryza sativa (leaves of 30 days old plants - 300mg), (E) Solanum lycopersicum (young leaves of 35 days old short day-grown plants - 300mg). The monosome peaks are indicated by arrows. Please click here to view a larger version of this figure.
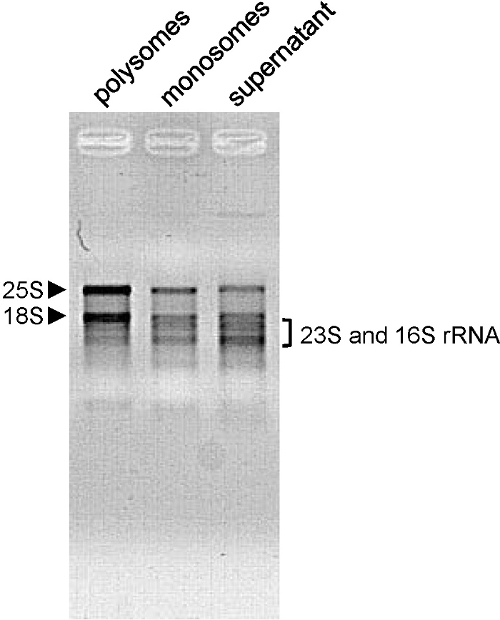 Figure 4. Assessment of RNA quality. RNA (500 ng extracted from shoots of 6 days old Arabidopsis thaliana seedlings) from the indicated fractions were loaded on a 1.2% agarose gel and separated by electrophoresis (100V - 15 min). Cytosolic (25S and 18S) rRNA are indicated by arrowheads and chloroplastic (23S and 16S) rRNA by a bracket. Please click here to view a larger version of this figure.
Figure 4. Assessment of RNA quality. RNA (500 ng extracted from shoots of 6 days old Arabidopsis thaliana seedlings) from the indicated fractions were loaded on a 1.2% agarose gel and separated by electrophoresis (100V - 15 min). Cytosolic (25S and 18S) rRNA are indicated by arrowheads and chloroplastic (23S and 16S) rRNA by a bracket. Please click here to view a larger version of this figure.
Discussion
The protocol we present here is an easy and cheap method for generating polysome profiles and isolating mRNAs associated with polysomes, single ribosomes or free of ribosomes. A wide range of different polysome fractionation methods is described in the literature. The method we have described here has been optimized to keep only the necessary compounds and has been adapted for plant material. In particular, we reduced the amount of detergent11 and added chloramphenicol to the buffer to fix the chloroplastic ribosomes to the mRNA (as cycloheximide does for the cytosolic ribosomes)12 . We have also reduced the total ultracentrifugation time7.
To obtain high quality polysome profiles, it is essential to use freshly collected plant material and to perform all steps at 4°C. When using tissues that are poorly translationally active, more plant material can be loaded on the gradient (up to 600 mg).
Analysis of polysome profiles can provide insights into both the overall translational status of cells13 and the translational status of a specific mRNA. We have used RNAs isolated by this method for different applications. Using the RNAs for microarrays allowed us to identify a class of cadmium stress response genes for which transcription and translation are uncoupled 14. We also used this method to identify small RNAs associated with polysomal fractions. This identification was made by northern blotting15 of RNA extracted from polysomal fractions, and provided biochemical evidence for a translational component in the miRNA pathway in plants. In another study, a cis-NAT RNA was identified by quantitative RT-PCR. This cis-NAT is associated to the phosphate homeostasis and promote translation of the PHO1;2 transcript 16.
The main limits of the polysome profiling approach are the lack of information concerning both the position of the ribosome on the mRNA and its progression along the mRNA. Ribosome profiling has emerged to address these limitations 17 and has been successfully used on plant tissues18. Ribosome profiling provides a global measurement of translation by taking advantage of the advances in sequencing technology. Nevertheless, as for any sequencing based assays, the quality of the results depends on the mapping of the sequences to the genome, therefore focusing on the small ribosome-protected fragments makes it difficult to deconvolute repetitive sequences. Moreover, the digestion of RNA not protected by the ribosomes leads to the loss of regulatory information contained in the 3' and 5' UTRs. It is then impossible to distinguish transcript variants that have different 3' or 5' UTRs and show different levels of translation19. The polysome profiling method described here is rapid and does not require specific technical skills. Altogether, these two methods represent complementary approaches to study translation regulation.
Disclosures
The authors have nothing to disclose
Acknowledgments
This work was supported by the French National Research Agency (ANR-14-CE02-0010). We thank Dr Benjamin Field and Dr. Elodie Lanet for critical reading of the manuscript. We thank Mr. Michel Terese for his help with video editing.
References
- Nelson CJ, Millar AH. Protein turnover in plant biology. Nat Plants. 2015;1(3):15017. doi: 10.1038/nplants.2015.17. [DOI] [PubMed] [Google Scholar]
- Warner JR. The economics of ribosome biosynthesis in yeast. Trends Biochem Sci. 1999;24(11):437–440. doi: 10.1016/s0968-0004(99)01460-7. [DOI] [PubMed] [Google Scholar]
- Amthor JS. The McCree-de Wit-Penning de Vries-Thornley Respiration Paradigms: 30 Years Later. Ann Bot. 2000;86(1):1–20. [Google Scholar]
- Preiss T, W Hentze M. Starting the protein synthesis machine: eukaryotic translation initiation. BioEssays news and reviews in molecular, cellular and developmental biology. 2003;25(12):1201–1211. doi: 10.1002/bies.10362. [DOI] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet. 2013;13(4):227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerenfaller K, et al. Genome-scale proteomics reveals Arabidopsis thaliana gene models and proteome dynamics. Science. 2008;320(5878):938–941. doi: 10.1126/science.1157956. [DOI] [PubMed] [Google Scholar]
- Zanetti E, Chang I, Gong F, Galbraith DW, Bailey-Serres J. Immunopurification of Polyribosomal Complexes of Arabidopsis for Global Analysis of Gene Expression. Plant physiol. 2005;138(2):624–635. doi: 10.1104/pp.105.059477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murashige T, Skoog F. A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol plant. 1962;15(3):473–497. [Google Scholar]
- del Prete MJ, Vernal R, Dolznig H, Müllner EW, Garcia-Sanz JA. Isolation of polysome-bound mRNA from solid tissues amenable for RT-PCR and profiling experiments. RNA. 2007;13(3):414–421. doi: 10.1261/rna.79407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas J, Sanchez-Serrano JJ, editors. Arabidopsis protocols. Clifton, N.J: 2006. (Methods in molecular biology; 323). [Google Scholar]
- Piques M, et al. Ribosome and transcript copy numbers, polysome occupancy and enzyme dynamics in Arabidopsis. Mol syst biol. 2009;5(314):314. doi: 10.1038/msb.2009.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Alain T, Topisirovic I, Sonenberg N. Polysome Profiling Analysis. bio-protocol. 2013;3(14):3–8. [Google Scholar]
- Yángüez E, Castro-Sanz AB, Fernández-Bautista N, Oliveros JC, Castellano MM. Analysis of genome-wide changes in the translatome of Arabidopsis seedlings subjected to heat stress. PloS One. 2013;8(8) doi: 10.1371/journal.pone.0071425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sormani R, et al. Sublethal cadmium intoxication in Arabidopsis thaliana impacts translation at multiple levels. Cell Physiol. 2011;52(2):436–447. doi: 10.1093/pcp/pcr001. [DOI] [PubMed] [Google Scholar]
- Lanet E, et al. Biochemical evidence for translational repression by Arabidopsis microRNAs. Plant cell. 2009;21(6):1762–1768. doi: 10.1105/tpc.108.063412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabnoune M, Secco D, Lecampion C, Robaglia C, Shu Q, Poirier Y. A Rice cis-Natural Antisense RNA Acts as a Translational Enhancer for Its Cognate mRNA and Contributes to Phosphate Homeostasis and Plant Fitness. Plant cell. 2013;25(10):4166–4182. doi: 10.1105/tpc.113.116251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT. Methods Enzymol. 10. Vol. 470. Elsevier Inc; 2010. Genome-wide translational profiling by ribosome footprinting. [DOI] [PubMed] [Google Scholar]
- Juntawong P, Girke T, Bazin J, Bailey-Serres J. Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. PNAS. 2013;111(1):203–212. doi: 10.1073/pnas.1317811111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT. Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet. 2014;15(3):205–213. doi: 10.1038/nrg3645. [DOI] [PubMed] [Google Scholar]